DOI:
10.1039/D0SC06258H
(Edge Article)
Chem. Sci., 2021,
12, 1924-1929
Intermediates involved in serotonin oxidation catalyzed by Cu bound Aβ peptides†
Received
13th November 2020
, Accepted 11th December 2020
First published on 22nd December 2020
Abstract
The degradation of neurotransmitters is a hallmark feature of Alzheimer's disease (AD). Copper bound Aβ peptides, invoked to be involved in the pathology of AD, are found to catalyze the oxidation of serotonin (5-HT) by H2O2. A combination of EPR and resonance Raman spectroscopy reveals the formation of a Cu(II)–OOH species and a dimeric, EPR silent, Cu2O2 bis-μ-oxo species under the reaction conditions. The Cu(II)–OOH species, which can be selectively formed in the presence of excess H2O2, is the reactive intermediate responsible for 5-HT oxidation. H2O2 produced by the reaction of O2 with reduced Cu(I)–Aβ species can also oxidize 5-HT. Both these pathways are physiologically relevant and may be involved in the observed decay of neurotransmitters as observed in AD patients.
Introduction
Alzheimer's disease (AD) is a terminal neurodegenerative disease which is characterized by the deposition of insoluble plaques in the hippocampus of an AD affected brain.1,2 It is clinically characterized by progressive dementia.3 The exact cause for this disease is still unclear. Over the past two decades extensive research has focused on determining the actual cause of this disease, resulting in important hypotheses relating to the pathology of AD. Among them, one of the more accepted hypothesis is the “amyloid hypothesis”.4–6 The amyloid β-peptide is the main constituent of this hypothesis and is found in high proportions in the plaques that are diagnostic of AD.5,6 Moreover transition metals like Cu(II) and Zn(II) and cofactors like heme are found in the plaques of an AD brain.7–9 Apart from the ability of these bivalent metals to aggregate the monomeric Aβ peptides, redox active metals like Cu and Fe are proposed to cause oxidative stress resulting in damage to the neuronal cell membrane making it “leaky”.7,9 The oxidative damage is proposed to be an upstream event which eventually leads to the formation of insoluble plaques which is a hallmark of the disease.7,10 The oxidative stress can be due to hydrogen peroxide (H2O2), which, apart from its natural availability, can also be produced by the reaction of the reduced redox active metals bound Aβ peptides with O2.9,11,12
The recent investigation of Cu bound Aβ peptides and their site-directed mutants using a combination of spectroscopic techniques and theoretical calculations has revealed a Cu active site coordinated to histidine residues and exchangeable ligands.13–18 The Cu–Aβ complexes exhibit peroxidase activity in the presence of H2O2.12 Presently several naturally occurring copper containing metalloenzymes are known which can oxidize/hydroxylate organic compounds in the presence of H2O2. Enzymes like amine oxidase participate in the breakdown of amines to produce an aldehyde and ammonia.19–21 Alternatively, copper enzymes like dopamine β-hydroxylase (DβM) are involved in the synthesis of a small-molecule neurotransmitter by catalyzing the hydroxylation of dopamine to convert it to norepinephrine.19,22–24 Note that the Cu active sites of these above-mentioned proteins also have histidine and water derived exchangeable ligands in their active sites similar to that of Cu–Aβ. Now oxidative degradation of neurotransmitters like serotonin generating neurotoxins like tryptamine-4,5-dione is a hallmark of AD, which can lead to impaired neuronal signaling.25–33 This raises the possibility of Cu bound Aβ reacting with H2O2 and catalyzing the degradation of neurotransmitters (Scheme 1). Previously Ming et al. have kinetically shown the catalytic hydroxylation/oxidation of substrates like dopamine, catechol derivatives, phenol and serotonin and proposed a side-on μ-peroxo dicopper(II) intermediate as the active species, though no spectroscopic proof was provided.34–37 Since the oxidation of neurotransmitters by Cu–Aβ in the presence of H2O2 is a likely physiological process occurring in the human brain, the identification and characterization of the reactive intermediates involved in this chemical oxidation process are crucial for the understanding of this disease.
 |
| | Scheme 1 Oxidation of the neurotransmitter serotonin (5-HT) by Cu–Aβ and H2O2. | |
In this manuscript, we experimentally determine the reaction intermediates involved in serotonin oxidation as well as in the peroxidase activity pathway of Cu(II) bound Aβ peptides in the presence of H2O2. Spectroscopic evidence reveals that two intermediates, Cu2O2-bis-μ-oxo and Cu(II)–OOH, are formed in the reaction of Cu–Aβ and H2O2.
Results and discussion
Neurotransmitter serotonin (5-HT) oxidation by Cu–Aβ with H2O2
The reaction of the neurotransmitter serotonin (5-HT) with Cu–Aβ in the presence of H2O2 produces absorption peaks at 289, 323, 355, 392, 488 and 535 nm (Fig. 1A and S1†). These peaks are indicative of serotonin oxidation. Note that, as Cu–Aβ (1–16) and Cu–Aβ (1–40) produce similar serotonin oxidized products, all the other experiments have been performed with Cu–Aβ (1–16). The oxidized products of serotonin are further characterized using HPLC as previously reported (Fig. S2 and Scheme S1†).38,39 The products are found to be tryptamine-4,5-dione (T-4,5-D), 5-hydroxy-3-ethylamino-2-oxindole (5-HEO) and 3,3′-bis(2-aminoethyl)-5-hydroxy-[3,7′-bi-1H-indole]-2,4′,5′(3H)-trione, which is the aerially oxidized dimer of T-4,5-D and 5-HEO. The pseudo 1st order rate for this reaction is found to be (4 ± 0.2) × 10−4 s−1 (Fig. 1B and S3†). Note that the slow kinetics of this oxidation matches the slow onset and progression of this disease. Control experiments without Cu–Aβ, i.e. H2O2 or Cu(II) (Fenton reaction conditions) alone, show barely any oxidation of 5-HT (Fig. 1B).
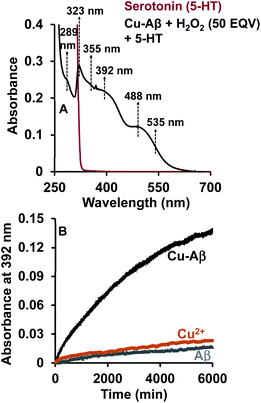 |
| | Fig. 1 (A) Absorption spectrum of serotonin (5-HT), red; 5-HT + Cu–Aβ + 50 eq. H2O2, black; (B) kinetics of serotonin oxidation monitored at 392 nm; 5-HT + Cu–Aβ + 50 eq. H2O2, black; 5-HT + CuSO4 + 50 eq. H2O2, brown; 5-HT + Aβ + 50 eq. H2O2, grey; in 100 mM HEPES buffer at pH 7. [Cu–Aβ = 0.055 mM, H2O2 = 2.77 mM and 5-HT = 2.77 mM]. | |
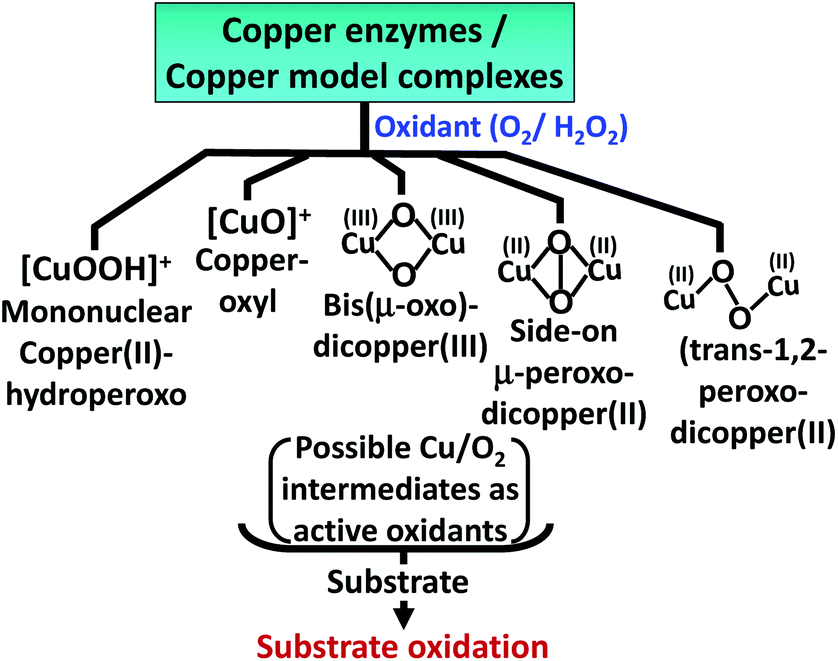
The oxidation of 5-HT by H2O2 catalyzed by Cu(II)–Aβ can proceed via several reactive intermediates similar to those which have been observed in different Cu enzymatic active sites and their synthetic analogues19,40–43 (Scheme 2). Hence the reaction of Cu–Aβ with H2O2 is monitored with the aim of trapping and characterizing the active oxidant responsible for oxidizing substrates like serotonin and other neurotransmitters.
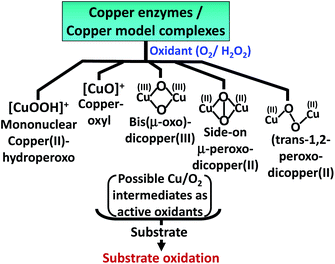 |
| | Scheme 2 Possible reactive Cu/O2 intermediates for substrate oxidation by different copper enzymes and copper model complexes in the presence of oxidants (O2 or H2O2).19,40–42 | |
Characterization of Cu/O2 intermediates formed in the reaction of Cu–Aβ with H2O2
Absorption spectroscopy.
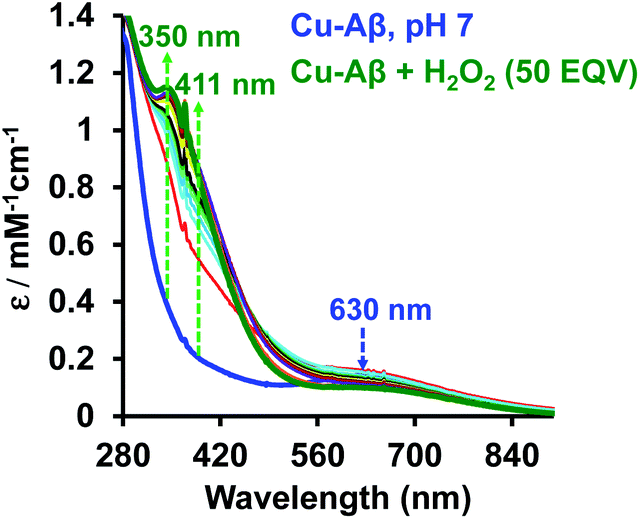
At physiological pH, Cu–Aβ shows a broad ligand field band at 630 nm. The addition of H2O2 to Cu–Aβ produces charge transfer (CT) bands at 350 nm (ε = 1200 M−1 cm−1) and 411 nm (ε = 850 M−1 cm−1) (Fig. 2). The energies of these CT bands are characteristic of peroxide  . CT bands in these regions hint at the plausible formation of either a side-on μ-peroxo-dicopper(II) complex or a bis(μ-oxo)dicopper(III) complex as depicted in Scheme 2.41,42,44–46 Previously Ming and coworkers proposed the formation of a side-on μ-peroxo dicopper(II) intermediate.34,36
. CT bands in these regions hint at the plausible formation of either a side-on μ-peroxo-dicopper(II) complex or a bis(μ-oxo)dicopper(III) complex as depicted in Scheme 2.41,42,44–46 Previously Ming and coworkers proposed the formation of a side-on μ-peroxo dicopper(II) intermediate.34,36
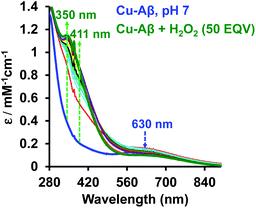 |
| | Fig. 2 Absorption spectrum of Cu–Aβ, blue; Cu–Aβ with 50 eq. of H2O2 at different times, green being the final spectrum after 120 minutes; data are collected in 100 mM HEPES buffer at pH 7. The arrows indicate the direction of the spectral changes. | |
EPR spectroscopy.
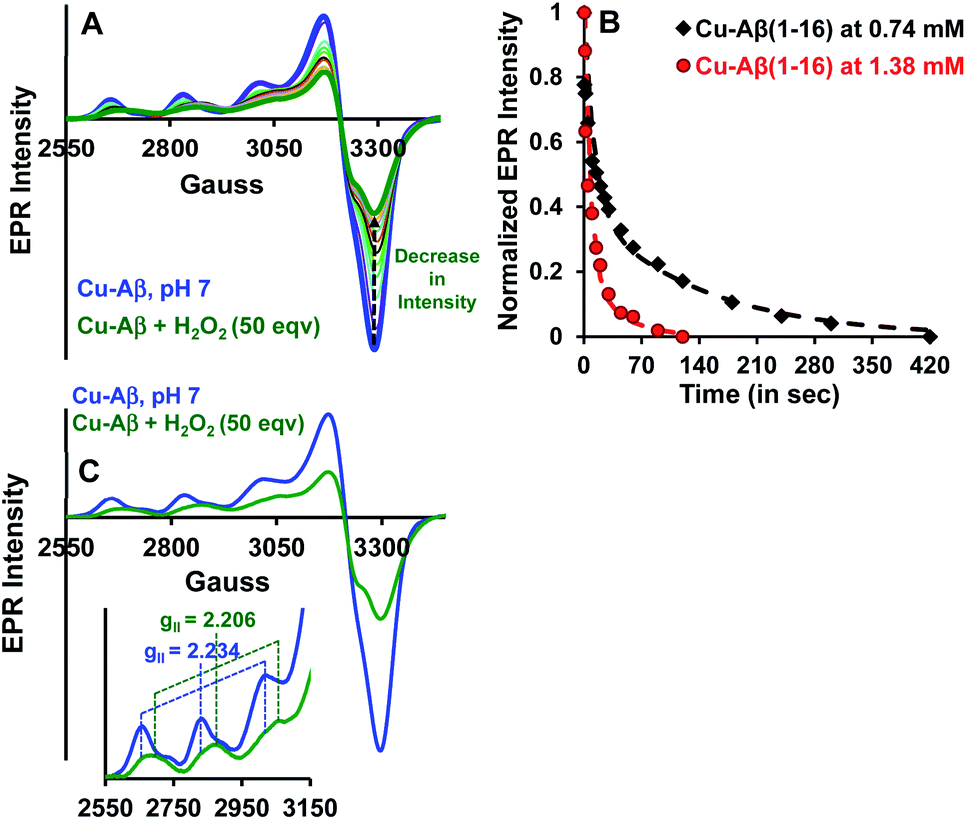
Both side-on peroxo and bis(μ-oxo) complexes are diamagnetic (S = 0) and hence are expected to be EPR silent.44,47,48 The native Cu–Aβ at pH 7 shows a characteristic (S = 1/2) axial EPR signal (predominantly component I with a trace of component II) (Fig. 3A and C).13,16 When H2O2 is added to Cu–Aβ, ∼40% loss of the EPR signal is observed relative to that of the starting Cu–Aβ signal (Fig. 3A and S4†). A loss in EPR signal intensity can be indicative of the formation of a diamagnetic side-on μ-peroxo- dicopper(II) or a bis(μ-oxo)dicopper(III) species. The formation of a dimeric species from mononuclear Cu–Aβ should result from the dimerization of two monomeric Cu species. Accordingly, as the Cu–Aβ concentration is doubled, the rate of spin loss of the Cu–Aβ EPR signal is doubled (Fig. 3B). This indicates that the side-on μ-peroxo-dicopper(II) or bis(μ-oxo)dicopper(III) bridging is intermolecular resulting in the formation of a diamagnetic species. Note that in this reaction, the EPR signal of the reaction mixture of Cu–Aβ and H2O2 did not disappear completely (Fig. 3A). Thus, the remaining spin on Cu–Aβ after the reaction is likely due to the presence of unreacted Cu–Aβ or the formation of another paramagnetic species. The EPR parameters of the residual paramagnetic species (S = 1/2, gII = 2.206) (Fig. 3A, C and Table 1) indicate the formation of a distinct species and is not consistent with residual unreacted Cu–Aβ. The new diamagnetic and paramagnetic species observed in absorption and EPR spectroscopy have characteristic vibrations and have been further probed using resonance Raman spectroscopy.
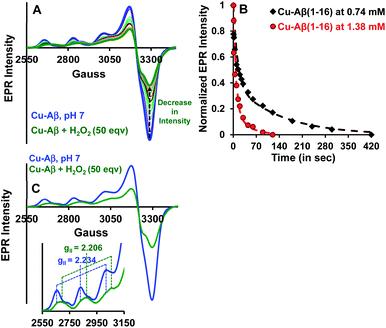 |
| | Fig. 3 (A) EPR spectra of Cu–Aβ, blue; Cu–Aβ with 50 eq. H2O2 at different times, green being the final spectrum after 120 minutes; (B) kinetics of the loss of the EPR signal for 50 eq. H2O2 with 0.74 mM Cu–Aβ, black and 1.38 mM Cu–Aβ, red; (C) EPR spectra of Cu–Aβ, blue; Cu–Aβ with 50 eq. H2O2, green, inset: hyperfine overlay of the data; data are obtained in 100 mM HEPES buffer at pH 7 and 77 K. | |
Table 1 EPR parameters of Cu–Aβ and a Cu–Aβ + 50 eq. H2O2 mixture at pH 7
| Complex |
A
ll
|
g
ll
|
| Cu–Aβ |
173 |
2.234 |
| Cu–Aβ + 50 eq. H2O2 |
166 |
2.206 |
Resonance Raman spectroscopy.
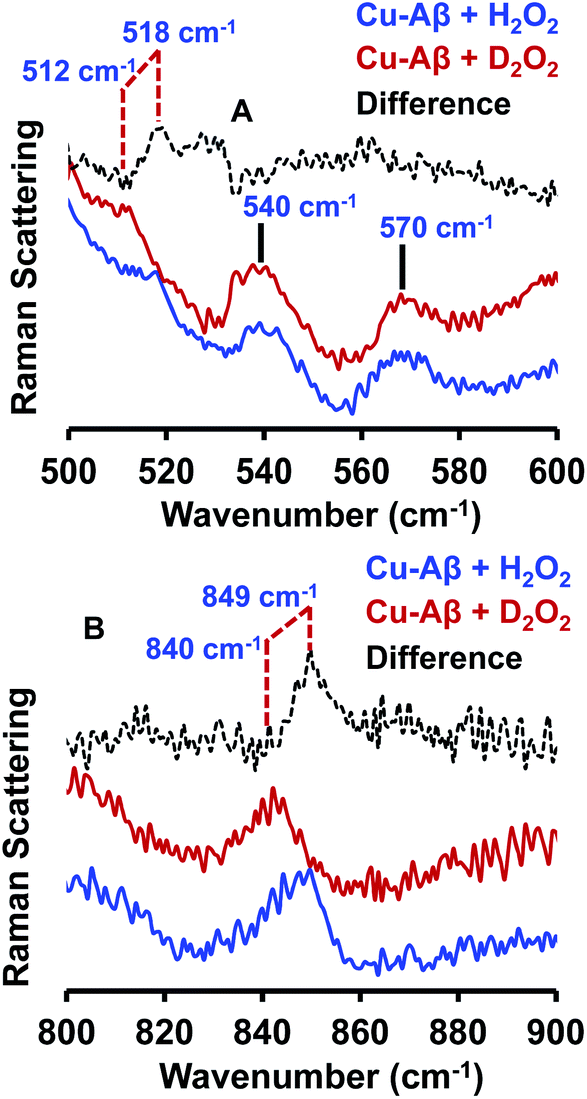
The resonance Raman spectra of the frozen sample of the reaction mixture of Cu–Aβ and H2O2 is obtained using an excitation wavelength of 415.4 nm at 77 K. The reaction mixture of Cu–Aβ and H2O2 exhibits resonance Raman bands at 518, 540, 570 and 849 cm−1 (Fig. 4). Bands in the range of 500–600 cm−1 are characteristic of Cu–O vibrations of either a bis(μ-oxo)copper(III) core or a Cu(II)–OOH species, whereas a band at around 849 cm−1 is characteristic of O–O vibration of a Cu(II)–OOH species.40–42,49–51 Note that the characteristic Cu–O and O–O vibrations of the side-on μ-peroxo-dicopper(II) complex are usually observed at ∼580 and ∼750 cm−1 respectively.41,44 These assignments can be verified by isotopic substitution of the oxygen or proton of H2O2. The reaction mixture of Cu–Aβ and H2O2 in a deuterated medium shows a shift of the bands from 849 and 518 cm−1 to 840 and 512 cm−1 respectively (Fig. 4B). However, the bands at 540 and 570 cm−1 remain unperturbed (Fig. 4A). This H/D shift of the 849 and 518 cm−1 bands is comparable with those reported for several Cu–OOH and Fe–OOH species.51–57 Therefore the 849 cm−1 band can be assigned to the O–O vibration and the 518 cm−1 band can be assigned to the Cu–O vibration of a Cu(II)–OOH species of Cu–Aβ, and represents the new paramagnetic species observed in the EPR spectrum (Fig. 3 and Table 1) when H2O2 is added to Cu–Aβ. The two bands at 540 and 570 cm−1 which do not show any deuterium shift arise from a Fermi resonance (Fig. 4A) and are characteristic Cu–O vibrations of a diamond core bis(μ-oxo) species.45,48,49 Hence, the doublet in the resonance Raman spectra, CT bands at 350 nm and 411 nm and EPR inactivity (S = 0) are all consistent with the formation of a bis(μ-oxo)dicopper(III) species. Thus, Cu–Aβ reacts with H2O2 to generate two different Cu/O2 species; a bis(μ-oxo)dicopper(III) species and a mononuclear Cu(II)–OOH species as shown in Scheme 3. Note that the trans-1,2-peroxo dicooper(II) (Cu–OOCu) could be another possibility but is excluded since the UV-Vis spectrum of Cu–Aβ + H2O2 does not possess any intense band in the range between 480 and 550 nm, which is characteristic of a trans-1,2-peroxo dicooper(II) species.40,41,58,59 Moreover, it has a characteristic O–O vibration in between 800 and 830 cm−1 which is very low as compared to a mononuclear Cu(II)–OOH species, which shows O–O stretch ranging from 830 to 880 cm−1.40,41,51,52 Most importantly the trans-1,2-peroxo species does not show any H/D shift, which is seen in a mononuclear Cu(II)–OOH species.40,41,51–53,59
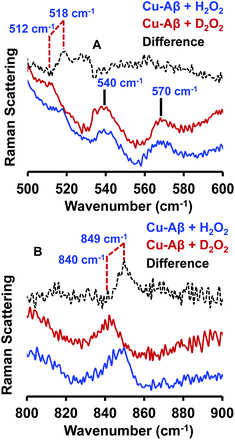 |
| | Fig. 4 Resonance Raman spectra of (A) Cu–Aβ + 50 eq. H2O2, blue and Cu–Aβ + 50 eq. D2O2 in, red; difference spectrum of H2O2 data from D2O2 data, dashed black; lower energy region; (B) Cu–Aβ + 50 eq. H2O2, blue and Cu–Aβ + 50 eq. D2O2 in, red; difference spectrum of H2O2 data from D2O2 data, dashed black; higher energy region. Data were obtained with an excitation wavelength of 415.4 nm (10 mW at the sample) at 77 K (full spectra in Fig. S5†). | |
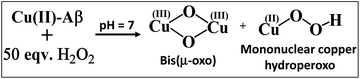 |
| | Scheme 3 Reaction of Cu–Aβ with 50 eq. H2O2 at pH 7. | |
Active oxidant.
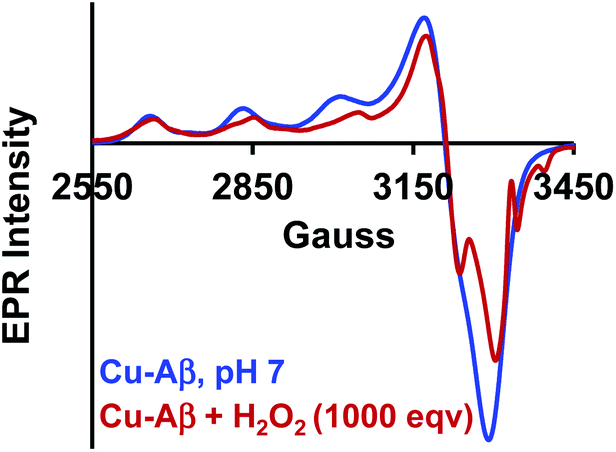
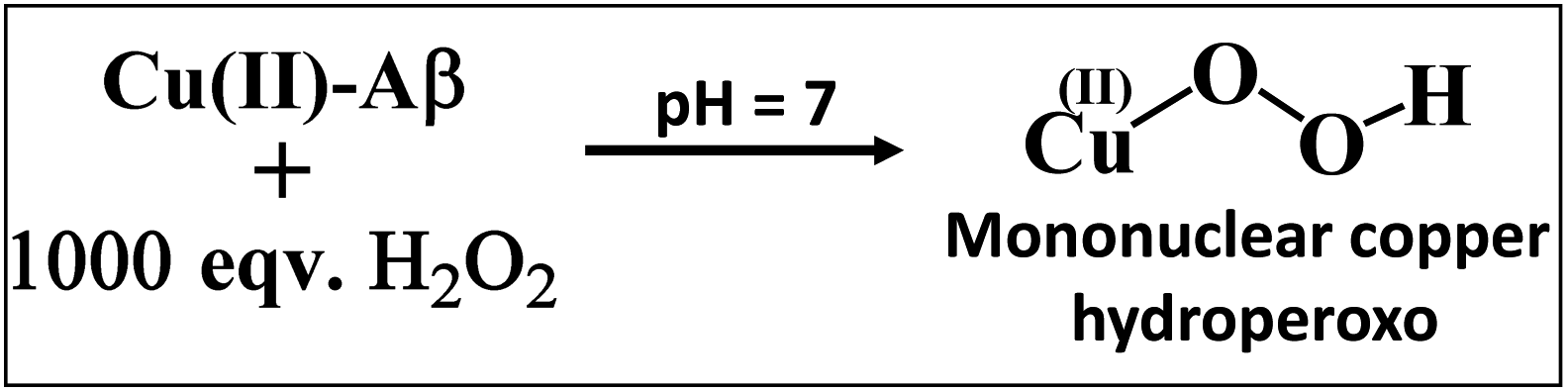
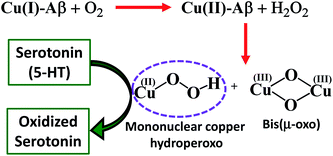
The Cu2O2 bis(μ-oxo)dicopper(III) dimer can be formed by a reaction between two equivalents of Cu–Aβ and one equivalent of H2O2. Alternatively, Cu–Aβ can react with H2O2 to form Cu(II)–OOH which then dimerizes to Cu2O2 bis(μ-oxo)dicopper(III) in the presence of another Cu–Aβ. We find that when Cu–Aβ reacts with an increased amount of H2O2 (1000 equivalents) keeping everything else the same, the EPR spectrum shows that there is no loss of spin during this reaction (Fig. 5) and parallel rR data do not reveal the formation of the dimeric Cu2O2 bis(μ-oxo)dicopper(III) species. Although the O–O vibration of the Cu(II)–OOH species is masked by the O–O vibration of free H2O2 in solution at 890 cm−1, the Cu–O vibration and its shift in D2O are clearly observed (Fig. S6†). Thus under these reaction conditions, Cu(II)–OOH is formed exclusively (Scheme 4). Interestingly, the oxidation of 5-HT continues to occur under these conditions (Fig. S7, S8 and Table S1†). Note that the use of 1000 eq. H2O2 is not physiologically relevant and the purpose of the experiment is to avoid dimerization and to generate mononuclear Cu(II)–OOH exclusively, to evaluate its reactivity towards the substrate. Moreover, the rate of 5-HT oxidation decreases with an increase in Cu–Aβ concentration from 0.74 mM to 1.38 mM (Fig. S9 and Table S1†). Note that the Cu–Aβ concentration dependence on substrate oxidation is consistent with the higher rate of dimerization of Cu(II)–OOH to produce bis-μ-oxo dicopper(III), as observed in EPR spectroscopy (Fig. 3B). These results unambiguously support that Cu(II)–OOH is the active oxidant responsible for the oxidation of 5-HT. This is in contrast to the report by Ming et al., in which they have proposed a side-on μ-peroxo dicopper(II) intermediate as the active oxidant for Cu–Aβ for substrate oxidation in the presence of H2O2.34,36 Our conclusion agrees very well with the mechanism of enzymes like DβM, where the Cu(II)–OOH species or species originating from it are responsible for the chemical oxidation of amino acids and neurotransmitters.60–62
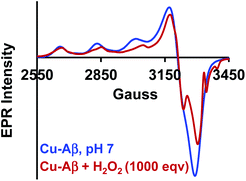 |
| | Fig. 5 EPR spectra of Cu–Aβ, blue; for the reaction of Cu–Aβ with 1000 eq. H2O2, red; in 100 mM HEPES buffer at pH 7, at 77 K. | |
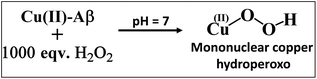 |
| | Scheme 4 Reaction of Cu–Aβ with 1000 eq. H2O2 at pH 7. | |
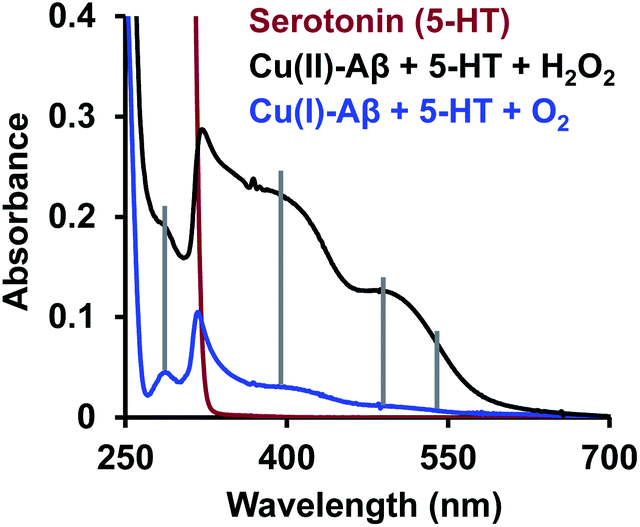
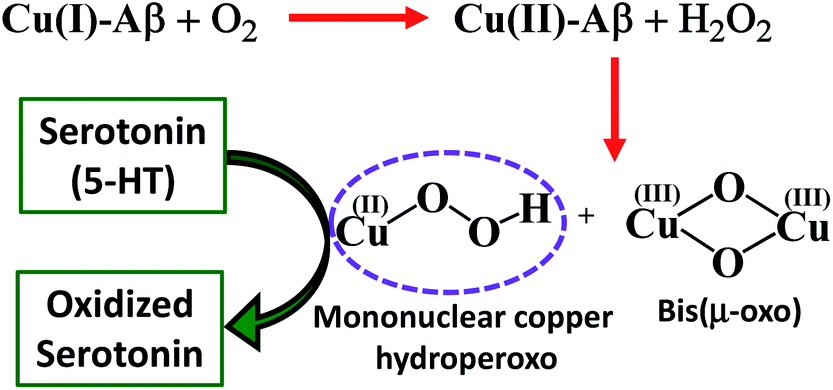
Finally, reduced Cu–Aβ can react with O2, where the Cu gets oxidized and H2O2 is generated via disproportionation of the O2− produced.11 The H2O2 generated from dissolved oxygen in blood can trigger the oxidation of serotonin. This is exactly the same case as indicated by the gradual appearance of the bands corresponding to the oxidized products of 5-HT when incubated with a solution of Cu–Aβ reduced with ascorbic acid in aerated buffer solutions (Fig. 6, blue), albeit the extent of oxidized HT produced is much less under these stoichiometric conditions. Ming et al. have previously observed a similar result as Cu–Aβ significantly accelerated the aerobic oxidation of the neurotransmitters.34Scheme 5 demonstrates the possible routes for the oxidation of serotonin.
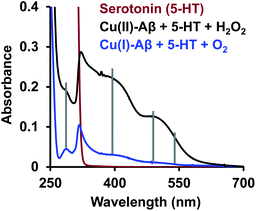 |
| | Fig. 6 Absorption spectra of serotonin (5-HT), red; oxidation of 5-HT by H2O2 catalyzed by Cu(II)–Aβ, black and by O2 catalyzed by Cu(I)–Aβ, blue; in 100 mM HEPES buffer at pH 7. | |
 |
| | Scheme 5 Serotonin oxidation catalyzed by a reactive oxidant. | |
Conclusion
Cu–Aβ oxidizes the neurotransmitter serotonin (5-HT) in the presence of H2O2. The combined absorption, EPR and resonance Raman data indicate that Cu(II)–Aβ reacts with H2O2 to produce bis(μ-oxo)dicopper and mononuclear copper hydroperoxo intermediates. This is the first experimental characterization of the active oxidants originating from Cu bound Aβ that can oxidize neurotransmitters like serotonin (5-HT) and generate neurotoxins like tryptamine-4,5-dione, which are also observed in an AD brain. Cu(II)–Aβ can be reduced by physiologically relevant reductants like ascorbic acid and this reduced Cu center can generate H2O2 by reacting with dissolved oxygen, which can then oxidize 5-HT catalyzed by the Cu(II)–Aβ produced. Both these pathways are accessible under physiological conditions and may account for the abnormal neurotransmission, a key pathological feature of AD.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank DST, SERB India for financial support (grants EMR/2014/000392 and EMR/2015/0008063). A. K. N. and A. G. are funded by the University Grants Commission Fellowship. The authors thank CSS and IACS for providing HPLC facility.
Notes and references
- T. Lührs, C. Ritter, M. Adrian, D. Riek-Loher, B. Bohrmann, H. Döbeli, D. Schubert and R. Riek, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17342 CrossRef.
- J. J. Balbach, A. T. Petkova, N. A. Oyler, O. N. Antzutkin, D. J. Gordon, S. C. Meredith and R. Tycko, Biophys. J., 2002, 83, 1205–1216 CrossRef CAS.
- C. Schmidt, M. Wolff, M. Weitz, T. Bartlau, C. Korth and I. Zerr, Arch. Neurol., 2011, 68, 1124–1130 CrossRef.
- J. Hardy and D. J. Selkoe, Science, 2002, 297, 353 CrossRef CAS.
- F. Kametani and M. Hasegawa, Front. Neurosci., 2018, 12, 25 CrossRef.
- L. M. F. Gomes, J. C. Bataglioli and T. Storr, Coord. Chem. Rev., 2020, 412, 213255 CrossRef CAS.
- A. Rauk, Chem. Soc. Rev., 2009, 38, 2698–2715 RSC.
- S. Bagheri, R. Squitti, T. Haertlé, M. Siotto and A. A. Saboury, Front. Aging Neurosci., 2018, 9, 446 CrossRef.
- C. Garza, Y. Posadas, L. Quintanar, M. Gonsebatt and R. Franco, Antioxid. Redox Signaling, 2018, 28, 1669–1703 CrossRef.
- C. Cheignon, M. Tomas, D. Bonnefont-Rousselot, P. Faller, C. Hureau and F. Collin, Redox Biol., 2018, 14, 450–464 CrossRef CAS.
- D. Pramanik and S. G. Dey, J. Am. Chem. Soc., 2011, 133, 81–87 CrossRef CAS.
- D. Pramanik, C. Ghosh and S. G. Dey, J. Am. Chem. Soc., 2011, 133, 15545–15552 CrossRef CAS.
- C. Ghosh and S. G. Dey, Inorg. Chem., 2013, 52, 1318–1327 CrossRef CAS.
- I. Pal, M. Roy and S. G. Dey, JBIC, J. Biol. Inorg. Chem., 2019, 24, 1245–1259 CrossRef CAS.
- S. C. Drew, C. J. Noble, C. L. Masters, G. R. Hanson and K. J. Barnham, J. Am. Chem. Soc., 2009, 131, 1195–1207 CrossRef CAS.
- P. Dorlet, S. Gambarelli, P. Faller and C. Hureau, Angew. Chem., Int. Ed., 2009, 48, 9273–9276 CrossRef CAS.
- B.-k. Shin and S. Saxena, Biochemistry, 2008, 47, 9117–9123 CrossRef CAS.
- S. Furlan, C. Hureau, P. Faller and G. La Penna, J. Phys. Chem. B, 2012, 116, 11899–11910 CrossRef CAS.
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659–3853 CrossRef CAS.
- R. H. Holm, P. Kennepohl and E. I. Solomon, Chem. Rev., 1996, 96, 2239–2314 CrossRef CAS.
- J. P. Klinman, Chem. Rev., 1996, 96, 2541–2562 CrossRef CAS.
- A. Beliaev, H. Ferreira, D. A. Learmonth and P. Soares-da-Silva, Curr. Enzyme Inhib., 2009, 5, 27–43 CrossRef.
- J. P. Evans, K. Ahn and J. P. Klinman, J. Biol. Chem., 2003, 278, 49691–49698 CrossRef CAS.
- J. P. Klinman, J. Biol. Chem., 2006, 281, 3013–3016 CrossRef CAS.
- R. Kandimalla and P. H. Reddy, J. Alzheimer's Dis., 2017, 57, 1049–1069 CAS.
- D. M. Bowen, AGE, 1988, 11, 104–109 CrossRef.
- S. A. Lyness, C. Zarow and H. C. Chui, Neurobiol. Aging, 2003, 24, 1–23 CrossRef CAS.
- S. G. Snowden, A. A. Ebshiana, A. Hye, O. Pletnikova, R. O'Brien, A. Yang, J. Troncoso, C. Legido-Quigley and M. Thambisetty, J. Alzheimer's Dis., 2019, 72, 35–43 CAS.
- H. Förstl and P. Fischer, Eur. Arch. Psychiatr. Clin. Neurosci., 1994, 244, 252–260 CrossRef.
- S. Kaur, G. DasGupta and S. Singh, Neurophysiology, 2019, 51, 293–309 CrossRef CAS.
- M. Z. Wrona and G. Dryhurst, Chem. Res. Toxicol., 1998, 11, 639–650 Search PubMed.
- L. Volicer and P. B. Crino, Neurobiol. Aging, 1990, 11, 567–571 CrossRef CAS.
- P. B. Crino, B. A. Vogt, J.-C. Chen and L. Volicer, Brain Res., 1989, 504, 247–257 CrossRef CAS.
- G. F. Z. da Silva and L.-J. Ming, Angew. Chem., Int. Ed., 2007, 46, 3337–3341 CrossRef CAS.
- G. F. Z. da Silva and L.-J. Ming, Angew. Chem., Int. Ed., 2005, 44, 5501–5504 CrossRef CAS.
- G. F. da Silva, V. Lykourinou, A. Angerhofer and L. J. Ming, Biochim. Biophys. Acta, 2009, 1792, 49–55 CrossRef CAS.
- G. F. da Silva, W. M. Tay and L. J. Ming, J. Biol. Chem., 2005, 280, 16601–16609 CrossRef CAS.
- M. Z. Wrona, Z. Yang, M. McAdams, S. O'Connor-Coates and G. Dryhurst, J. Neurochem., 1995, 64, 1390–1400 CrossRef CAS.
- V. F. Ximenes, G. J. Maghzal, R. Turner, Y. Kato, C. C. Winterbourn and A. J. Kettle, Biochem. J., 2009, 425, 285–293 CrossRef.
- C. E. Elwell, N. L. Gagnon, B. D. Neisen, D. Dhar, A. D. Spaeth, G. M. Yee and W. B. Tolman, Chem. Rev., 2017, 117, 2059–2107 CrossRef CAS.
- L. M. Mirica, X. Ottenwaelder and T. D. P. Stack, Chem. Rev., 2004, 104, 1013–1046 CrossRef CAS.
- E. I. Solomon, P. Chen, M. Metz, S.-K. Lee and A. E. Palmer, Angew. Chem., Int. Ed., 2001, 40, 4570–4590 CrossRef CAS.
- F. Nastri, M. Chino, O. Maglio, A. Bhagi-Damodaran, Y. Lu and A. Lombardi, Chem. Soc. Rev., 2016, 45, 5020–5054 RSC.
- J. A. Halfen, S. Mahapatra, E. C. Wilkinson, S. Kaderli, V. G. Young, L. Que, A. D. Zuberbühler and W. B. Tolman, Science, 1996, 271, 1397 CrossRef CAS.
- M. J. Henson, M. A. Vance, C. X. Zhang, H.-C. Liang, K. D. Karlin and E. I. Solomon, J. Am. Chem. Soc., 2003, 125, 5186–5192 CrossRef CAS.
- S. Mahapatra, J. A. Halfen, E. C. Wilkinson, G. Pan, C. J. Cramer, L. Que Jr and W. B. Tolman, J. Am. Chem. Soc., 1995, 117, 8865–8866 CrossRef CAS.
- E. I. Solomon, J. W. Ginsbach, D. E. Heppner, M. T. Kieber-Emmons, C. H. Kjaergaard, P. J. Smeets, L. Tian and J. S. Woertink, Faraday Discuss., 2011, 148, 11–108 RSC.
- D. J. E. Spencer, A. M. Reynolds, P. L. Holland, B. A. Jazdzewski, C. Duboc-Toia, L. Le Pape, S. Yokota, Y. Tachi, S. Itoh and W. B. Tolman, Inorg. Chem., 2002, 41, 6307–6321 CrossRef CAS.
- P. L. Holland, C. J. Cramer, E. C. Wilkinson, S. Mahapatra, K. R. Rodgers, S. Itoh, M. Taki, S. Fukuzumi, L. Que and W. B. Tolman, J. Am. Chem. Soc., 2000, 122, 792–802 CrossRef CAS.
- N. Lehnert, K. Fujisawa and E. I. Solomon, Inorg. Chem., 2003, 42, 469–481 CrossRef CAS.
- Y. J. Choi, K.-B. Cho, M. Kubo, T. Ogura, K. D. Karlin, J. Cho and W. Nam, Dalton Trans., 2011, 40, 2234–2241 RSC.
- P. Chen, K. Fujisawa and E. I. Solomon, J. Am. Chem. Soc., 2000, 122, 10177–10193 CrossRef CAS.
- B. Kim, D. Jeong, T. Ohta and J. Cho, Commun. Chem., 2019, 2, 81 CrossRef.
- P. J. Mak, W. Thammawichai, D. Wiedenhoeft and J. R. Kincaid, J. Am. Chem. Soc., 2015, 137, 349–361 CrossRef CAS.
- N. Lehnert, F. Neese, R. Y. N. Ho, L. Que and E. I. Solomon, J. Am. Chem. Soc., 2002, 124, 10810–10822 CrossRef CAS.
- R. Y. N. Ho, G. Roelfes, B. L. Feringa and L. Que, J. Am. Chem. Soc., 1999, 121, 264–265 CrossRef CAS.
- A. Ghatak, S. Bhunia and A. Dey, ACS Catal., 2020, 10, 13136–13148 CrossRef CAS.
- C. Citek, S. Herres-Pawlis and T. D. P. Stack, Acc. Chem. Res., 2015, 48, 2424–2433 CrossRef CAS.
- M. T. Kieber-Emmons, J. W. Ginsbach, P. K. Wick, H. R. Lucas, M. E. Helton, B. Lucchese, M. Suzuki, A. D. Zuberbühler, K. D. Karlin and E. I. Solomon, Angew. Chem., Int. Ed., 2014, 53, 4935–4939 CrossRef CAS.
- P. Chen and E. I. Solomon, J. Am. Chem. Soc., 2004, 126, 4991–5000 CrossRef CAS.
- A. Crespo, M. A. Martí, A. E. Roitberg, L. M. Amzel and D. A. Estrin, J. Am. Chem. Soc., 2006, 128, 12817–12828 CrossRef CAS.
- P. Chen and E. I. Solomon, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 13105–13110 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc06258h |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  * and
Somdatta Ghosh
Dey
* and
Somdatta Ghosh
Dey

 . CT bands in these regions hint at the plausible formation of either a side-on μ-peroxo-dicopper(II) complex or a bis(μ-oxo)dicopper(III) complex as depicted in Scheme 2.41,42,44–46 Previously Ming and coworkers proposed the formation of a side-on μ-peroxo dicopper(II) intermediate.34,36
. CT bands in these regions hint at the plausible formation of either a side-on μ-peroxo-dicopper(II) complex or a bis(μ-oxo)dicopper(III) complex as depicted in Scheme 2.41,42,44–46 Previously Ming and coworkers proposed the formation of a side-on μ-peroxo dicopper(II) intermediate.34,36