
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic asymmetric synthesis of N-substituted tetrahydroquinoxalines via regioselective Heyns rearrangement and stereoselective transfer hydrogenation in one pot†
Jin
Huang
ab,
Guang-xun
Li
 *a,
Gao-feng
Yang
a,
Ding-qiang
Fu
a,
Xiao-kang
Nie
a,
Xin
Cui
a,
Jin-zhong
Zhao
c and
Zhuo
Tang
*a
*a,
Gao-feng
Yang
a,
Ding-qiang
Fu
a,
Xiao-kang
Nie
a,
Xin
Cui
a,
Jin-zhong
Zhao
c and
Zhuo
Tang
*a
aNatural Products Research Center, Chengdu Institution of Biology, Chinese Academy of Science, Chengdu, Sichuan 610041, China. E-mail: ligx@cib.ac.cn; tangzhuo@cib.ac.cn
bUniversity of Chinese Academy of Sciences, China
cCollege of Art and Sciences, Shanxi Agricultural University, Taigu, Shanxi 030800, China
First published on 19th February 2021
Abstract
N-Substituted tetrahydroquinoxalines (37 examples) were step-economically obtained in good yield (<97%) and ee (<99%) with readily available substrates. The reaction proceeds through an interesting regioselective Heyns rearrangement/enantioselective transfer hydrogenation in one pot. The substrate scope and the reaction mechanism were systematically investigated.
Introduction
N-Substituted tetrahydroquinoxalines (THQ) represent an important class of N-heterocycles, which are present in numerous biologically active substances,1 including the prostaglandin D2 receptor antagonist I,2 the cholesteryl ester transfer protein (CETP) inhibitor II,3 and the selective bromodomain protein inhibitor III4 (Fig. 1). Therefore, extensive efforts have been devoted to the enantioselective synthesis of this class of heterocycles. Among the different strategies, asymmetric hydrogenation5 or transfer hydrogenation6 of quinoxalines represents one of the most straightforward choices (Scheme 1a). Many elegant catalytic systems have been reported using transition-metal catalysis or organocatalysis. However, despite these advances, synthesis of multi-substituted THQ, especially N-substituted THQ is still difficult,7 owing to the prior preparation and purification of the quinoxaline substrate, the limited regioselectivity, or multi-step procedures. Consequently, the step-economic synthesis of THQ via the in situ generation of quinoxalines from readily available starting materials in a regioselective,5g,6d diastereoselective, and enantioselective way is both challenging and meaningful. | ||
| Fig. 1 N-Substituted THQ with biological activities. | ||
 | ||
| Scheme 1 The background and the proposal of this research: (a) Previous works via catalytic reduction of quinoxalines. (b) Previous work for preparation of quinoxalines via Heyns rearrangement. (c) Our proposal for obtaining chiral THQ via regioselective Heyns rearrangement and stereoselective reduction in one pot. | ||
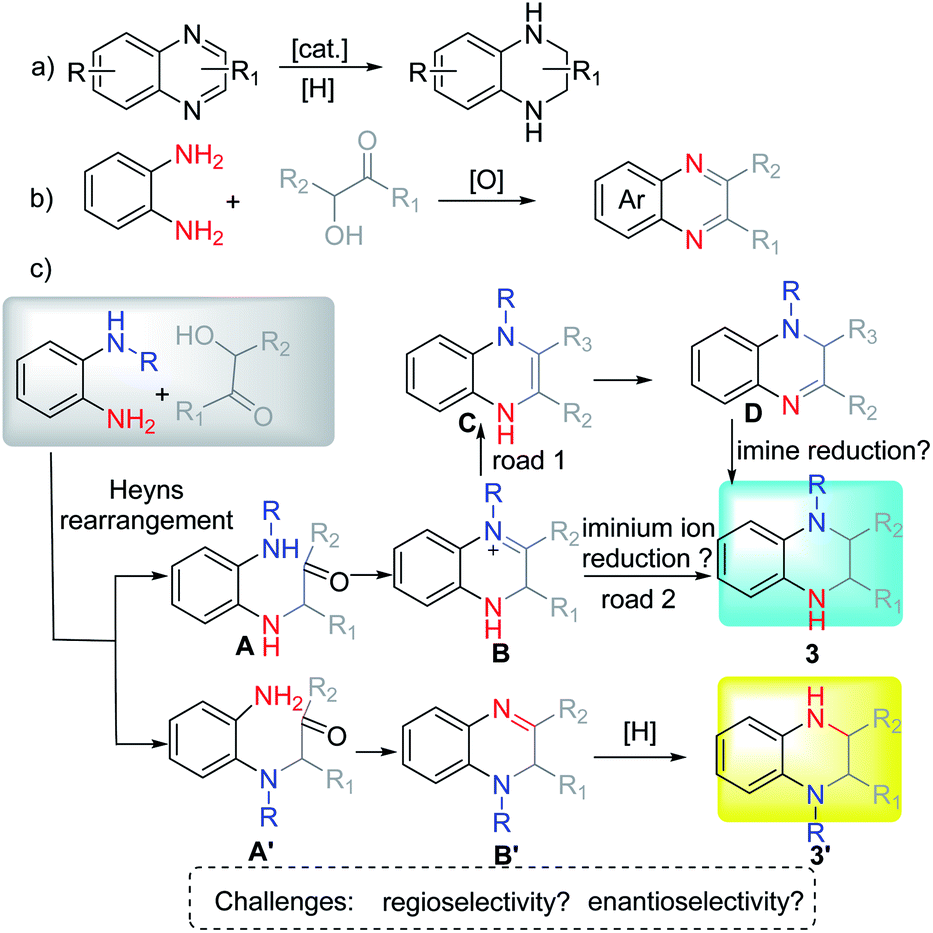
Amadori and Heyns rearrangements have been famous among carbohydrate chemists over the decades.8 Both reactions suffer from a variety of preparative shortcomings such as separation drawbacks, side reactions, further degradation entering into the Maillard reaction cascade, etc. Therefore, this rearrangement appears to be highly underrated as a useful method for synthetic chemists.9 Due to the significance of the reaction as well as the importance of the corresponding products, our group has paid much attention to its application.10 Recently, we investigated the production and application of the in situ formed α-amino aldehyde intermediate and synthesized different N-heterocycles by using different types of nucleophiles.10a
For example, symmetric o-phenylenediamine (o-PDA) reacted smoothly with α-hydroxyl ketones to afford the corresponding quinoxaline (Scheme 1b). Due to the important application of N-substituted THQ as well as our interest in Heyns rearrangement, we proposed that the substituted unsymmetric o-PDA could react with α-hydroxyl ketone via Heyns rearrangement to afford the corresponding α-amino ketone A or A′ (Scheme 1c). Then the reaction continues to form the iminium ion B or imine B′ by cyclization. Next, the reaction could proceed in three possible pathways: (1) B could isomerize to enamine C which would be isomerized again to stable imine D. Then D would be reduced to afford N-substituted THQ. (2) Iminium ion B might be reduced directly to afford the corresponding product. (3) Imine B′ might be reduced to get product 3′. According to the possible reaction process, the regioselectivity of the reaction might be controlled by the Heyns rearrangement, while the enantioselectivity might be controlled in different ways: enantioselective protonation and enantioselective reduction of iminium ions or imine. These properties pose great challenges for us to obtain N-substituted THQ in one pot.
Results and discussion
Luckily, THQ 3a was obtained exclusively in 70% yield and 68% ee using Hantzsch ester (HEH) and chiral phosphoric acid CPA 1 at 70 °C after optimizing the reaction conditions (Scheme 2a, for the optimization experiments, see Table S1 in the ESI†). However, further improvement of the reaction yield and the ee value was very hard. We therefore investigated the reaction process via the addition of D2O into the reaction (Scheme 2b and c). When the reaction was run with the addition of HEH at the beginning, the corresponding product 3a was deuterated at C2–H and C3–H (Scheme 2b). This means that 3a might be formed by the reduction of intermediates B and D. In contrast, only the C2–H was deuterated when the reaction was run in two steps (Scheme 2c). This means that only the intermediate D was reduced.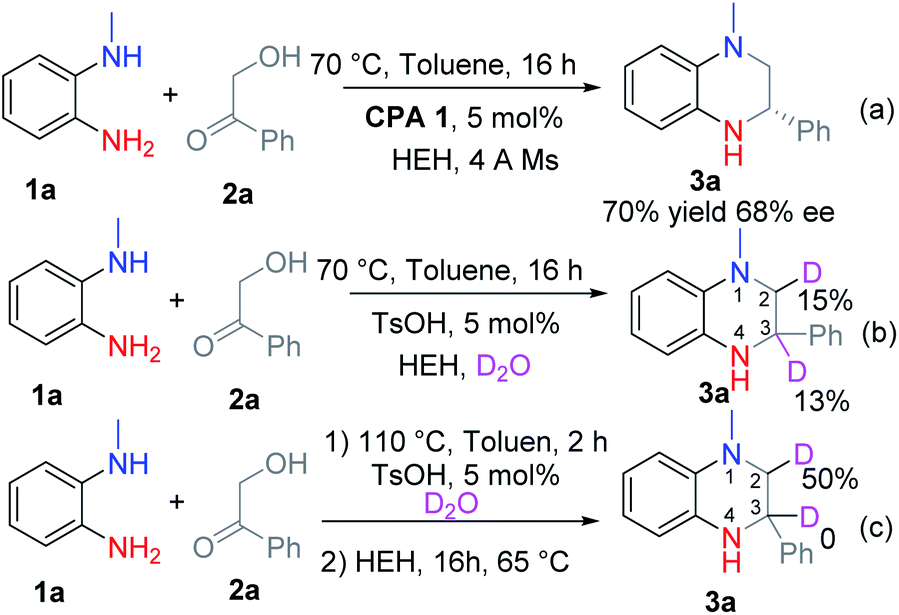 | ||
| Scheme 2 Investigation of the reaction process: (a) moderate ee was obtained if HEH was added to the reaction at the very beginning. (b) D2O and HEH were added to the reaction at the beginning. (c) D2O was added at the beginning while HEH was added after 2 hours. | ||
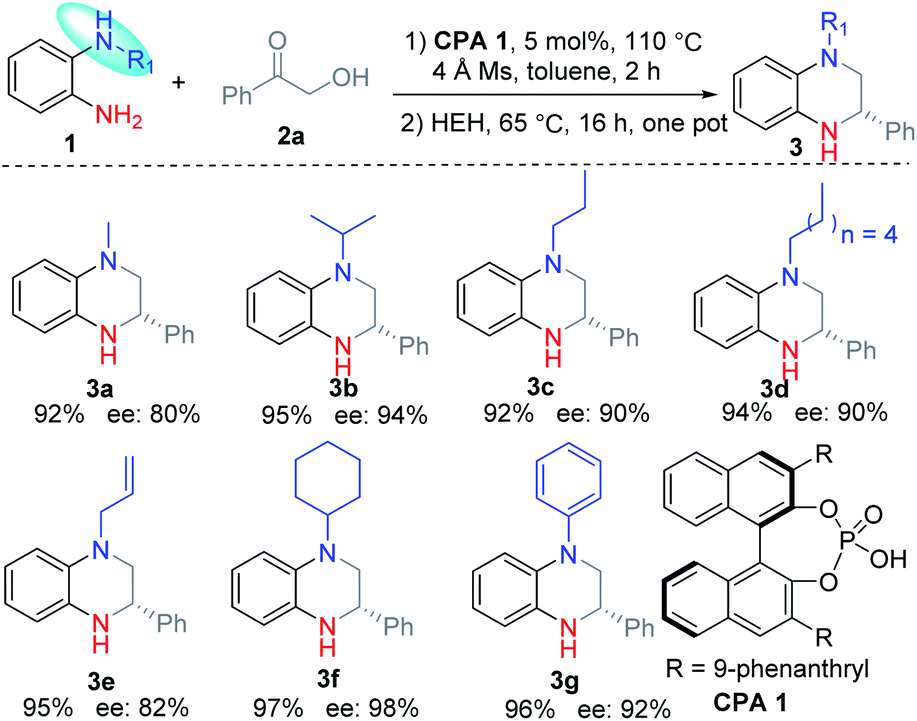
According to this result, we changed the addition sequence of HEH to ensure that the THQ 3 was formed from the transfer hydrogenation of imine D. To our delight, both the reaction yield and the ee value could be improved in this way (Table S1, see ESI†). The optimal reaction conditions were obtained as follows: equal equiv. of 1a and 2a reacted under a nitrogen atmosphere with 5 mol% CPA 1 in toluene for 2 h, then HEH was added and the reaction continued to proceed at 65 °C for 16 h in one pot. The corresponding product 3a was obtained in 92% yield and 80% ee (Table 1).
|
Then the substituent of the nitrogen was investigated under the optimal conditions. The results revealed that the reaction proceeded smoothly with good yields (92–97%) and high regioselectivities. Moreover, the ee value of 3 increased greatly for substituents with better steric hindrance (3b and 3f). Meanwhile, the long alkyl substituents (3c and 3d) and aromatic ring substituent (3g) are proved to be feasible substrates with good ee values. Substrates with allyl afforded the corresponding product 3e with a moderate ee value due to the small steric hindrance.
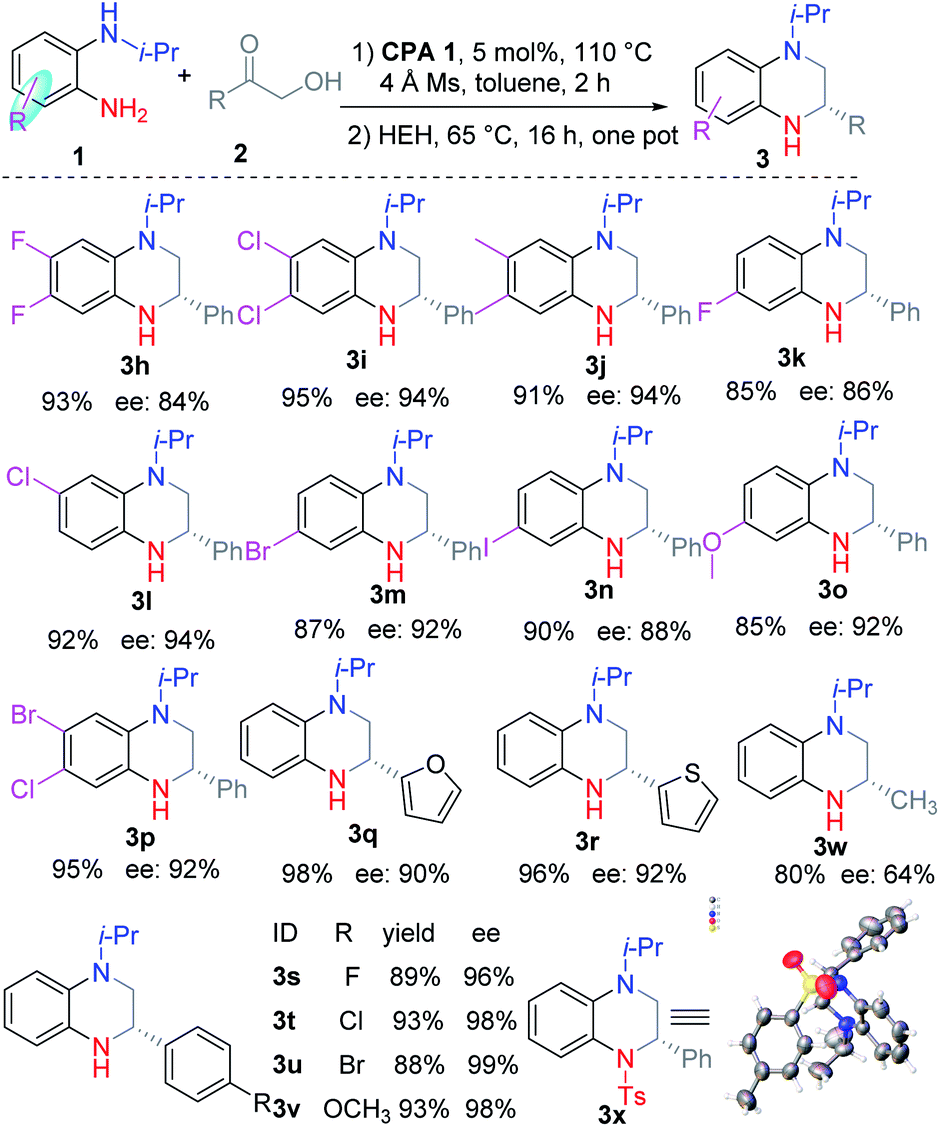
Next, we investigated the substituents of the aromatic ring of o-PDA (Table 2). Generally, the reaction proceeded smoothly with good regioselectivity. Firstly, symmetric o-PDAs were investigated, which smoothly reacted and afforded the corresponding products 3h–3j in high yield (91–95%) and ee (84–94%). Then unsymmetric o-PDAs were evaluated. The results revealed that o-PDAs with single halogen (3k–3n) or with two halogens (3p), with an electron donating group (3o), could be efficiently reacted to obtain the corresponding products in good yields (85–95%) and ees (86–94%). Then the primary α-hydroxyl ketones were investigated. Generally, they were efficiently converted to the corresponding products 3q–3v in good yields (88–98%), excellent ees (84–99%), and good regioselectivities. The substituent of the phenyl ring was studied, which demonstrated that halogen substituents such as F, Cl, and Br (3s–3u), and electron donating groups such as methoxyl (3v), were efficiently converted to the corresponding products. Moreover, α-hydroxyl ketones with aromatic heterocycles such as furan and thiophene were easily converted to the corresponding products 3q and 3r effectively. Alkyl substituted α-hydroxyl ketone was investigated and the corresponding THQ 3w was obtained in moderate yield and ee. The absolute configuration of the products was confirmed according to the single crystal diffraction result of 3x, which was obtained from 3b.
|
Then we turned to investigate the reaction with racemic α-hydroxyl ketone 2′a as the substrate (see ESI†). The corresponding product 5a was obtained in 46% yield and 90% ee under the optimal reaction conditions (Scheme 3a). Further attempts to increase the reaction yield were very hard. Therefore, we tried to investigate the reaction process based on the deuteration experiment.
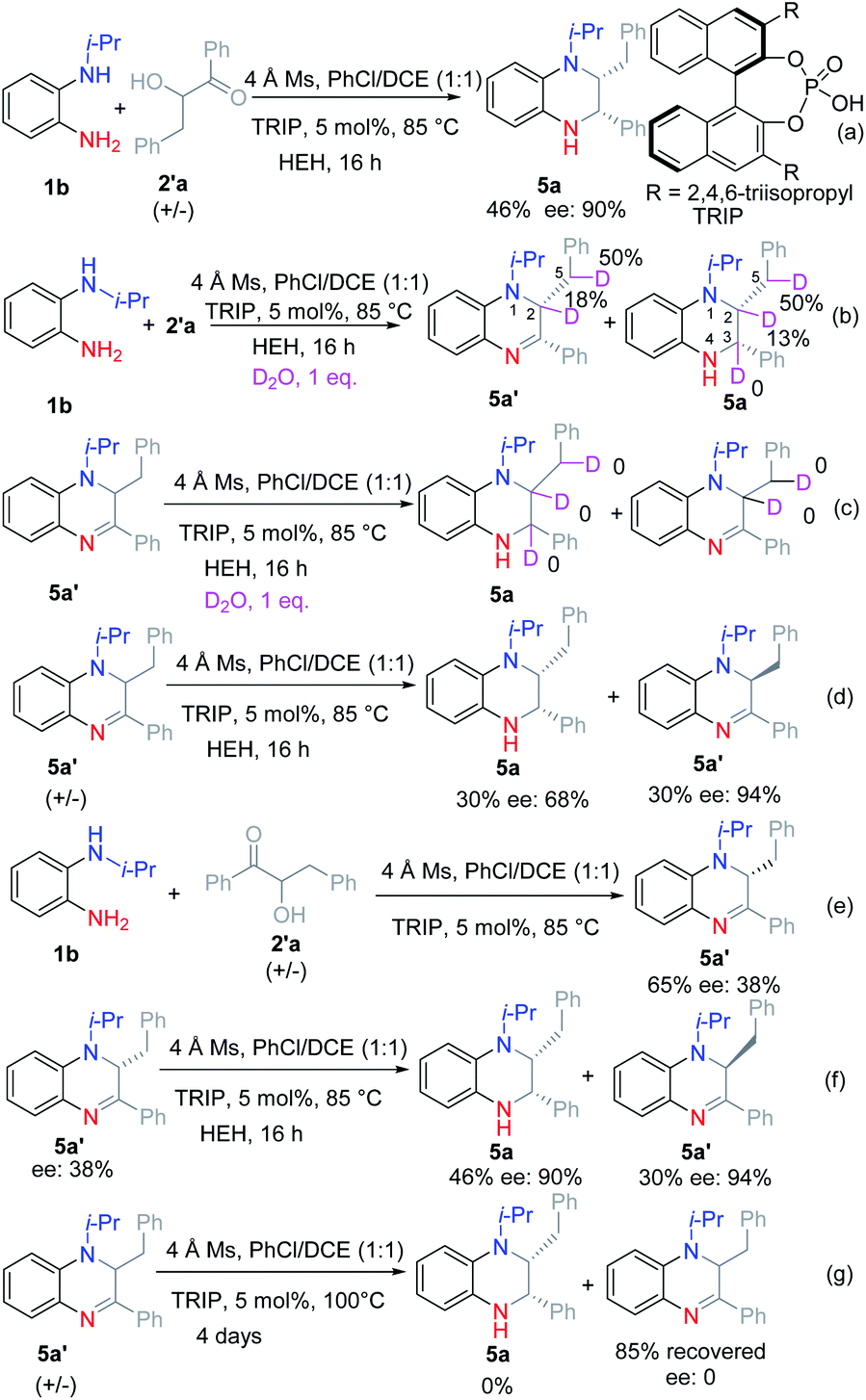 | ||
| Scheme 3 Investigation of the reaction for obtaining di-substituted THQ: (a) the optimal reaction condition for obtaining di-substituted THQ. (b) D2O was added to investigate the reaction. (c) DHQ 5a′ was reduced at the optimal reaction condition with the addition of D2O. (d) Racemic DHQ 5a′ was reduced under the optimal reaction condition. (e) No reducing reagent was added. (f) DHQ 5a′ with low ee value was reduced under the optimal reaction condition. (g) Racemic DHQ 5a′ was reacted without the addition of HEH. | ||
Firstly, 1 equiv. D2O was added to the reaction mixture under the optimal reaction conditions (Scheme 3b). According to the result, the C2–H at the α-position of the tertiary amine was deuterated, while C3–H at the α-position of the secondary amine was not deuterated. Moreover, the benzylic hydrogen was deuterated. Based on this result, we guess that the reaction proceeded selectively by the reduction of ketone imine rather than the iminium ion. Then dihydroquinoxaline (DHQ) 5a′ was used as a substrate with D2O to check if the ketone imine would isomerize into enamine or iminium ions (Scheme 3c). According to the result, no deuterated product was found, which proved that the ketone imine was stable and could not isomerize into enamine under the reaction conditions. Finally, the racemic DHQ 5a′ was reduced under the optimal reaction conditions, and the corresponding product 5a was obtained in 30% yield and 68% ee (Scheme 3d). To our surprise, the recovered substrate was obtained in 94% ee. Therefore, we could conclude that the reaction proceeded through a kinetic resolution process. To figure out the source of the enantioselectivities we also investigated the reaction without the addition of reduction reagents (Scheme 3e). According to the result, 5a′ was obtained with moderate yield and low ee (38%). Moreover, 5a′ with 38% ee could be reduced under the optimal reaction conditions to afford 5a with similar ee (Scheme 3f). When racemic 5a′ was stirred under the optimal reaction conditions without adding HEH, no product was found even after raising the reaction temperature to 100 °C for 4 days (Scheme 3g). Therefore, we could preclude that 5a′ might act as a reductant.11
Based on the above results, we proposed a reasonable reaction mechanism (Scheme 4). Firstly, the reaction selectively proceeded via Heyns rearrangement to form α-amino ketone A. Then the iminium ion B was formed by cyclization. Meanwhile, enamine intermediates C and D might be formed by isomerization. Next, the enantioselective protonation occurred to form the stable DHQ 5a′ in low ee. Finally, 5a was formed by enantioselective reduction via kinetic resolution.
 | ||
| Scheme 4 Plausible reaction process for obtaining disubstituted THQ. | ||
Then we turned to investigate the substrate scope for obtaining disubstituted THQ (Table 3). Generally, racemic α-hydroxyl ketones could easily react with substituted o-PDAs to afford the corresponding THQ (5a–5m) in moderate yields (25–46%) and good ees (86–96%). Compared with symmetric o-PDAs as substrates, the use of unsymmetric o-PDAs as substrates afforded the corresponding THQ (5f–5g, 5m) in slightly lower yields. Interestingly, increasing the carbon chain also afforded the corresponding product 5h in moderate yield (35%) and ee (82%). The absolute configuration of the products was confirmed according to the single crystal diffraction result of 5a.
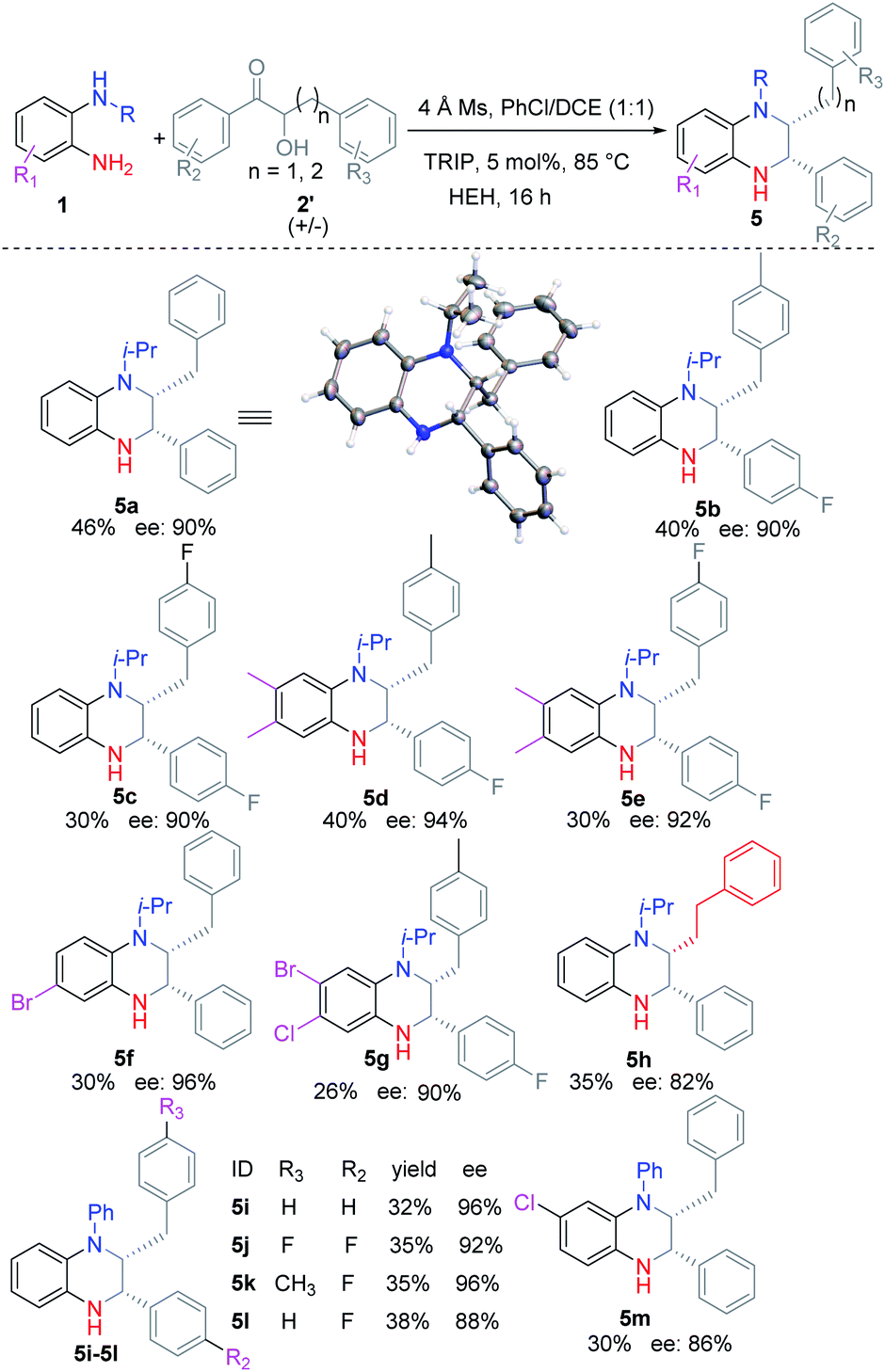
|
Then we tried to show the advantages of the method. DHQ 5′a could be obtained from the reaction mixture in moderate yield and high ee, which might explain the slightly low yield of the reaction for obtaining 2,3-disubstituted THQ 5. Interestingly the other enantiomer of 5a could be easily obtained by reducing DHQ 5′a with NaBH4 (Scheme 5). Meanwhile, o-PDA 1i was synthesized and used under the optimal reaction conditions to provide the N-substituted product 3y in good yield and ee, which could be used for the preparation of useful CETP inhibitor 6.11
 | ||
| Scheme 5 Application of the method in the synthesis of useful compounds. | ||
Conclusions
In summary, we have developed an interesting tandem reaction for conveniently obtaining N-substituted THQ with readily available o-PDA and α-hydroxyl ketones as starting materials. A series of monosubstituted and disubstituted THQ (37 examples) were obtained in good yields (up to 97%) and high ees (up to 99%). Moreover, good regioselectivity was achieved for reactions with unsymmetric o-PDA as the substrate, which was seldom used as a substrate due to its low regioselectivity. The substituents of the amino group of o-PDA were investigated, and it was demonstrated that the reaction could tolerate alkyl, allyl, and aromatic substituents for high yield and ee. Meanwhile, different types of primary α-hydroxyl ketones and racemic α-hydroxyl ketones could be efficiently reacted to afford the corresponding monosubstituted and disubstituted THQ. Generally, high regioselectivities of the reaction were realized via selective Heyns rearrangement. The high enantioselectivities of the reactions were investigated. For reactions with primary α-hydroxyl ketones as substrates, the high enantioselectivities were obtained from enantioselective transfer hydrogenation, while for reactions with racemic secondary α-hydroxyl ketones, the enantioselectivities came from the combination of enantioselective protonation and kinetic resolution.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported financially by the Youth Innovation Promotion Association CAS (2018402) and the West Light Foundation of the Chinese Academy of Sciences (25E0C304).Notes and references
- (a) M. Patel, R. J. McHugh, B. C. Cordova, R. M. Klabe, S. Erickson-Viitanen, G. L. Trainor and J. D. Rodgers, Bioorg. Med. Chem. Lett., 2000, 10, 1729–1731 CrossRef CAS; (b) S. Li, X. Tian, D. M. Hartley and L. A. Feig, J. Neurosci., 2006, 26, 1721–1729 CrossRef CAS; (c) M. Fantin, M. Marti, Y. P. Auberson and M. Morari, J. Neurosci., 2007, 103, 2200–2211 CAS.
- K. Torisu, K. Kobayashi, M. Iwahashi, Y. Nakai, T. Onoda, T. Nagase, I. Sugimoto, Y. Okada, R. Matsumoto, F. Nanbu, S. Ohuchida, H. Nakai and M. Toda, Bioorg. Med. Chem., 2004, 12, 5361–5378 CrossRef CAS.
- (a) J. A. Sikorski, J. Med. Chem., 2006, 49, 1–22 CrossRef CAS; (b) C. T. Eary, Z. S. Jones, R. D. Groneberg, L. E. Burgess, D. A. Mareska, M. D. Drew, J. F. Blake, E. R. Laird, D. Balachari, M. O'Sullivan, A. Allen and V. Marsh, Bioorg. Med. Chem. Lett., 2007, 17, 2608–2613 CrossRef CAS; (c) J. E. Wilson, R. Kurukulasuriya, M. Reibarkh, M. Reiter, A. Zwicker, K. Zhao, F. Zhang, R. Anand, V. J. Colandrea, A.-M. Cumiskey, A. Crespo, R. A. Duffy, B. A. Murphy, K. Mitra, D. G. Johns, J. L. Duffy and P. Vachal, ACS Med. Chem. Lett., 2016, 7, 261–265 CrossRef CAS.
- R. P. Law, S. J. Atkinson, P. Bamborough, C.-w. Chung, E. H. Demont, L. J. Gordon, M. Lindon, R. K. Prinjha, A. J. B. Watson and D. J. Hirst, J. Med. Chem., 2018, 61, 4317–4334 CrossRef CAS.
- (a) C. Bianchini, P. Barbaro, G. Scapacci, E. Farnetti and M. Graziani, Organometallics, 1998, 17, 3308–3310 CrossRef CAS; (b) N. Mršić, T. Jerphagnon, A. J. Minnaard, B. L. Feringa and J. G. de Vries, Adv. Synth. Catal., 2009, 351, 2549–2552 CrossRef; (c) W. Tang, L. Xu, Q.-H. Fan, J. Wang, B. Fan, Z. Zhou, K.-h. Lam and A. S. C. Chan, Angew. Chem., Int. Ed., 2009, 48, 9135–9138 CrossRef CAS; (d) D. Cartigny, T. Nagano, T. Ayad, J.-P. Genêt, T. Ohshima, K. Mashima and V. Ratovelomanana-Vidal, Adv. Synth. Catal., 2010, 352, 1886–1891 CrossRef CAS; (e) S. Urban, N. Ortega and F. Glorius, Angew. Chem., Int. Ed., 2011, 50, 3803–3806 CrossRef CAS; (f) T. Nagano, A. Iimuro, R. Schwenk, T. Ohshima, Y. Kita, A. Togni and K. Mashima, Chem.–Eur. J., 2012, 18, 11578–11592 CrossRef CAS; (g) D. Cartigny, F. Berhal, T. Nagano, P. Phansavath, T. Ayad, J.-P. Genêt, T. Ohshima, K. Mashima and V. Ratovelomanana-Vidal, J. Org. Chem., 2012, 77, 4544–4556 CrossRef CAS; (h) S. Fleischer, S. Zhou, S. Werkmeister, K. Junge and M. Beller, Chem.–Eur. J., 2013, 19, 4997–5003 CrossRef CAS; (i) N. Arai, Y. Saruwatari, K. Isobe and T. Ohkuma, Adv. Synth. Catal., 2013, 355, 2769–2774 CrossRef CAS; (j) Z. Zhang and H. Du, Angew. Chem., Int. Ed., 2015, 127, 633–636 CrossRef; (k) S. Sun and P. Nagorny, Chem. Commun., 2020, 56, 8432–8435 RSC.
- (a) M. Rueping, F. Tato and F. R. Schoepke, The First General, Efficient and Highly Enantioselective Reduction of Quinoxalines and Quinoxalinones, Chem.–Eur. J., 2010, 16, 2688–2691 CrossRef CAS; (b) D.-W. Wang, D.-S. Wang, Q.-A. Chen and Y.-G. Zhou, Chem.–Eur. J., 2010, 16, 1133–1136 CrossRef CAS; (c) Q.-A. Chen, D.-S. Wang, Y.-G. Zhou, Y. Duan, H.-J. Fan, Y. Yang and Z. Zhang, J. Am. Chem. Soc., 2011, 133, 6126–6129 CrossRef CAS; (d) F. Shi, W. Tan, H.-H. Zhang, M. Li, Q. Ye, G.-H. Ma, S.-J. Tu and G. Li, Adv. Synth. Catal., 2013, 355, 3715–3726 CrossRef CAS.
- (a) C. J. Abraham, D. H. Paull, M. T. Scerba, J. W. Grebinski and T. Lectka, J. Am. Chem. Soc., 2006, 128, 13370–13371 CrossRef CAS; (b) C. L. Hamblett, J. L. Methot, D. M. Mampreian, D. L. Sloman, M. G. Stanton, A. M. Kral, J. C. Fleming, J. C. Cruz, M. Chenard, N. Ozerova, A. M. Hitz, H. Wang, S. V. Deshmukh, N. Nazef, A. Harsch, B. Hughes, W. K. Dahlberg, A. A. Szewczak, R. E. Middleton, R. T. Mosley, J. P. Secrist and T. A. Miller, Bioorg. Med. Chem. Lett., 2007, 17, 5300–5309 CrossRef CAS; (c) M. K. Ghorai, A. K. Sahoo and S. Kumar, Org. Lett., 2011, 13, 5972–5975 CrossRef CAS; (d) L. Gavara, E. Saugues, F. Anizon and P. Moreau, Tetrahedron, 2011, 67, 1633–1639 CrossRef CAS; (e) B. A. Hopkins and J. P. Wolfe, Chem. Sci., 2014, 5, 4840–4844 RSC; (f) A.-G. A. El-Helby, R. R. A. Ayyad, K. El-Adl and A. Elwan, Med. Chem. Res., 2017, 26, 2967–2984 CrossRef CAS; (g) R. Huang, X. Chen, C. Mou, G. Luo, Y. Li, X. Li, W. Xue, Z. Jin and Y. R. Chi, Org. Lett., 2019, 21, 4340–4344 CrossRef CAS.
- Z. Wang, Heyns Rearrangement, in Comprehensive Organic Name Reactions and Reagents, ed. Z. Wang, 2010, pp. 1403–1407 Search PubMed.
- (a) T. J. Guzi and T. L. Macdonald, Tetrahedron Lett., 1996, 37, 2939–2942 CrossRef CAS; (b) A. Frongia, F. Secci, F. Capitta, P. P. Piras and M. L. Sanna, Chem. Commun., 2013, 49, 8812–8814 RSC; (c) A. Frongia, N. Melis, I. Serra, F. Secci, P. P. Piras and P. Caboni, Asian J. Org. Chem., 2014, 3, 378–381 CrossRef CAS; (d) D. J. Aitken, P. Caboni, H. Eijsberg, A. Frongia, R. Guillot, J. Ollivier, P. P. Piras and F. Secci, Adv. Synth. Catal., 2014, 356, 941–945 CrossRef CAS; (e) N. Melis, L. Ghisu, R. Guillot, P. Caboni, F. Secci, D. J. Aitken and A. Frongia, Eur. J. Org. Chem., 2015, 2015, 4358–4366 CrossRef CAS.
- (a) G. Li, L. Tang, H. Liu, Y. Wang, G. Zhao and Z. Tang, Org. Lett., 2016, 18, 4526–4529 CrossRef CAS; (b) L.-y. Li, Q.-l. Zeng, G.-x. Li and Z. Tang, Synlett, 2019, 30, 694–698 CrossRef CAS; (c) J.-x. Liang, G.-b. Yang, Y.-p. Zhang, D.-d. Guo, J.-z. Zhao, G.-x. Li and Z. Tang, Org. Chem. Front., 2020, 7, 3242–3246 RSC.
- R. Anand, V. J. Colandrea, M. Reiter, P. Vachal, A. Zwicker, J. E. Wilson, F. Zhang and R. Zhao, Benzopiperazine Derivatives as CETP Inhibitors, Patent application WO2013/028382, Feb 28, 2013.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc06264b |
| This journal is © The Royal Society of Chemistry 2021 |