
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentification of the peptide epimerase MslH responsible for D-amino acid introduction at the C-terminus of ribosomal peptides†
Zhi
Feng
a,
Yasushi
Ogasawara
 *b and
Tohru
Dairi
*b
*b and
Tohru
Dairi
*b
aGraduate School of Chemical Sciences and Engineering, Hokkaido University, Sapporo, Hokkaido 060-8628, Japan
bGraduate School of Engineering, Hokkaido University, Sapporo, Hokkaido 060-8628, Japan. E-mail: yogasawa@eng.hokudai.ac.jp; dairi@eng.hokudai.ac.jp
First published on 29th December 2020
Abstract
A lasso peptide MS-271 is a ribosomally synthesized and post-translationally modified peptide (RiPP) consisting of 21 amino acids with a D-tryptophan (Trp) at its C terminus. The presence of D-amino acids is rare in RiPPs and few mechanisms of D-amino acid introduction have been characterized. Here, we report the identification of MslH, previously annotated as a hypothetical protein, as a novel epimerase involved in the post-translational epimerization of the C-terminal Trp residue of the precursor peptide MslA. MslH is the first epimerase that catalyzes epimerization at the Cα center adjacent to a carboxylic acid in a cofactor-independent manner. We also demonstrate that MslH exhibits broad substrate specificity toward the N-terminal region of the core peptide, showing that MslH-type epimerases offer opportunities in peptide bioengineering.
Introduction
Lasso peptides are a group of natural products that have a characteristic lariat topology in which a macrocyclic ring with 7–9 amino acid residues is formed by an isopeptide bond between the N-terminal amino group and the carboxylic acid side chain of an aspartate or a glutamate, and the C-terminal tail threads through the ring.1–3 Lasso peptides exhibit high stability against heat treatment and proteases due to the interlocked lasso topology and show diverse biological activities such as antimicrobial, antitumor, antiviral, and enzyme inhibitory activities. Thus, lasso peptides have attracted increasing attention from natural product chemists in recent years. Because of the lasso topology, chemical synthesis of lasso peptides is challenging and had not been reported until the recent first total synthesis of BI-32169 using a cryptand–imidazolium complex as a multi-linker support.4 In terms of their biosynthesis, lasso peptides belong to a group of ribosomally synthesized and post-translationally modified peptides (RiPPs).5 Lasso peptide biosynthesis begins with the translation of the gene encoding the precursor peptide, which comprises an N-terminal leader peptide and a C-terminal core peptide. The precursor peptide is then modified by at least two enzymes, a cysteine protease called the “B” enzyme and a macrolactam synthetase called the “C” enzyme.6,7 The “B” enzyme consists of an N-terminal precursor peptide recognition element (PRE) domain and a C-terminal protease domain, and cleaves the amide bond between the leader peptide and the core peptide of the precursor by expending ATP to provide the core peptide with a free N-terminal amino group.8 “B” enzymes from actinobacteria are generally split into the N- and C-terminal domains of the full-length B protein and are called “B1” and “B2”, respectively. The “C” enzyme then catalyzes ATP-dependent isopeptide bond formation to complete biosynthesis of the lasso peptide.MS-271, originally isolated from Streptomyces sp. M-271, is a lasso peptide natural product with potent inhibitory activity toward calmodulin-activated myosin light-chain kinase (Fig. 1).9,10 MS-271 is composed of 21 amino acids and contains a D-tryptophan (Trp) at its C terminus. Considering that lasso peptides are ribosomal peptides, the mechanism of D-Trp introduction in MS-271 biosynthesis is of great interest. Recently, we identified the biosynthetic gene cluster of MS-271 (msl), which contained genes for the B1 enzyme (MslB1), B2 enzyme (MslB2), C enzyme (MslC), disulfide oxidoreductases (MslE, MslF), ABC transporters, regulators, and a protein of unknown function (MslH) besides the gene encoding a precursor peptide (MslA) ending with the C-terminal L-Trp residue.11 Heterologous expression experiments of the msl cluster spanning 11 kbp (mslR2–mslH) revealed that it contained all necessary genes for MS-271 biosynthesis. However, the msl cluster lacked obvious candidate genes for the epimerization. Because MslH was the only protein of unknown function in the msl cluster and was conserved among all msl-like gene clusters identified in the genome database (Fig. S1†), we hypothesized that MslH is responsible for the epimerization of the C-terminal tryptophan. In the present study we performed in vivo and in vitro functional characterization of MslH and demonstrated that MslH is a novel peptide epimerase catalyzing epimerization of the nascent precursor peptide in the early stage of MS-271 biosynthesis.
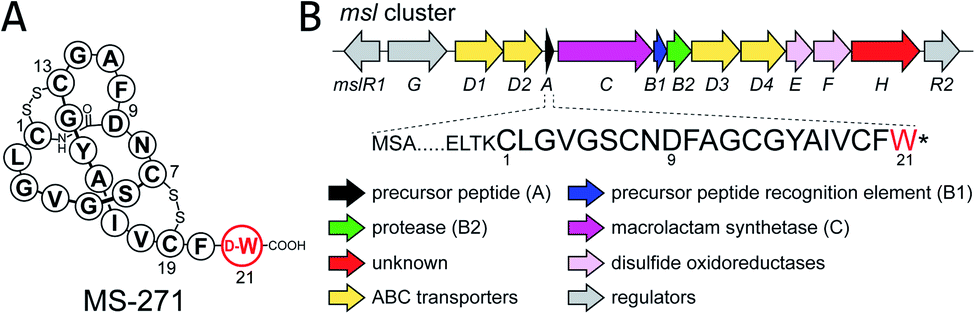 | ||
| Fig. 1 (A) Structure and (B) biosynthetic gene cluster (msl) of MS-271. | ||
Results
In vivo characterization of MslH as a novel peptide epimerase
To obtain initial insight into the epimerization, we employed an in vivo heterologous expression strategy using Escherichia coli as the host. However, no expression of proteins encoded by some msl genes, including mslA and mslH, from Streptomyces sp. M-271 was observed. We thus used msl genes from Streptomyces griseorubiginosus NBRC 12899, which possess the same enzymes (86–92% identities) as those from Streptomyces sp. M-271 and produced a large amount of MS-271 (Fig. S1 and S2†). In this case, expression of most of the enzymes was observed, but mslA did not yield a detectable amount of MslA on SDS-PAGE. Recently, Ojima-Kato et al. reported that the addition of a DNA sequence coding for Ser-Lys-Ile-Lys (SKIK tag) just after the start codon remarkably improved the expression of recombinant proteins in E. coli and Saccharomyces cerevisiae.12 Using the same strategy, we successfully expressed MslA fused with SKIK and His tags at its N-terminus although the recombinant MslA was insoluble (Fig. S3†).Considering that many modification enzymes involved in RiPP biosynthesis require leader peptides for their substrate recognition,5 we speculated that the epimerization occurs prior to the cleavage of the precursor peptide by MslB2 in MS-271 biosynthesis. Consequently, mslA was expressed in E. coli in the presence or absence of mslH and the precursor peptide recognition element gene mslB1. As shown in Fig. S3,† MslA was expressed as a soluble peptide in the presence of MslB1 in contrast to when it was expressed alone. Hydrophobic MslA may be solubilized by MslB1 through the interaction between them. The MslA product was purified by tricine–SDS-PAGE (16% gel) and the chirality of the C-terminal Trp, which is the sole Trp residue in MslA, was analyzed by a modified Marfey's method after acid hydrolysis followed by HPLC purification of Trp. When mslH was coexpressed with mslA and mslB1, D-Trp was observed in the purified MslA (Fig. 2). The formation of D-Trp was also observed, albeit a small amount, when mslB1 was omitted. In contrast, D-Trp was not detected in the purified MslA products prepared without expression of mslH. These results clearly indicated that MslH catalyzed the epimerization of the nascent precursor peptide to generate its epimer, epi-MslA, and that MslB1 enhanced the activity of MslH. In addition, we examined whether expression of MslB2 or MslC as well resulted in more formation of D-Trp in MslA. However, no effect was observed with either enzyme.
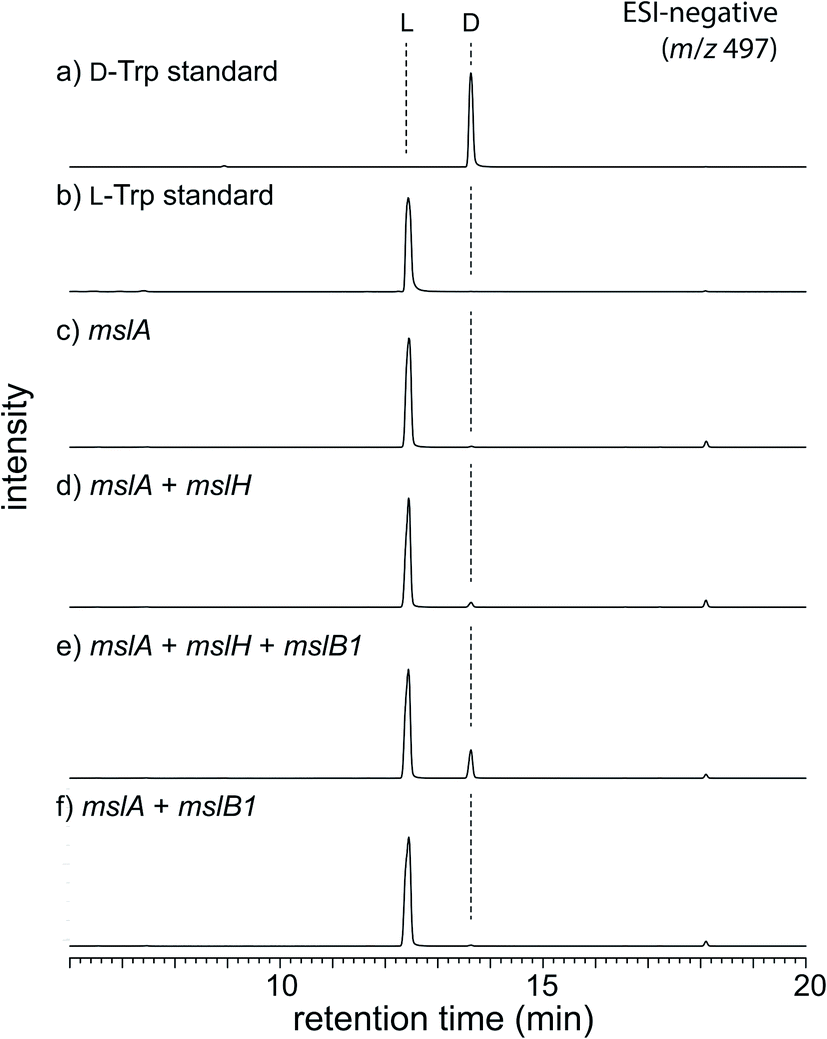 | ||
| Fig. 2 LC-MS analysis (ESI negative ion mode, y-axis is signal intensity monitored at m/z 497 and the same scale applies for all chromatograms) of in vivo assay. L-FDLA derivatives of (a) D-Trp standard and (b) L-Trp standard. L-FDLA derivatives of Trp in MslA by heterologous expression of (c) mslA alone, (d) mslA and mslH, (e) mslA, mslH, and mslB1, and (f) mslA and mslB1. | ||
MslH is a metal- and cofactor-independent epimerase
We next investigated the MslH-catalyzed epimerization by in vitro experiments. To prepare MslA, we first coproduced MslA (SKIK-His-MslA) and MslB1 (without tags) in E. coli and purified MslA by Ni–NTA agarose affinity chromatography. MslB1 was unexpectedly co-purified with MslA even though a buffer containing a high salt concentration (2.0 M NaCl) was used during purification, indicating tight binding of MslB1 to MslA (Fig. S4†). We thus purified MslA under denaturing conditions (8 M urea) on a Ni–NTA column to near homogeneity. MslH and MslB1 were independently purified as His-tagged proteins. When MslH was incubated with MslA, the formation of D-Trp in MslA was observed in a time-dependent manner and, as expected, the reaction was accelerated upon the addition of MslB1 (Fig. 3A–D). Prolonged incubation (16 h) resulted in about 50% conversion of MslA to epi-MslA, indicating that MslH generated an equilibrium mixture of the epimers (Fig. S5†). Because MS-271 derivatives containing L-Trp at the C-terminus have never been identified in the producers, the next enzyme of the pathway likely recognizes epi-MslA exclusively as a substrate. Furthermore, we examined the MslH reaction using a chemically synthesized core peptide of MslA (MslA-core). However, only a small amount of D-Trp was observed after the MslH reactions (Fig. 3F–I). These results clearly indicated that the leader peptide is important for substrate recognition by MslH. Because Phyre2 (ref. 13) and I-TASSER (ref. 14) (web-based programs to predict protein structure) analysis and InterProScan analysis suggested that MslH belongs to the metallo-dependent phosphatase family (Fig. S6–S8†), we next examined the metal dependency of MslH using reaction conditions without MslB1. However, its activity was not affected by the addition of divalent metal ions (Mg2+, Zn2+, Co2+, Fe2+, Mn2+, and Ca2+) or ethylenediaminetetraacetic acid (EDTA), indicating that a divalent metal is not required for MslH activity (Fig. S9†). In addition, we analysed the purified MslH by UV-vis and protein mass analysis to examine whether organic cofactors were co-purified with MslH. However, neither covalently bound nor non-covalently bound organic cofactor was detected (Fig. S10†). These results suggested that MslH-catalyzed epimerization occurred in metal- and cofactor-independent manner.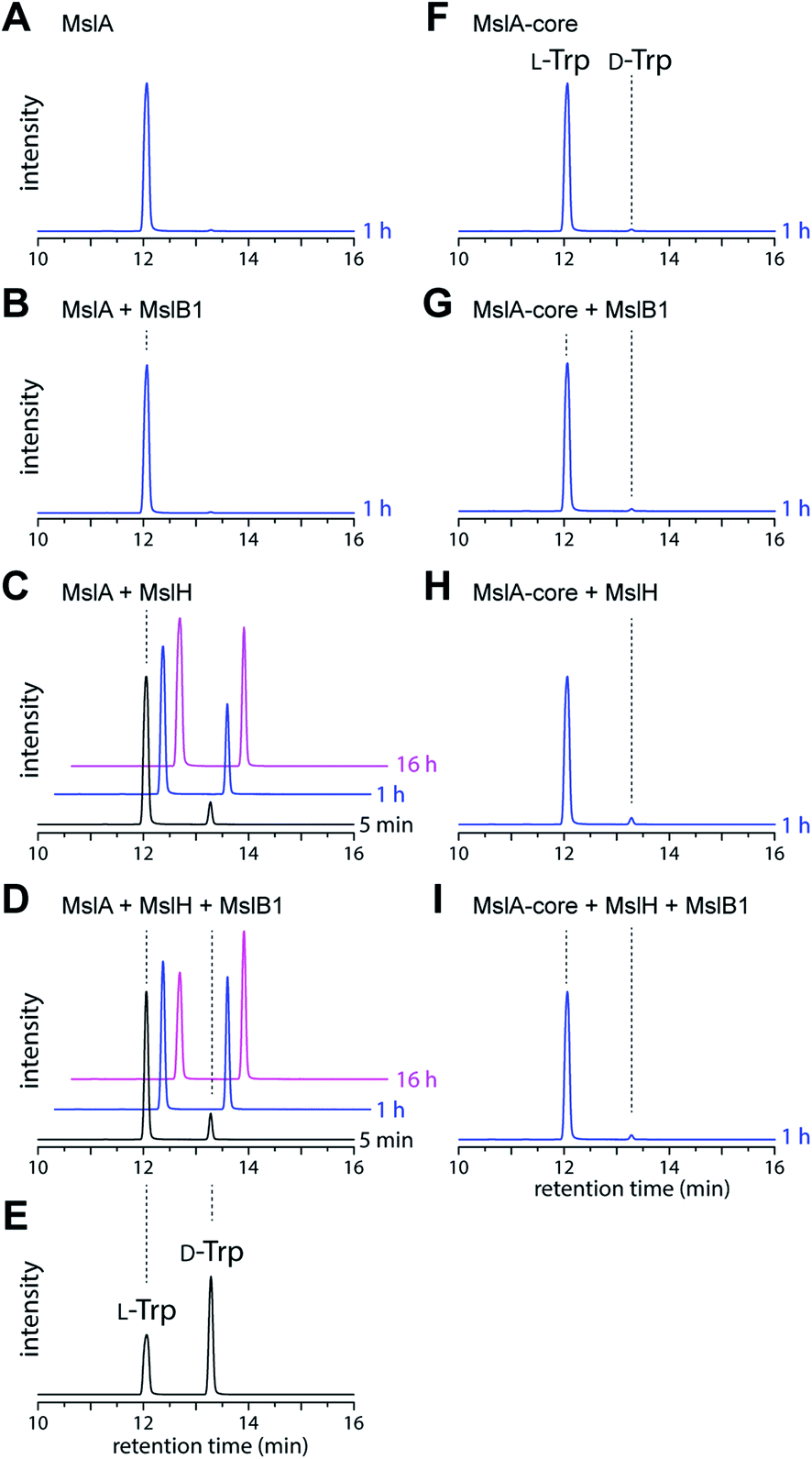 | ||
| Fig. 3 LC-MS analysis (ESI negative ion mode, y-axis is signal intensity monitored at m/z 497 and the same scale applies for all chromatograms) of L-FDLA–Trp derived from in vitro reactions. The reaction time is shown next to each trace. (A) MslA only, (B) MslA + MslB1, (C) MslA + MslH, (D) MslA + MslH + MslB1, (E) L- and D-Trp standards, (F) MslA-core only, (G) MslA-core + MslB1, (H) MslA-core + MslH, and (I) MslA-core + MslH + MslB1. | ||
Heterologous production of D-amino acid containing unnatural lasso peptides
To probe the substrate specificity of the modification enzymes involved in MS-271 biosynthesis, we next carried out heterologous expression of the msl cluster to produce MS-271 derivatives by altering the core peptide sequences of the mslA gene using the same method described previously.11 The constructed plasmids were individually introduced into Streptomyces lividans:mslR2 and the metabolites were analyzed by LC-MS (Table S2† and Fig. 4 and S11†). When the C-terminal Trp was replaced with Phe or Tyr, the corresponding MS-271 derivatives, MS-271-W21F and MS-271-W21Y, were produced. Chiral analysis by a modified Marfey's method confirmed that MS-271-W21F and MS-271-W21Y contained D-Phe and D-Tyr, respectively (Fig. S12†). In contrast, a small amount of MS-271-W21V was detected by LC-MS analysis. These results suggested that an aromatic amino acid residue at the C-terminus of the precursor peptide is important for the epimerization reaction. We next examined the production of derivatives with 20 amino acid residues by eliminating the C-terminal Trp (MS-271-ΔW21) or an internal Ile (MS271-ΔI17) in MslA. However, production of the expected peptides was not observed in either case. Considering that the C-terminal four amino acid sequence of MS271-ΔI17 was identical to that of MS-271, the length of the precursor peptide may also be important.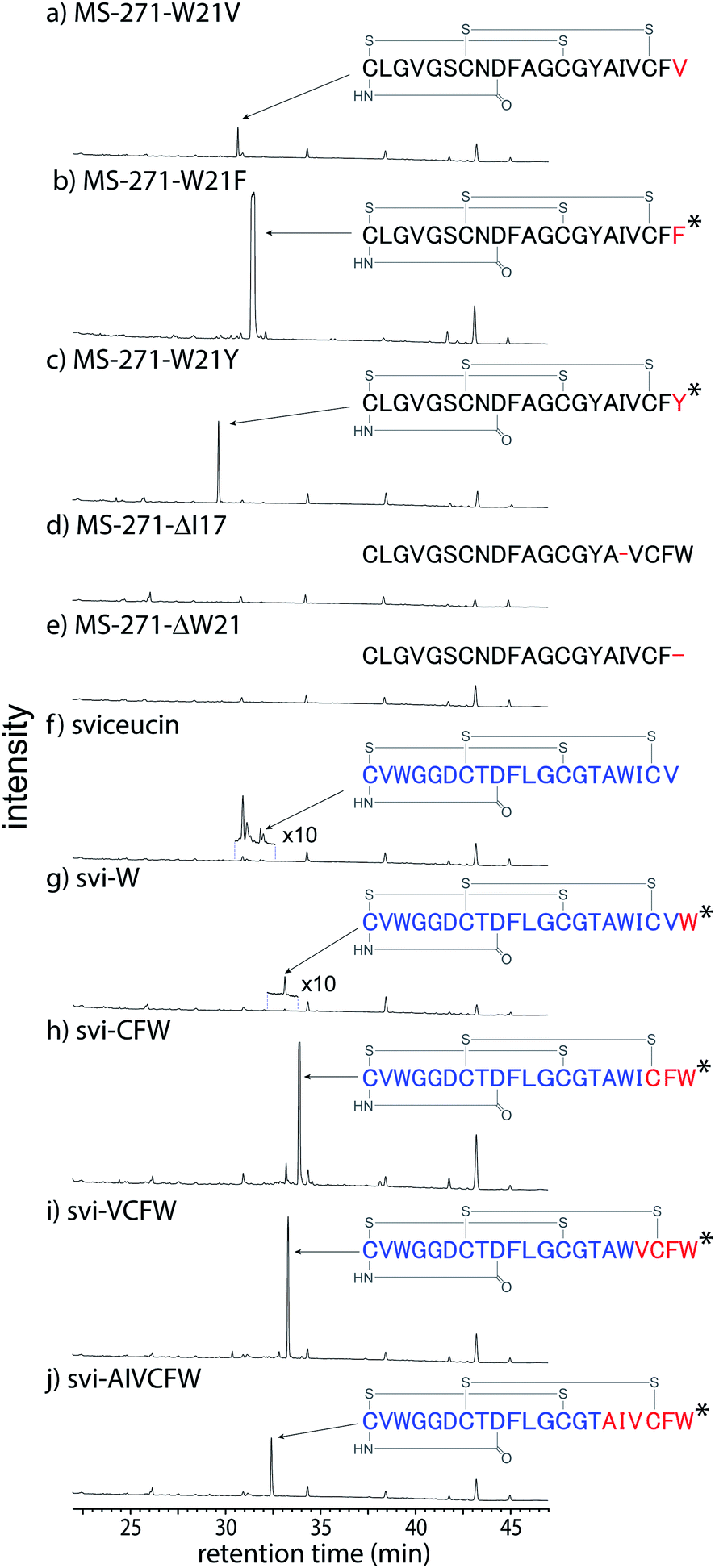 | ||
| Fig. 4 LC-MS analysis (y-axis is UV absorbance monitored at 210 nm and the same scale applies for all chromatograms) of metabolites produced by heterologous production of the msl cluster with mslA mutant plasmids, (a) pWHM3-msl-MS-271-W21V, (b) pWHM3-msl-MS-271-W21F, (c) pWHM3-msl-MS-271-W21Y, (d) pWHM3-msl-MS-271-ΔI17, (e) pWHM3-msl-MS-271-ΔW21, (f) pWHM3-msl-svi, (g) pWHM3-msl-svi-W, (h) pWHM3-msl-svi-CFW, (i) pWHM3-msl-svi-VCFW, and (j) pWHM3-msl-svi-AIVCFW. The mutated amino acids in MS-271 derivative are shown in red. Sviceucin- and MS-271-derived sequences in the hybrid lasso peptides are shown in blue and red, respectively. The D-amino acid-containing products confirmed by chiral analysis are marked with an asterisk. | ||
We next investigated the MS-271 biosynthetic tolerance using sviceucin, a non-cognate lasso peptide with 20 amino acid residues, as a core peptide. The structure of sviceucin contains two disulfide bridges, Cys1–Cys13 and Cys7–Cys19, in the same manner as MS-271, although its amino acid sequence is quite different from that of MS-271 (Fig. 4 and Table S2†).15 We prepared a chimeric precursor peptide gene by fusing the leader peptide of MslA to the core peptide of sviceucin and then the sequence of the C-terminal region in sviceucin core peptide was changed to generate a series of sviceucin derivatives. HPLC analysis revealed successful production of some derivatives, especially svi-CFW, svi-VCFW, and svi-AIVCFW (Fig. 4 and S11†), while minute amounts of native sviceucin and svi-W were detected. In addition, chiral analysis indicated that the C-terminal Trp in all MS-271/sviceucin hybrid lasso peptides had the D-configuration (Fig. S12†). These results indicated that the MS-271 biosynthetic enzymes exhibit broad substrate specificities toward the N-terminal region of core peptides while the C-terminal “CFW” sequence is important for substrate recognition.
Discussion
The presence of D-amino acids is generally the hallmark of peptides biosynthesized via non-ribosomal peptide synthetases (NRPSs). NRPSs are modular-type large enzyme complexes in which the formation of polypeptides occurs in accordance with the embedded catalytic domains.16,17 During chain elongation, epimerization (E) domains epimerize the amino acid residue directly connected to the phosphopantetheine arm of the carrier protein via thioester linkage. X-ray structural analysis of an E-domain revealed that the reaction mechanism involves an abstraction of the α-proton of the thioester and stabilization of an enolate anion intermediate by conserved Glu and His residues.18 In RiPP biosynthesis, epimerization must occur post-translationally by an abstraction of the α-proton of an amide, which is less acidic than those of thioesters. To date, only two types of epimerases have been identified in RiPP biosynthesis. One is radical S-adenosylmethionine (rSAM)-dependent epimerases (PoyD,19,20 YydG21), which catalyze unidirectional epimerization using radical chemistry to abstract the α-hydrogen (Fig. S13A†). The other is an α/β-hydrolase family enzyme (BotH)22 that catalyzes epimerization at the Cα of an Asp residue adjacent to a thiazolinyl group. In this case, the α-proton is acidic enough for spontaneous epimerization, albeit slowly, and BotH just enhances the epimerization by stabilizing the enamine intermediate (Fig. S13B†). In addition, two-step process to introduce D-Ala has also been reported in lanthipeptide biosynthesis. In this process, a dehydroalanine residue, which is generated by a dehydratase from L-Ser residue, undergoes reduction by an NADPH/zinc-dependent reductase,23 a flavin-dependent reductase,24 or an F420H2 dependent reductase25 to form D-Ala residue (Fig. S13C†).Importantly, the MslH-catalyzed epimerization is chemically more challenging because the abstraction of the α-proton adjacent to the carboxylic acid is unfavorable. To the best of our knowledge, MslH is the first epimerase that catalyzes epimerization at the Cα center adjacent to a carboxylic acid in a cofactor-independent manner. Further studies such as crystal structure analysis are necessary to understand the reaction mechanism.
Our results also provided insight into the timing of modification reactions in lasso peptide biosynthesis. The MslH-catalyzed epimerization occurs on the full-length MslA prior to proteolytic cleavage by MslB2 (Fig. 5). This observation is common for all lasso peptide modification enzymes characterized in vitro, including a methyltransferase (StspM),26 kinases (ThcoK and SyanK),27,28 and an iron/2-oxoglutarate-dependent hydroxylase (CanE).29 Together with the fact that MslH exhibited broad substrate specificity toward the N-terminal region of the core peptide, this feature is potentially useful in peptide bioengineering.
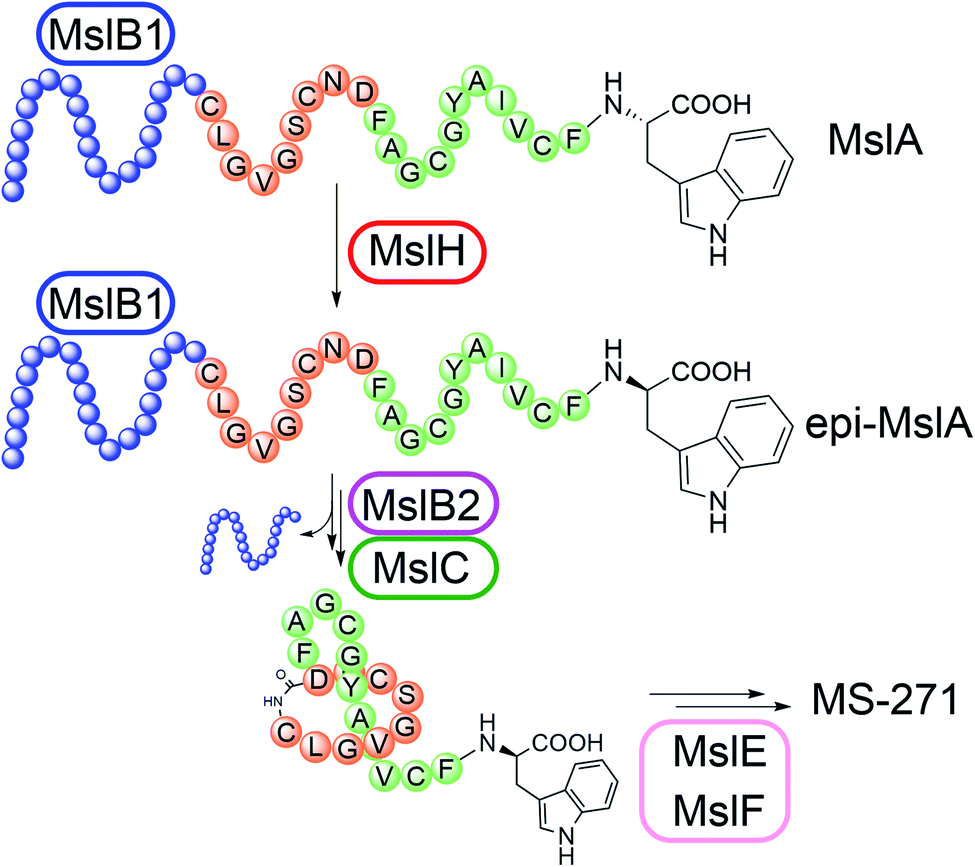 | ||
| Fig. 5 Biosynthesis of MS-271. MslB1 tightly binds to the leader peptide region (blue) in MslA and enhances the MslH reaction. | ||
Interestingly, MslH exhibited homology (∼52% identity) to CapA, a function-unknown protein responsible for DL-poly-γ-glutamic acid (PGA) synthesis. PGA is a natural polymer of D- and L-glutamic acid linked by isopeptide bonds.30 Although the detailed biosynthesis of PGA has never been characterized due to the instability of the membrane-bound biosynthetic enzymes, we recently demonstrated the involvement of peptide epimerization in PGA biosynthesis by isotope tracer experiments.31 Our characterization of MslH suggests that CapA is an epimerase that introduces D-Glu residues into a homopolymer of L-Glu in PGA biosynthesis. Future functional characterization of CapA will provide further insight into MslH-type epimerases.
Experimental
General
All chemicals were purchased from Fujifilm Wako Pure Chemical Corporation (Osaka, Japan), Sigma-Aldrich Japan (Tokyo, Japan), or Tokyo Chemical Industry Co. Ltd. (Tokyo, Japan) unless specified otherwise. The custom synthetic peptide (Msl-core 21: CLGVGSCNDFAGCGYAIVCFW) was purchased from Sigma-Aldrich Japan. Enzymes and kits for DNA manipulations were purchased from Takara Bio (Shiga, Japan), Nippon Gene Co. Ltd. (Tokyo, Japan) or New England Biolabs Japan Inc. (Tokyo, Japan). Polymerase chain reaction (PCR) were carried out using a GeneAmp PCR System 9700 thermal cycler and Tks Gflex DNA polymerase (Takara Bio). Oligonucleotide synthesis and DNA sequencing were performed in Fasmac (Kanagawa, Japan). General genetic manipulations of E. coli were performed according to standard protocols.32 Plasmids in E. coli were maintained using appropriate antibiotics with the following concentrations; ampicillin (100 μg mL−1), kanamycin (25 μg mL−1), chloramphenicol (30 μg mL−1), and/or streptomycin (20 μg mL−1). The MS-271-producing bacterium Streptomyces sp. M-271 was kindly provided by Kyowa Hakko Bio Co., Ltd. Streptomyces olivochromogenes NBRC 3561, Streptomyces nodosus NBRC 12895, S. griseorubiginosus NBRC 12899 (=Streptomyces phaeopurpureus DSM 40125) and Streptomyces diastatochromogenes NBRC 13389 were obtained from NITE Biological Resource Center, the National Institute of Technology and Evaluation (Tokyo, Japan).Production of MS-271 and its derivatives
To produce MS-271, Streptomyces sp. M-271, S. olivochromogenes, S. nodosus, S. griseorubiginosus or S. diastatochromogenes was first grown in 10 mL of seed medium (1% glucose, 1% soluble starch, 0.5% BD Bacto tryptone, 0.5% BD Bacto yeast extract, 0.3% Ehrlich's fish extract (Kyokuto, Tokyo, Japan), 0.5% CaCO3, pH 7.2 by NaOH) for 2 days at 30 °C on a rotary shaker (200 rpm). A 1 mL portion of the culture was inoculated on an agar plate containing 25 mL of production medium (4% soluble starch, 1% soybean meal, 0.5% corn steep liquor (Sigma-Aldrich), 0.5% dry yeast, 0.05% KH2PO4, 0.05% Mg3(PO4)2·8H2O, 0.001% ZnSO4·7H2O, 0.0001% CoCl2·6H2O, 0.0001% NiSO4, 1.5% agar, pH 7.0 by NaOH) and the culture was incubated at 30 °C for 5 days. The cells on the agar plate were collected by spatula and transferred into a vial containing 10 mL MeOH. After soaking the cells in MeOH at 4 °C for 16 h, the MeOH extract was concentrated and re-dissolved in 80% aqueous MeOH (1 mL) for analysis. HPLC analysis was performed with a Shimadzu Prominence HPLC system equipped with a photo diode array (PDA) Detector. The analytical conditions were as follows: column, Mightysil RP-18GP Aqua column (250 × 4.6 mm, 5 mm, Kanto Chemical); column temperature, 40 °C; detection, PDA (190–350 nm); mobile phase, A: water with 0.05% trifluoroacetic acid (TFA), B: acetonitrile with 0.05% TFA, 100% solvent A for 0–10 min, a linear gradient to 80% solvent B for 10–35 min, and 80% solvent B for 35–45 min; flow rate, 1.0 mL min−1. Heterologous production of MS-271 derivatives and MS-271/sviceucin hybrid lasso peptides were performed using the plasmids listed in Table S2.† The resulting metabolites were analyzed by LC-MS (Waters ACQUITY UPLC system equipped with a SQ Detector2) under the following conditions: column: Mightysil RP-18GP Aqua column (150 × 2.0 mm, 3 mm); column temperature, 40 °C; detection, ESI-negative mode and PDA; mobile phase, A: water with 0.05% TFA, B: acetonitrile with 0.05% TFA, 5% solvent B for 0–10 min and a linear gradient to 85% solvent B for an additional 30 min; flow rate, 0.2 mL min−1. High resolution (HR)-MS was recorded on Bruker microTOF-HS at the Open Facility, Global Facility Center, Creative Research Institution, Hokkaido University.Chiral analysis of the peptides
The chirality of the amino acid of interest (Trp, Tyr, or Phe) in peptides was analyzed using Marfey's method. The purified peptide product was first hydrolyzed at 110 °C for 10 h with 3 M 2-mercaptoethanesulfonic acid (MESA). After neutralization with 1 M aqueous NaOH, the amino acid of interest was purified by HPLC using the conditions described above and dissolved in 15 μL of water. To the sample solution was added 20 μL of 0.5 M sodium bicarbonate and 50 μL of 1% 1-fluoro-2,4-dinitrophenyl-5-L-leucinamide (L-FDLA) in acetone. After incubation for 2 h at 37 °C, the reaction was quenched by the addition of 10 μL of 1 M HCl, and then diluted with 300 μL of acetonitrile. The sample containing L-FDLA derivatives was analyzed by Waters ACQUITY UPLC system equipped with a SQ Detector2 under the following conditions: column: Mightysil RP-18GP Aqua column (150 × 2.0 mm, 3 μm); column temperature, 40 °C; detection, ESI-negative mode, single ion monitoring and PDA; mobile phase, A: water with 0.05% TFA, B: acetonitrile with 0.05% TFA, 30% solvent B for 0–2 min and a linear gradient to 95% solvent B for an additional 18 min; flow rate, 0.2 mL min−1.Construction of plasmids
Oligo nucleotides used for PCR in this study are summarized in Table S1.† All plasmids were confirmed by DNA sequencing to ensure all gene sequences were correct.In vivo peptide epimerization assay
E. coli BL21(DE3) transformants harboring various combinations of plasmids (pET21-SKIK-His-mslA, pSTV-His-mslH, pCDF-His-mslB1, pCola-MBP-His-mslC, and pCola-MBP-His-mslB2) were grown at 200 rpm at 37 °C in LB medium (50 mL in a 250 mL Erlenmeyer flask) supplied with appropriate antibiotics and were induced by adding 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) when the optical density at 600 nm reached about 1. The cultivation at 200 rpm was continued for an additional 16 h at 16 °C. The cells were resuspended in a 3.5 mL wash buffer I (50 mM sodium phosphate, 300 mM NaCl, 25 mM imidazole, pH 8.0) and disrupted by sonication using an ultrasonic disruptor (TOMY, UD-200). After centrifugation at 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 30 min, the MslA was purified from either the supernatant or the precipitate as follows. When MslA was produced in an insoluble form (MslA alone and MslA + H), the precipitate was resuspended in 900 μL of buffer I (50 mM sodium phosphate, 300 mM NaCl, pH 8) and an aliquot (300 μL) was treated with an equal volume of 2× SDS-PAGE sample buffer (10% 2-mercaptoethanol, 4% SDS, 10% sucrose, and 0.01% bromophenol blue in 0.125 M Tris–HCl, pH 6.8) at 100 °C for 10 min. The entire sample (600 μL) was then separated by tricine–SDS-PAGE (16% T, 3% C polyacrylamide gel).34 After reverse staining using EzStain reverse (ATTO) to visualize proteins, a gel slice containing MslA was excised from the gel and MslA was extracted from the gel with an Attoprep filter unit (ATTO). The buffer was exchanged to 10 mM Tris–HCl (pH 8.0) by ultrafiltration (Amicon Ultracel-3, 0.5 mL, Merck). The purified MslA (concentrated in ca. 100 μL buffer) was lyophilized and used for chiral analysis. When MslA was produced in a soluble form (MslA + B1, MslA + H + B1, MslA + H + B1 + B2, and MslA + H + B1 + B2), the supernatant was subjected to Ni–NTA affinity chromatography. The fractions containing MslA were collected and concentrated to 300 μL by Amicon (Ultracel-3, 0.5 mL) and further purified by tricine–SDS-PAGE as described above.
000 × g for 30 min, the MslA was purified from either the supernatant or the precipitate as follows. When MslA was produced in an insoluble form (MslA alone and MslA + H), the precipitate was resuspended in 900 μL of buffer I (50 mM sodium phosphate, 300 mM NaCl, pH 8) and an aliquot (300 μL) was treated with an equal volume of 2× SDS-PAGE sample buffer (10% 2-mercaptoethanol, 4% SDS, 10% sucrose, and 0.01% bromophenol blue in 0.125 M Tris–HCl, pH 6.8) at 100 °C for 10 min. The entire sample (600 μL) was then separated by tricine–SDS-PAGE (16% T, 3% C polyacrylamide gel).34 After reverse staining using EzStain reverse (ATTO) to visualize proteins, a gel slice containing MslA was excised from the gel and MslA was extracted from the gel with an Attoprep filter unit (ATTO). The buffer was exchanged to 10 mM Tris–HCl (pH 8.0) by ultrafiltration (Amicon Ultracel-3, 0.5 mL, Merck). The purified MslA (concentrated in ca. 100 μL buffer) was lyophilized and used for chiral analysis. When MslA was produced in a soluble form (MslA + B1, MslA + H + B1, MslA + H + B1 + B2, and MslA + H + B1 + B2), the supernatant was subjected to Ni–NTA affinity chromatography. The fractions containing MslA were collected and concentrated to 300 μL by Amicon (Ultracel-3, 0.5 mL) and further purified by tricine–SDS-PAGE as described above.
Preparation of MslA for in vitro assay
An E. coli BL21(DE3) transformant harboring pET21-SKIK-His-mslA and pCDF-mslB1 was grown in LB medium as described above. After harvest, the cells were resuspended in a 3.5 mL wash buffer I and disrupted by sonication. After centrifugation at 25000 × g for 30 min, the supernatant was loaded onto a column containing Ni–NTA agarose resin. The column was washed with wash buffer I followed by wash buffer II (50 mM sodium phosphate, 8 M urea, 300 mM NaCl, 25 mM imidazole, pH 8.0). MslA was then eluted from the column using elution buffer (50 mM sodium phosphate, 300 mM NaCl, 250 mM imidazole, pH 8.0). The buffer was exchanged to buffer II (100 mM Tris–HCl, 300 mM NaCl, pH 8.0) and was reduced by Amicon ultrafiltration units (Ultracel-3, 0.5 mL).
Protein purifications for in vitro assay
E. coli BL21(DE3) transformants harboring pET28-His-mslH and pCDF-His-mslB1 were used to produce MslH and MslB1, respectively. After cell lysate preparation as described above, the protein was purified on Ni–NTA resin using wash buffer I and elution buffer. Finally, the protein was prepared in buffer II using Amicon filter units (Ultracel-3 for MslB1 and Ultracel-30 for MslH) for in vitro reactions.Spectroscopic analysis of MslH
UV-vis spectrum of MslH (10 μM in buffer II) was recorded using NanoDrop 2000c (Thermo Scientific) with a micro cuvette (volume: 100 μL, path length: 1 cm). Protein mass was recorded with a Maxis Plus (Bruker) connected with HPLC (Agilent Technologies Inc.) using following conditions. Column: Sunshell C8-30 HT 3.4 μm (150 × 2.1 mm, ChromaNik Technologies Inc.) at 70 °C, detection: ESI-positive mode (capillary voltage: 4500 V, nebulizer: 1.5 bar, dry gas: 7.0 L min−1, dry temperature: 200 °C), mobile phase, A: water with 0.1% TFA, B: acetonitrile with 0.1% TFA, a linear gradient from 30% B (0 min) to 60% B (30 min), flow rate: 0.3 mL min−1, injection volume: 10 μL of 20 μM MslH in buffer II.In vitro assay of MslH
A reaction mixture (300 μL) containing 470 μM MslA (SKIK- and His-tagged), 500 μM MslB1, 5 μM MslH, and 10 mM DTT in buffer II was incubated at 30 °C. Control reactions omitting MslB1 and/or MslH were also performed. The reactions were terminated by incubating with an equal volume of 2× SDS-PAGE sample buffer at 100 °C for 10 min. MslA was purified by tricine–SDS-PAGE and subjected to chiral analysis as described above. Reactions with the synthetic MslA core peptide contained 900 μM MslA-core, 600 μM MslB1, 10 μM MslH, and 10 mM DTT in buffer II (total 150 μL) and were incubated at 30 °C for 1 h. Control reactions omitting MslB1 and/or MslH were also performed. The resulting MslA-core peptides were purified by tricine–SDS-PAGE (16% T, 6% C polyacrylamide gel) and the extracted MslA was concentrated by lyophilization after the removal of small molecules using a Tube-O-Dialyzer Medi 1 k MWCO dialysis unit (Geno Technology).Metal requirements of MslH
A reaction mixture (230 μL) containing 430 μM MslA, 20 μM MslH, and 5 mM metal chloride (CoCl2, MnCl2, FeCl2, CaCl2, ZnCl2, or MgCl2) or 5 mM EDTA in buffer (100 mM Tris–HCl, pH 8.0) was incubated at 30 °C. After 2 h, MslA was purified by tricine–SDS-PAGE and subjected to chiral analysis as described above.Conclusions
In conclusion, we discovered a novel peptide epimerase, MslH, involved in the biosynthesis of the D-amino acid-containing lasso peptide MS-271. In vivo and in vitro experiments revealed that MslH catalyzed cofactor-independent epimerization on full-length MslA and that the reaction was accelerated by the precursor peptide recognition element MslB1. Furthermore, we showed that MslH exhibited broad substrate specificity toward the N-terminal region of the core peptide and produced several MS-271 derivatives and MS-271/sviceucin hybrid lasso peptides containing a D-amino acid residue. While the details of substrate recognition by MslH await further study, MslH-type epimerases could be useful to produce D-amino acid-containing peptides with improved proteolytic stability and/or biological activities.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported in part by Grants-in-Aid for Research on Innovative Areas from MEXT, Japan (JSPS KAKENHI Grant Number JP16H06452 to T. D.), Grants-in-Aid for Scientific Research from JSPS (JP18H03937 to T. D. and JP18K05449 to Y. O.), and Hokkaido University through Program for Leading Graduate Schools (Hokkaido University “Ambitious Leader's Program” to Z. F.). We are grateful to Kyowa Hakko Bio Co. Ltd., Tokyo, Japan, for providing Streptomyces sp. MS-271. We also thank Prof. Yoshimitsu Hamano and Prof. Chitose Maruyama at Fukui Prefectural University for protein mass analysis of MslH. We thank Robbie Lewis, MSc, from Edanz Group (https://en-author-services.edanzgroup.com/ac) for editing a draft of this manuscript.Notes and references
- M. O. Maksimov, S. J. Pan and A. James Link, Nat. Prod. Rep., 2012, 29, 996–1006 RSC.
- J. D. Hegemann, M. Zimmermann, X. Xie and M. A. Marahiel, Acc. Chem. Res., 2015, 48, 1909–1919 CrossRef CAS.
- W. L. Cheung-Lee and A. J. Link, J. Ind. Microbiol. Biotechnol., 2019, 46, 1371–1379 CrossRef CAS.
- M. Chen, S. Wang and X. Yu, Chem. Commun., 2019, 55, 3323–3326 RSC.
- P. G. Arnison, M. J. Bibb, G. Bierbaum, A. A. Bowers, T. S. Bugni, G. Bulaj, J. A. Camarero, D. J. Campopiano, G. L. Challis, J. Clardy, P. D. Cotter, D. J. Craik, M. Dawson, E. Dittmann, S. Donadio, P. C. Dorrestein, K. D. Entian, M. A. Fischbach, J. S. Garavelli, U. Goransson, C. W. Gruber, D. H. Haft, T. K. Hemscheidt, C. Hertweck, C. Hill, A. R. Horswill, M. Jaspars, W. L. Kelly, J. P. Klinman, O. P. Kuipers, A. J. Link, W. Liu, M. A. Marahiel, D. A. Mitchell, G. N. Moll, B. S. Moore, R. Muller, S. K. Nair, I. F. Nes, G. E. Norris, B. M. Olivera, H. Onaka, M. L. Patchett, J. Piel, M. J. Reaney, S. Rebuffat, R. P. Ross, H. G. Sahl, E. W. Schmidt, M. E. Selsted, K. Severinov, B. Shen, K. Sivonen, L. Smith, T. Stein, R. D. Sussmuth, J. R. Tagg, G. L. Tang, A. W. Truman, J. C. Vederas, C. T. Walsh, J. D. Walton, S. C. Wenzel, J. M. Willey and W. A. van der Donk, Nat. Prod. Rep., 2013, 30, 108–160 RSC.
- S. Duquesne, D. Destoumieux-Garzon, S. Zirah, C. Goulard, J. Peduzzi and S. Rebuffat, Chem. Biol., 2007, 14, 793–803 CrossRef CAS.
- K. P. Yan, Y. Li, S. Zirah, C. Goulard, T. A. Knappe, M. A. Marahiel and S. Rebuffat, Chembiochem, 2012, 13, 1046–1052 CrossRef CAS.
- B. J. Burkhart, G. A. Hudson, K. L. Dunbar and D. A. Mitchell, Nat. Chem. Biol., 2015, 11, 564–570 CrossRef CAS.
- K. Yano, S. Toki, S. Nakanishi, K. Ochiai, K. Ando, M. Yoshida, Y. Matsuda and M. Yamasaki, Bioorg. Med. Chem., 1996, 4, 115–120 CrossRef CAS.
- R. Katahira, M. Yamasaki, Y. Matsuda and M. Yoshida, Bioorg. Med. Chem., 1996, 4, 121–129 CrossRef CAS.
- Z. Feng, Y. Ogasawara, S. Nomura and T. Dairi, Chembiochem, 2018, 19, 2045–2048 CrossRef CAS.
- T. Ojima-Kato, S. Nagai and H. Nakano, J. Biosci. Bioeng., 2017, 123, 540–546 CrossRef CAS.
- L. A. Kelley, S. Mezulis, C. M. Yates, M. N. Wass and M. J. Sternberg, Nat. Protoc., 2015, 10, 845–858 CrossRef CAS.
- J. Yang and Y. Zhang, Nucleic Acids Res., 2015, 43, W174–W181 CrossRef CAS.
- Y. Li, R. Ducasse, S. Zirah, A. Blond, C. Goulard, E. Lescop, C. Giraud, A. Hartke, E. Guittet, J. L. Pernodet and S. Rebuffat, ACS Chem. Biol., 2015, 10, 2641–2649 CrossRef CAS.
- T. Stachelhaus and C. T. Walsh, Biochemistry, 2000, 39, 5775–5787 CrossRef CAS.
- C. T. Walsh, Nat. Prod. Rep., 2016, 33, 127–135 RSC.
- S. A. Samel, P. Czodrowski and L. O. Essen, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2014, 70, 1442–1452 CrossRef CAS.
- M. F. Freeman, C. Gurgui, M. J. Helf, B. I. Morinaka, A. R. Uria, N. J. Oldham, H. G. Sahl, S. Matsunaga and J. Piel, Science, 2012, 338, 387–390 CrossRef CAS.
- B. I. Morinaka, A. L. Vagstad, M. J. Helf, M. Gugger, C. Kegler, M. F. Freeman, H. B. Bode and J. Piel, Angew. Chem., Int. Ed., 2014, 53, 8503–8507 CrossRef CAS.
- A. Benjdia, A. Guillot, P. Ruffie, J. Leprince and O. Berteau, Nat. Chem., 2017, 9, 698–707 CrossRef CAS.
- A. Sikandar, L. Franz, S. Adam, J. Santos-Aberturas, L. Horbal, A. Luzhetskyy, A. W. Truman, O. V. Kalinina and J. Koehnke, Nat. Chem. Biol., 2020, 16, 1013–1018 CrossRef.
- X. Yang and W. A. van der Donk, J. Am. Chem. Soc., 2015, 137, 12426–12429 CrossRef CAS.
- L. Huo and W. A. van der Donk, J. Am. Chem. Soc., 2016, 138, 5254–5257 CrossRef CAS.
- M. Xu, F. Zhang, Z. Cheng, G. Bashiri, J. Wang, J. Hong, Y. Wang, L. Xu, X. Chen, S. X. Huang, S. Lin, Z. Deng and M. Tao, Angew. Chem., Int. Ed., 2020, 59, 18029–18035 CrossRef CAS.
- Y. Su, M. Han, X. Meng, Y. Feng, S. Luo, C. Yu, G. Zheng and S. Zhu, Appl. Microbiol. Biotechnol., 2019, 103, 2649–2664 CrossRef CAS.
- S. Zhu, C. D. Fage, J. D. Hegemann, D. Yan and M. A. Marahiel, FEBS Lett., 2016, 590, 3323–3334 CrossRef CAS.
- S. Zhu, J. D. Hegemann, C. D. Fage, M. Zimmermann, X. Xie, U. Linne and M. A. Marahiel, J. Biol. Chem., 2016, 291, 13662–13678 CrossRef CAS.
- C. Zhang and M. R. Seyedsayamdost, ACS Chem. Biol., 2020, 15, 890–894 CrossRef CAS.
- M. H. Sung, C. Park, C. J. Kim, H. Poo, K. Soda and M. Ashiuchi, Chem. Rec., 2005, 5, 352–366 CrossRef CAS.
- Y. Ogasawara, M. Shigematsu, S. Sato, H. Kato and T. Dairi, Org. Lett., 2019, 21, 3972–3975 CrossRef CAS.
- J. Sambrook and W. Russel, Molecular cloning: A Laboratory Manual, Cold Spring Harbor, NY, 3rd edn, 2001 Search PubMed.
- S. Hayashi, Y. Satoh, T. Ujihara, Y. Takata and T. Dairi, Sci. Rep., 2016, 6, 35441 CrossRef CAS.
- H. Schagger, Nat. Protoc., 2006, 1, 16–22 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: ESI Tables S1 and S2 and Fig. S1–S13. See DOI: 10.1039/d0sc06308h |
| This journal is © The Royal Society of Chemistry 2021 |