
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSustainable existence of solid mercury (Hg) nanoparticles at room temperature and their applications†
Villa Krishna
Harika‡
a,
Tirupathi Rao
Penki‡
 a,
Boddapati
Loukya
b,
Atanu
Samanta
a,
Gui-Liang
Xu
c,
Cheng-Jun
Sun
c,
Ilya
Grinberg
a,
Francis Leonard
Deepak
b,
Khalil
Amine
c,
Doron
Aurbach
*a and
Aharon
Gedanken
*a
a,
Boddapati
Loukya
b,
Atanu
Samanta
a,
Gui-Liang
Xu
c,
Cheng-Jun
Sun
c,
Ilya
Grinberg
a,
Francis Leonard
Deepak
b,
Khalil
Amine
c,
Doron
Aurbach
*a and
Aharon
Gedanken
*a
aBar-Ilan Institute for Nanotechnology and Advanced Materials (BINA), Department of Chemistry, Bar-Ilan University, Ramat-Gan, 5290002, Israel. E-mail: Aharon.Gedanken@biu.ac.il; Doron.Aurbach@biu.ac.il
bNanostructured Materials Group, Department of Advanced Electron Microscopy Imaging and Spectroscopy, International Iberian Nanotechnology Laboratory (INL), Avenida Mestre Jose Veiga, Braga, 4715-330, Portugal
cChemical Sciences and Engineering Division, Argonne National Laboratory, 9700 South Cass Avenue, Lemont, IL 60439, USA
First published on 6th January 2021
Abstract
Although liquid mercury (Hg) has been known since antiquity, the formation of stable solid nano forms of Hg at room temperature has not been reported so far. Here, for the first time, we report a simple sonochemical route to obtain solid mercury nanoparticles, stabilized by reduced graphene oxide at ambient conditions. The as-formed solid Hg nanoparticles were found to exhibit remarkable rhombohedral morphology and crystallinity at room temperature. Extensive characterization using various physicochemical techniques revealed the unique properties of the solid nanoparticles of Hg compared to its bulk liquid metal phase. Furthermore, the solid nature of the Hg nanoparticles was studied electrochemically, revealing distinctive properties. We believe that solid Hg nanoparticles have the potential for important applications in the fields of electroanalytical chemistry and electrocatalysis.
Introduction
Mercury is one of the few metal elements in nature that exist as liquids under ambient conditions, challenging material scientists to explore Hg nanoparticles (NPs).1 Nanoparticles usually have different properties from the bulk material and often lead to significant technological applications.2–6 Previously, there were a few reports on the synthesis of colloidal amalgams of silver and gold nanoparticles, which revealed the UV-absorption spectrum of nano-mercury and is responsible for the theoretically calculated surface-plasmon absorption in colloids.7–10 Although nano-mercury colloids have been synthesized using laser irradiation and chemical reductions methods, stable nano-forms of mercury were not observed in these studies. Radhakrishnan et al. reported the fabrication of stable mercury nanodrops by in situ reduction of Hg-ions inside a polymer film.11 However, the stabilized nanodrops did not show any nanocrystalline characteristics under ambient conditions, unless frozen to −120 °C.11 Previously, we reported the formation of core–shell particles of mercury by thermolysis of dimethyl mercury in which liquid mercury drops were encapsulated in a carbon shells using a RAPET (Reaction under Autogenic Pressure at Elevated Temperatures) synthesis.12 However, these studies were limited to micro-level mercury core–shell particles and the mercury did not solidify. The large surface area and absence of counteractive repulsive forces between any two metal nanoparticles always leads to coalescence that minimizes the surface energy, thermodynamically favouring a bulk phase. Nanoparticles can be stabilized by either electrostatic or steric stabilization using capping agents.13,14 Graphene-based nanocomposites have been synthesized for various potential applications, due to their remarkable properties such as high thermal and electrical conductivity, high surface area and inertness. In addition, they can serve as nucleation sites for nanoparticle growth and serve as substrates for depositing metal nanoparticles.15,16 Thus, grafting of nanomaterials onto graphene sheets can reduce agglomeration without affecting the inherent properties of the metal nanoparticles.15,16 It should be also noted that extensive research on the sonochemistry of low-melting-point metals has been conducted in our laboratory.17,18Here, for the first time, we report a facile synthetic route that forms highly stable crystalline mercury nanoparticles supported on solid carbon supports such as reduced graphene oxide (RGO), graphene, graphite oxide, graphite, etc., using ultra-sonication under ambient conditions. Unlike previous reports on the synthesis of nanoparticles,11,12 we utilized liquid mercury as the starting material thereby avoiding the use of reducing agents for the formation of mercury metal nanoparticles. Similarly, no molecular linkers were used to bridge between the Hg nanoparticles and the graphene matrices. The Hg nanoparticles supported by RGO were characterized by various physicochemical techniques. Due to their remarkable sensitivity and selectivity, liquid Hg and its amalgams have been extensively used in the fields of basic and applied electrochemistry.19–21 To understand the electrochemical behaviour of solid mercury nanoparticles, we performed comparative electrochemical measurements with liquid Hg electrodes using ferro/ferricyanide ([Fe(CN)6]4−/3−) couple as a red-ox probe.22 Similarly, possible electrocatalytic activity of the composite structures containing Hg solid nanoparticles was explored using the hydrogen evolution reaction (HER) as a probe.23 As the surface of mercury electrodes usually impedes HER, it was interesting to explore to what extent the solid structure changes the behaviour of mercury nanoparticles towards HER.
This paper provides several proofs and an explanation for the new finding, namely, apparently stable solid state mercury particles at ambient conditions, based on a comprehensive analytical work. The preliminary characterization of their unique electrochemical properties presented herein, promotes further intensive studies on the practical/analytical consequences of this discovery.
Experimental
Materials
Analytical high pure grade chemicals, namely, mercury (Hg, 99.9%), potassium hexacyanoferrate(II) trihydrate (K4[Fe(CN)6]·3H2O, ≥99.0%), potassium hexacyanoferrate(III) (K3[Fe(CN)6], ≥99.0%), potassium chloride (KCl, 99.9%), zinc nitrate hexahydrate (ZnNO3)2·6H2O ≥99.0%, 20 wt% Nafion solution, conc. sulphuric acid (H2SO4, 98.0%), sodium nitrate (Na(NO3), ≥99.9%), hydrogen peroxide (H2O2, 30% (w/w) in H2O), and hydrogen chloride (HCl, 99.99%) were received from Sigma Aldrich™ and used without further purification. In addition, Graphite KS-25 (LONZA), potassium permanganate (KMnO4, ≥99.9%) (Bio-Lab. Ltd.,) and double distilled (DD) water prepared in the laboratory was used.Synthesis of RGO
Reduced graphene oxide (RGO) used for the synthesis of solid Hg nanoparticles in this paper was prepared by the following two stages as shown in Scheme S1.† In the initial stage, graphite oxide (GO) was synthesized from graphite by the well-known modified Hummers method.24 For the synthesis, 92 ml of H2SO4 (98 wt%) was transferred into a 500 ml conical flask and placed in an ice bath. 2 g of graphite powder and 2 g of NaNO3 were then added. The conical flask was then removed from ice bath and naturally allowed to reach room temperature. 6 g of KMnO4 was then slowly added to the mixture and continuously stirred for 2 h at room temperature. To the above mixture, 92 ml of DD water followed by 9 ml of H2O2 was added carefully. The formed yellow product of graphite oxide (GO) was separated by filtration and washed with 5% HCl and DD water to remove impurities. The solid GO obtained after drying in a vacuum oven at 60 °C for 24 h was subjected to thermal exfoliation in the later stage by keeping it in a preheated furnace at 450 °C temperature under ambient conditions.25,26 We can observe about a tenfold increase in volume of the sample within a few seconds, indicating the formation of thermally exfoliated reduced graphene. The formed RGO was removed from the oven and used as is for the further synthesis of solid Hg nanoparticles. The formation of RGO was confirmed by XRD, FTIR, ESEM, XPS and Raman spectroscopy as shown in Fig. S11–S13, S19 and S20,† respectively.Fabrication of Hg solid nanoparticles
To prepare the stable solid Hg nanoparticles at ambient conditions, metallic liquid Hg and RGO were taken in weight ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, respectively. In the typical synthesis, 75 mg of liquid mercury was placed in a clean beaker to which a well dispersed 15 ml aqueous solution containing 25 mg of RGO was added. The above mixture was sonicated for 1 h in a water bath. A black precipitate of Hg nanoparticles stabilized by RGO was obtained at the end of the sonochemical reaction with complete disappearance of the liquid mercury. The obtained black solid product was washed with DD water followed by ethanol and dried in a desiccator under vacuum at room temperature and was further investigated using different physicochemical characterization techniques.
:1, respectively. In the typical synthesis, 75 mg of liquid mercury was placed in a clean beaker to which a well dispersed 15 ml aqueous solution containing 25 mg of RGO was added. The above mixture was sonicated for 1 h in a water bath. A black precipitate of Hg nanoparticles stabilized by RGO was obtained at the end of the sonochemical reaction with complete disappearance of the liquid mercury. The obtained black solid product was washed with DD water followed by ethanol and dried in a desiccator under vacuum at room temperature and was further investigated using different physicochemical characterization techniques.
Characterization techniques
Ultrasonic transducer model no. 51-05-290, Ultrasonic Power Corporation, Free Port, Illinois was operated using an amplitude of 36%. The X-ray diffraction (XRD) of all the samples was examined using Bruker Inc. (Germany) AXS D8 ADVANCE diffractometer (reflection q–q geometry, 40 kV and 30 mA using a Cu Kα (λ = 1.5418 Å) radiation source, receiving slit of 0.2 mm, and a high-resolution energy-dispersive detector). The morphology of the samples was characterized by high-resolution scanning electron microscopy (HRSEM) with an FEI Megallen 400 L microscope, environmental scanning electron microscopy (ESEM) with INSPECT-FEI microscope. The elemental composition of the samples was examined by EDAX supported in the ESEM and HRSEM. Elemental analysis of the synthesized HgNPs (two different batches) were carried out using the ICP technique (ICP-AES, spectrometer Ultima-2 from Jobin Yvon Horiba). Images of RGO and Hg nanoparticles distributed and incorporated into RGO were measured using high-resolution transmission electron microscopy (HRTEM) JEOL 2100 microscope at an acceleration voltage of 200 kV. The aberration corrected scanning transmission electron microscopy (AC-STEM) Z-contrast imaging using high-angled annular dark field (HAADF) detector is performed in FEI Titan G2 80–200 kV ChemiSTEM operating at 80 kV. The high-resolution TEM imaging was performed at 60 kV using gun monochromated electron beam in a Titan Themis 60–300 kV TEM/STEM microscope. STEM elemental mapping was performed using Super-X EDX system. EELS measurements were carried out using Gatan Energy Filter Quantum. Optical absorption studies were carried out using Ultraviolet-visible (UV-Vis) model Cary Bio100 spectrophotometer with the integration sphere (Varian), operated by Lab Sphere software. Differential scanning calorimetry (DSC) measurements were performed using NETZSCH instrument model 200 F3-MAIA using liquid nitrogen as a cooling system. XANES spectra of Hg-based samples were measured in transmission mode at 20-BM beamline of Advanced Photon Source (APS). The liquid Hg XANES was measured in fluorescence using the single element Canberra under grazing angle conditions on a liquid mercury surface under reducing atmosphere (He/3.5% H2) at 20-ID beamline of APS. Electrochemical studies were carried out using a Solartron model SI-1287 electrochemical interface and a 1255 HF frequency response analyser and Autolab PGSTAT302N (AUT83352) electrochemical workstation with conventional three-electrode cells in Swagelok systems (as shown in Fig. S2†). Saturated Ag/AgCl in KCl and Pt foil and graphite rod electrodes were used as reference and counter-electrodes, respectively.Calculations of ab initio density functional theory
The total energies calculations are performed using ab initio density functional theory within numerical atomic basis set as implemented in the SIESTA package.27 The valence and core electrons are represented within pseudopotential approximation and norm-conserving pseudopotentials28 were used which were obtained from PseudoDojo.29 The exchange-correlation energy of the electrons is approximated by the PBE functional within the generalized gradient approximation and van-der Waals (vdW) interactions are included within the DFT-D2 method of Grimme.30 The change density cut off of 300 Ry and double-ς and single polarization function (DZP) basis functions of the electron were used. The atomic relaxation was carried out using the conjugate gradient (CG) method until the forces on each atom and the total energy are less than 0.04 eV Å−1 and <10−5 eV, respectively. To study the interaction between the Hg-particle and graphene sheet, we have considered 32 and 122 atoms of Hg-particle on top of 7 × 4 × 1 (112 atoms of C) and 10 × 6 × 1 (240 atoms of C) rectangular supercell of the graphene (CG) structure (as shown in Fig. 3 and S8†). To avoid self-interaction from periodic images of the structure, a vacuum of 15 Å is applied to the simulation system. The Brillion zone was sampled with 3 × 3 × 1 and single points Γ-centered Monkhorst-Pack k-grid31 for structure consist of 32 atoms of Hg particles and graphene comprised of 112 atoms of C (32Hg+112CG) and 122 atoms of Hg particle and graphene sheet constitute of 240 atoms of C. To model RGO, 12 oxygen and 3 hydrogen atoms were added to the graphene sheets comprised of 240 atoms of C: 9 epoxy and 3 OH groups per 240 carbon atoms.Electrochemical studies
A three-electrode cell configuration (Swagelok configuration, Fig. 3c and Scheme S2a†) was used for the electrochemical characterization (CV, EIS) including the GCE, RGO/GCE, Hg/GCE, and HgNPs/GCE working electrodes, Ag/AgCl/KCl and Pt foil served as reference and counter electrodes, respectively in the aqueous solutions containing [Fe(CN)6]4−/3− red-ox moieties (or) Zn(NO3)2 solution. The voltammograms of the four working electrodes were recorded at different scan rates from 5 to 100 mV s−1 within the potential window −0.1 to 0.5 V vs. Ag/AgCl, as shown Fig. S14–S17.† These peak current values are measured by extrapolation of the baselines of anodic and cathodic peaks (see Fig. S21†) for the calculation of diffusion coefficient.§ For all four electrodes, the electrochemical impedance spectra (EIS) were measured under similar conditions (cells, solutions, equilibrium at OCV) in the frequency range 1000 kHz to 0.01 Hz with 5 mV amplitude in the aqueous solutions containing [Fe(CN)6]4−/3− red-ox moieties.
All of the electrochemical measurements (LSV) for hydrogen evolution are carried out in acidic medium of 0.5 M H2SO4 on bare GC, liquid Hg, RGO and HgNPs in conventional three-electrode cell (shown in Fig. 4d, Scheme S2b and Videos S4–S8†) under ambient conditions at a scan rate of 10 mV s−1vs. Ag/AgCl. Here, saturated Ag/AgCl electrode and graphite rod are used as reference and counter-electrodes, respectively. The electrolyte of electrochemical cell was purged with N2 gas for 15 min to remove dissolved air and all the studies are carried out under air-conditioned room temperatures of 22 ± 2 °C (as shown in Video S4†). The recorded video of HER reaction for all the electrodes are given in supplementary information (Videos S4–S8).† All the potentials are referred to the reference hydrogen electrode (RHE) potential, by an appropriate translation of the potentials actually measured vs. Ag/AgCl RE.¶
Handling Hg during the experimentation
Throughout the study, extra precautionary measures were taken during handling of Hg and HgNPs. All the synthetic procedures and heating experiments were carried out in a fume hood.Results and discussion
Molten metals that undergo sonication in liquid media experience pronounced shear forces after the collapse of the cavitation bubbles, that rapidly disperse the liquid metal drops into small spheres, resulting in the formation of micro or nano-spheres.17,18 For metals such as Ga, In, Pb, Bi, Sn and Zn, the as-formed microspheres do not coalesce to give bulk metal, even under temperatures much higher than its melting point. Instead, nano–micro solid particles precipitate at these high temperatures. This is due to the presence of a carbon layer that coats the spheres as a result of the reaction of metal with the non-aqueous liquid (such as dodecane, ethylene glycol, etc.,) under the extreme local conditions of pressure and temperature that develop during the short time of the bubbles collapse. In the case of liquid Hg, we observed that after sonication in water for a long time, tiny spheres ultimately recombine to give bulk liquid Hg once the sonication is stopped (Scheme S3a and Video S1†). On the other hand, sonication in the presence of non-aqueous solvents such as hexane and dodecane results in the formation of mercury microspheres at the end of reaction (Scheme S3b and Video S2†). The microspheres were around 10 to 50 μm in size and the reason they do not coalesce was that the spheres were separated from each other by layers of carbon which coat the liquid Hg spheres (core–shell structure), as shown in Fig. S1 and S2.† The possibility of in situ carbon coating that restrict recombination of micro and even nano droplets of mercury was inspiring, leading to the work reported herein, in which the precursors for carbon coating were graphene moieties. In order to increase possible interactions between micro/nano droplets of mercury and graphene, reduced graphene oxide (RGO) sheets were used.We discovered that sonicating mercury in an aqueous medium containing RGO sheets forms solid mercury nanoparticles (HgNPs) as displayed in Scheme 1a. It is well known that sonication is one of the most effective synthetic routes for the formation of nanosized crystalline materials by breaking chemical bonds and for depositing metals, metal oxides onto various substrates.32–34 A possible mechanism for HgNPs formation consisting of the stages is shown in Scheme 1b (further substantiated below). In the initial stage, liquid Hg metal in an aqueous medium containing RGO sheets undergoes fine dispersion upon ultra-sonication during which the silvery liquid metal is emulsified and surrounded by layers of RGO. In the later stage, shockwaves and micro-jets generated after the collapse of the acoustic bubble induce structural changes in the nanoscale dispersions of Hg, forming solid crystals through a process known as sonocrystallization.32,35,36 Suslick argues that sonication generally yields small crystals with a narrower size distribution than conventional crystallization.32,35 Following this step, the micro-jets move at a very high speed towards the solid RGO, “throwing” the crystalline solid mercury nanoparticles onto the RGO sheets, thereby embedding the solid particles in RGO. The solid nano-Hg particles thus formed are adsorbed onto the surface of RGO and a few particles are even entrapped between the RGO layers and stabilized as shown in Scheme 1b. Here RGO acts as a 2D solid template and stabilizes the dispersed solid Hg nanoparticles without aggregation.
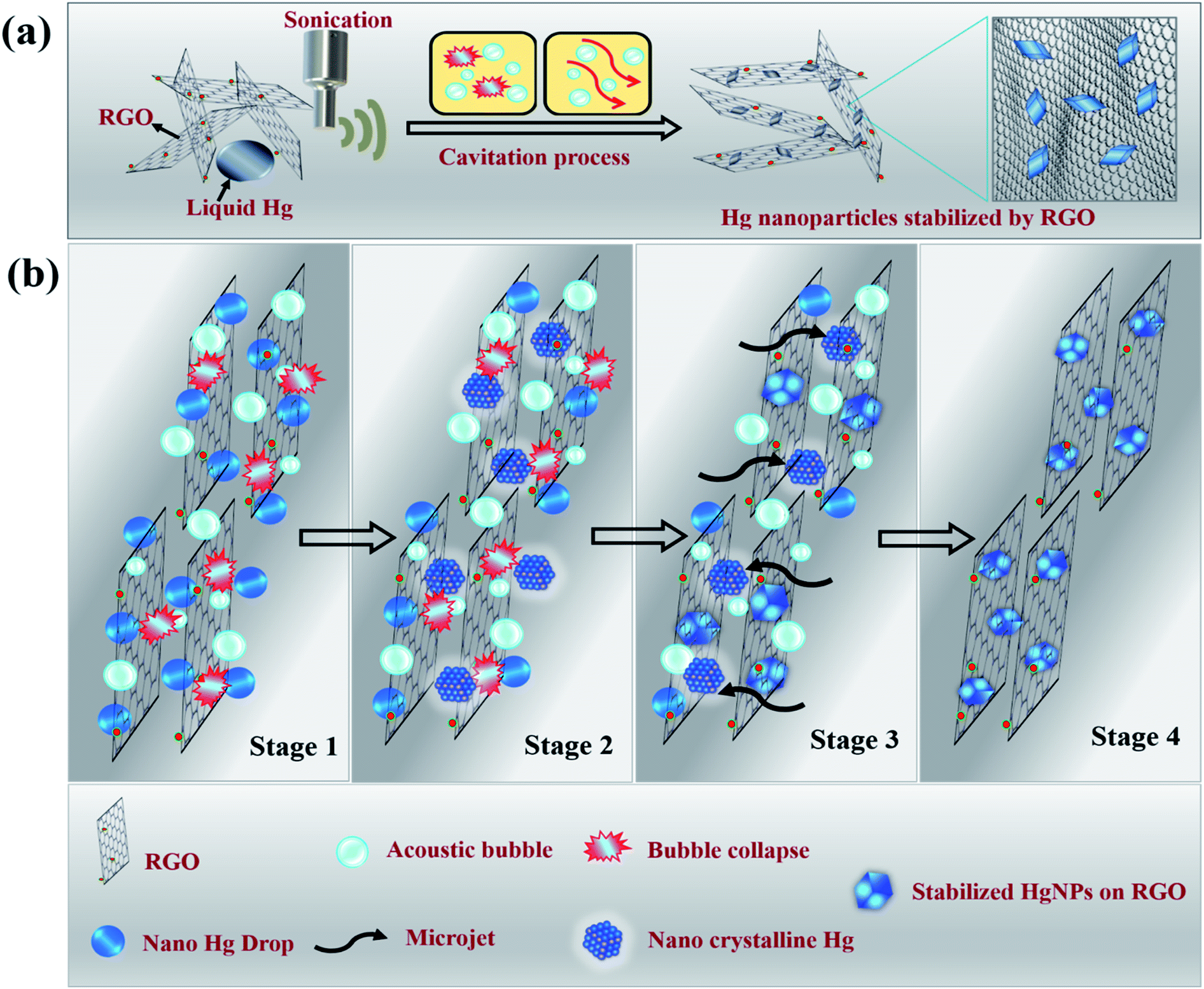 | ||
| Scheme 1 Illustrative (a) synthetic procedure and (b) mechanism of formation of solid Hg nanoparticles under sonication. | ||
Fig. 1a depicts the X-ray diffraction (XRD) patterns of bare RGO and HgNPs. The XRD pattern of RGO (black-line, (i)) obtained after the reduction of graphene oxide (GO) is confirmed by (002) reflection at 2θ = 23.8°.24–26 The red line (ii) corresponds to diffraction pattern of HgNPs with reflections at 2θ = 32.7, 40.3, 53.1, 63.3, 68.5 77.9 and 86.6°, with corresponding planes (101), (003), (110) (104), (113), (015), and (211), respectively, indicative of the rhombohedral crystalline phase of mercury. The obtained peaks are in agreement with the standard pattern (PDF No. 01-077-2681) of Hg rhombohedral phase (blue line, (iii)) with diffraction angels at 2θ = 32.6, 40.2, 52.7, 63.3, 68.3, 77.7 and 86.9°. Bulk Hg metal at ambient temperature exists in the alpha phase (liquid) and so does not provide any diffraction pattern. However, when Hg metal is frozen to −38 °C, it undergoes a phase change from alpha to rhombohedral phase (solid) providing crystalline diffraction patterns.12,17,37 Interestingly, in the present study, we were able to obtain rhombohedral diffraction peaks for Hg nanoparticles entrapped in RGO at ambient temperature. This provides strong evidence for the formation of solid Hg nanoparticles (rhombohedral structure) supported by RGO. The broadness of diffraction peaks obtained for HgNPs may be due to the small size of the solid mercury particles as well as their semi-crystalline nature. The morphology of the HgNPs samples was determined using basic and advanced microscopy imaging tools. Fig. 1b1 displays a representative environmental scanning electron microscope (ESEM) image of HgNPs (circled), visualizing that the nanosized mercury particles are entrapped and supported on the RGO sheets. The mercury particles entrapped on the RGO sheets range from 50–250 nm in size, showing a rhombohedral morphology (Fig. 1b2). These particles are stable under the electron beam of the ESEM (HV – 20 keV). In addition, the quantification and uniform distribution of Hg nanoparticles among RGO layers was confirmed by elemental mapping and energy-dispersive X-ray spectroscopy (EDS) supported by high-resolution scanning electron microscopy (HRSEM) (Fig. S3†). It was confirmed by ICP analysis that the properties of the solid HgNPs were not affected due to a formation of amalgams with sodium and potassium (see the ESI file and Fig. S3(i)†).
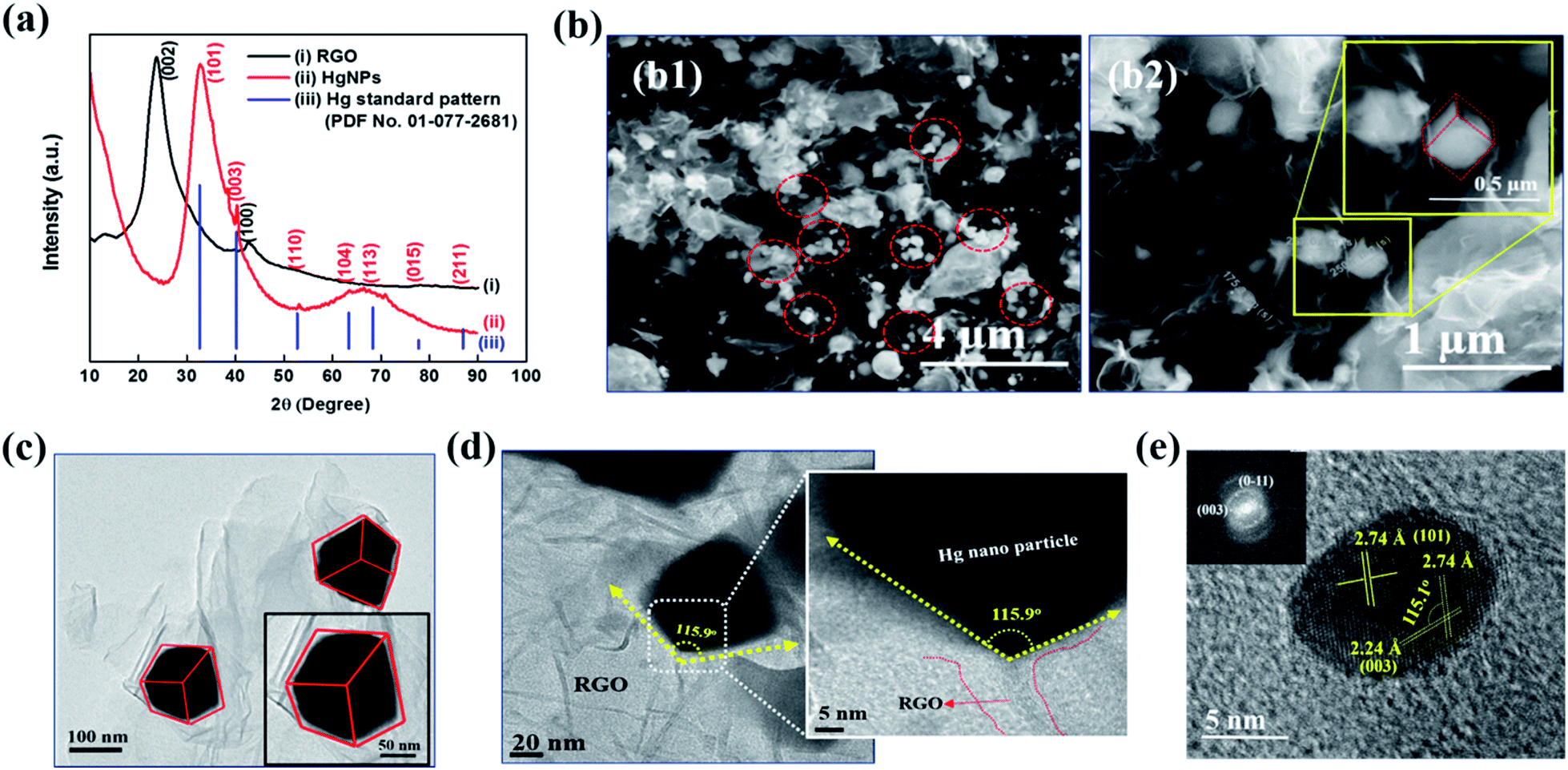 | ||
| Fig. 1 (a) XRD patterns of a typical RGO sample (black line, i), HgNPs (red line, ii) and standard Hg patterns (PDF No. 01-077-2681) (blue line, iii), (b) ESEM images of Hg nanoparticles on RGO at different magnifications, (c) TEM image of HgNPs (magnified rhombohedral Hg particle in the inset), (d) HR-TEM images of HgNPs showing zoomed edge of the particle and (e) HR-TEM image of Hg cluster along with the Fourier-filtered transform in the inset. | ||
TEM and HRTEM imaging (Fig. 1c–e) are fully in line with the above description (concluded by HRSEM). Fig. 1d shows a high-magnification image of one edge of a Hg nanoparticle entrapped in RGO; the 115.9° angle indicating a rhombohedral shape. Crystalline Hg particles are clearly resolved in Fig. 1e, representing (101) and (003) planes with a d-spacing of 2.74 and 2.24 Å, respectively, and the corresponding Fourier-filtered transform (FFT) is shown in the inset. In Fig. 1e, the angle between (101) and (003) plane is 115.1°, which is very close to the angle calculated near the edge of the Hg particles. These results are in a good agreement with the obtained XRD patterns of HgNPs showing a rhombohedral phase. Further morphological studies were carried out using AC-HAADF-STEM Z-contrast imaging, STEM elemental mapping and electron energy-loss spectra (EELS), as presented and discussed in Fig. S4–S6.†
All of these structural and morphological studies by HR electron microscopy and XRD clearly confirm the solid state of the HgNPs in contact with RGO sheets and do not fit a possibility of a liquid state (with a droplet, round shape morphology). According to the literature mercury has four solid phases under high pressure, such as α, β, γ and δ.38 The α phase (rhombohedral structure) is obtained from the liquid mercury either by pressurizing above 1.2 GPa at room temperature or cooling it below 234 K at atmospheric pressure. The transformation of β phase (body-centred tetragonal structure) from α phase occurs only at 3.4 GPa. The other two phases, γ-quasi close-packed (16 GPa) and δ-hexagonal-close-packed (>37 GPa), only exist under very high pressure. Thus, it is evident that simple rhombohedral structure favours at low pressures compared to the other solid phases of mercury. In the current study the formation of the rhombohedral phase of HgNPs under acoustic pressure in an aqueous medium is well correlated with the theoretically calculated pressure39 created during the propagation of ultrasonic waves in a liquid medium (water) where the pressure is in the order of 1 GPa, achievable for air nano bubbles collapsing close to solid surfaces. Thus, from the above explanation, the possible conclusion that we could draw is that there is high probability of formation of rhombohedral phase (alpha) at the acoustic pressures developed during sonication, rather than other phases, which requires much higher pressures.
To reveal the unique physical properties of the as-formed HgNPs at room temperature, thermal behaviour studies were performed by DSC measurements. In all the DSC measurements, the melting and freezing temperatures were considered at the onset of the signals.40 The DSC thermogram shown in Fig. 2a reveals that, upon heating from room temperature to 400 °C, the as-synthesized sample of HgNPs shows two endothermic peaks. The first endothermic peak is detected at 215 °C (onset signal started at 155 °C), which may correspond to the melting of the Hg nanoparticles entrapped in RGO. Another peak around 313 °C (onset signal started at 257 °C) is observed, which may correspond to the boiling point of liquid mercury.17,37Fig. 2b represents the DSC thermogram with heat-freeze cycles of a solid HgNP sample. In segment 1 (Fig. 2b, black curve), samples containing solid HgNPs were heated from room temperature to 250 °C, which revealed an endothermic peak around 215 °C. In the process marked as segment 2, upon cooling from 250 °C to −60 °C (Fig. 2b, red curve), a small exothermic peak at −40.7 °C was observed, which corresponds to the crystallization/solidification of the Hg microspheres formed by the escape of melted Hg nanoparticles from the RGO moiety in the first heating segment. Later, after cooling the samples to −60 °C they were reheated to 250 °C in segment 3 (Fig. 2b, blue curve) during which an endothermic peak around −35.2 °C was observed, corresponding to melting of crystallized mercury microspheres obtained by the cooling process of segment 2. The exo- and endo-peaks can be assigned to crystallization and melting of mercury at −40.7 °C and −35.2 °C respectively and is in agreement with the freezing and melting temperatures of Hg liquid microspheres formed in organic solvents, as shown in Fig. S2.† This thermal analysis provides further strong evidence for the formation of solid mercury nanoparticles stabilized by RGO at room temperature. It is important to note that the size distribution of the HgNPs should affect the sharpness of the DSC response. The microscopic measurements seem to show that the size distribution of the HgNPs reported herein is relatively narrow. Indeed, the DSC response includes sharp peaks, coherent with a relatively narrow size distribution.
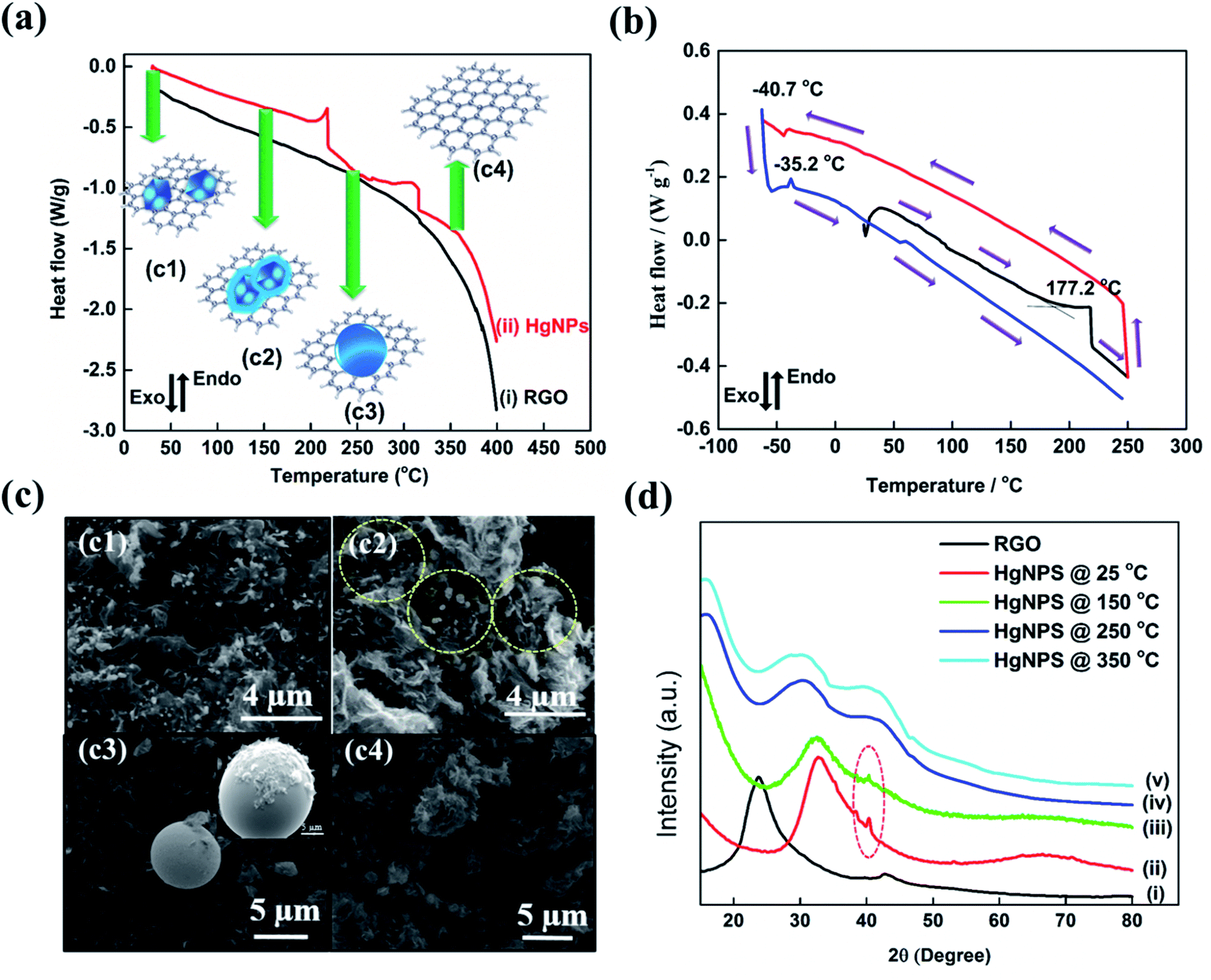 | ||
| Fig. 2 DSC thermogram of (a) RGO (i) and HgNPs (ii) heated to 400 °C and (b) DSC curve of HgNPs segment 1, heating from 27 to 250 °C (black curve), segment 2, cooling from 250 to −60 °C (red curve), segment 3, heating from −60 to 250 °C (blue curve) with temperature scan rate of 5 K min−1. Note that positive values in the ordinate of these charts means heat flow into the system (i.e. endothermic processes). (c) ESEM images of HgNPs, heated at 27 °C (c1), 150 °C (c2), 250 °C (c3) and 350 °C (c4), and (d) Powder XRD of RGO (i), HgNPs at 27 °C (ii), 150 °C (iii), 250 °C (iv), and 350 °C (v). | ||
As additional support to the DSC analysis (Fig. 2a), HgNPs were heated from room temperature (27 °C) to various higher temperatures (150, 250 and 350 °C) under ambient conditions. The heated samples were analyzed using ESEM supported by elemental mapping and XRD patterns (Fig. 2c and d and S7†). Fig. 2c2 shows the microscopic image of HgNPs heated to 150 °C, revealing the formation of a few nano-sphere droplets of Hg within RGO, along with stable solid HgNPs, indicating the initiation of melting of HgNPs. On heating the sample to 250 °C, Fig. 2c3, bigger spherical micro-droplets of Hg were observed as a result of melting of the mercury particles are indeed released from its cage in the RGO, which then coalesce. At that temperature, the Hg moieties escape from the graphene layers, as shown in Fig. S7c1–c6.† On further heating to 350 °C, the disappearance of the Hg microspheres was observed, indicating their evaporation since that temperature is close to the boiling point of metallic mercury, Fig. 2c4. In addition, the XRD patterns obtained from the HgNPs samples heated to various temperatures provide strong validation of the scenario described above, which involves melting of the solid Hg nanoparticles. In curve (iii) of Fig. 2d, the appearance of crystalline peaks at 2θ = 41° demonstrates the crystalline nature of the HgNPs samples, even after heating up to 150 °C. After further heating to 250 and 350 °C, the characteristic crystalline peaks of the solid Hg nanoparticles disappear and, simultaneously, a broad peak starts to appear towards lower 2θ = 29°. Therefore, the thermal study of HgNPs by DSC and XRD, SEM analysis at different temperatures reveal significant stability of the solid Hg nanoparticles, even at elevated temperatures up to 150 °C. Furthermore, the HgNPs are highly stable under ambient conditions in the laboratory and there is no noticeable change even after one and a half year time (Fig. S7(e)†).
Synchrotron based X-ray absorption near-edge structure (XANES) spectroscopy measurements were performed in order to understand the reason for the existence of the solid nature as well as the chemical state of the as synthesized HgNPs dispersed on RGO.41,42Fig. 3a and b shows significant variations in Hg LIII-edge XANES spectra and the inflection point of difference (IPD) at their first derivative curves. The inflection points related to the mercury pre-adsorption edge are less pronounced for the Hg+ species (Hg2Cl2), and more pronounced for the Hg2+ species (HgO, HgCl2 and Hg(AC)2). None of the XANES data for the Hg/RGO composites displays the obvious pre-edge features of the reference standards, indicating the significant different Hg local structure and bonding chemistry compared to any of the reference species. The HgNPs spectra looks more or less similar to elemental liquid Hg0, but with a slight deviation in the energy shift and more oscillation at the post-edge than liquid Hg0, indicating partial oxidation of Hg due to charge transfer between RGO and Hg. Such a partial charge transfer between the Hg and the RGO sheets is clearly supported by the extended theoretical study (see below). As shown in Fig. 3b and Table S1,† the magnitude of the IPD for the HgNPs (8.2 eV) is lower when compared to HgO (13.3 eV) and other oxidized mercury species, which confirms that during the sonochemical synthesis there is no formation of HgO in the aqueous media (more details are provided in the ESI†).
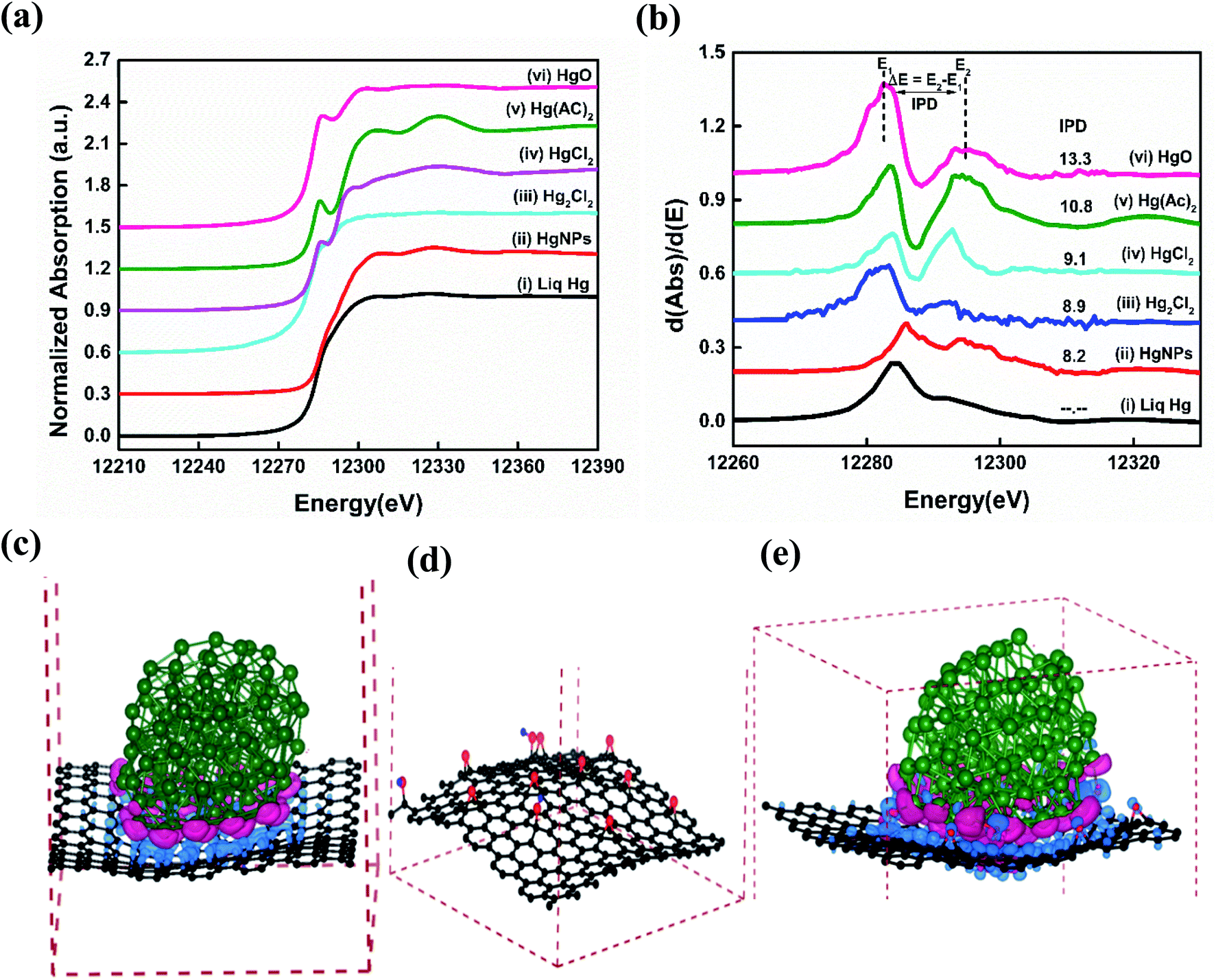 | ||
| Fig. 3 Hg LIII-edge (a) XANES spectra and (b) their corresponding first-derivative curves of as synthesized HgNPs (curve (ii)) and reference mercury compounds (liq Hg (curve (i)), Hg2Cl2 (curve (iii)), HgCl2 (curve (iv)), Hg(OOCCH3)2 (Hg(AC)2) (curve (v)), HgO (curve (vi))). (c) Charge accumulation (shown by cyan colour) and depletion (represented by magenta colour) of relaxed structure of Hg particles comprised of 122 atoms and graphene sheets containing 240 carbon atoms, (d) relaxed structure of RGO containing 12 oxygen and 3 hydrogen atoms to the graphene sheets for 9 epoxy and 3 OH groups per 240 carbon atoms and (e) charge accumulation (shown by cyan colour) and depletion (represented by magenta colour) of relaxed structure consisting of Hg particles comprised of 122 atoms, 9 epoxy and 3 OH groups per 240 carbon atoms. The iso-surface value is set to 0.003 e Å−3. Hg, C, O and H atoms are shown by green, black, red and blue spheres, respectively. Doted solid maroon colour lines represent cell boundaries. | ||
To further examine the atomistic origin of the Hg/RGO composites stability, ab initio density functional theory calculations (fully discussed in the ESI† and Experimental section) were carried out as summarized in Fig. 3c–e and S8.† We used models of Hg particles of 122 atoms and RGO sheets consisting of graphene sheet of 240 C atoms, and 12 oxygen and 3 hydrogen atoms added to the graphene sheets to obtain 9 epoxy and 3 OH groups per 240 carbon atoms. It was found that 10.95 e (0.042 e per atom (RGO)) is transferred to the RGO from the Hg particle in the model. The interactions between Hg and RGO composite moieties lead to the stabilization of the system (relative to isolated Hg and RGO) of −0.22 eV per atom for each Hg/RGO cluster. The nano-dispersed mercury droplets formed during the sonication process are compressed onto the RGO surfaces and undergo electronic interactions with the RGO. There are obvious electronic interactions with a slight charge transfer between the mercury (donor) and the graphene (acceptor), as obtained from the experimental work (XANES) and the calculations. These ionic interactions give rise to electrical polarization and together with the van der Waals interactions explain the additional stabilization, the solid-state formation of these HgNPs and explain the relatively high melting point. The whole synthesis of such moieties should be facilitated by the nature of sonication: dispersing materials, forming nano-droplets in solution phase and the cavitation phenomena that can promote highly energetic, high pressure, local interactions on the nano-scale. More on the mechanism of the formation and embedment of the solid HgNPsin RGO and their stabilization is explained in the ESI.†
The optical properties of HgNPs were identified by measuring their absorbance using UV-Vis reflectance spectrometry. The spectrum (Fig. S9,† curve (i)) displays an absorption peak at λmax ∼ 284 nm assigned to nanoscale solid mercury particles, and agrees well with the calculated UV-absorbance values and the measured plasmon resonance of Hg.7,11 Additional spectral characterizations of bare RGO and HgNPs samples were performed using FTIR, XPS and Raman spectroscopies, presented in Fig. S10–S13.†
Electrochemical measurements
The electrochemical behaviour of electrodes comprising HgNPs were explored for reversible oxidation and reduction reactions, electrochemical hydrogen reduction, and reversible electro-deposition, and were compared with bare GC electrodes and GC electrodes loaded with RGO or liquid mercury (electrochemical cell setups shown in Scheme S2†). Fig. 4 describes the comparative electrochemical response (CV, EIS) of the electrodes in aqueous solutions containing [Fe(CN)6]4−/3− red-ox moieties. The Fe(II)/Fe(III) red-ox couple serves as a best electrochemical benchmark probe for characterization of electrodes, due to its close-to-ideal outer sphere reversible behaviour. Measuring these electrodes with the appropriate probe provides qualitative information about heterogeneous electron-transfer rates, the nature of red-ox behaviour of the electrodes and their electrochemical interactions with solution species.22,43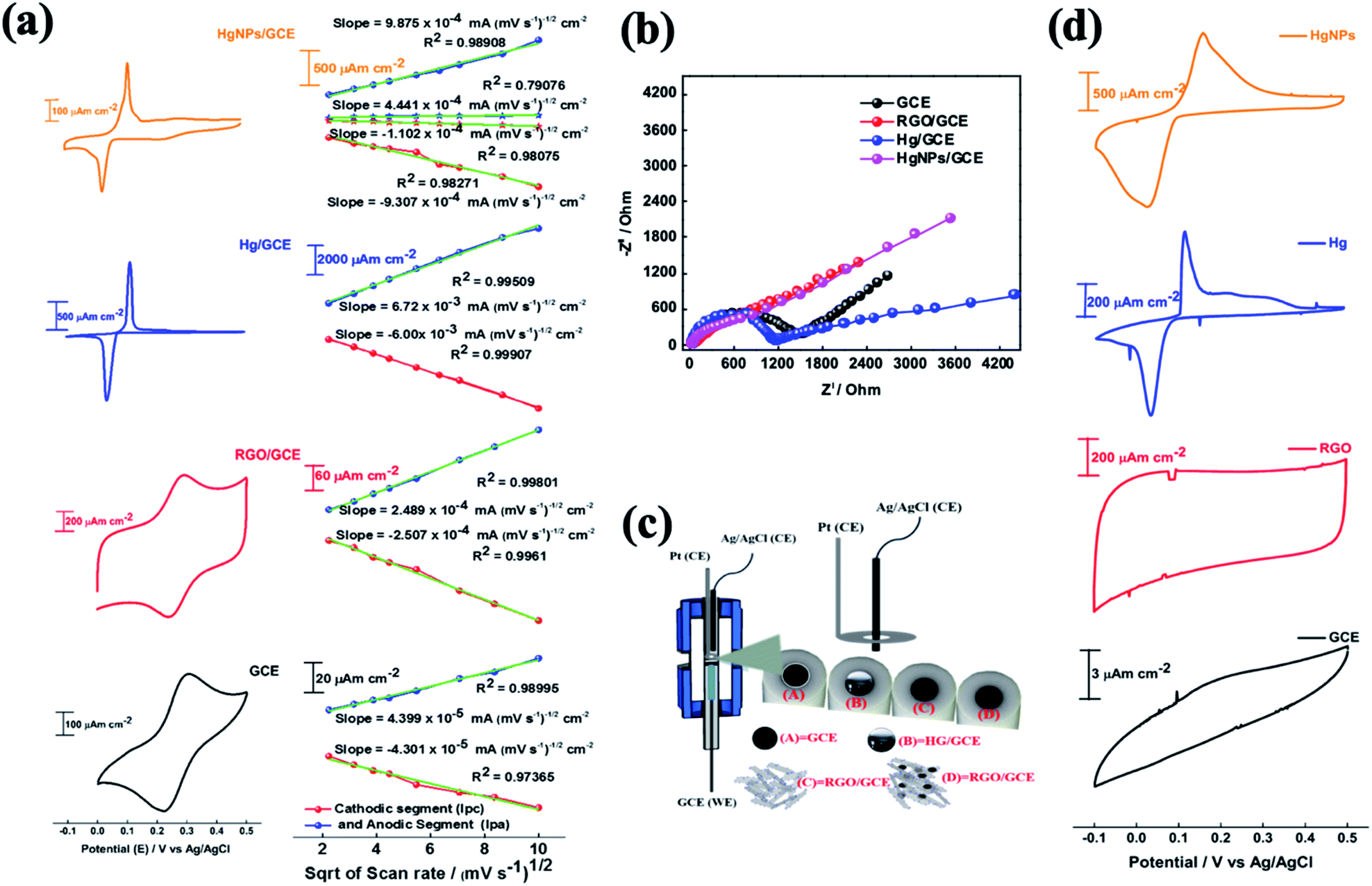 | ||
| Fig. 4 (a) Comparative voltammograms of Fe(II)/Fe(III) red-ox reactions on electrodes (GC, RGO/GC. Hg/GC and HgNPs/GC) at scan rate of 5 mV s−1vs. Ag/AgCl, and their corresponding Randles–Sevcik plots of linear dependence of the anodic and cathodic peak current values vs. the square root of the scan rate, (b) corresponding electrochemical impedance spectra (EIS) presented as Nyquist plots and (c) presentation of the three-electrode cell configuration with GC rods as the substrates for the working electrodes, Ag/AgCl/KCl and Pt foils reference and counter electrodes, respectively and 1 M aqueous KCl solutions containing 2 mM [Fe(CN)6]4−/3− (1:1). (d) Comparative voltammograms of Zn deposition and dissolution for these electrodes at scan rate of 5 mV s−1vs. Ag/AgCl: bare GCE and GCE-modified electrodes in 1 M aqueous KCl solutions containing 10 ml of Zn(NO3)2. Ag/AgCl/KCl and Pt wire served as reference and counter electrodes respectively. | ||
The cyclic voltammograms (CV) of the four working electrodes (GCE, RGO/GCE, Hg/GCE and HgNPs/GCE) were recorded at various scan rates from 5 to 100 mV s−1 within the potential window −0.1 to 0.5 V vs. Ag/AgCl, as shown in Fig. S14–S17.† The RGO/GC electrode in Fig. 4a shows excellent redox behaviour (oxidation peak potential Epa = 0.29 V, reduction peak potential Epc = 0.24 V, Δ[Epa − Epc] = 0.053 V of the Fe(II)/Fe(III) couple compared to the bare GC electrode (Δ[Epa − Epc] = 0.086 V), probably due to their high specific surface area and fast charge and diffusion kinetics (see data in Tables S2, S3, Fig. S14 and S15†). The liquid Hg/GC electrode shows oxidation and reduction peaks at 0.115 V and 0.036 V respectively, thus exhibiting a pronounced cathodic shift (>0.2 V) in the electrochemical reaction potentials, with Δ[Epa − Epc] = 0.079 V (Fig. 4, see also data in Fig. S16†) and Table S4† which complete the comparison among the various electrodes). HgNPs/GC electrodes (Fig. 4a, S17 and Table S5†) show red-ox peaks at nearly the same potentials as the liquid Hg/GC electrodes. The cathodic peak appears at around 0.025 V, 0.01 V lower than the Hg/GC electrodes. Despite the very small amount of mercury in the HgNPs/GC electrodes, the electrochemical behaviour is dominated by Hg nanoparticles rather than RGO. RGO/GC electrodes show the red-ox response of the probe at higher potentials, but this response is very small compared to that of the Hg nano-particles in these electrodes. It is also significant that the peak currents of the HgNPs/GC electrodes are only five times smaller than that of the Hg/GC electrodes, while each electrode typically contains 5–10 μg of mercury compared to around 0.5–1.0 g of mercury in the Hg/GC electrodes. The four types of electrodes exhibit somewhat different Δ[Epa − Epc] values, however, they are comparable and reflect nearly reversible behaviour of the Fe(II)/(III) red-ox couple.
The effect of scan rate on the electrodes' voltammetric behaviour is presented in Fig. S14–S17,† suggests a diffusion-controlled kinetics. This is clearly demonstrated by the linear dependence of the CV peaks currents on the square root of the scan rates, as shown in Fig. 4a. The diffusion coefficient for all the samples was calculated using the Randles–Sevcik (RS) equation22 (for more detailed calculations, see materials and methods). The diffusion coefficients obtained for the GC, RGO/GC, Hg/GC, HgNPs/GC electrodes for the anodic reaction (8.7 × 10−7, 2.8 × 10−6, 1.95 × 10−5 and 4.2 × 10−5 cm2 s−1, respectively) and for the cathodic reaction (8.3 × 10−7, 2.9 × 10−6, 1.55 × 10−5, 3.7 × 10−5, respectively) (see Tables S2–S6 in the ESI†) are comparable and demonstrate the general reversible character of the Fe(CN)64−/3− red-ox reaction. The significance of the calculated diffusion coefficient values cannot be discussed on the quantitative level because the actual relevant active surface area of these electrodes is not clear. Hence, these values can be discussed only on the qualitative level (as presented in the ESI†). The calculated diffusion coefficient for HgNP electrodes is very high and distinct, because it may demonstrate a fast kinetics due to the possible unique electro-catalytic behaviour of the solid mercury nano-particles which influences the response. The catalytic and electrocatalytic properties of the solid HgNPs confined in RGO sheets reported in the present work is aimed at their general characterization and proof of their solid state as its main purpose.
The electrochemical impedance spectra (EIS) of these electrodes provide a further information about the dominant time constants of their reactions.44Fig. 4b shows their comparative Nyquist plots, measured under the same conditions (cells, solutions, equilibrium at OCV) in the frequency range 1000 kHz to 0.01 Hz with 5 mV amplitude. The spectra of the GC and Hg/GC electrodes are similar, showing well developed and separated semicircles and linear domains (Fig. 4b and S18†), reflecting the fact that the electrodes' surface for the electrochemical reaction is nearly their plane geometric area. Hence, the electrochemical response for the classic probe we used is simple, reflecting a nice separation between the major reaction time constants (interfacial charge transfer and diffusion). There are interesting differences between the impedance spectra of the GC and Hg/GC, however, they cannot be discussed here. The spectra of RGO/GC and the HgNPs (confined in RGO)/GC are also similar but differ from the spectra of the GC and Hg/GC electrodes. They exhibit a linear shape and do not show a developed semi-circle at the high frequency domain, reflecting dominating mass transport control, probably due to the very high surface area and catalytic properties. As shown in Fig. S18,† the low-frequency domains in all the impedance spectra can be considered as Warburg-type impedances which reflect mass transport (diffusion) behaviour. We further demonstrate the electrochemical uniqueness of the HgNPs electrodes using voltammograms of the Zn2+/Zn couple in solutions containing 10 mM Zn(NO3)2 in the potential range −0.1 to 0.5 vs. Ag/AgCl/KCl, at scan rates range between 5 and 100 mV s−1 (Fig. 4d). The Hg-containing electrodes show an electrochemical response related to a reversible deposition and dissolution of Zn, while the GC and RGO/GC electrodes do not show any pronounced electrochemical response. The response of the HgNPs/GC electrodes is very pronounced, showing currents three times higher and total reaction charges nearly ten times higher than the Hg/GC electrodes. This comparative response seems to confirm the electro-catalytic properties of the solid HgNPs and encourages further detailed electrochemical investigation.
Furthermore, the unique electrochemical response of the solid Hg nanoparticles was confirmed by preliminary studies of hydrogen evolution reactions (HER). Inspired by the pioneering work of Frumkin and Bockris,45,46 comparative HER studies were conducted for the electro-reduction of protons (H+) at bare GC and modified GC electrodes with liquid Hg, RGO and HgNPs. In 1950, Bockris46 demonstrated the effect of the Hg substrates on the rate's nature of HER, working at −70 °C and +20 °C (hence with both solid and liquid states of mercury). These studies demonstrated a clear difference in over-potential for HER between solid and liquid Hg. The over-potential at a current density of 10−4 A cm−2 decreased by about 15 mV at the freezing point of mercury. In turn, α in the Tafel equations related to these measurements increased pronouncedly at the melting point of the Hg electrodes.
Fig. 5 presents preliminary results of comparative studies of HER by linear sweep voltammetry (LSV) in 0.5 M H2SO4 solution at a potential scanning rate of 10 mV s−1 using GC, Hg/GC, RGO/GC and HgNPs/GC working electrodes. Ag/AgCl and graphite rod were used as quasi-reference and counter-electrode in the three-electrode cell shown in Fig. 5d. Fig. 5a–c display the potentiodynamic behaviour, relevant Tafel plots of HER and comparative bar graph showing the over-potentials required to obtain HER at five different current densities with the four electrode systems. The results presented in Fig. 5a–c demonstrate the kinetic superiority, indicating pronounced electro-catalytic properties of the active mass comprising solid Hg nano-particles confined in RGO sheets, in terms of lowest HER onset potentials, better Tafel plot and I vs. E response. The onset potential for bare GCE, Hg, RGO and HgNPs are −900, −550, −306 and −210 mV vs. RHE respectively. These onset potentials were measured at the points where the current density reaches 1 mA cm−2 (Fig. 5a and S22†). The over-potentials are 1120, 872, 670 and 472 mV vs. RHE for bare GCE, Hg, RGO and HgNPs. Their values at different current densities are shown in Fig. S22.† These values are in good agreement with literature values for GCE, Hg and RGO.
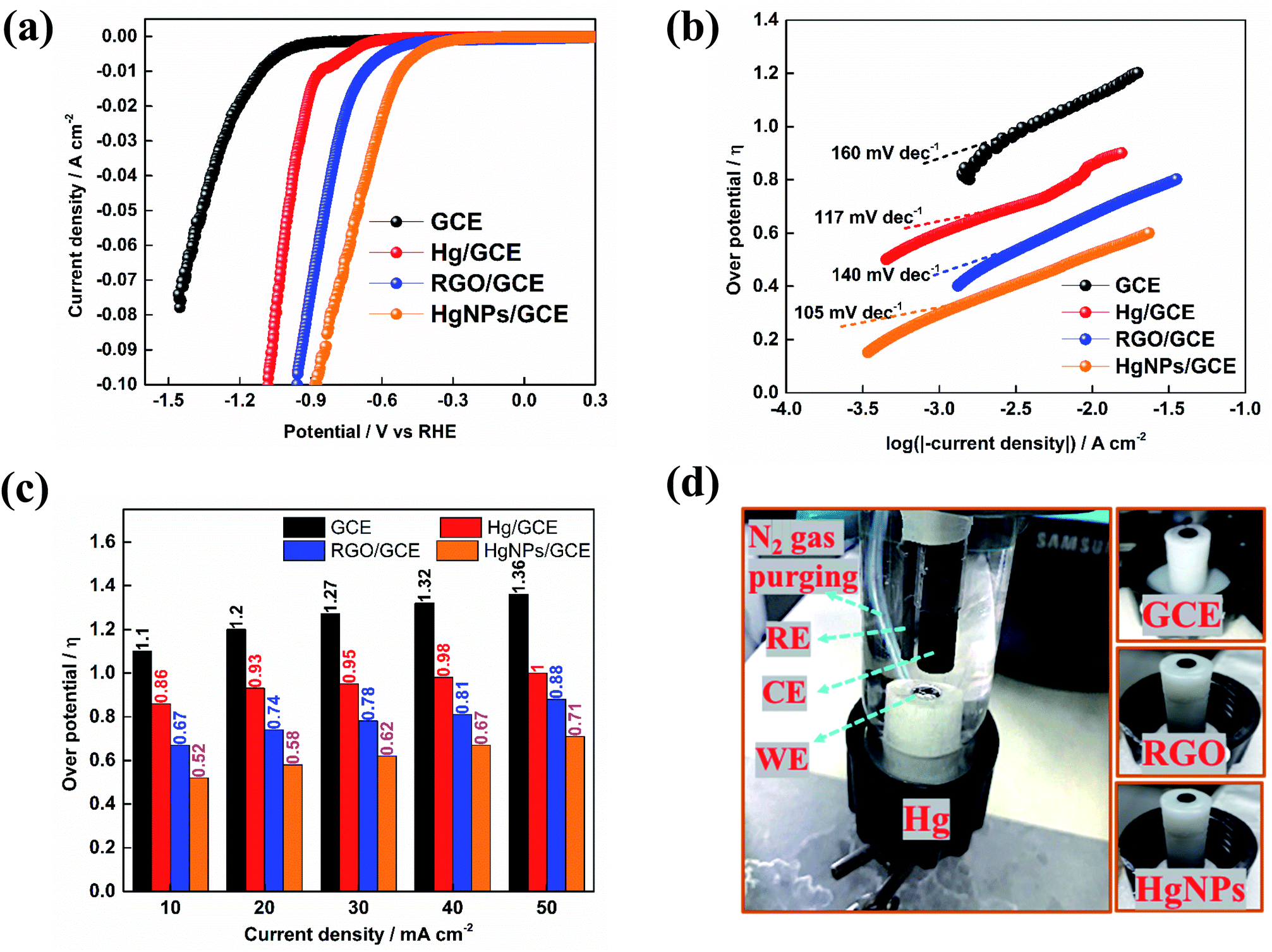 | ||
| Fig. 5 Results of HER study by linear sweep voltammetry using GC, Hg/GC, RGO/GC and HgNPs/GC electrodes in 0.5 M H2SO4 solution at 10 mV s−1. (a) Potentio-dynamic behavior of HER with the four electrodes (b) Tafel plots, (c) histograms comparing the onset overpotential and (d) photograph of the electrochemical cell configuration for HER study. | ||
In the present study, the possible electro-catalytic mechanism of the HER process was revealed using Tafel slope analysis.45,47–50 Tafel plots of the over-potentials versus logarithmic current density are shown in Fig. 5b and are useful for the quantitative kinetic analysis of the HER. The Tafel slopes were determined by fitting the linear portions of the Tafel plots to the Tafel equation (η = blogj + a, where j is the current density, a is the empirical coefficient and b is the Tafel slope). In theory, the mechanism of (conversion of H+ to H2) HER process in aqueous acidic solutions follows three basic steps, two electrochemical and one chemical. In the first step, the electrochemical hydrogen adsorption reaction takes place (H3O+ + e− → Hads + H2O) and is referred to as the Volmer reaction. The second step follows the electrochemical desorption path (Hads + H3O+ + e− → H2 + H2O) and is referred to as the Heyrovsky reaction. The final step involves the recombination path Hads + Hads → H2 and is referred to as the Tafel reaction. The evolution of hydrogen follows either the Volmer–Heyrovsky or the Volmer–Tafel reaction mechanisms. In light of previous reports, the Tafel slope values of ≈30 mV dec−1, ≈40 mV dec−1 and ≈120 mV dec−1 represents the Tafel, Heyrovsky and Volmer reactions, respectively as the rate determining steps of HER. The Tafel value obtained for Hg and HgNPs at higher negative potentials is close to the theoretical value of 118 mV dec−1, which indicates that HER proceeds via a Volmer–Heyrovsky type mechanism, while the Tafel step is negligible.45–48,50
In Fig. 5b, a straight line with slope of 117 and 105 mV dec−1 is obtained for Hg and HgNPs, respectively, at 30 to 40 mV above the onset potentials. The Tafel value obtained for Hg and HgNPs at higher negative potentials is close to the theoretical value of 118 mV dec−1, which indicates that HER proceeds via a Volmer–Heyrovsky mechanism, while the Tafel step is negligible.23,48,50 These kinetic studies indicate that the rate of HER is controlled by the Volmer step mechanism at low negative potentials, a discharge step that converts protons into adsorbed hydrogen atoms on a catalytic surface. The remarkable electrocatalytic activity of hydrogen evolution of HgNPs (onset 472 mV and Tafel slope of 105 mV dec−1) are comparable with metal nanoparticles (Pd, Ru, Au, Co, No, Cu, etc.) loaded in carbon in 0.5 M H2SO4 solutions, as shown in Table S7 of the ESI.† It is important to note that the extent of HER catalysis by the HgNPs described herein requires further intensive studies in order to quantify the effect and examine the practical horizons of these new composite matrices in electro-catalysis.
Hence, three independent studies of electrochemical reactions: the Fe3+/Fe2+ couple, reversible electrodeposition of zinc, and proton reduction to hydrogen gas, clearly showed unique, favorable and promising behavior of the new electro-active composite materials containing solid mercury nanoparticles developed in this work.
Conclusions
In the present study, we were successful in preparing for the first time solid Hg nanoparticles stabilized by RGO sheets which are stable at room temperature, using a relatively simple synthesis by applying ultrasonic radiation. The analytical work presented herein fully proves this conclusion. XANES measurements confirmed a partial charge transfer between the HgNPs and the RGO sheets, what explains their solid state at RT and their high melting point. The possible charge transfer between Hg and RGO and the stabilizing partial ionic interactions between them are substantiated by ab initio calculations that clearly confirm the possibility of stabilization by electrons transfer from the HgNPs and the RGO sheets. Hence, the sonication and resulting cavitation phenomenon lead to a very fine dispersion of the mercury in the aqueous solutions and promote collisions between nano-droplets of mercury and RGO sheets. A partial charge transfer during such collisions forms stabilized Hg–RGO clusters that enables the crystallization of the partially ionized HgNPs. Various characterisation techniques were employed to study the properties of the newly synthesized nanomaterials. The as-formed solid Hg nanoparticles show X-ray diffraction patterns with crystalline peaks at room temperature revealing a rhombohedral phase of mercury. The solid mercury nanoparticles exhibit a clear rhombohedral morphology with a particle size ranging from 50–100 nm characterised by SEM and HRTEM. The optical properties of the as-synthesized solid Hg nanoparticles reveal UV plasmon absorption at 284 nm wavelength. The DSC results related to the HgNPs are well correlated to the XRD and SEM analyses of the HgNPs measured at different elevated temperatures, revealing significant stability of mercury nanoparticles up to 150 °C. It is important to note that while we provide herein clear evidences about the formation of a new, apparently stable, solid state form of mercury at ambient conditions, there are still several interesting open mechanistic questions. A most important point left for close examination is the effect of particles size on the stability and thermal response of the Hg nanoparticles. Also, it is important to understand the impact of the substrate's nature on the possible stabilization of SSHgNPs. Work in these directions are in progress, but its description is beyond the scope of this paper, which main focus is to provide strong enough evidences for the phenomenon we report herein. Electrochemical studies in three independent routes: exploring a simple reversible outer sphere reaction of the Fe3+/Fe2+ red-ox couple, reversible Zn deposition/dissolution reaction and hydrogen evolution reaction in an acidic solution, reveal unique electrochemical behaviour of the solid-state Hg nanoparticles stabilized by the RGO nano-sheets with clear electrocatalytic properties. Since this paper is aimed at the first presentation of these new composite materials, the electrochemical studies were limited to preliminary qualitative analyses. However, they indicate that these materials may have very important and useful catalytic activity, what makes them very promising and should promote further intensive basic scientific and practical studies.Author contributions
Study concept, experimental work, data analysis and paper writing were equally done by V. H. K., T. R. P, D. A. and A. G. AC-HAADF-STEM measurements and data processing were carried out by B. L. and F. L. D. XANES spectra measurements and analysis were conducted by G.-L. X, C.-J. S. and K. A. Ab initio density functional theoretical studies were performed by A. S. and I. G.Conflicts of interest
There are no conflicts to declare.Acknowledgements
V. K. Harika and T. R. Penki thank Dr Ilana Perelshtein, Dr Merav Tsubery, Dr Yuri Koltypin, Dr Netanel Shpigel for their helpful discussions. B. Loukya and F. L. Deepak acknowledges financial support by the N2020: Nanotechnology based functional solutions (NORTE-45-2015-02). Research at the Argonne National Laboratory was funded by the U.S. Department of Energy (DOE), Vehicle Technologies Office. This research used resources of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, and was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357, and the Canadian Light Source and its funding partners. The authors thank Drs E. D. Crozier and R. A Gordon at Simon Fraser University Canada for providing the Hg XANES reference data.References
- J. D. Blum, Nat. Chem., 2013, 5, 1066 CrossRef CAS.
- J. Jeevanandam, A. Barhoum, Y. S. Chan, A. Dufresne and M. K. Danquah, Beilstein J. Nanotechnol., 2018, 9, 1050–1074 CrossRef CAS.
- W. Lin, Chem. Rev., 2015, 115, 10407–10409 CrossRef CAS.
- K. Watanabe, D. Menzel, N. Nilius and H. J. Freund, Chem. Rev., 2006, 106, 4301–4320 CrossRef CAS.
- F. J. Heiligtag and M. Niederberger, Mater. Today, 2013, 16, 262–271 CrossRef CAS.
- C. Wang and D. Astruc, Prog. Mater. Sci., 2018, 94, 306–383 CrossRef CAS.
- J. A. Creighton and D. G. Eadon, J. Chem. Soc., Faraday Trans., 1991, 87, 3881–3891 RSC.
- A. Henglein and M. Giersig, J. Phys. Chem. B, 2000, 104, 5056–5060 CrossRef CAS.
- A. Henglein and C. Brancewicz, Chem. Mater., 1997, 9, 2164–2167 CrossRef CAS.
- L. Katsikas, M. Gutiérrez and A. Henglein, J. Phys. Chem., 1996, 100, 11203–11206 CrossRef CAS.
- G. V. Ramesh, M. D. Prasad and T. P. Radhakrishnan, Chem. Mater., 2011, 23, 5231–5236 CrossRef CAS.
- A. Gedanken, E. Luvchik, J. M. Calderon-Moreno, N. Veglio and J. L. I. Tamarit, Adv. Mater., 2008, 20, 1000–1002 CrossRef.
- J. Polte, CrystEngComm, 2015, 17, 6809–6830 RSC.
- H. Kang, J. T. Buchman, R. S. Rodriguez, H. L. Ring, J. He, K. C. Bantz and C. L. Haynes, Chem. Rev., 2019, 119, 664–699 CrossRef CAS.
- W. C. Lee, K. Kim, J. Park, J. Koo, H. Y. Jeong, H. Lee, D. A. Weitz, A. Zettl and S. Takeuchi, Nat. Nanotechnol., 2015, 10, 423–428 CrossRef CAS.
- Z. Wang, Y. Ping, Q. Fu and C. Pan, MRS Adv., 2018, 3, 849–854 CrossRef CAS.
- V. K. Harika, V. B. Kumar and A. Gedanken, Ultrason. Sonochem., 2018, 40, 157–165 CrossRef CAS.
- V. B. Kumar, Y. Koltypin, A. Gedanken and Z. Porat, J. Mater. Chem. A, 2014, 2, 1309–1317 RSC.
- J. Gajdar, E. Horakova, J. Barek, J. Fischer and V. Vyskocil, Electroanalysis, 2016, 28, 2659–2671 CrossRef CAS.
- J. Barek, Port. Electrochim. Acta, 2013, 291–295 CrossRef CAS.
- J. Barek and J. Zima, Electroanalysis, 2003, 15, 467–472 CrossRef CAS.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS.
- P. Quaino, F. Juarez, E. Santos and W. Schmickler, Beilstein J. Nanotechnol., 2014, 5, 846–854 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- C. Zhang, W. Lv, X. Xie, D. Tang, C. Liu and Q. H. Yang, Carbon, 2013, 62, 11–24 CrossRef CAS.
- N. M. S. Hidayah, W. W. Liu, C. W. Lai, N. Z. Noriman, C. S. Khe, U. Hashim and H. C. Lee, AIP Conf. Proc., 2017, vol. 1892, 150002 CrossRef.
- J. M. Soler, E. Artacho, J. D. Gale, A. García, J. Junquera, P. Ordejón and D. Sánchez-Portal, J. Phys.: Condens. Matter, 2002, 14, 2745–2779 CrossRef CAS.
- N. Troullier and J. L. Martins, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 43, 1993–2006 CrossRef CAS.
- M. J. van Setten, M. Giantomassi, E. Bousquet, M. J. Verstraete, D. R. Hamann, X. Gonze and G. M. Rignanese, Comput. Phys. Commun., 2018, 226, 39–54 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- J. R. G. Sander, B. W. Zeiger and K. S. Suslick, Ultrason. Sonochem., 2014, vol. 21, 1908–1915 CrossRef.
- V. G. Pol, H. Grisaru and A. Gedanken, Langmuir, 2005, 21, 3635–3640 CrossRef CAS.
- V. G. Pol, M. Motiei, A. Gedanken, J. Calderon-Moreno and Y. Mastai, Chem. Mater., 2003, 15, 1378–1384 CrossRef CAS.
- H. Kim and K. Suslick, Crystals, 2018, 8, 280 CrossRef.
- N. Mirsaleh-Kohan, A. Fischer, B. Graves, M. Bolorizadeh, D. Kondepudi and R. N. Compton, Cryst. Growth Des., 2017, 17, 576–581 CrossRef CAS.
- M. R. Baren, J. Phase Equilib., 1996, 17, 122–128 CrossRef CAS.
- K. Takemura, S. Nakano, Y. Ohishi, Y. Nakamoto and H. Fujihisa, Mater. Res. Express, 2015, 2, 16502 CrossRef.
- K. Yasui, T. Tuziuti and W. Kanematsu, Phys. Rev. E, 2016, 94, 1–13 CrossRef.
- V. A. Drebushchak, J. Therm. Anal. Calorim., 2012, 109, 545–553 CrossRef CAS.
- M. Takaoka, T. Yamamoto, N. Takeda, K. Oshita, T. Tanaka and T. Uruga, AIP Conf. Proc., 2007, 882, 283–285 CrossRef CAS.
- F. E. Huggins, N. Yap, G. P. Huffman and C. L. Senior, Fuel Process. Technol., 2003, 82, 167–196 CrossRef CAS.
- E. P. Randviir and C. E. Banks, Anal. Methods, 2013, 5, 1098–1115 RSC.
- B.-Y. Chang and S.-M. Park, Annu. Rev. Anal. Chem., 2010, 3, 207–229 CrossRef CAS.
- A. Frumkin, Faraday Discuss., 1947, 1, 57–67 RSC.
- J. O. M. Bockris, R. Parsons and H. Rosenberg, Trans. Faraday Soc., 1951, 47, 766–772 RSC.
- A. Eftekhari, Int. J. Hydrogen Energy, 2017, 42, 11053–11077 CrossRef CAS.
- H. Ogihara, M. Fujii and T. Saji, RSC Adv., 2014, 4, 58660–58663 RSC.
- J. O. M. Bockris and R. Parsons, Trans. Faraday Soc., 1949, 45, 916–928 RSC.
- M. Jafarian, M. Behazin, I. Danaee and F. Gobal, Res. J. Chem. Sci., 2013, 3, 56–63 CAS.
Footnotes | |||||||||
| † Electronic supplementary information (ESI) available: Experimental schematic diagrams, several analytical, theoretical, electrochemical characterisations and their detailed analysis. Live movies of sonochemical synthesis and electrochemical live experimental measurements. See DOI: 10.1039/d0sc06139e | |||||||||
| ‡ Equal contribution. | |||||||||
§ Calculation of diffusion coefficient. The diffusion coefficient for all the samples was calculated using the Randles–Sevcik (RS) equation as follows:22
Substituting the values in eqn (2), we obtain
| |||||||||
¶ Standard potential conversion in hydrogen evaluation reaction. All the potentials are referred to the reference hydrogen electrode (RHE) potential, by an appropriate translation of the potentials actually measured vs. Ag/AgCl RE as follows.
In 0.5 M H2SO4 solution, pH = 0, then
The onset potentials were measured at the point where the current density reaches 1 mA cm−2 (Fig. S22†). |
| This journal is © The Royal Society of Chemistry 2021 |