
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A 2,2′-diphosphinotolane as a versatile precursor for the synthesis of P-ylidic mesoionic carbenes via reversible C–P bond formation†
Hannah K.
Wagner
,
Hubert
Wadepohl
and
Joachim
Ballmann
 *
*
Anorganisch-Chemisches Institut, Universität Heidelberg, Im Neuenheimer Feld 276, D-69120 Heidelberg, Germany. E-mail: joachim.ballmann@uni-heidelberg.de
First published on 29th January 2021
Abstract
A metal-templated synthetic route to cyclic (aryl)(ylidic) mesoionic carbenes (CArY-MICs) featuring an endocyclic P-ylide is presented. This approach, which requires metal templates with two cis-positioned open coordination sites, is based on the controlled cyclisation of a P,P′-diisopropyl-substituted 2,2′-diphosphinotolane (1) and leads to chelate complexes coordinated by a phosphine donor and the CArY-MIC carbon atom. The C–P bond formation involved in the former partial cyclisation of 1 proceeds under mild conditions and was shown to be applicable all over the d-block. In the presence of a third fac-positioned open coordination site, the P–C bond formation was found to be reversible, as shown for a series of molybdenum complexes. DFT modelling studies are in line with an interpretation of the target compounds as CArY-MICs.
Introduction
N-Heterocyclic carbenes (NHC),1 mesoionic carbenes (MIC)2 and cyclic (alkyl)(amino) carbenes (CAAC)3 are among the most widely used carbene ligands. Their stability mainly arises from the presence of one or more endocyclic nitrogen atoms stabilising the formally divalent carbon atom via donation into its empty p-orbital.4 Although spatial protection of the carbene centres certainly plays a role for isolating the free carbenes, the introduction of steric bulk is occasionally not required as metal-templated synthetic routes and in situ trapping protocols are well-established nowadays.5 The emergence of these procedures opened-up new possibilities to modulate the electronic properties of N-containing carbene ligands, for example by replacing the alkyl groups in CAACs for aryl moieties or by introducing an exocyclic ylide as a replacement for one of the nitrogens in NHCs. This approach led to cyclic (amino)(aryl) carbenes (CAArC)6 and cyclic (amino)(ylide) carbenes (CAYC)7 (see Scheme 1), respectively, which have been generated in situ and trapped as their metal complexes. By formally combining CAArCs and P-ylidic CAYCs, a new motif in carbene chemistry comes into reach, namely cyclic (aryl)(ylidic) carbenes (CArYCs), which do not contain an endocyclic nitrogen atom. In these CArYC (see Scheme 1), the quaternary R2C-carbon in Zeng's and Bertrand's CAArC6a is replaced by an ylidic R2P-moiety, which results in a structure with an endocyclic P-ylide rather than an exocyclic P-ylide (cf. Kawashima's CAYC7d shown in Scheme 1). In 2010, Bertrand pointed out that free CArYCs with an endocyclic P-ylide are expected to be unstable,5a which is easily understood considering the corresponding CArY-MIC resonance structure (see Scheme 1). A CArY-MIC of that type is expected to ring-open to afford the unstrained phosphino alkyne (see Scheme 1). This notion, however, provides a blueprint for the metal-templated assembly of CArY-MICs, namely by reversing the ring-opening process. For electron-withdrawing Ph2P-substituted tolanes, such ring-closure reactions are known to afford phosphindolium-based organic materials, usually under harsh reaction conditions.8 The use of more nucleophilic alkyl phosphines has not been explored yet, although these phosphines are expected to attack the alkyne more readily. This attack may be further promoted by using a metal template,8d,8e which is kept in place by means of an appropriately positioned chelate. As explained in the following, we reasoned that iPr2P-substituted 2,2′-diphosphinotolanes are well-suited precursors for the synthesis of the desired CArY-MICs.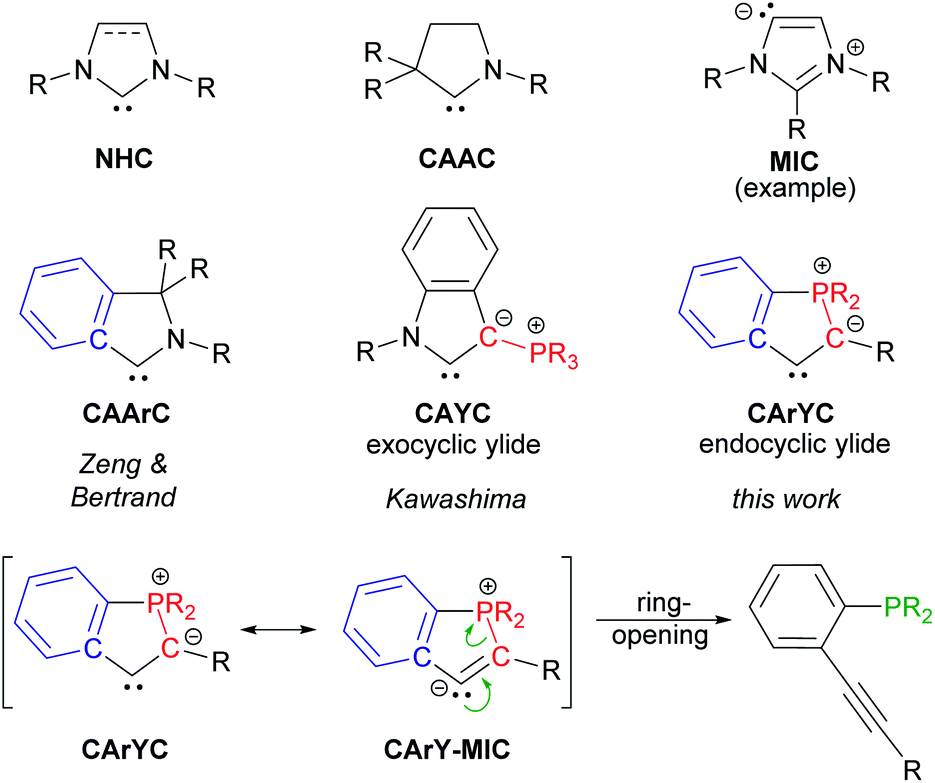 | ||
| Scheme 1 Selected carbene ligands together with the ring-opening of CArY-MICs. | ||
In previous studies, 2,2′-diphosphinotolanes, such as 1 (see Scheme 2) were shown to readily form [PCCP]-pincer type complexes (2), namely via reaction of 1 (or its Ph2P-derivative) with octahedral (or square planar) metal precursors with three accessible mer-arranged coordination sites.9 We have also shown that 1 is prone to a twofold cyclisation to selectively afford the diylide cyclo-1 (see Scheme 2).10 In view of these findings, it seemed possible that the reaction between 1 and a metal template with two cis-positioned open coordination sites may afford the desired κ2-P,C-chelating CArY-MIC complexes via attack of only one phosphine at the central alkyne unit.11 As reported herein, CArY-MICs may indeed be trapped this way.
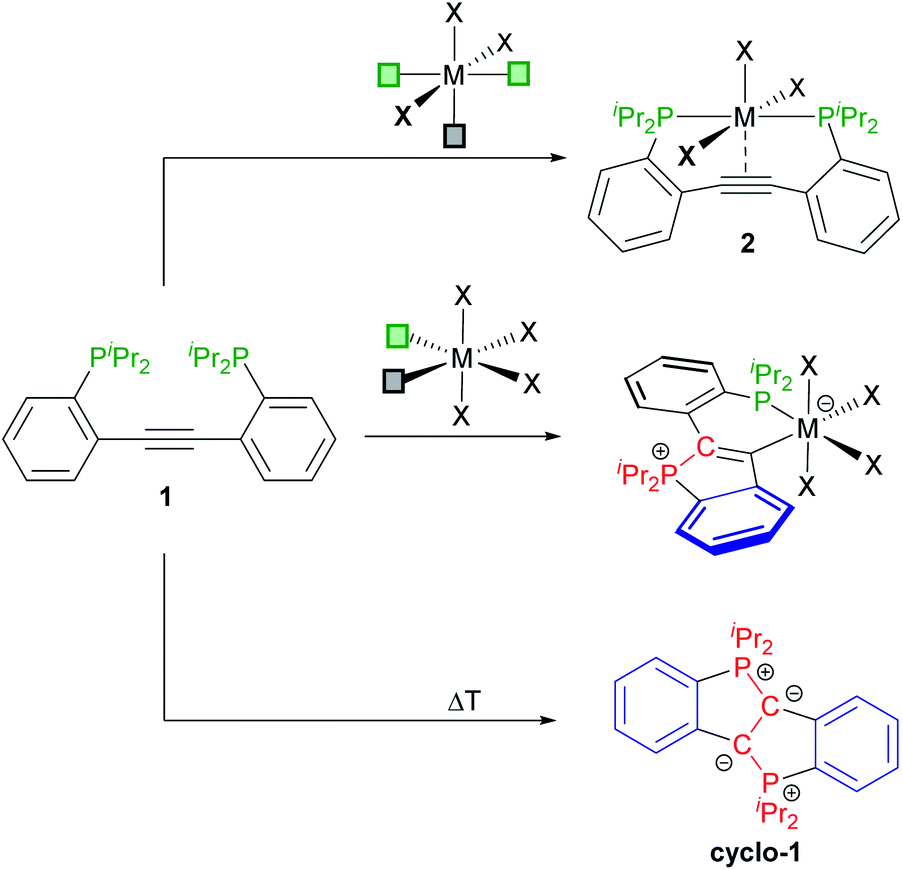 | ||
| Scheme 2 Known routes to [PCCP]-pincer complexes 2 (top) and to the doubly-cyclised diylide cyclo-1 (bottom) together with the κ2-P,C-coordinated CArY-MIC complexes targeted herein (middle).‡ | ||
Results and discussion
Following the approach outlined above, numerous transition metal complexes with two accessible coordination sites were reacted with 1 without limiting ourselves to one specific group of the d-block. This screening led to the expected cyclisation of 1 to afford the desired CArY-MICs under mild reaction conditions (all reactions were conducted at or below r.t.). As illustrated in Scheme 3, one example was isolated for each group in the d-block, either as cationic (3, 8–11) or neutral (4–7, 12) derivative (compounds are numbered according to the group in the periodic table). For the successful synthesis of these CArY-MICs, the use of a non-coordinating solvent (chlorobenzene or CH2Cl2) was found to be mandatory to avoid the formation of coordinatively saturated solvent adducts (e.g. HfCl4(thf)2, which was found to be unreactive towards 1). To ascertain that the C–P bond formation occurred in the anticipated manner, all complexes were characterised by single crystal X-ray diffraction (see Scheme 3, see ESI† for details, see Table 1 for selected metrical parameters). With exception of the yttrium derivative 3, a κ2-P,C-chelate was formed in each case, independent of the coordination environment (tetrahedral, square planar or octahedral). In the case of 3, the phosphine is not bound to the metal. Instead, an agostic ortho-interaction with one aryl-C–H of the CArY-MIC ligand was found (see Scheme 3). This is consistent with the 31P{1H} NMR spectrum of 3, which exhibits a yttrium-coupled resonance at δ = +52.8 ppm and a singlet at δ = −7.7 ppm (cf. δ(31P{1H}) (1) = +4.9 ppm). For the zinc complex 12, a similar situation with one uncoordinated phosphine (31P NMR resonance at δ(31P{1H}) = −7.9 ppm) is observed in solution as judged by NMR spectroscopy. For all other complexes, two (mutually coupled) 31P{1H} NMR resonances are observed between +10 and +63 ppm (see Table 1). For the tantalum derivative 5, the κ1-C- and the κ2-C,P-derivatives are both observed in solution (cf.31P{1H} NMR shifts in Table 1), while two stereoisomers (differing in the orientation of the bromide) were found for the manganese complex 7. In the latter case, only one set of 31P{1H} NMR signals was observed after treatment with AgBF4, but the resulting tetrafluoroborate derivative was not isolated due to its instability. | ||
| Scheme 3 Synthesis of CArY-MIC complexes starting from 1.‡ Each group in the d-block is represented by one crystallographically characterised example. | ||
| Compound (dn) | 3 (d0) | 4 (d0) | 5 (d0) | 6 (d6) | 7 (d6) | 8 (d6) | 9 (d6) | 10 (d8) | 11 (d8) | 12 (d10) |
|---|---|---|---|---|---|---|---|---|---|---|
| a Values in ppm. b In o-Cl2C6D4. c In CD2Cl2. d Two isomers (see text). e In C6D5NO2. f Not detected due to signal broadening. g Not detected unambiguously due to the presence of two isomers. h Not detected due to decomposition over the time of the measurement at −40 °C. i Values in Å, rounded to two digits (see ESI for additional metrical parameters/standard deviations). j Four independent molecules are present in the asymmetric unit (see ESI for details). k Two intrinsic bond orbitals (reminiscent of two d-orbitals) are involved in back-bonding. l Internal binding energy from energy decomposition analysis. m Values at the (3,–1) bond critical point of the C–M bond. | ||||||||||
| δ(31P{1H})a (P1) | 52.8b | 53.9c | 54.6/54.5c,d | 43.2c | 44.1/45.8c,d | 49.8c | 48.2c | 53.2c | 55.3c | 55.0e |
| δ(31P{1H})a (P2) | −7.7b | 10.3c | 32.2/−3.4c,d | 69.2c | 57.5/62.4c,d | 46.5c | 47.7c | 29.6c | 43.2c | −7.9e |
| δ(13C{1H})a (C2) | n.d.f | 220.8c | n.d.g | 245.5c | n.d.g | 214.1c | 208.3c | 198.8c | 195.6c | n.d.h |
| d(P1–C1)i | 1.83 | 1.82 | 1.82 | 1.79 | 1.80 | 1.79 | 1.81 | 1.80 | 1.82 | 1.83–1.84j |
| d(C1–C2)i | 1.37 | 1.37 | 1.37 | 1.38 | 1.37 | 1.39 | 1.37 | 1.37 | 1.36 | 1.36–1.37j |
| d(C2–M)i | 2.47 | 2.36 | 2.26 | 2.11 | 2.08 | 2.07 | 2.05 | 2.06 | 2.08 | 2.04–2.04j |
| IBO σC–M e−(C)/e−(M) | 1.76/0.15 | 1.78/0.12 | 1.78/0.13 | 1.53/0.30 | 1.51/0.34 | 1.36/0.57 | 1.35/0.54 | 1.52/0.36 | 1.49/0.32 | 1.86/0.06 |
| IBO πM–C e−(M)/e−(C) | —/— | —/— | —/— | 1.18,![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1.24k/0.06, 0.07k 1.24k/0.06, 0.07k |
1.46,1.51k/0.04, 0.04k |
1.86/0.05 | 1.87/0.05 | 1.86/0.06 | 1.95/0.02 | —/— |
| ΔEint(EDA)l | −92.6 | −107.7 | −111.5 | −114.2 | −122.7 | −190.2 | −182.1 | −199.1 | −167.5 | 101.6 |
| |Vbcp|/Gbcp(C–M)m | 1.277 | 1.456 | 1.552 | 1.192 | 1.278 | 1.522 | 1.504 | 1.555 | 1.574 | 1.275 |
| H bcp/ρbcp(C–M)m | −0.176 | −0.251 | −0.322 | −0.200 | −0.289 | −0.428 | −0.423 | −0.494 | −0.455 | −0.281 |
| ∇2ρbcp(C–M)m | 0.096 | 0.101 | 0.108 | 0.282 | 0.272 | 0.208 | 0.217 | 0.214 | 0.170 | 0.259 |
With the molecular structures of complexes 3–12 ascertained, their electronic structures were elucidated, in particular with respect to the nature of the C–M interactions and with respect to the bonding situation within the five-membered P-heterocycles. For true ylides (without resonance stabilisation), significantly shortened P–C bonds in the range of 1.63–1.75 Å are commonly observed.12 In the case of complexes 3–12, this shortening is not observed, i.e. the lengths of the P1–C1 bonds (1.81 ± 0.03 Å, see Table 1) are in line with an interpretation as single bonds, which was further confirmed by DFT modelling studies (vide infra). The bonds between the carbenoid carbon atoms (C2) and the (formally) ylidic carbon atoms (C1) are best interpreted as partially delocalised C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds (1.37 ± 0.02 Å, see Table 1, the corresponding CC bond lengths in 2-aryl 1H-indene derivatives range from 1.32–1.42 Å (ref. 13)). Taken together, these crystallographic data support a description of complexes 3–12 as CArY-MICs, which may be denoted with a no-bond and a betaine resonance structure (see Scheme 4). For the betaine resonance structure, a certain degree of backbonding or delocalisation is expected, at least for d1- to d9-configured metals.14 For d0- and d10-configured metals, backbonding is not possible (empty d-shell) or commonly not observed (closed d-shell),15 while hyperconjugative X → C2 interactions are frequently found in these cases (X = metal-bound co-ligand). Keeping in mind that differences between d1- to d9-configured complexes and d0/d10-configured complexes have to be expected, we set out to model all complexes by DFT-calculations.
C double bonds (1.37 ± 0.02 Å, see Table 1, the corresponding CC bond lengths in 2-aryl 1H-indene derivatives range from 1.32–1.42 Å (ref. 13)). Taken together, these crystallographic data support a description of complexes 3–12 as CArY-MICs, which may be denoted with a no-bond and a betaine resonance structure (see Scheme 4). For the betaine resonance structure, a certain degree of backbonding or delocalisation is expected, at least for d1- to d9-configured metals.14 For d0- and d10-configured metals, backbonding is not possible (empty d-shell) or commonly not observed (closed d-shell),15 while hyperconjugative X → C2 interactions are frequently found in these cases (X = metal-bound co-ligand). Keeping in mind that differences between d1- to d9-configured complexes and d0/d10-configured complexes have to be expected, we set out to model all complexes by DFT-calculations.
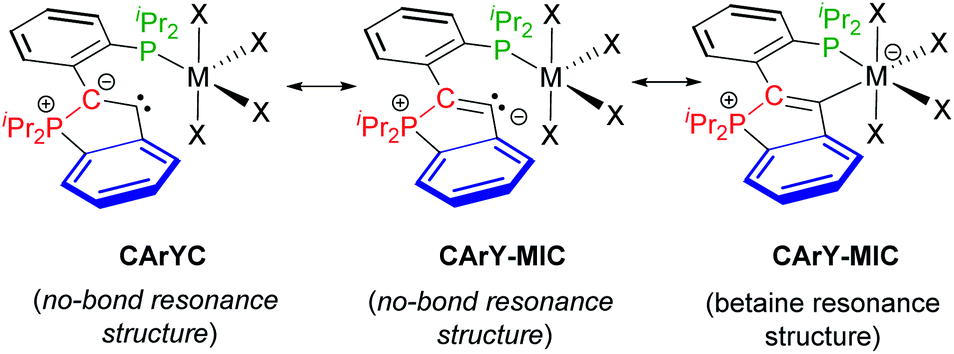 | ||
| Scheme 4 Resonance structures for complexes 3–12. The CArYC no-bond resonance structure with its C1–C2 single bond is considered less important on basis of the observed metrical parameters. The CArY-MIC notation (no-bond and betaine resonance structure) is in line with the crystallographic data and the computational results. Note that a dative C→M bond is implied by the superimposition of both CArY-MIC resonance structures. | ||
The geometry of each complex was optimised on the PBE1PBE/Def2-TZVP level of theory and the bonding situation was examined by means of IBO, EDA-NOCV (re-optimised on the BP86/TZ2P-D3 level) and QTAIM analysis. In all complexes, a σ-symmetric P1–C1 single bond (cf. example shown in Fig. 1-A) and a π-symmetric C1C2 double bond (cf. example shown in Fig. 1-B) were found by IBO analysis (see ESI† for full details), which is in line with our interpretation of the crystallographic data (vide supra).
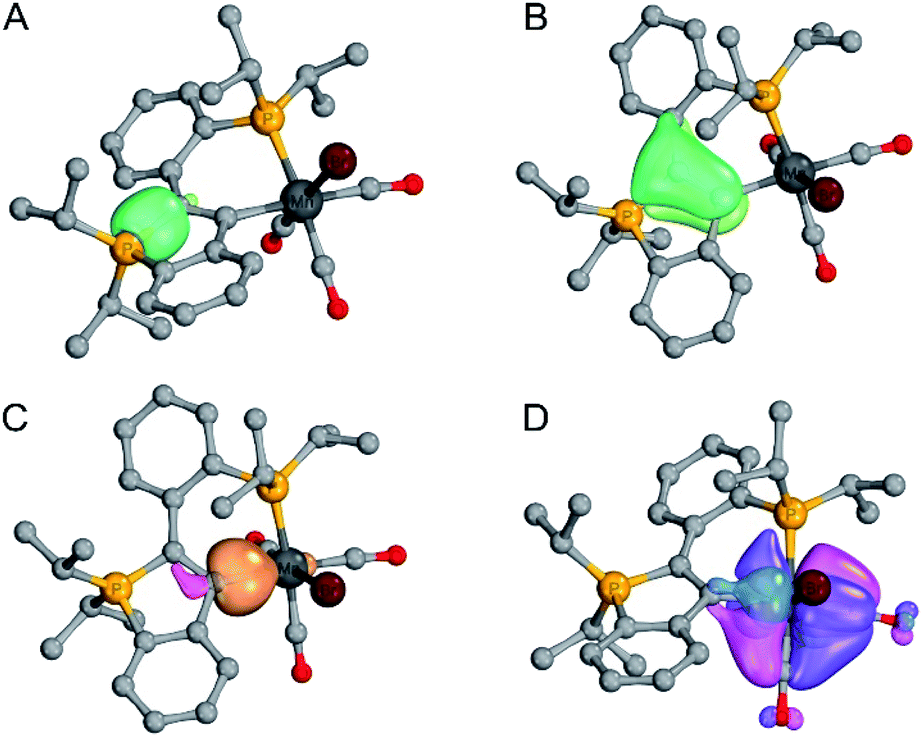 | ||
| Fig. 1 Selected IBO plots for complex 7 ((A) P–C σ-bond, (B) CC π-bond, (C) dative C→Mn σ-bond, (D) Mn–C π-backbonding via two orthogonal IBOs). Threshold value for printing: 75.0 for (A–C), 85.0 for (D). | ||
The IBO charge distributions e−(C2)/e−(M) in the σ-symmetric C2–M bonds (see Table 1) are indicative of dative C→M bonds, i.e. the electrons in these bonds are mainly located at the C2 centres (as represented by the CArY-MIC no-bond resonance structure shown in Scheme 4). In the case of the d6-configured (6, 7, [8]+, [9]+) and the d8-configured ([10]+, [11]+) complexes, backbonding interactions between the respective metals and the carbene centres were identified by IBO analysis (see Table 1), suggesting that a delocalized betaine resonance structure is also suited to describe these complexes. For the four complexes with π-acceptor ligands in trans-position to the carbene (6, 7, 10 and 11), delocalised 3c-(4σ + kπ)e− bonds (with k = number of delocalised π-electrons) are equally suited to represent the bonding situation, given that backbonding interactions between the respective metals and the trans-positioned π-acceptors were also found in these complexes. As an example, the most important IBOs of 7 are shown in Fig. 1 (see ESI† for analogous IBO plots for all other complexes).
As expected (vide supra), π-backbonding IBO interactions were only detected for complexes 6–11, but not for the d0- and d10-configured derivatives ([3]+, 4, 5 and 12). To gain further insights into the electronic structures of the latter four complexes, energy decomposition analysis (EDA-NOCV) was used to fragment each complex into a closed-shell ligand part and a closed-shell metal-containing part. Due to the chelating nature of our ligand, a cautionary note is required, given that EDA-NOCV is commonly used for monodentate ligands, which affords a clean picture of the fragmented metal–ligand bond.16 In our case, however, the individual contributions from the phosphine and the carbene donors are not separated, which requires a visual inspection of all EDA-NOCV deformation densities and interferes with a quantitative analysis. Nevertheless, a coarse picture of the bonding situation in complexes 3–12 was obtained using this methodology and the expected π-backbonding interactions were found for complexes 6–11. The respective NOCV deformation densities for complexes 4, 5 and 12 are in line with the presence of hyperconjugative interactions between one of the metal-bound co-ligands and the carbene centres (Hf–Cl → C2 for 4, TaNPh → C2 for 5 and Zn–SMes → C2 for 12). Given that similar interactions are often found for d0-configured NHC-complexes,4b,15 this analysis argues for an interpretation of 4, 5 and 12 as carbenoid species. Selected deformation densities of 12 are shown in Fig. 2 as an example (see ESI† for full detail on EDA-NOCV calculation for 3–12). For the yttrium derivative 3, EDA-NOCV analysis indicated that hyperconjugative interactions between the [Cp*2Y]+ fragment and the ligand play a minor role, while a dative C→Y bond is found upon inspection of the NOCV deformation densities (see ESI, Fig. S56†).
 | ||
| Fig. 2 Plots of the EDA-NOCV deformation densities Δρn for 12 (charge flow: red → blue). The sum Δρ1 + Δρ2 (left) is interpreted as combined σ-donation (C → Zn(SMes)2 and P → Zn(SMes)2), Δρ3 (right) is interpreted as MesS → C hyperconjugative stabilisation. The eigenvalues |νn| indicate the relative magnitude of the charge flow. | ||
To assess the covalency of the metal carbon σ-bond, the electron densities and energy densities at the (3,–1) bond critical points (bcp) of the M–C bonds were calculated for all complexes 3–12 by means of topological wavefunction analysis (Bader's QTAIM methodology).17
The ratio |Vbcp|/Gbcp (with Vbcp = virial potential energy at the bcp, Gbcp = kinetic energy density at the bcp) is expected to be >2 for covalent bonds and <1 for ionic bonds.18 For dative and highly polarised bonds, values in the regime 1 < |Vbcp|/Gbcp < 2 are expected and indeed found for 3–12 (see Table 1). Together with the bond degrees (defined as Hbcp/ρbcp: expected to be negative for dative bonds; Hbcp = total energy density at the bcp, ρbcp = electron density at the bcp) and the laplacian of electron densities at the bcp (∇2ρbcp: expected to be positive for dative bonds),19,18a it is concluded that the C→M σ-bonds in all complexes 3–12 (see Table 1) are best described as dative bonds as implied by an overlay of both CArY-MIC resonance structures shown in Scheme 4. As examples, ∇2ρ contour plots for [3]+ and 8 are shown in Fig. 3 (see ESI† for the corresponding plots for complexes 3–12). Notably, the 13C{1H} NMR shifts of the carbenoid C2 centres (approx. 200 ppm or higher) agree with this notion as similar chemical shift values are commonly found for N-stabilised heterocyclic carbenes.14d
 | ||
| Fig. 3 Counter plot of the laplacian of the electron densities (∇2ρ) of [3]+ (left) and 8 (right). Positive and negative values are shown in red and blue respectively. | ||
In the case of 3 and 12, which are both κ1-C-coordinated in solution (vide supra), ligand substitution reactions were considered conceivable, also as these two complexes were found to exhibit the smallest interaction energies ΔEint of the series 3–12 (according to energy decomposition analysis, see Table 1). Upon dissolution of 3 or 12 in pyridine-d5, deep green solutions containing cyclo-1 were obtained within seconds at room temperature. For the formation of cyclo-1 in the absence of a metal complex, solution of 1 need to be kept at 60 °C for several days.10 Therefore, it is assumed that pyridine-d5 coordinates to the metal centres in 3 and 12, which induces the second cyclisation event, either with the CArY-MIC still within the coordination sphere of the metal or via displacement and liberation of the free carbene. The free carbene is then expected to either rapidly cyclise to cyclo-1 or to ring-open to 1. NMR analysis revealed that cyclo-1 is the only product formed in the case of 12. For 3, however, the deep green solution obtained after reaction with pyridine-d5 was found to contain only small amounts of cyclo-1 (3–5%), while 1 was produced in more than 90% conversion. Evidently, the C–P bond, which is formed during the synthesis of 3 was re-opened, at least in this case (Scheme 5).
 | ||
| Scheme 5 Reaction of 3 and 12 with pyridine-d5. In the case of 12, cyclo-1 is produced exclusively, while 1 (major) is generated along with cyclo-1 (minor) in the case of 3.‡ | ||
To elucidate whether similar C–P bond cleavage reactions are also possible within the coordination sphere of a metal, a CArY-MIC complex with one free coordination site was sought-after. In such a complex, the endocyclic phosphine, which is liberated during ring-opening may be trapped by the metal ion. However, the free coordination site must be fac-positioned with respect to the κ2-C,P-bound CArY-MIC to circumvent the formation of a [PCCP]-pincer complex (cf.Scheme 2). Hence, 1 was reacted with fac-(MeCN)3Mo(CO)3 in MeCN and the expected CArY-MIC complex 13 was isolated (see Scheme 6). Two MeCN ligands in fac-(MeCN)3Mo(CO)3 were substituted, i.e. one MeCN ligand was left behind in fac-position to the P,C-chelate. Upon dissolution of 13 in thf, ring-opening of the P-heterocycle and dissociation of the MeCN ligand occurred over 24 h to afford the fac-coordinated complex 14 (see Scheme 6). This process was found to be fully reversible as shown by interconverting 13 and 14 several times, simply by changing the solvent (and keeping the thus obtained solutions for 24 h at r.t.). In this context, it needs to be noted that 13 precipitates from solution upon dissolution of 14 in MeCN, which may add its share to the driving force of the reverse reaction. Compound 14 may also be prepared directly via reaction between 1 and fac-(MeCN)3Mo(CO)3 in thf at room temperature. Upon heating (60 °C in thf), 14 was found to rearrange irreversibly to the mer-coordinated [PCCP]-pincer complex 15 (see Scheme 6). The finding that no reverse reaction between 15 and MeCN was observed, suggested that a local minimum on the potential energy surface was reached upon pincer formation. This was further elaborated by examining the reaction of 13–15 with excess CO. While 13 and 14 both reacted instantaneously with CO to afford 1620 (see Scheme 6, see Table 2 for characterisation data for 13–16), no reaction was noticed upon pressurising samples of 15 with carbon monoxide, not even under forcing reaction conditions (10 bar CO, 110 °C in toluene-d8). According to DFT calculations (PBE1PBE/Def2-TZVP-GD3), the reaction of 15 with CO to afford 16 is thermodynamically favoured (ΔG = 13.0 kcal mol−1, see ESI† for details), indicative of a high kinetic barrier prohibiting this reaction in our experiments. With the different reaction pathways interconnecting 13–16 now established, the focus was set on the most exciting reactivity pattern, namely the reversible P–C bond formation interconnecting 13 and 14. As the low solubility of 13 in MeCN (vide supra) precluded a meaningful kinetic analysis, a DFT modelling study (PBE1PBE/Def2-TZVP-GD3) was carried out to further elucidate this transformation.
 | ||
| Scheme 6 P–C bond cleavage and re-formation in a series of molybdenum complexes (13–16).‡ | ||
| Compound | 13 | 14 | 15 | 16 |
|---|---|---|---|---|
a Recorded in different solvents (13: thf-d8/MeCN-d3 = 4/1, 14: thf-d8, 15: C6D6, 16: CD2Cl2) values in ppm.
b Carbonyl stretching frequencies (solid state, ATR-IR, some vibrations are coupled to vibrations of the alkyne or the MeCN ligand in 13), values in cm−1.
c CC distance in 13 and 16, C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C distance in 14 and 15.
d Rounded to two digits (see ESI for additional metrical parameters and standard deviations), values in Å.
e Two independent molecules are present in the asymmetric unit (second molecule: d(C–C) = 1.23 Å, d(P–Mo) = 2.56/2.57 Å, d(C–Mo) = 2.37/2.40 Å). C distance in 14 and 15.
d Rounded to two digits (see ESI for additional metrical parameters and standard deviations), values in Å.
e Two independent molecules are present in the asymmetric unit (second molecule: d(C–C) = 1.23 Å, d(P–Mo) = 2.56/2.57 Å, d(C–Mo) = 2.37/2.40 Å).
|
||||
| δ(31P{1H})a | 43.0/49.6 | 54.3 | 87.5 | 46.0/51.0 |
| ν(CO)b | 1994, 1887, 1790, 1751 | 1930, 1847, 1829, 1818 | 1974, 1946, 1865, 1837 | 1987, 1892, 1855, 1826 |
| d(C–C)c,d | 1.38 | 1.24e | 1.25 | 1.38 |
| d(P–Mo)d | 2.51 | 2.54/2.60e | 2.50/2.50 | 2.50 |
| d(C–Mo)d | 2.25 | 2.38/2.40e | 2.30/2.30 | 2.26 |
Three different scenarios for the key step of the forward reaction 13 → 14 (exp. conditions: thf, r.t., approx. 95% conversion in 24 h) were inspected in silico: (a) initial dissociation of the MeCN ligand of 13 to afford a five-coordinate molybdenum centre and subsequent ring-opening of the CArY-MIC (P–C bond cleavage), (b) initial replacement of the MeCN ligand of 13 for a thf ligand and consecutive P–C bond cleavage once the thf-analogue of 13 has been formed, and (c) initial ring-opening of the CArY-MIC in 13 followed by dissociation of MeCN. The possibilities (a) and (b) had to be excluded (see ESI† for details) due to barriers of 34.7 and 31.8 kcal mol−1 respectively, which cannot be overcome under the employed reaction conditions.
For the third scenario, a barrier of 26.8 kcal mol−1 was calculated (PBE1PBE/Def2-TZVP-GD3, solvent corrected for thf) for the forward reaction 13 → 14 (cf.TS1 in Fig. 4), which is slightly higher than expected (≈25 kcal mol−1)21 on basis of the reaction conditions. This discrepancy, however, is well within the error (up to 4 kcal mol−1)22 of our DFT model. We also noticed that the calculated energies for TS1 were found to be nearly independent of the applied functional, but significantly affected by dispersion and solvent correction terms (see ESI† for details). A considerably lower barrier of only 21.8 kcal mol−1 was found for TS1 when dispersion effects were neglected, suggesting that small geometric distortions may lead to large errors, in particular with respect to TS1. Hence, the reaction pathway shown in Fig. 4 was considered the most plausible scenario. In the first step of the forward reaction 13 → 14, ring-opening of the CArY-MIC in 13 takes place, which gives rise to intermediate INT1. Throughout this step, the MeCN ligand in 13 remains coordinated to the molybdenum centre. Upon dissociation of the MeCN ligand in INT1, a spontaneous (barrier-free) coordination of the phosphine was found to occur, i.e. the experimentally observed product 14 is formed without an additional activation barrier. Once again, dispersion effects were found to significantly influence the free energy associated with this second step, i.e. upon neglecting dispersion effects, an activation barrier of approximately 10 kcal mol−1 was calculated for this step (see Fig. 4, see ESI† for details). Overall, the forward reaction 13 → 14 was found to be exergonic by ΔG = −0.6 kcal mol−1 (PBE1PBE/Def-2-TZVP-GD3, solvent corrected for thf), which agrees with our experiments. For the reverse reaction 14 → 13 (PBE1PBE/Def-2-TZVP-GD3, solvent corrected for MeCN), a nearly identical barrier of 27.1 kcal mol−1 (referenced against 14) was found for TS1. The overall reaction was found to be exergonic by ΔG = −1.0 kcal mol−1, suggesting that the ground state energies are well-described with the employed functional.
 | ||
| Fig. 4 Calculated (PBE1PBE/Def2-TZVP-GD3) energy profile for the conversion of 13 (set to 0.0 kcal mol−1) to 14 in thf (PCM solvent correction for thf, black lines) and for the backward reaction in MeCN (PCM solvent correction for MeCN, blue lines).‡ Free energies are corrected for the liberated (13 → 14) or incoming (13 → 14) MeCN ligand. | ||
In ongoing work, various modifications23 at the CArY-MIC backbone and at the co-ligands in 13 and 14 are examined in vitro and in silico to identify similar interconversions between facially coordinated phosphinoalkyne complexes and CArY-MIC complexes. These efforts are directed towards a deeper understanding of this new ligand-cooperative24 reactivity pattern.
Conclusions
In summary, we have shown that a new class of CArY-MIC complexes is accessible under mild conditions via reaction between 1 and a variety of metal precursors, each featuring two cis-positioned open coordination sites. This methodology was found to be applicable all over the d-block as shown by the preparation of 3–12. The P–C bond formation involved in this transformation was shown to be reversible, at least under certain circumstances, as demonstrated for a series of molybdenum complexes (13–16). This new ligand-cooperative reactivity pattern is further explored in our laboratory.Author contributions
H. K. Wagner carried out the experimental work and parts of the computational work. The crystallographic work was carried out by H. Wadepohl. J. Ballmann wrote the manuscript and carried out parts of the computational work. The ESI† was compiled by all three authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding from the DFG (BA 4859/2) is gratefully acknowledged. The authors acknowledge support by the state of Baden-Württemberg through bwHPC and the DFG through grant INST 40/467-1 FUGG (JUSTUS cluster). We thank P. W. Antoni, F. Braun and J. Stracke for help with preparing some of the required metal precursors during research internships. We also thank Prof. L. H. Gade for generous support over the past years. We would like to thank an unknown reviewer for pointing out that an overlay of the betaine and the no-bond resonance structure (cf.Scheme 4) infers the presence of a dative bond.‡Notes and references
- (a) D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39–92 CrossRef CAS PubMed; (b) T. Dröge and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 6940–6952 CrossRef PubMed; (c) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed; (d) A. Gómez-Suárez, D. J. Nelson and S. P. Nolan, Chem. Commun., 2017, 53, 2650–2660 RSC; (e) H. V. Huynh, Chem. Rev., 2018, 118, 9457–9492 CrossRef CAS PubMed; (f) A. Doddi, M. Peters and M. Tamm, Chem. Rev., 2019, 119, 6994–7112 CrossRef CAS PubMed.
- (a) O. Schuster, L. Yang, H. G. Raubenheimer and M. Albrecht, Chem. Rev., 2009, 109, 3445–3478 CrossRef CAS PubMed; (b) G. Guisado-Barrios, J. Bouffard, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 4759–4762 CrossRef CAS PubMed; (c) R. H. Crabtree, Coord. Chem. Rev., 2013, 257, 755–766 CrossRef CAS; (d) J. B. Waters, Q. Chen, T. A. Everitt and J. M. Goicoechea, Dalton Trans., 2017, 46, 12053–12066 RSC; (e) G. Guisado-Barrios, M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2018, 51, 3236–3244 CrossRef CAS PubMed; (f) Á. Vivancos, C. Segarra and M. Albrecht, Chem. Rev., 2018, 118, 9493–9586 CrossRef PubMed; (g) F. M. Chadwick, B. F. E. Curchod, R. Scopelliti, F. Fadaei Tirani, E. Solari and K. Severin, Angew. Chem., Int. Ed., 2019, 58, 1764–1767 CrossRef CAS PubMed; (h) A. Merschel, D. Rottschäfer, B. Neumann, H.-G. Stammler and R. S. Ghadwal, Organometallics, 2020, 39, 1719–1729 CrossRef CAS; (i) D. Rottschäfer, T. Glodde, B. Neumann, H.-G. Stammler and R. S. Ghadwal, Chem. Commun., 2020, 56, 2027–2030 RSC; (j) S. C. Sau, P. K. Hota, S. K. Mandal, M. Soleilhavoup and G. Bertrand, Chem. Soc. Rev., 2020, 49, 1233–1252 RSC.
- (a) M. Melaimi, R. Jazzar, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10046–10068 CrossRef CAS PubMed; (b) U. S. D. Paul and U. Radius, Eur. J. Inorg. Chem., 2017, 3362–3375 CrossRef CAS; (c) C. M. Weinstein, G. P. Junor, D. R. Tolentino, R. Jazzar, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2018, 140, 9255–9260 CrossRef CAS PubMed; (d) D. Pichon, M. Soleilhavoup, J. Morvan, G. P. Junor, T. Vives, C. Crévisy, V. Lavallo, J.-M. Campagne, M. Mauduit, R. Jazzar and G. Bertrand, Chem. Sci., 2019, 10, 7807–7811 RSC.
- (a) D. M. Andrada, N. Holzmann, T. Hamadi and G. Frenking, Beilstein J. Org. Chem., 2015, 11, 2727–2736 CrossRef CAS PubMed; (b) D. Munz, Organometallics, 2018, 37, 275–289 CrossRef CAS.
- (a) M. Melaimi, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 8810–8849 CrossRef CAS PubMed; (b) J. Ruiz, L. García, M. Vivanco, Á. Berros and J. F. Van der Maelen, Angew. Chem., Int. Ed., 2015, 54, 4212–4216 CrossRef CAS PubMed; (c) S. Kuwata and F. E. Hahn, Chem. Rev., 2018, 118, 9642–9677 CrossRef CAS PubMed.
- (a) B. Rao, H. Tang, X. Zeng, L. Liu, M. Melaimi and G. Bertrand, Angew. Chem., Int. Ed., 2015, 54, 14915–14919 CrossRef CAS PubMed; (b) M. B. Gildner and T. W. Hudnall, Chem. Commun., 2019, 55, 12300–12303 RSC; (c) J. Lorkowski, M. Krahfuß, M. Kubicki, U. Radius and C. Pietraszuk, Chem.–Eur. J., 2019, 25, 11365–11374 CrossRef CAS PubMed; (d) P. R. Sultane, G. Ahumada, D. Janssen-Müller and C. W. Bielawski, Angew. Chem., Int. Ed., 2019, 58, 16320–16325 CrossRef CAS PubMed; (e) M. Gernert, L. Balles-Wolf, F. Kerner, U. Müller, A. Schmiedel, M. Holzapfel, C. M. Marian, J. Pflaum, C. Lambert and A. Steffen, J. Am. Chem. Soc., 2020, 142, 8897–8909 CrossRef CAS PubMed; (f) R. Jazzar, M. Soleilhavoup and G. Bertrand, Chem. Rev., 2020, 120, 4141–4168 CrossRef CAS PubMed.
- (a) M. Asay, B. Donnadieu, A. Baceiredo, M. Soleilhavoup and G. Bertrand, Inorg. Chem., 2008, 47, 3949–3951 CrossRef CAS PubMed; (b) A. Fürstner, M. Alcarazo, K. Radkowski and C. W. Lehmann, Angew. Chem., Int. Ed., 2008, 47, 8302–8306 CrossRef PubMed; (c) J. Kobayashi, S.-y. Nakafuji, A. Yatabe and T. Kawashima, Chem. Commun., 2008, 6233–6235 RSC; (d) S.-y. Nakafuji, J. Kobayashi and T. Kawashima, Angew. Chem., Int. Ed., 2008, 47, 1141–1144 CrossRef CAS PubMed; (e) M. Alcarazo, T. Stork, A. Anoop, W. Thiel and A. Fürstner, Angew. Chem., Int. Ed., 2010, 49, 2542–2546 CrossRef CAS PubMed; (f) G. Ung, D. Mendoza-Espinosa, J. Bouffard and G. Bertrand, Angew. Chem., Int. Ed., 2011, 50, 4215–4218 CrossRef CAS PubMed; (g) E. González-Fernández, J. Rust and M. Alcarazo, Angew. Chem., Int. Ed., 2013, 52, 11392–11395 CrossRef PubMed; (h) G. Ung, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 758–761 CrossRef CAS PubMed; (i) H. Steinert, C. Schwarz, A. Kroll and V. H. Gessner, Molecules, 2020, 25, 796 CrossRef CAS.
- (a) B. Godoi, R. F. Schumacher and G. Zeni, Chem. Rev., 2011, 111, 2937–2980 CrossRef CAS PubMed; (b) S. Arndt, M. M. Hansmann, P. Motloch, M. Rudolph, F. Rominger and A. S. K. Hashmi, Chem.–Eur. J., 2017, 23, 2542–2547 CrossRef CAS PubMed; (c) S. Arndt, M. M. Hansmann, F. Rominger, M. Rudolph and A. S. K. Hashmi, Chem.–Eur. J., 2017, 23, 5429–5433 CrossRef CAS PubMed; (d) Q. Ge, J. Zong, B. Li and B. Wang, Org. Lett., 2017, 19, 6670–6673 CrossRef CAS PubMed; (e) S. Arndt, J. Borstelmann, R. Eshagh Saatlo, P. W. Antoni, F. Rominger, M. Rudolph, Q. An, Y. Vaynzof and A. S. K. Hashmi, Chem.–Eur. J., 2018, 24, 7882–7889 CrossRef CAS PubMed; (f) J.-J. Hou, Y.-Z. Xu, Z.-J. Gan, X. Zhao, Z. Duan and F. Mathey, J. Organomet. Chem., 2019, 879, 158–161 CrossRef CAS; (g) Y. Song, L. Wang, Z. Duan and F. Mathey, Chin. Chem. Lett., 2020, 31, 329–332 CrossRef CAS.
- (a) K. Okamoto, Y. Omoto, H. Sano and K. Ohe, Dalton Trans., 2012, 41, 10926–10929 RSC; (b) K. Sasakura, K. Okamoto and K. Ohe, Organometallics, 2018, 37, 2319–2324 CrossRef CAS; (c) P. Federmann, T. Richter, H. Wadepohl and J. Ballmann, Organometallics, 2019, 38, 4307–4318 CrossRef CAS.
- P. Federmann, H. K. Wagner, P. W. Antoni, J.-M. Mörsdorf, J. L. Pérez Lustres, H. Wadepohl, M. Motzkus and J. Ballmann, Org. Lett., 2019, 21, 2033–2038 CrossRef CAS PubMed.
- In difference to the reactions between nucleophiles and terminal CO or isonitrile ligands, which also lead to carbene complexes, the reactions described herein proceed via nucleophilic attack on a side-on coordinated internal alkyne. For a more detailed discussion on metal-templated synthetic routes to carbene complexes starting from terminal CO or isonitrile complexes, see; (a) M. Tamm and F. Ekkehardt Hahn, Coord. Chem. Rev., 1999, 182, 175–209 CrossRef; (b) R. A. Michelin, A. J. L. Pombeiro and M. F. C. Guedes da Silva, Coord. Chem. Rev., 2001, 218, 75–112 CrossRef CAS; (c) K. H. Dötz and J. Stendel, Chem. Rev., 2009, 109, 3227–3274 CrossRef PubMed; (d) A. Flores-Figueroa, T. Pape, K.-O. Feldmann and F. E. Hahn, Chem. Commun., 2010, 46, 324–326 RSC; (e) A. S. K. Hashmi, C. Lothschütz, C. Böhling, T. Hengst, C. Hubbert and F. Rominger, Adv. Synth. Catal., 2010, 352, 3001–3012 CrossRef CAS; (f) D. I. Bezuidenhout, S. Lotz, D. C. Liles and B. van der Westhuizen, Coord. Chem. Rev., 2012, 256, 479–524 CrossRef CAS; (g) M. Schmidtendorf, T. Pape and F. E. Hahn, Angew. Chem., Int. Ed., 2012, 51, 2195–2198 CrossRef CAS PubMed; (h) V. Blase, A. Flores-Figueroa, C. Schulte to Brinke and F. E. Hahn, Organometallics, 2014, 33, 4471–4478 CrossRef CAS; (i) M. Knorn, E. Lutsker and O. Reiser, Chem. Soc. Rev., 2020, 49, 7730–7752 RSC.
- (a) D. G. Gilheany, Chem. Rev., 1994, 94, 1339–1374 CrossRef CAS PubMed; (b) L. T. Scharf and V. H. Gessner, Inorg. Chem., 2017, 56, 8599–8607 CrossRef CAS PubMed; (c) A. Sarbajna, V. S. V. S. N. Swamy and V. H. Gessner, Chem. Sci., 2021 10.1039/D0SC03278F.
- Based on a CSD search (v5.41, update May 2020): 41 hits with a mean CC bond length of 1.360 Å (standard deviation: 0.025 Å).
- (a) B. N. Haerizade, M. Z. Kassaee, H. Zandi, M. Koohi and A. A. Ahmadi, J. Phys. Org. Chem., 2014, 27, 902–908 CrossRef CAS; (b) B. Borthakur and A. K. Phukan, Chem.–Eur. J., 2015, 21, 11603–11609 CrossRef CAS PubMed; (c) E. Kleinpeter and A. Koch, Eur. J. Org. Chem., 2018, 3114–3121 CrossRef CAS; (d) E. Kleinpeter and A. Koch, Tetrahedron, 2019, 75, 1548–1554 CrossRef CAS.
- J. Cheng, L. Wang, P. Wang and L. Deng, Chem. Rev., 2018, 118, 9930–9987 CrossRef CAS PubMed.
- (a) R. Tonner, G. Heydenrych and G. Frenking, Chem.–Asian J., 2007, 2, 1555–1567 CrossRef CAS PubMed; (b) P. Jerabek, P. Schwerdtfeger and G. Frenking, J. Comput. Chem., 2019, 40, 247–264 CrossRef CAS PubMed.
- (a) R. F. W. Bader and H. Essén, J. Chem. Phys., 1984, 80, 1943–1960 CrossRef CAS; (b) R. F. W. Bader, Acc. Chem. Res., 1985, 18, 9–15 CrossRef CAS; (c) C. Lepetit, P. Fau, K. Fajerwerg, M. L. Kahn and B. Silvi, Coord. Chem. Rev., 2017, 345, 150–181 CrossRef CAS.
- (a) R. Bianchi, G. Gervasio and D. Marabello, Inorg. Chem., 2000, 39, 2360–2366 CrossRef CAS PubMed; (b) E. Espinosa, I. Alkorta, J. Elguero and E. Molins, J. Chem. Phys., 2002, 117, 5529–5542 CrossRef CAS.
- (a) D. Cremer and E. Kraka, Angew. Chem., Int. Ed., 1984, 23, 627–628 CrossRef; (b) E. Kraka and D. Cremer, J. Mol. Struct.: THEOCHEM, 1992, 255, 189–206 CrossRef.
- Compound 16 was also prepared independently via reaction of (piperidine)2Mo(CO)4 with 1 (see ESI† for details).
- H. Ryu, J. Park, H. K. Kim, J. Y. Park, S.-T. Kim and M.-H. Baik, Organometallics, 2018, 37, 3228–3239 CrossRef CAS.
- T. Weymuth, E. P. A. Couzijn, P. Chen and M. Reiher, J. Chem. Theory Comput., 2014, 10, 3092–3103 CrossRef CAS PubMed.
- In preliminary experiments, one of the most profound modifications has been explored already, namely the replacement of molybdenum for tungsten. Unfortunately, these efforts led to inseparable mixtures of the expected tungsten complexes 13-W, 14-W and 15-W (see ESI† for details). Starting from 1 and (piperidine)2W(CO)4, 16-W may be prepared as well (see ESI† for details).
- (a) J. I. van der Vlugt, Eur. J. Inorg. Chem., 2012, 363–375 CrossRef CAS; (b) J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed; (c) D. G. A. Verhoeven and M.-E. Moret, Dalton Trans., 2016, 45, 15762–15778 RSC.
- J. Brecher, Pure Appl. Chem., 2008, 80, 277–410 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic data, computational data and crystallographic data. CCDC 2036100–2036115. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc06128j |
| ‡ Throughout the manuscript, neutral CArY-MIC complexes are denoted as betaines, while cationic CArY-MIC complexes are denoted with a positive formal charge on the endocyclic phosphine. This notation avoids the use of dative bond arrows, which is in line with the IUPAC recommendations.25 |
| This journal is © The Royal Society of Chemistry 2021 |