
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of N-aryl amines enabled by photocatalytic dehydrogenation†
Jungwon
Kim
 *a,
Siin
Kim
abc,
Geunho
Choi
a,
Geun Seok
Lee
a,
Donghyeok
Kim
abc,
Jungkweon
Choi
abc,
Hyotcherl
Ihee
*abc and
Soon Hyeok
Hong
*a
*a,
Siin
Kim
abc,
Geunho
Choi
a,
Geun Seok
Lee
a,
Donghyeok
Kim
abc,
Jungkweon
Choi
abc,
Hyotcherl
Ihee
*abc and
Soon Hyeok
Hong
*a
aDepartment of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea. E-mail: kikj11@snu.ac.kr; hyotcherl.ihee@kaist.ac.kr; soonhyeok.hong@kaist.ac.kr
bKI for the BioCentury, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea
cCenter for Nanomaterials and Chemical Reactions, Institute for Basic Science, Daejeon 34141, Republic of Korea
First published on 8th December 2020
Abstract
Catalytic dehydrogenation (CD) via visible-light photoredox catalysis provides an efficient route for the synthesis of aromatic compounds. However, access to N-aryl amines, which are widely utilized synthetic moieties, via visible-light-induced CD remains a significant challenge, because of the difficulty in controlling the reactivity of amines under photocatalytic conditions. Here, the visible-light-induced photocatalytic synthesis of N-aryl amines was achieved by the CD of allylic amines. The unusual strategy using C6F5I as an hydrogen-atom acceptor enables the mild and controlled CD of amines bearing various functional groups and activated C–H bonds, suppressing side-reaction of the reactive N-aryl amine products. Thorough mechanistic studies suggest the involvement of single-electron and hydrogen-atom transfers in a well-defined order to provide a synergistic effect in the control of the reactivity. Notably, the back-electron transfer process prevents the desired product from further reacting under oxidative conditions.
Introduction
Catalytic dehydrogenation (CD) offers a useful strategy for constructing unsaturated systems from readily available sp3 carbon-rich scaffolds.1 For example, the CD of hydrocarbons provides valuable olefin feedstocks,1c and alcohol dehydrogenation can promote the formation of carbonyl functional groups.1a By exploiting the thermodynamic preference towards aromatization, a number of transformations to obtain various aromatic compounds have been developed.2 Nevertheless, despite the significant advances in the CD process, the requirement for harsh reaction conditions, such as high reaction temperatures and the use of strong oxidants still limits the practical application of the strategy.Utilization of visible-light in organic transformations via photoredox catalysis has led to novel and practical synthesis of target molecules under relatively mild reaction conditions.3 Visible-light photoredox catalysis has also been applied to CD reactions (Scheme 1).4 Since the initial development of a photocatalyst and the use of an external oxidant such as O2,4atert-butyl peroxybenzoate,4b and sulfonyl chloride,4h the combination of a visible-light photocatalyst (Ir, Ru, organocatalyst) and a hydrogen-evolution metal catalyst (Co, Pd) has afforded the aromatization of saturated N-heterocycles such as tetrahydroquinoline and indoline via an acceptorless CD process to efficiently produce the corresponding aromatic heterocycles (Scheme 1A).4c–f,i–k,m Further elaboration of CD strategies using an additional hydrogen-atom transfer (HAT) catalyst enabled direct access to more challenging substrates. For example, Kanai designed a triple hybrid catalysis (photoredox, HAT, and metal catalysts) to achieve the acceptorless photoredox CD of tetrahydronaphthalene derivatives (Scheme 1A).4e,g Very recently, the developed strategies were applied to CD of acyclic alcohols or secondary amines, providing carbonyl or imine compounds.4l,n,o
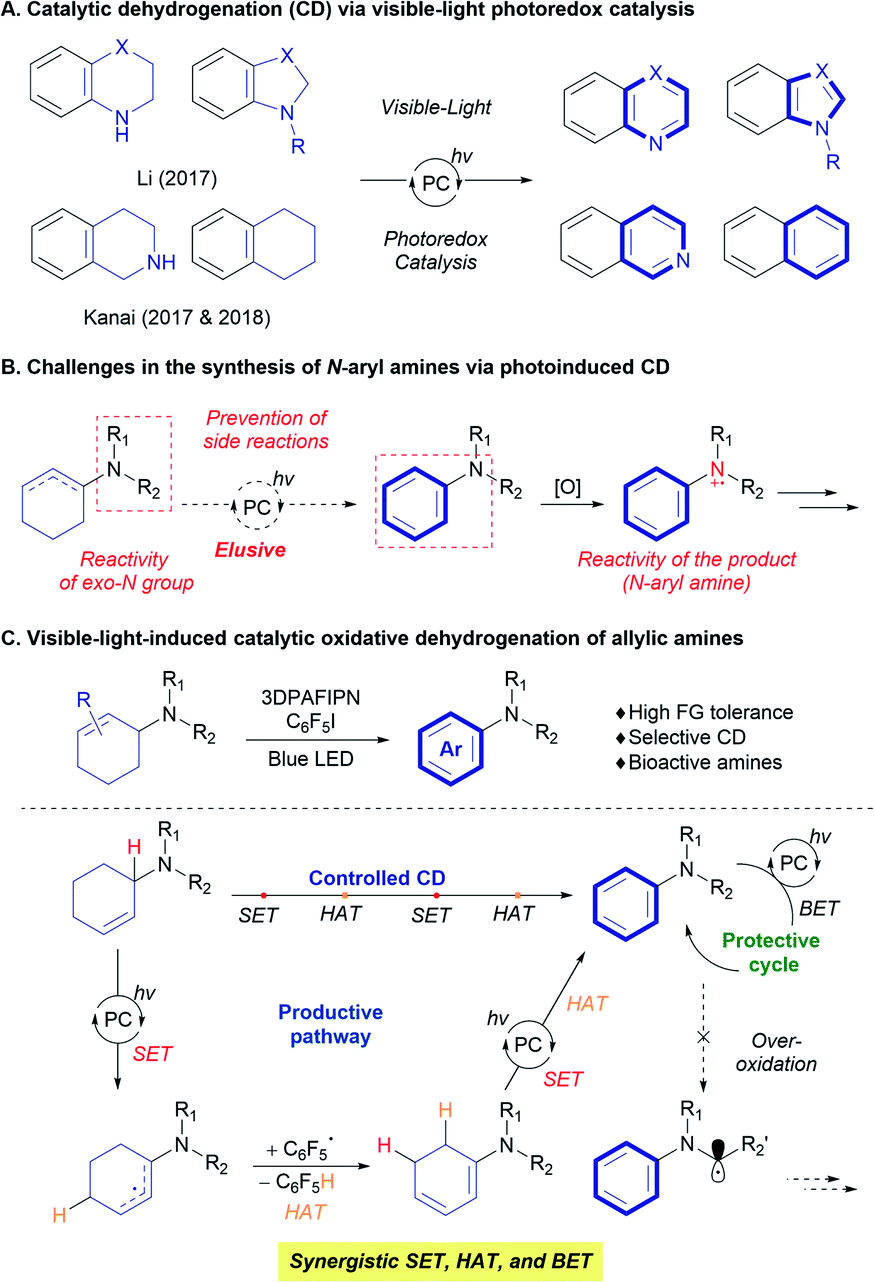 | ||
| Scheme 1 Visible-light-induced catalytic dehydrogenation. | ||
Despite these advances, the synthesis of functionalized benzenes, such as N-aryl amines, has been challenging, despite the widespread use of these moieties in organic, materials, and biological chemistries. Direct access to N-aryl amines via visible-light photocatalytic dehydrogenation is difficult (Scheme 1B) because the strategy previously applied in photocatalytic dehydrogenation relies on the single-electron transfer (SET) of a nitrogen atom to produce reactive amine radical cation species.5 Saturated N-heterocyclic systems, which contain nitrogen atoms inside the ring system, allow control of the reaction to promote the desired dehydrogenation; however, systems with an exo-nitrogen functionality are more prone to unwanted side-reactions (Scheme 1B).6 These possibilities raise the question of how the reactivity of the exo-nitrogen atom is transferred to the target cyclic system to selectively achieve the desired dehydrogenation reaction. Moreover, the produced N-aryl amine would become a competing and more reactive substrate in the course of the reaction. This has the potential to transform N-aryl amine products into unwanted α-amino radical, iminium, and enamine species under visible-light photoredox catalysis (Scheme 1B).5,6 Very recently, Leonori and co-workers successfully overcame the above-mentioned hurdles via photocatalytic dehydrogenation of in situ generated enamines with photoredox/cobalt dual catalytic system.7
Herein, we demonstrate the synthesis of N-aryl amines from a 2-cyclohexenyl amine precursor via visible-light photoredox CD (Scheme 1C). The use of perfluoroarene as a hydrogen-atom acceptor is key to achieving selective aromatization on the cyclohexenyl moiety, and wide ranges of functional groups and bioactive motifs are tolerated under the reaction conditions. The operating mechanism was revealed to involve the synergistic relay of SET and HAT processes mediated by the organic photoredox catalyst and a pentafluorophenyl radical, in combination with the protective back-electron transfer (BET) process, based on the nature of the amine substrates.
Results and discussion
The 2-cyclohexenyl amine A1 was initially selected as a model substrate to explore the target reaction. After extensive screening (Tables S1–S5†), 3DPAFIPN was identified as the photocatalyst, iodopentafluorobenzene (C6F5I) as the hydrogen-atom acceptor, and K2CO3 as the base to induce the desired oxidative aromatization in high yield (95%). It is noteworthy that no imine compounds derived from either A1 or B1 were observed, although the CD of secondary amines under visible-light photoredox catalytic conditions was reported in the synthesis of imines.4o Further variation of the standard reaction conditions was implemented to check the effect of each component (Table 1). Changing the photocatalyst to a well-known iridium photocatalyst ([Ir(dF(CF3)ppy)2(dtbbpy)](PF6)) significantly decreased the reactivity (52%, entry 2), and photocatalyst-free conditions yielded only a small amount of the desired product (4%, entry 10). Replacing C6F5I with C6F5Br (entry 3) completely shut down the reaction, although it was previously reported that C6F5Br can be converted to a pentafluorophenyl radical under visible-light irradiation in the presence of Eosin Y.8 In addition, no pentafluorophenylation of the aniline aromatic ring as a side reaction was observed in all entries. The developed reaction is highly sensitive to the solvent (entries 4–6, Table S3†), and dichloromethane can only be replaced with 1,2-dichloroethane (1,2-DCE) without loss of the reactivity (91%, entry 6). Potassium carbonate was essential for obtaining a high yield of the product (entries 7, 8, 12), and a control experiment under dark conditions (entry 13) proved that the reaction proceeded under visible-light irradiation.|
|
||
|---|---|---|
| Entry | Variation of reaction conditions | Yield B1b (%) |
| a Reaction conditions: A1 (0.2 mmol), 3DPAFIPN (0.012 mmol), C6F5I (0.8 mmol), K2CO3 (0.6 mmol), CH2Cl2 (1 mL) in a 4 mL reaction vial, irradiated with a 34 W Kessil blue LED for 24 h under fan cooling. b Determined by GC using dodecane as an internal standard. THF = tetrahydrofuran. N. D. = not detected. | ||
| 1 | None (standard) | 95 |
| 2 | [Ir(dF(CF3)ppy)2(dtbbpy)](PF6) instead of 3DPAFIPN | 52 |
| 3 | C6F5Br instead of C6F5I | N. D. |
| 4 | CH3CN instead of CH2Cl2 | 3 |
| 5 | THF instead of CH2Cl2 | 12 |
| 6 | 1,2-DCE instead of CH2Cl2 | 91 |
| 7 | K3PO4 instead of K2CO3 | 4 |
| 8 | Triethylamine instead of K2CO3 | N. D. |
| 9 | 18 h reaction time | 49 |
| 10 | No 3DPAFIPN | 4 |
| 11 | No C6F5I | N. D. |
| 12 | No K2CO3 | 32 |
| 13 | Dark condition | N. D. |

With the optimal conditions in hand, the substrate scope of the reaction was evaluated (Table 2). Several benzylamine derivatives were initially tested for this reaction. Notably, various functional groups, including halides (B3, B4, B8), ester (77%, B5), trifluoromethyl (61%, B6), and boronate ester (44%, B7), were tolerated under the reaction conditions and gave the desired N-aryl amines in good yields. A piperonylamine derivative (77%, B9) and other heteroaromatic methyl amine derivatives (85%, B10 and 32%, B11) were reactive under the reaction conditions. Other secondary amine precursors, such as α-methyl benzylamine (84%, B12), linear (70%, B14), or cyclic (70%, B15) aliphatic amines, and even a sterically bulky 1-adamantylamine derivative (74%, B16) could be applied in the reaction. Notably, the cyclohexyl ring of B15 remains intact under the dehydrogenative conditions, implying the necessity of an allylic α-amino C–H bond for the desired reaction. The efficiency of the reaction was maintained even with different types of tertiary allyl amines (B17–B21). To our delight, no oxidation at the benzyl,4o,9 tetrahydronaphthyl,4e,g,7 or α-amino10 positions was observed (e.g., reactions with A22 or A23, highlighted in orange), demonstrating the advantage of the mild reaction conditions compared to previously reported oxidative aromatization conditions.
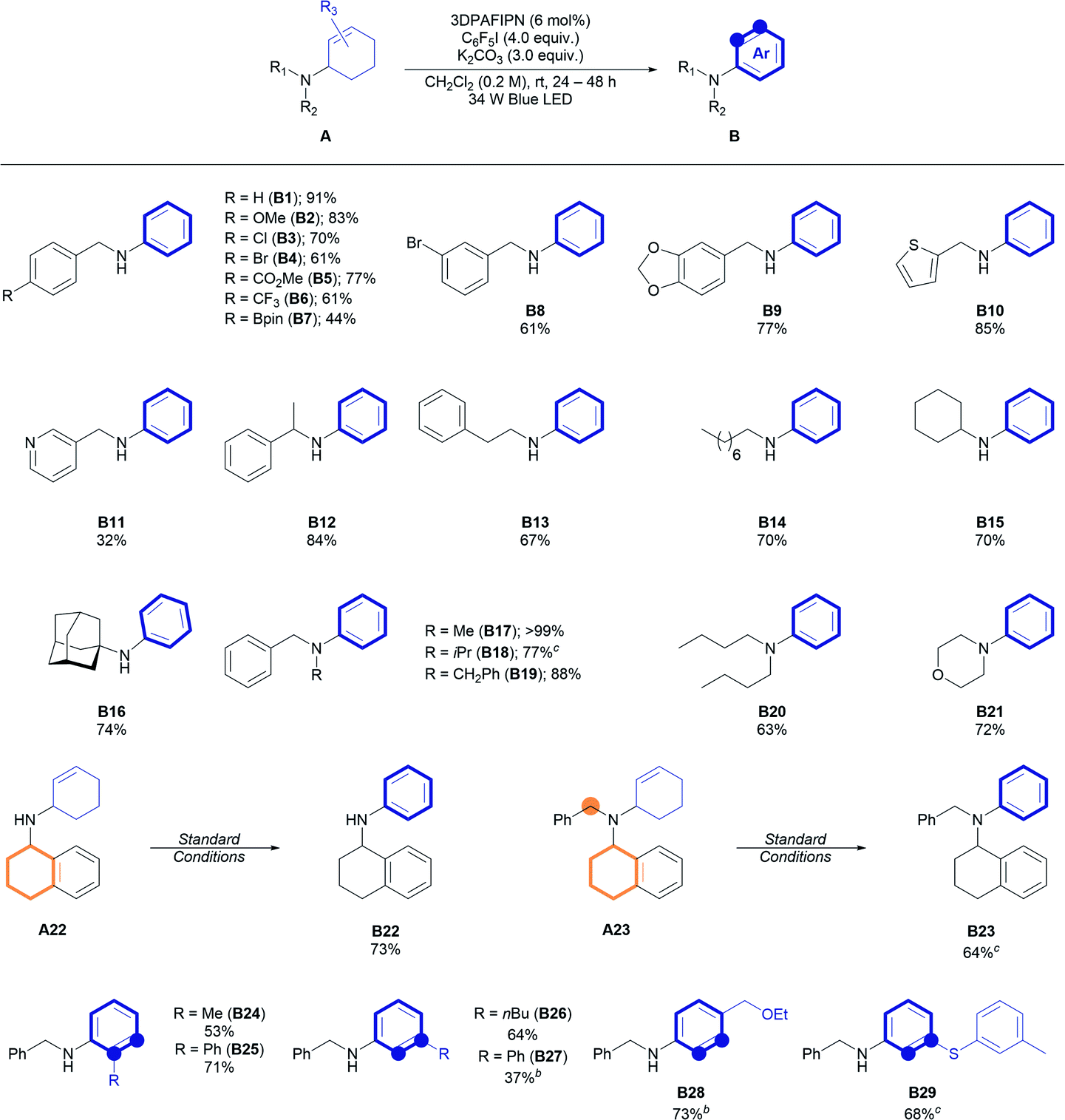
The cyclohexenyl scaffold was then varied and the synthesis of ortho-/meta-/para-substituted N-aryl amines was achieved using the developed method, albeit with a reduced yield of the product in some cases (B24–B29). It is noteworthy that sulfide (A29) is compatible with the reaction, producing a sulfur-containing N-aryl amine in good yield (68%, B29), considering that sulfides are known to be readily oxidizable under oxidation conditions and tend to inhibit transition metal-mediated CD.11
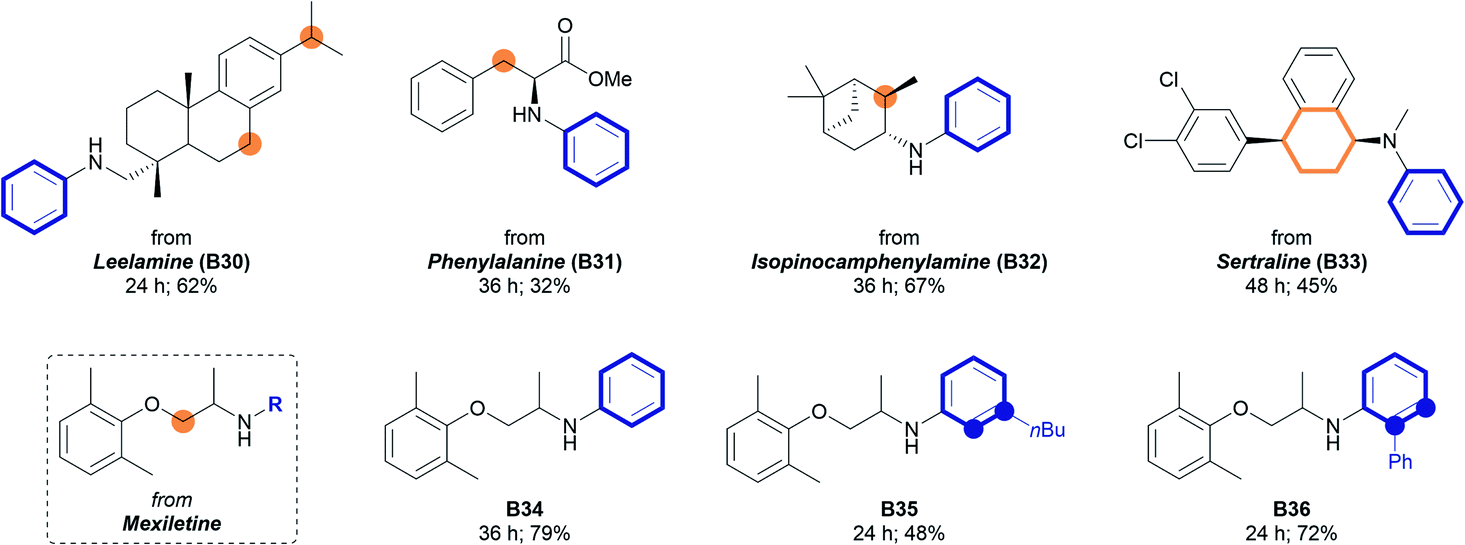
The developed method was further applied to bioactive amines (Table 3). Amines containing polycyclic structures, amino acid moieties, and terpenoid structures all remained intact to form corresponding N-aryl amines in moderate to good yields. Various types of reactive positions, such as benzylic (62%, B30), tertiary (67%, B32), and α-oxygen carbons (79%, B34), were tolerated under the developed reaction conditions. When the Sertraline derivative (A33) containing both cyclohexenyl and tetrahydronaphthyl groups was used, selective aromatization of the cyclohexenyl group was achieved to form the corresponding N-aryl amine (45%, B33) with the tetrahydronaphthyl group remaining intact. This unique chemoselectivity may be beneficial for the synthesis of bioactive N-aryl amines while maintaining a specific structure. Further elaborations with different cyclohexenyl moieties were conducted using Mexiletine as a starting amine. As depicted in Table 3, the introduction of a simple phenyl group (B34) and substituted phenyl groups (48% for B35, 72% for B36) was realized under the standard reaction conditions with good efficiency.
| a Reaction conditions: A (0.2 mmol), 3DPAFIPN (0.012 mmol), C6F5I (0.8 mmol), K2CO3 (0.6 mmol), CH2Cl2 (1 mL) in a 4 mL reaction vial, irradiated with a 34 W Kessil blue LED for the indicated time under fan cooling. Yields of the isolated products were described. |
|---|
|
The developed protocol for photocatalytic dehydrogenation provides a high degree of reactivity and selectivity for a wide range of allylic amines. This raises fundamental questions regarding the mechanism of the reaction (Fig. 1). First, the origin of the selective dehydrogenation leading to aromatization needs to be addressed to understand the underlying principle of the reaction. Several reactions with A containing a reactive benzylic position did not show any sign of activation of the benzylic position, which is in clear contrast to the reported functionalizations of benzylic amines.5,12 In addition, the role of C6F5I as a hydrogen-atom acceptor in the reaction must be further investigated. Lastly, the preservation of the synthesized N-aryl amines under photocatalytic conditions (Eoxp (B1) = 0.88 V vs. SCE in CH3CN)13 is the most noticeable aspect of the reaction, which enables controlled dehydrogenation with high efficiency.
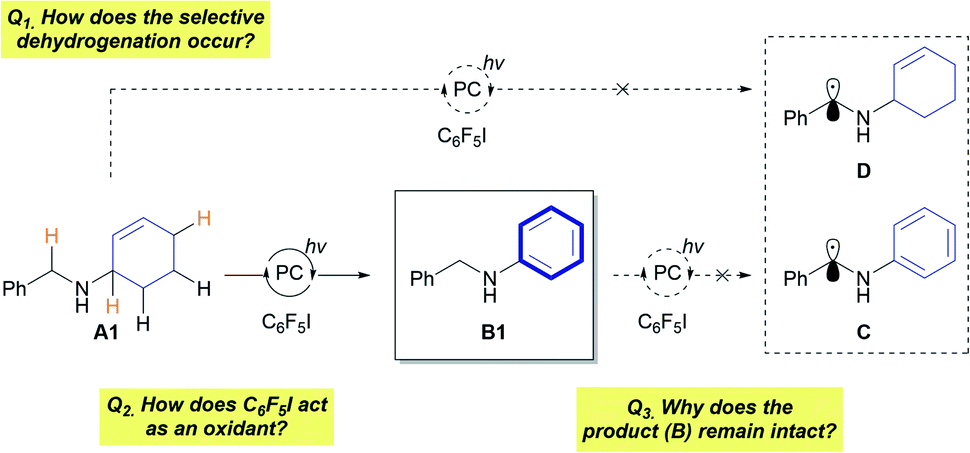 | ||
| Fig. 1 Major mechanistic questions regarding the reaction. | ||
To answer the questions about the mechanism of the developed reaction, a number of control experiments were conducted (Table 4). The reactions with C6F5Br as the sole reagent did not provide the desired amine B1 (entry 2; Table 1, entry 3), although C6F5Br can provide a C6F5 radical and promote perfluoroarylation under visible-light photocatalytic conditions.8 Noticeably, the existence of both C6F5I and C6F5Br exhibited a comparable reactivity, and the conversion of only C6F5I to C6F5H was observed by 19F NMR spectroscopy (72%, entry 3, see Fig. S2†). The effect of the counteranion was not observed in either case (Table 4, entries 4 and 5), suggesting that halide anions did not affect the reactivity. CV measurements revealed that the single-electron reduction of C6F5I (Eredp = −1.36 V vs. SCE in CH3CN) is more facile than that of C6F5Br (Eredp = −1.64 V vs. SCE in CH3CN),14 which may account for the difference in the reactivity of the two C6F5 radical precursors. The addition of a radical scavenger ((2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) or galvinoxyl) unambiguously hampered the reaction, indicating the involvement of radical intermediates in the reaction (Table 4, entries 6 and 7).
|
|
||
|---|---|---|
| Entry | Additive | Yield B1b (%) |
| a Reaction conditions: A1 (0.2 mmol), 3DPAFIPN (0.012 mmol), additive, K2CO3 (0.6 mmol), CH2Cl2 (1 mL) in a 4 mL reaction vial, irradiated with a 34 W Kessil blue LED for 24 h under fan cooling. b Measured by GC using dodecane as an internal standard. | ||
| 1 | C6F5I (4.0 equiv.) | 95 |
| 2 | C6F5Br (4.0 equiv.) | N. D. |
| 3 | C6F5I (4.0 equiv.) + C6F5Br (4.0 equiv.) | 72 |
| 4 | C6F5I (4.0 equiv.) + KBr (3.0 equiv.) | 82 |
| 5 | C6F5Br (4.0 equiv.) + nBu4NI (3.0 equiv.) | N. D. |
| 6 | C6F5I (4.0 equiv.) + TEMPO (1.0 equiv.) | <2% |
| 7 | C6F5I (4.0 equiv.) + galvinoxyl (1.0 equiv.) | N. D. |
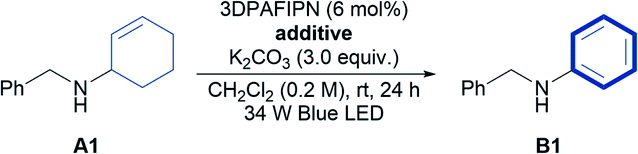
Evaluation of the effect of 3DPAFIPN (Table 1, entries 1 and 10) indicated the formation of a radical intermediate by photoexcited 3DPAFIPN. Stern–Volmer quenching experiments with A1 and C6F5I (Fig. S6†) provided insight into the reductive quenching process (Scheme 2A, left). In addition, the involvement of HAT of the C6F5 radical was substantiated by the observation of C6F5H after the reaction (Fig. S1†). However, the detailed aspects of HAT should be discussed by considering a number of possible scenarios. In particular, the target of HAT by the C6F5 radical should be addressed. First, it is possible for the reactive C6F5 radical to abstract activated hydrogen-atom from the neutral amine (A) (Scheme 2A, right). When amine A1 deuterated at the allylic position (A1-D) was applied in the reaction, only a trace amount of C6F5D was observed in the reaction mixture (Scheme 2B, Fig. S7 and S8†), which is in clear contrast with the results of significant deuterium incorporation in the hydrodefluorination of perfluoroarene with an α-deuterated aliphatic amine.15 This result strongly suggests that the direct HAT of A1 by the C6F5 radical is not operative, considering that HAT with A1 would occur at an allylic α-amino C–H bond due to the cumulative stabilization effect of the nitrogen atom and the olefin (Scheme S1†). Next, the standard reaction was conducted with A1 using CD2Cl2 as the solvent (Scheme 2C). Notably, no C6F5D was detected by 2H NMR spectroscopy, suggesting that the formation of a dichloromethyl radical by HAT between the C6F5 radical and CH2Cl2 does not occur in the reaction mixture.16
 | ||
| Scheme 2 Mechanistic studies of HAT process. | ||
From the experimental observations, together with the reductive quenching between 3DPAFIPN and A1, it is expected that the generated amine radical cation (A1-radcat) would expel the allylic proton (deprotonation) to generate the α-amino radical, which undergoes HAT at the C4 position with the C6F5 radical to generate the cyclic dienamine intermediate (A1-diene) (Scheme 2D). The involvement of A1-radcat as an intermediate would result in selective functionalization of the allylic structures, which can provide a more stable allylic α-amino radical (A1-rad) than the radical intermediate (A1-rad′) derived from deprotonation of the benzylic position. The thermodynamic preference of A1-rad to A1-rad′ in 3 kcal mol−1 would result in a >150 times higher concentration of A1-rad in equilibrium. The same trend is expected in the corresponding transition states of the deprotonation steps.17 The observed solvent effect in Table 1 could be derived from the thermodynamically preferred deprotonation, which is favoured in dichloromethane, as recently reported by Liu and Ready.17 The formation of double bonds via HAT in radical species has been demonstrated with a Co complex,18 a tert-butoxy radical,4b and TEMPO.19 Because the reactive C6F5 radical can be generated only after the initial reductive quenching of the photoexcited 3DPAFIPN by A1, such a HAT event occurs in a highly selective manner between the C6F5 radical and A1-rad. This order of the elementary steps would be key to driving an array of oxidation steps toward the desired aromatization process. HAT at the C6 position, which provides a different type of diene (A1-diene′), is a thermodynamically less favoured pathway (Scheme S2†), and SET between the C6F5 radical and A1-radcat would not be viable, based on the calculated SET barrier of the reaction (31.8 kcal mol−1, Scheme S2†). Unfortunately, a number of attempts to trap the proposed diene via Diels–Alder trapping with dienophiles were not successful (Scheme S6†), presumably due to the facile oxidation of A1-diene (Eox (calc.) = 0.43 V vs. SCE in CH3CN) by 3DPAFIPN for further transformations and deconstruction of the catalytic cycle by the external dienophiles. The reaction with in situ generated imine species (A1-imine) gave only trace amount of B1,20 implying that the formation of the imine intermediate from A1-radvia HAT or A1-radcatvia halogen-atom transfer and deprotonation is not operating in this reaction.21 The synergistic SET and HAT processes could proceed again on the generated A1-diene, performing deprotonation on C5 position and HAT on C6 position, to provide the aromatized product (B1). The involvement of the proposed HAT pathway was further supported by the reaction with A1-D3 (Scheme 2E), which provided about 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of C6F5H and C6F5D as a result of sequential HAT processes on C4 position of A1-D3 and C6 position of the corresponding diene intermediate.
:1 ratio of C6F5H and C6F5D as a result of sequential HAT processes on C4 position of A1-D3 and C6 position of the corresponding diene intermediate.
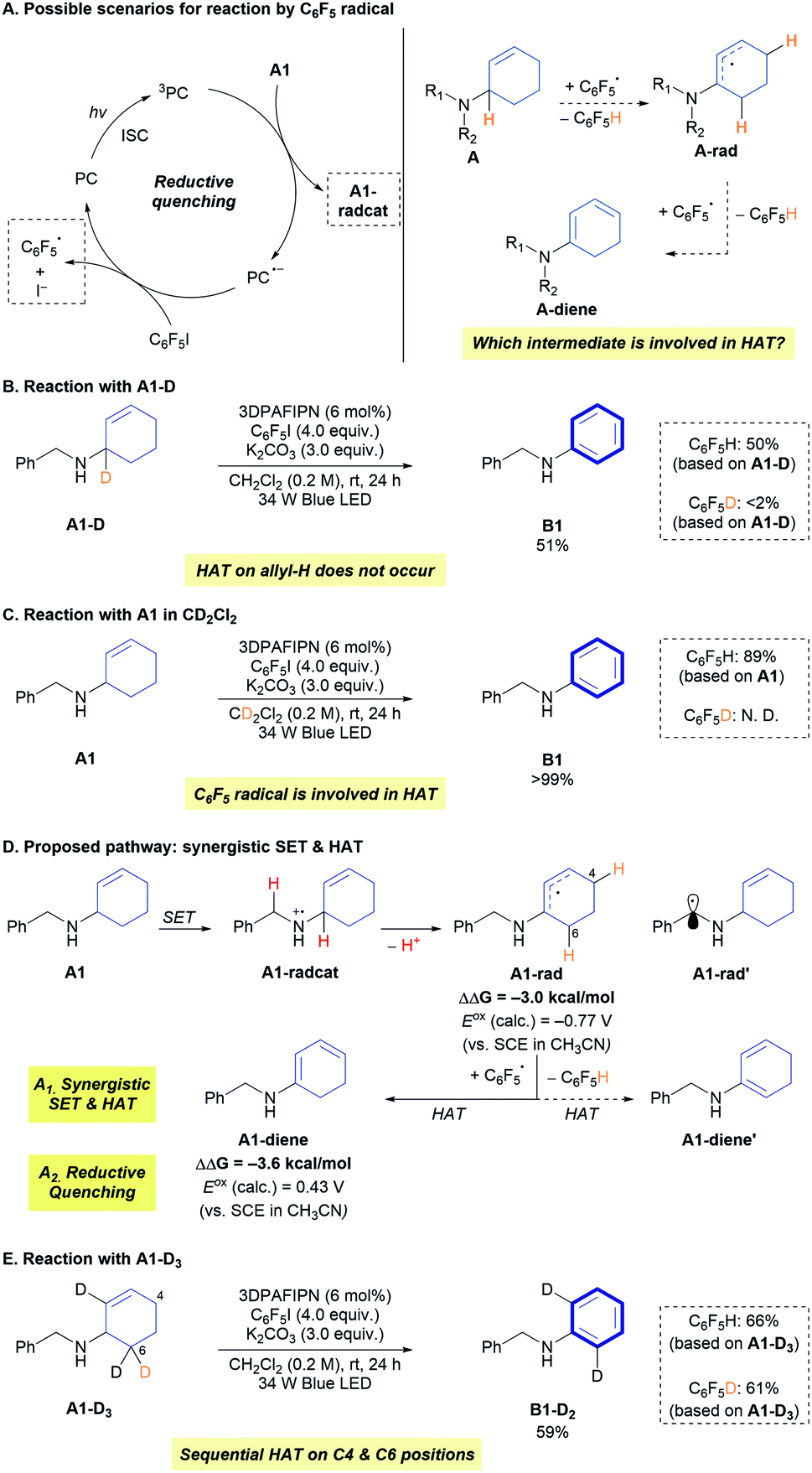
The developed reaction provides an N-aryl amine as a product, but over-reactions of the generated N-aryl amine, such as the formation of corresponding imines, were not observed. Because the proposed mechanisms involving SET and HAT are also possible with N-aryl amines as reactants (Fig. S6†), it was envisaged that an additional mechanism that discriminates the starting alkyl allyl amines (A) and the products (B) may be operative in the reaction. Insight into this aspect of the mechanism was gained from the reactivity of an aniline-derived allylic amine substrate (A37), which has a lower oxidation barrier (Eoxp = 0.88 V vs. SCE in CH3CN) than that of the model amine substrate (A1, Eoxp = 1.29 V vs. SCE in CH3CN) (Scheme 3A). The conversion of A37 and the formation of C6F5H were very low under the optimized reaction conditions (Fig. S3†), implying that the productive catalytic cycle (Scheme 3A, orange arrow) did not operate efficiently. From the proposed mechanism that includes deprotonation of the amine radical cation, it was hypothesized that the stability of the amine radical cation would affect the reaction kinetics. Specifically, we focused on the possibility of back-electron transfer (BET) between the reduced form of 3DPAFIPN (PC˙−) and the amine radical cation (Scheme 3A, green arrow).
 | ||
| Scheme 3 Involvement of back-electron transfer in the reaction. | ||
BET affects the reactivity of a number of visible-light photoredox catalysis, and the control of this elementary step has been elaborated to improve the reactivity and selectivity.22 When the triplet-state photoexcited 3DPAFIPN is quenched by the amine substrate, the radical ion-pair is immediately generated from two neutral species. Immediate and facile deprotonation of the amine radical cation then occurs via the desired catalytic cycle (Scheme 3A, orange arrow) under basic conditions (K2CO3).23 However, in the case of the N-aryl amine radical cation, which is known to have higher stability than the aliphatic amine radical cation and hence undergoes slower deprotonation,24 BET can regenerate the starting amine and 3DPAFIPN (Scheme 3A, green arrow). This unproductive catalytic cycle clearly excludes the N-aryl substrate from the productive cycle, while no consumption of any reagents or the catalyst happens. The concept of BET in product protection is well established in the photocatalytic oxygenation of benzene to phenol, where the electron-rich phenol does not undergo oxidation due to the facile BET between the aryl radical cation and the photocatalyst.22a,22b
To verify the effect of the rate of deprotonation of various amine radical cations on the reaction, an array of N-aryl amines bearing ortho-substituents and N-substituents were prepared. Dinnocenzo and co-workers demonstrated the stereoelectronic effect of N–Ar substituents in the deprotonation of N-aryl amine radical cations.24c Incorporation of a substituent at the ortho-position of the aryl group led to structural distortion through the N–C1 bond, and the structurally-disfavoured conjugation induced destabilization of the amine radical cation and thus faster deprotonation (Scheme 3B).24c Inspired by this stereoelectronic effect on the rate of deprotonation of amine radical cations, the correlation between the amine reactivity and the dihedral angle (∠C2C1NRN) of the amine radical cation was investigated with independently prepared N-aryl amine substrates (Scheme 3C). Notably, aryl allyl amines with large dihedral angles in the corresponding amine radical cations (A38-Me and A39-Me) were reactive toward CD, producing N,N-diaryl amines (B38-Me and B39-Me) in noticeable yields (28% and 34%, respectively). These results support the involvement of BET in the reaction by demonstrating that preventing BET could initiate the productive catalytic cycle (Scheme 3A, orange arrow). In the synthesis of N-aryl amines from aliphatic allylic amines, BET is highly advantageous for achieving the targeted synthesis in an efficient and selective manner by preventing further undesired transformation of the product B (Scheme 3D).
Based on mechanistic studies and previous literature, we propose an overall mechanism for the photoredox CD of 2-cyclohexenyl amines (Fig. 2A). Transformation of the allylic amine substrate (A) is initiated by single-electron oxidation by the triplet-state photocatalyst (3PC) to generate the amine radical cation (A-radcat), which undergoes facile deprotonation to produce the α-amino radical species (A-rad). The pentafluorophenyl radical 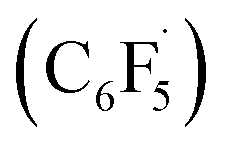 , which is generated by the subsequent reduction of C6F5I by the reduced form of the photocatalyst (PC˙−), abstracts a hydrogen-atom from another allylic position of A-rad, forming the dieneamine intermediate (A-diene). With this intermediate, a similar mechanistic scenario is possible, considering the redox potential of the dienamine derived from amine A1 (A1-diene, Eox (calc.) = 0.43 V vs. SCE in CH3CN). The generated dienamine radical cation (A-dieneradcat) undergoes the facile deprotonation at the C5 position (ΔΔG (C5 and C6) = −6.5 kcal mol−1, Scheme S3†) to provide the 7π-enaminyl radical, as previously reported in the production of the 5π-enaminyl radical.25 Further HAT by the C6F5 radical of the highly conjugated enaminyl radical intermediate (A-dienerad) provides an aromatized product (B). The involvement of a chain-propagating mechanism for the generation of radical intermediates was excluded, based on the low quantum yield of 0.08 (Fig. 2B). Throughout the photoredox catalytic cycle, both the productive pathway (Fig. 2B, black arrow) and the non-productive pathway (Fig. 2B, green arrow) are operative, leading to discriminatory activity where the substrates react and the product is preserved in the reaction media. The possibility of halogen bonding between the substrate amine (A) and C6F5I was excluded by the absence of noticeable interaction in the 19F NMR spectra of the mixture (Fig. S15†),26 along with the UV-Vis observation of that the direct SET via the donor–acceptor complexation and photoinduced electron transfer (PET) do not likely operate in the reaction (Fig. S16†).27
, which is generated by the subsequent reduction of C6F5I by the reduced form of the photocatalyst (PC˙−), abstracts a hydrogen-atom from another allylic position of A-rad, forming the dieneamine intermediate (A-diene). With this intermediate, a similar mechanistic scenario is possible, considering the redox potential of the dienamine derived from amine A1 (A1-diene, Eox (calc.) = 0.43 V vs. SCE in CH3CN). The generated dienamine radical cation (A-dieneradcat) undergoes the facile deprotonation at the C5 position (ΔΔG (C5 and C6) = −6.5 kcal mol−1, Scheme S3†) to provide the 7π-enaminyl radical, as previously reported in the production of the 5π-enaminyl radical.25 Further HAT by the C6F5 radical of the highly conjugated enaminyl radical intermediate (A-dienerad) provides an aromatized product (B). The involvement of a chain-propagating mechanism for the generation of radical intermediates was excluded, based on the low quantum yield of 0.08 (Fig. 2B). Throughout the photoredox catalytic cycle, both the productive pathway (Fig. 2B, black arrow) and the non-productive pathway (Fig. 2B, green arrow) are operative, leading to discriminatory activity where the substrates react and the product is preserved in the reaction media. The possibility of halogen bonding between the substrate amine (A) and C6F5I was excluded by the absence of noticeable interaction in the 19F NMR spectra of the mixture (Fig. S15†),26 along with the UV-Vis observation of that the direct SET via the donor–acceptor complexation and photoinduced electron transfer (PET) do not likely operate in the reaction (Fig. S16†).27
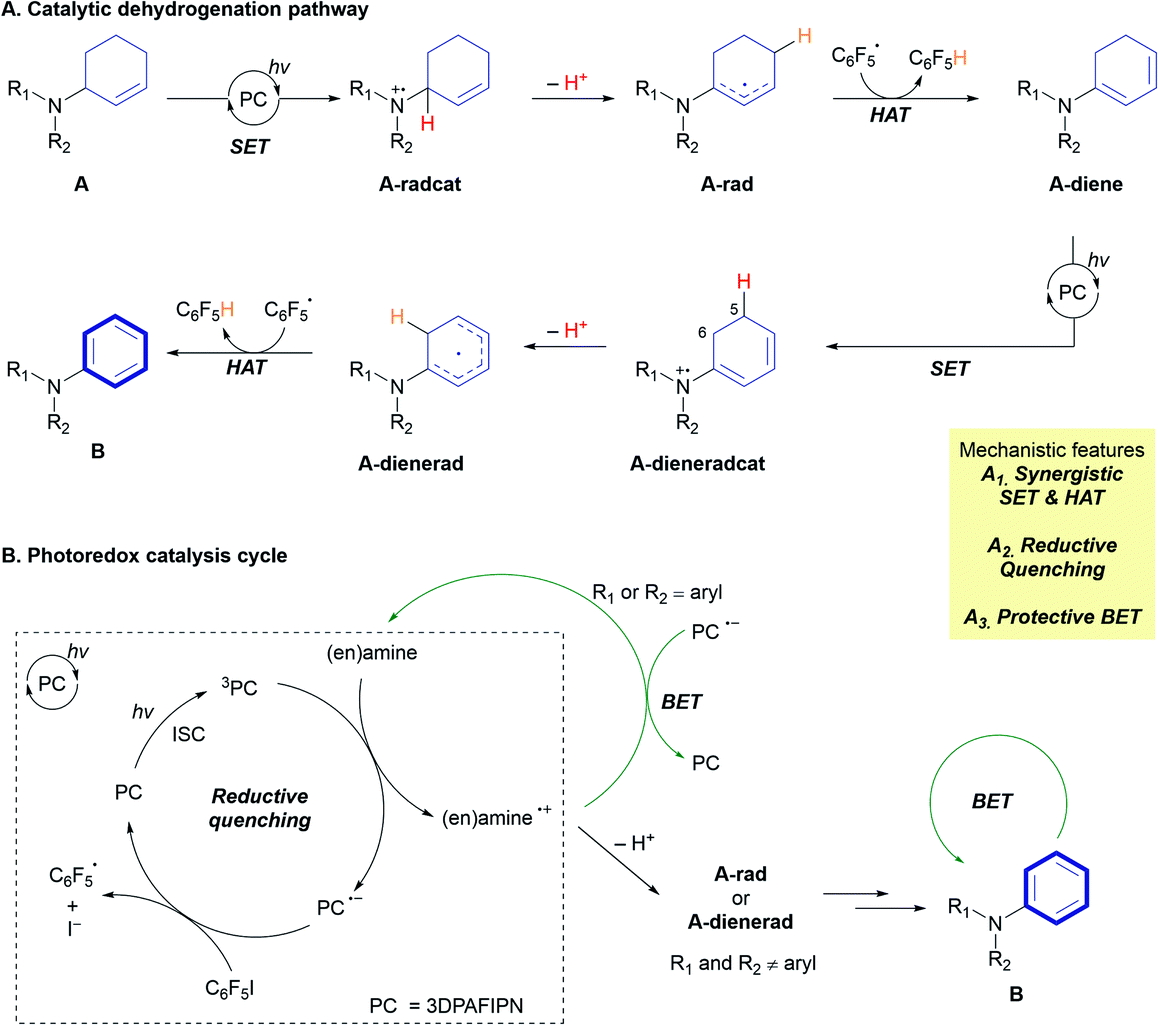 | ||
| Fig. 2 Proposed mechanism. | ||
Conclusions
In conclusion, the visible-light photoredox catalytic dehydrogenation of allylic amines was developed to synthesize invaluable N-aryl amines. Synergistic single-electron transfer and hydrogen-atom transfer enabled controlled dehydrogenation of the 2-cyclohexenyl amines bearing several reactive sites, thus affording a wide range of N-aryl amines under mild reaction conditions. Notably, the protective back-electron transfer pathway plays a crucial role in achieving the photoredox catalytic dehydrogenation protocol for the synthesis of N-aryl amines by preventing unwanted side-reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was gratefully supported by the National Research Foundation of Korea (NRF-2019R1A2C2086875; NRF-2014R1A5A1011165, Center for New Directions in Organic Synthesis) and the Institute for Basic Science (IBS-R004).Notes and references
- (a) C. Gunanathan and D. Milstein, Science, 2013, 341, 1229712 CrossRef; (b) R. H. Crabtree, Chem. Rev., 2017, 117, 9228–9246 CrossRef CAS; (c) A. Kumar, T. M. Bhatti and A. S. Goldman, Chem. Rev., 2017, 117, 12357–12384 CrossRef CAS; (d) D. L. J. Broere, Phys. Sci. Rev., 2018, 3, 20170029 Search PubMed; (e) G. A. Filonenko, R. van Putten, E. J. M. Hensen and E. A. Pidko, Chem. Soc. Rev., 2018, 47, 1459–1483 RSC; (f) S. Hati, U. Holzgrabe and S. Sen, Beilstein J. Org. Chem., 2017, 13, 1670–1692 CrossRef CAS; (g) C. Gunanathan and D. Milstein, Chem. Rev., 2014, 114, 12024–12087 CrossRef CAS; (h) G. E. Dobereiner and R. H. Crabtree, Chem. Rev., 2010, 110, 681–703 CrossRef CAS.
- (a) J. Cossy and D. Belotti, Org. Lett., 2002, 4, 2557–2559 CrossRef CAS; (b) H. Neumann, A. Jacobi von Wangelin, S. Klaus, D. Strübing, D. Gördes and M. Beller, Angew. Chem., Int. Ed., 2003, 42, 4503–4507 CrossRef CAS; (c) R. Yamaguchi, C. Ikeda, Y. Takahashi and K.-I. Fujita, J. Am. Chem. Soc., 2009, 131, 8410–8412 CrossRef CAS; (d) S. A. Girard, X. Hu, T. Knauber, F. Zhou, M.-O. Simon, G.-J. Deng and C.-J. Li, Org. Lett., 2012, 14, 5606–5609 CrossRef CAS; (e) J. Wu, D. Talwar, S. Johnston, M. Yan and J. Xiao, Angew. Chem., Int. Ed., 2013, 52, 6983–6987 CrossRef CAS; (f) K.-I. Fujita, Y. Tanaka, M. Kobayashi and R. Yamaguchi, J. Am. Chem. Soc., 2014, 136, 4829–4832 CrossRef CAS; (g) A. E. Wendlandt and S. S. Stahl, J. Am. Chem. Soc., 2014, 136, 11910–11913 CrossRef CAS; (h) S. Chakraborty, W. W. Brennessel and W. D. Jones, J. Am. Chem. Soc., 2014, 136, 8564–8567 CrossRef CAS; (i) W. Yao, Y. Zhang, X. Jia and Z. Huang, Angew. Chem., Int. Ed., 2014, 53, 1390–1394 CrossRef CAS; (j) X. Cui, Y. Li, S. Bachmann, M. Scalone, A.-E. Surkus, K. Junge, C. Topf and M. Beller, J. Am. Chem. Soc., 2015, 137, 10652–10658 CrossRef CAS; (k) S. A. Girard, H. Huang, F. Zhou, G.-J. Deng and C.-J. Li, Org. Chem. Front., 2015, 2, 279–287 RSC; (l) A. V. Iosub and S. S. Stahl, J. Am. Chem. Soc., 2015, 137, 3454–3457 CrossRef CAS; (m) D. Talwar, A. Gonzalez-de-Castro, H. Y. Li and J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 5223–5227 CrossRef CAS; (n) A. V. Iosub and S. S. Stahl, ACS Catal., 2016, 6, 8201–8213 CrossRef CAS; (o) H. Wang, Y. Li, Q. Lu, M. Yu, X. Bai, S. Wang, H. Cong, H. Zhang and A. Lei, ACS Catal., 2019, 9, 1888–1894 CrossRef CAS.
- (a) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC; (b) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS; (c) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS; (d) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS.
- (a) S. Chen, Q. Wan and A. K. Badu-Tawiah, Angew. Chem., Int. Ed., 2016, 55, 9345–9349 CrossRef CAS; (b) H. G. Yayla, F. Peng, I. K. Mangion, M. McLaughlin, L.-C. Campeau, I. W. Davies, D. A. DiRocco and R. R. Knowles, Chem. Sci., 2016, 7, 2066–2073 RSC; (c) K.-H. He, F.-F. Tan, C.-Z. Zhou, G.-J. Zhou, X.-L. Yang and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 3080–3084 CrossRef CAS; (d) M. K. Sahoo, G. Jaiswal, J. Rana and E. Balaraman, Chem.–Eur. J., 2017, 23, 14167–14172 CrossRef CAS; (e) S. Kato, Y. Saga, M. Kojima, H. Fuse, S. Matsunaga, A. Fukatsu, M. Kondo, S. Masaoka and M. Kanai, J. Am. Chem. Soc., 2017, 139, 2204–2207 CrossRef CAS; (f) M. Zheng, J. Shi, T. Yuan and X. Wang, Angew. Chem., Int. Ed., 2018, 57, 5487–5491 CrossRef CAS; (g) H. Fuse, M. Kojima, H. Mitsunuma and M. Kanai, Org. Lett., 2018, 20, 2042–2045 CrossRef CAS; (h) K. Muralirajan, R. Kancherla and M. Rueping, Angew. Chem., Int. Ed., 2018, 57, 14787–14791 CrossRef CAS; (i) X. He, Y.-W. Zheng, T. Lei, W.-Q. Liu, B. Chen, K. Feng, C.-H. Tung and L.-Z. Wu, Catal. Sci. Technol., 2019, 9, 3337–3341 RSC; (j) Z. Jia, Q. Yang, L. Zhang and S. Luo, ACS Catal., 2019, 9, 3589–3594 CrossRef CAS; (k) D. Chao and M. Zhao, Dalton Trans., 2019, 48, 5444–5449 RSC; (l) L. n. Unkel, S. Malcherek, E. Schendera, F. Hoffmann, J. Rehbein and M. Brasholz, Adv. Synth. Catal., 2019, 361, 2870–2876 CrossRef CAS; (m) M. K. Sahoo and E. Balaraman, Green Chem., 2019, 21, 2119–2128 RSC; (n) H. Fuse, H. Mitsunuma and M. Kanai, J. Am. Chem. Soc., 2020, 142, 4493–4499 CrossRef CAS; (o) F. Stanek, R. Pawlowski, P. Morawska, R. Bujok and M. Stodulski, Org. Biomol. Chem., 2020, 18, 2103–2112 RSC.
- J. Hu, J. Wang, T. H. Nguyen and N. Zheng, Beilstein J. Org. Chem., 2013, 9, 1977–2001 CrossRef.
- (a) S. A. Morris, J. Wang and N. Zheng, Acc. Chem. Res., 2016, 49, 1957–1968 CrossRef CAS; (b) J. W. Beatty and C. R. Stephenson, Acc. Chem. Res., 2015, 48, 1474–1484 CrossRef CAS.
- S. U. Dighe, F. Juliá, A. Luridiana, J. J. Douglas and D. Leonori, Nature, 2020, 584, 75–81 CrossRef.
- A. U. Meyer, T. Slanina, C.-J. Yao and B. König, ACS Catal., 2016, 6, 369–375 CrossRef CAS.
- J.-R. Wang, Y. Fu, B.-B. Zhang, X. Cui, L. Liu and Q.-X. Guo, Tetrahedron Lett., 2006, 47, 8293–8297 CrossRef CAS.
- M. Sutter, M.-C. Duclos, B. Guicheret, Y. Raoul, E. Métay and M. Lemaire, ACS Sustainable Chem. Eng., 2013, 1, 1463–1473 CrossRef CAS.
- T. Kondo and T. A. Mitsudo Ta, Chem. Rev., 2000, 100, 3205–3220 CrossRef CAS.
- (a) C. K. Prier and D. W. C. MacMillan, Chem. Sci., 2014, 5, 4173–4178 RSC; (b) A. Noble and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 11602–11605 CrossRef CAS; (c) T. Ide, J. P. Barham, M. Fujita, Y. Kawato, H. Egami and Y. Hamashima, Chem. Sci., 2018, 9, 8453–8460 RSC.
- P. F. Driscoll, E. Deunf, L. Rubin, O. Luca, R. Crabtree, C. Chidsey, J. Arnold and J. B. Kerr, ECS Trans., 2011, 35, 3–17 CrossRef CAS.
- A. Muthukrishnan and M. V. Sangaranarayanan, J. Electrochem. Soc., 2009, 156, F23 CrossRef CAS.
- S. M. Senaweera, A. Singh and J. D. Weaver, J. Am. Chem. Soc., 2014, 136, 3002–3005 CrossRef CAS.
- For the generation of dichloromethyl radical from dichloromethane and aryl radical, see: (a) Y. Liu, J.-L. Zhang, R.-J. Song and J.-H. Li, Org. Chem. Front., 2014, 1, 1289–1294 RSC; (b) Y. Liu, J.-L. Zhang, R.-J. Song and J.-H. Li, Eur. J. Org. Chem., 2014, 2014, 1177–1181 CrossRef CAS.
- Analysis of the transition state of the deprotonation step in this reaction was not viable. For the related work, see: L. Leng, Y. Fu, P. Liu and J. M. Ready, J. Am. Chem. Soc., 2020, 142, 11972–11977 CrossRef CAS.
- (a) J. G. West, D. Huang and E. J. Sorensen, Nat. Commun., 2015, 6, 10093 CrossRef; (b) D. J. Abrams, J. G. West and E. J. Sorensen, Chem. Sci., 2017, 8, 1954–1959 RSC.
- X.-Q. Hu, X. Qi, J.-R. Chen, Q.-Q. Zhao, Q. Wei, Y. Lan and W.-J. Xiao, Nat. Commun., 2016, 7, 11188 CrossRef.
- See ESI† for the detailed experimental procedure and the results.
- T. Constantin, M. Zanini, A. Regni, N. S. Sheikh, F. Juliá and D. Leonori, Science, 2020, 367, 1021–1026 CrossRef CAS.
- (a) K. Ohkubo, T. Kobayashi and S. Fukuzumi, Angew. Chem., Int. Ed., 2011, 50, 8652–8655 CrossRef CAS; (b) K. Ohkubo, A. Fujimoto and S. Fukuzumi, J. Am. Chem. Soc., 2013, 135, 5368–5371 CrossRef CAS; (c) S. Ruccolo, Y. Qin, C. Schnedermann and D. G. Nocera, J. Am. Chem. Soc., 2018, 140, 14926–14937 CrossRef CAS; (d) Q.-Q. Zhao, X.-S. Zhou, S.-H. Xu, Y.-L. Wu, W.-J. Xiao and J.-R. Chen, Org. Lett., 2020, 22, 2470–2475 CrossRef CAS; (e) N. G. W. Cowper, C. P. Chernowsky, O. P. Williams and Z. K. Wickens, J. Am. Chem. Soc., 2020, 142, 2093–2099 CrossRef CAS.
- S. F. Nelsen and J. T. Ippoliti, J. Am. Chem. Soc., 1986, 108, 4879–4881 CrossRef CAS.
- (a) J. P. Dinnocenzo and T. E. Banach, J. Am. Chem. Soc., 1989, 111, 8646–8653 CrossRef CAS; (b) X. Zhang, S.-R. Yeh, S. Hong, M. Freccero, A. Albini, D. E. Falvey and P. S. Mariano, J. Am. Chem. Soc., 1994, 116, 4211–4220 CrossRef CAS; (c) G. W. Dombrowski, J. P. Dinnocenzo, P. A. Zielinski, S. Farid, Z. M. Wosinska and I. R. Gould, J. Org. Chem., 2005, 70, 3791–3800 CrossRef CAS.
- M. T. Pirnot, D. A. Rankic, D. B. C. Martin and D. W. C. MacMillan, Science, 2013, 339, 1593–1596 CrossRef CAS.
- (a) X. Q. Yan, X. R. Zhao, H. Wang and W. J. Jin, J. Phys. Chem. B, 2014, 118, 1080–1087 CrossRef CAS; (b) D. Hauchecorne, B. J. van der Veken, W. A. Herrebout and P. E. Hansen, Chem. Phys., 2011, 381, 5–10 CrossRef CAS; (c) G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS; (d) Y. Liu, X.-L. Chen, K. Sun, X.-Y. Li, F.-L. Zeng, X.-C. Liu, L.-B. Qu, Y.-F. Zhao and B. Yu, Org. Lett., 2019, 21, 4019–4024 CrossRef CAS.
- (a) C. G. S. Lima, T. de M. Lima, M. Duarte, I. D. Jurberg and M. W. Paixão, ACS Catal., 2016, 6, 1389–1407 CrossRef CAS; (b) Y.-Q. Yuan, S. Majumder, M.-H. Yang and S.-R. Guo, Tetrahedron Lett., 2020, 61, 151506 CrossRef CAS; (c) E. Arceo, I. D. Jurberg, A. Alvarez-Fernandez and P. Melchiorre, Nat. Chem., 2013, 5, 750–756 CrossRef CAS; (d) Y. Cheng, X. Yuan, J. Ma and S. Yu, Chem.–Eur. J., 2015, 21, 8355–8359 CrossRef CAS; (e) L. Marzo, S. Wang and B. König, Org. Lett., 2017, 19, 5976–5979 CrossRef CAS; (f) C.-W. Hsu and H. Sundén, Org. Lett., 2018, 20, 2051–2054 CrossRef CAS; (g) T. Morack, C. Mück-Lichtenfeld and R. Gilmour, Angew. Chem., Int. Ed., 2019, 58, 1208–1212 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, computational details, and spectroscopic data for all new compounds. See DOI: 10.1039/d0sc04890a |
| This journal is © The Royal Society of Chemistry 2021 |