
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocatalyst-independent photoredox ring-opening polymerization of O-carboxyanhydrides: stereocontrol and mechanism†
Yongliang
Zhong
a,
Quanyou
Feng
 ab,
Xiaoqian
Wang
a,
Lei
Yang
b,
Andrew G.
Korovich
c,
Louis A.
Madsen
c and
Rong
Tong
*a
ab,
Xiaoqian
Wang
a,
Lei
Yang
b,
Andrew G.
Korovich
c,
Louis A.
Madsen
c and
Rong
Tong
*a
aDepartment of Chemical Engineering, Virginia Polytechnic Institute and State University, 635 Prices Fork Road, Blacksburg, Virginia, 24061, USA. E-mail: rtong@vt.edu
bKey Laboratory for Organic Electronics and Information Displays, Jiangsu Key Laboratory for Biosensors, Institute of Advanced Materials, Jiangsu National Synergetic Innovation Center for Advanced Materials, Nanjing University of Posts and Telecommunications, 9 Wenyuan Road, Nanjing, 210023, China
cDepartment of Chemistry, Virginia Polytechnic Institute and State University, 1040 Drillfield Drive, Blacksburg, Virginia, 24061, USA
First published on 25th January 2021
Abstract
Photoredox ring-opening polymerization of O-carboxyanhydrides allows for the synthesis of polyesters with precisely controlled molecular weights, molecular weight distributions, and tacticities. While powerful, obviating the use of precious metal-based photocatalysts would be attractive from the perspective of simplifying the protocol. Herein, we report the Co and Zn catalysts that are activated by external light to mediate efficient ring-opening polymerization of O-carboxyanhydrides, without the use of exogenous precious metal-based photocatalysts. Our methods allow for the synthesis of isotactic polyesters with high molecular weights (>200 kDa) and narrow molecular weight distributions (Mw/Mn < 1.1). Mechanistic studies indicate that light activates the oxidative status of a CoIII intermediate that is generated from the regioselective ring-opening of the O-carboxyanhydride. We also demonstrate that the use of Zn or Hf complexes together with Co can allow for stereoselective photoredox ring-opening polymerizations of multiple racemic O-carboxyanhydrides to synthesize syndiotactic and stereoblock copolymers, which vary widely in their glass transition temperatures.
Introduction
Polyesters have long been considered as environmentally friendly alternatives to petrochemical-based polyolefins because of their degradability and biocompatibility.1–5 Among many degradable polyesters, poly(α-hydroxy acids) (PAHAs) have been regarded as a type of industry applicable, degradable, and biocompatible polyester, and a few of them (e.g., poly(lactic-co-glycolic acid)) have been approved by the FDA (U.S. Food and Drug Administration) for clinical applications. However, the utility of PAHAs for applications that demand physico-mechanical and thermal properties, such as high stiffness and high glass transition temperatures (Tgs), is greatly limited by the lack of side-chain functionality in PAHAs and in their lactone monomers.6–8 Early work by the Baker group shows that PAHAs synthesized from functionalized lactides (LAs) present a wide range of Tgs from −46 °C to 100 °C.9–11 Unfortunately, the multi-step synthesis of functionalized LAs is challenging; monomers are afforded in low yields; while the polymerization reactivity significantly drops upon the introduction of pendant groups (Scheme 1, route i).9,12,13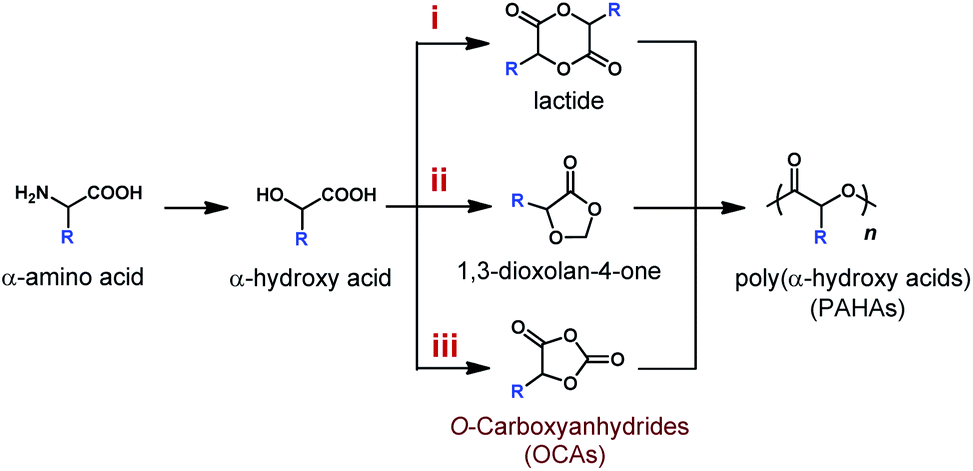 | ||
| Scheme 1 Synthetic routes of poly(α-hydroxy acids) (PAHAs) from α-amino acids and α-hydroxy acids via different monomers. | ||
Alternative strategies have been developed to access monomers that can be easily synthesized and polymerized. Noticeably, a five-membered heterocycle 1,3-dioxolan-4-one that bears both ester and acetal groups has been recently developed by Miller14 and Shaver groups (Scheme 1, route ii).15,16 Either through copolymerization with LAs for acetal retention,14 or ring-opening polymerizations (ROPs) via the deliberation of formaldehyde, this monomer provides an inexpensive strategy to prepare PAHAs. However, the ROP strategy for 1,3-dioxolan-4-one requires further development as the obtained polymers had relatively low molecular weights (MWs, <20 kDa) due to side reactions, and the ROP procedures demand constant removing of formaldehyde from the reaction solution.15,16
Since 2006, O-carboxyanhydrides (OCAs) have emerged as an alternative class of highly active monomers with pendant functional groups for the synthesis of functionalized PAHAs (Scheme 1, route iii).6,17,18 OCAs are readily prepared from amino acids or hydroxyl acids; however, the current ROP methods using organocatalysts cannot synthesize stereoregular and high-MW polyesters (Scheme 2A), and they cannot mediate stereoselective ROP of OCAs to prepare functionalized polyesters with different tacticities.19–23 Note that tacticity is one of the most critical determinants of the physical and mechanical properties of a polymer.24,25 For example, the stereocontrolled ROP of rac- and meso-lactide can result in a wide range of poly(lactide) microstructures with different Tgs.24 On the other hand, many well-defined metal catalysts (e.g., Zn and Hf complexes) that mediate stereoselective living and polymerization of lactones have been evaluated for stereoselective ROP of OCAs (Scheme 2B).24,26–30 Nevertheless, extensive utilization of metal-catalyzed ROP of OCAs was limited because of their complicated reactivity and inability to prepare high-MW polyesters.21,22 We have recently reported the controlled stereoselective ROPs of OCAs by using Ni/Zn/Ir-mediated photoredox ROP method,22,31 and Co/Zn-mediated electrochemical ROP (eROP) of OCAs32 that all provide high MW (over 140 kDa) functionalized polyesters with narrow MW distributions (Ð < 1.1). Importantly, both photoredox ROP and eROP allow for tuning the polymerization kinetics by turning the light or electricity on or off.31,32
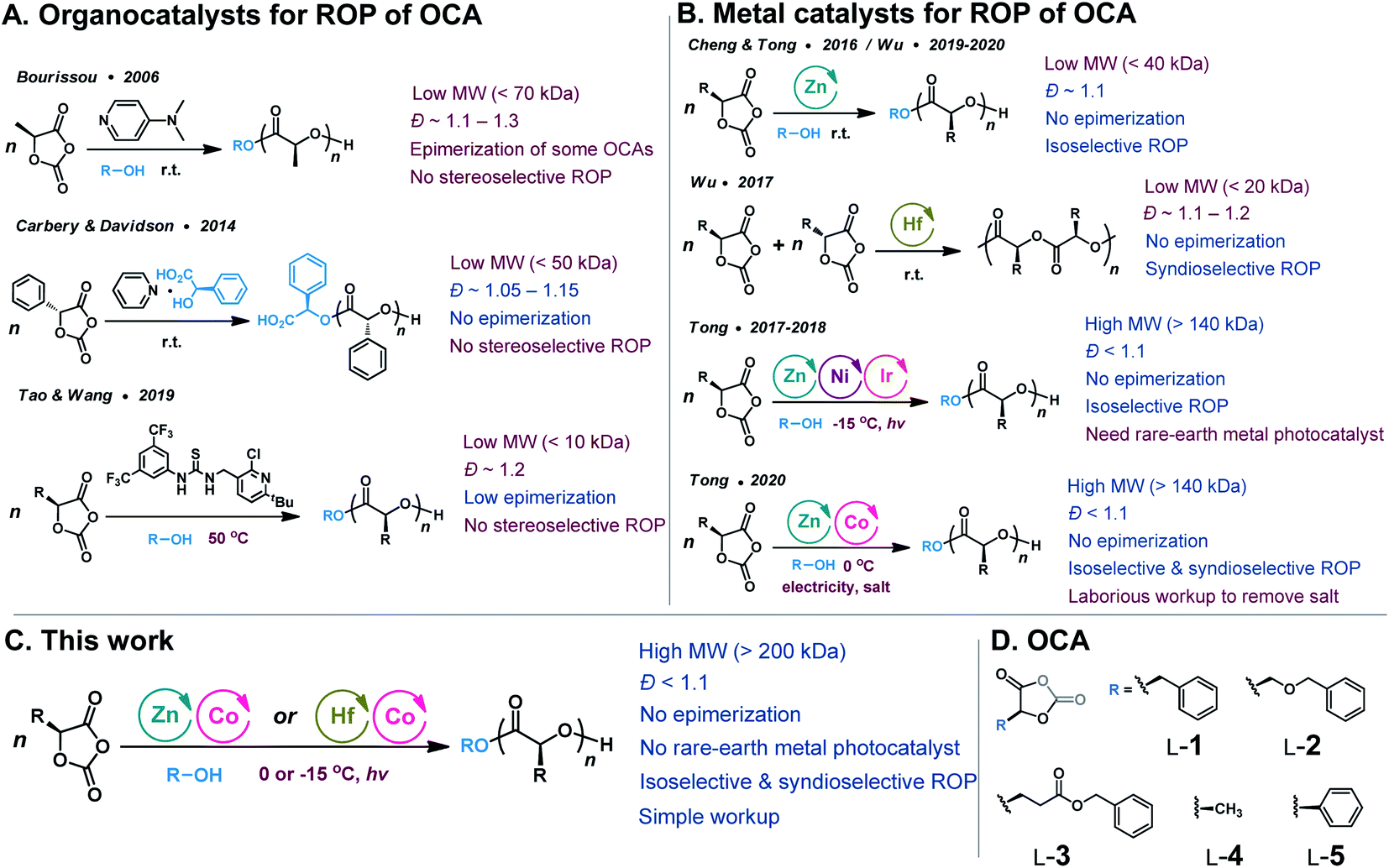 | ||
| Scheme 2 Ring-opening polymerization (ROP) methods of O-carboxyanhydrides (OCAs) for accessing functionalized polyesters: (A) the use of organocatalysts; (B) the use of metal catalysts; (C) Co/Zn or Co/Hf-mediated stereoselective photoredox ROPs in this work; (D) chemical structures of OCAs used in this work. MW, molecular weight. Colors in (A–C) highlight pros (blue) and cons (red) of different synthetic methods. | ||
Despite the high efficiency, the photoredox ROP protocol requires expensive precious metal photocatalysts for decarboxylation, and only isoselective polymerization was achieved.22,31 Precious metal-based photocatalysts, such as Ru and Ir complexes, impose significant environmental footprints due to terrestrial scarcity.33,34 The utilization of earth-abundant metals as visible-light photocatalysts has recently gained great interest in order to develop sustainable photocatalysts.35 Recently reported light-induced metal catalysts include Cu,36–39 Co,40–43 Ni,34,44–46 Mn,47–49 Ce50–53 and Fe54,55 complexes. Such metal complexes usually involve photoinduced single-electron transfer between the substrate and the catalyst, and allow for reactions that occur directly from their photoexcited states, without the assistance of exogenous photocatalysts that harvest light to transfer electron or energy.56,57 However, the use of earth-abundant photocatalysts in photo-polymerization could be hampered by the short excited-state lifetime, which is usually picoseconds to nanoseconds for first-row transition metals.58,59 In our initial report on the use of Co/Zn-mediated eROP of OCAs, we found that Co complexes could replace Ni/Ir in the photoredox ROP.32 To our knowledge, the use of photoactive organometallic complexes as polymerization catalysts has been rarely achieved.60 Considering our eROP method usually involves laborious workup to separate electrolytes from the solvent after electrochemical reactions,32 it is therefore crucial to develop viable earth-abundant metal catalysts that can mediate controlled polymerization to prepare high-MW polyesters with different tacticities.
Herein, we report the development of photoactive Co catalysts that are amenable to efficiently polymerize enantiopure OCAs in the presence of Zn or Hf complexes, resulting in isotactic polymers with unprecedented high MWs (>200 kDa) and narrow MW distributions (Ð < 1.1, Scheme 2C). Mechanistic studies implicate our Co complexes as underexplored alternatives to precious metal photocatalysts. We also demonstrate that these metal complexes can be used to prepare various syndiotactic and stereoblock polyesters from different racemic OCA monomers, with Tg values ranging over ∼100 °C (Scheme 2C and D).
Results and discussion
Discovery and optimization of Co/Zn complexes for photoredox ring-opening polymerization
In our prior studies on Co/Zn-mediated eROP,32 we found that CoII complexes could replace both the Ni catalyst and the Ir photocatalyst, and mediated photoredox ROP of OCA at 0 °C when irradiated with light from a blue LED (300–500 nm). Several CoII complexes could lead to controlled photoredox ROP with Mn values close to MWcal of 103.7 kDa at the [L-1]/[Zn-1] ratio of 700/1 (Fig. S1;†L-1, phenyl O-carboxyanhydride, see Scheme 2D; Mn, number-average MW; MWcal, calculated MW based on feeding ratios). However, when the [L-1]/[Zn-1] ratio was elevated to 800/1, only 3 CoII complexes led to controlled photoredox ROP with Mn values close to MWcal of 118.5 kDa at 0 °C (Fig. S1†). Specifically, Co-1 (Co-1 = (bpy)Co[N(SiMe3)2]2, bpy = 2,2′-bipyridine), Zn-1 (Zn-1 = Zn[N(SiMe3)2]2), and BnOH are all necessary for this controlled photoredox ROP (Table S1†). However, when the initial [L-1]/[Co-1]/[Zn-1] ratio was 900/1/1, monomer conversion was only 43.7% (Fig. 1A, green line), indicating the occurrence of side reactions. When the photoredox reaction temperature was decreased to −15 °C (Scheme 3), the Mn values of the poly(L-1) products increased linearly with initial [L-1]/[Co-1]/[Zn] ratio up to 1100/1/1 and were slightly higher than the MWcal values (Fig. 1A, blue line). Note that at this temperature, the Ð values of all of the obtained polymers were <1.1 (Fig. 1A; gel-permeation chromatography traces in Fig. S2†). Additionally, the MWs of poly(L-1) showed linear correlation with the conversion of L-1 at the initial [L-1]/[Co-1]/[Zn-1] ratio of 1000/1/1 (Fig. S3†). No epimerization of the α-methine hydrogen was observed in the homodecoupled 1H NMR spectra of the polymers, including high-MW poly(L-1) (Mn = 215.2 kDa, Fig. S4†), suggesting that the Co complex did not affect the chirality of L-1 during the ROP. Moreover, electrospray ionization mass spectrometry (ESI-MS) analysis of oligo(L-1) confirmed attachment of the BnO – group to the oligomer (Fig. S5†), indicating that a Zn–alkoxide was involved in ring-opening and chain propagation. Note that nearly all metal complexes can be readily removed by simply washing the polymer with methanol, as indicated by inductively coupled plasma mass spectrometry (Table S2†). | ||
| Fig. 1 Photoredox controlled ring-opening polymerization of L-1 mediated by Co/Zn complexes. (A) Plots of Mn and molecular weight distribution (Mw/Mn) of poly(L-1) versus [L-1]/[Zn-1] at various reaction temperatures ([Co-1]/[Zn-1]/[BnOH] = 1/1/1); MWcal, molecular weight calculated from monomer/catalyst ratio; Mn, number-average molecular weight; Mw, weight-average molecular weight. * and ** indicated incomplete monomer conversions. (B) Logarithmic plots of L-1 conversion versus time at various Zn-1 concentrations, showing first-order kinetics. [L-1] = 150 mM; [Co-1]/[Zn-1]/[BnOH] = 1/1/1; reaction temperature, −15 °C. (C) Dependence of the rate of L-1 conversion on the presence or absence of light irradiation onto the reaction at −15 °C ([L-1]/[Co-1]/[Zn-2]/[BnOH] = 800/1/1/1; [L-1] = 208.4 mM). The dashed lines indicate periods during which no light was applied; while solid lines indicate the period during which light was applied. | ||
 | ||
| Scheme 3 Photoredox controlled ring-opening polymerization of L-1 mediated by Co-1/Zn-1. | ||
We then examined the kinetics of photoredox ROP of L-1 at −15 °C by varying the concentration of each reaction component. The reaction was first-order with respect to L-1 (Fig. 1B). To explore whether Co and Zn catalysts formed a coordinate complex to mediate the polymerization, we fixed the Co/Zn ratio at 1/1 in the kinetic study and found the reaction order was 5.06 ± 0.21 (Fig. S5†); whereas the reaction orders with respect to Co-1, Zn-1 and BnOH were 2.33 ± 0.20, 2.64 ± 0.03, and 0 respectively (Fig. S6†). These results indicate that no formation of Co–Zn complex during the photoredox ROP, and the rate of chain propagation was independent of BnOH concentration. Therefore the kinetics of the photoredox ROP of L-1 follows an overall kinetic law of the form:
| −d[L-1]/dt = kp[Co-1]2.33[Zn-1]2.64[L-1]1 | (1) |
We then examined whether Co/Zn-mediated photoredox ROP could allow for the temporal modulation of polymerization rates by turning the light on or off. Similar to our previous studies using Ni/Zn/Ir catalysts,31 we found that in the presence of Zn-2 (Fig. 1C), a Zn complex with a tridentate ligand, irradiation of the reaction solution for 15 min resulted in 47% conversion of L-1 ([L-1]/[Co-1]/[Zn-2]/[BnOH] = 800/1/1/1). When the light was then turned off, the conversion of L-1 slightly increased to 50% over 30 min. Once the current was resumed, the polymerization revived with a conversion of L-1 to 97% over 30 min (Fig. 1C, blue line). In contrast, in a similar reaction mediated by Zn-1, consumption of L-1 continued even after the light was turned off (Fig. 1C, green line). Such “light on–off” ROP could be repeated twice: each time the light was turned off, the ROP almost stopped, and it proceeded again rapidly when the irradiation was resumed (Fig. 1C, red line).
With a set of optimized conditions in hand, we explored the generality of the Co/Zn-mediated photoredox polymerization by carrying out reactions of other OCAs (L-2, L-3, L-4, and L-5, and their structures in Scheme 2D; Table S3†). In all instances, polymerization proceeded smoothly, as was the case for the formation of poly(L-1); the Mn values of the obtained polymers were close to the MWcal values, most of Ð values were <1.1, and the α-methine hydrogens did not epimerize (Fig. S8–S11†). Note that considerable epimerization of L-5, which has an acidic α-methine proton, is often observed during ROP,20,21,61 but this was not the case in our system (Fig. S11†). Moreover, diblock copolymers and a triblock copolymer could be readily prepared by sequential addition of the monomers, and excellent control of the Mn and Ð values was achieved (Table S4 and Fig. S13–S21†).
Electrochemistry and photophysics studies of Co complex in photoredox ring-opening polymerization
Photoredox catalysts such as Ir- or Ru-based polypyridyl complexes are often used in the presence of an exogenous reductant or oxidant.33,62–64 Unlike in nearly all photo-polymerizations that use one photocatalyst to affect redox chemistry and a different catalyst to re-generate free radicals or radical ions for chain propagation,65–70 in our method a single Co-1 complex is responsible both for ring-opening of the OCAs and for photoredox decarboxylation to accelerate chain propagation. Given that no obvious oxidant or reductant was present in the reaction mixture and that light was required, we carried out electrochemistry and photophysics experiments to probe the mode of action of the Co catalyst.The cyclic voltammogram of Co-1 exhibited a reversible redox wave with an Ered1/2[CoIII/CoII] of 0.746 V versus SCE (SCE, saturated calomel electrode, the value equals 0.366 V vs. Fc+/Fc; Fc, ferrocene; Fig. 2A). Notably, the addition of Ir-1 (Ered1/2[*IrIII/IrII] = 1.21 V versus SCE)71 resulted in incomplete monomer conversion (Scheme 4; Table S5,† entry 2), presumably because electron transfer between Co-1 and Ir-1 led to inefficient chain propagation. This was also observed when adding another highly oxidizing photocatalyst, Ru(bpz)32+ (bpz = 2,2′-bipyrazyl; Ered1/2[*RuIII/RuII] = 1.45 V versus SCE).72 In contrast, photocatalysts that have lower excited-state reduction potentials (e.g., Ru(bpy)32+ and fac-Ir(ppy)3; ppy = 2-phenylpyridine)62 than those of Ir-1 did not affect the polymerization results (entries 3–4). The results indicated that the use of highly oxidative photocatalysts (Ir-1 and Ru-2 in Scheme 4) could disturb the electron transfer process in the photoexcited CoIII intermediate and resulted in observed inefficient polymerization.
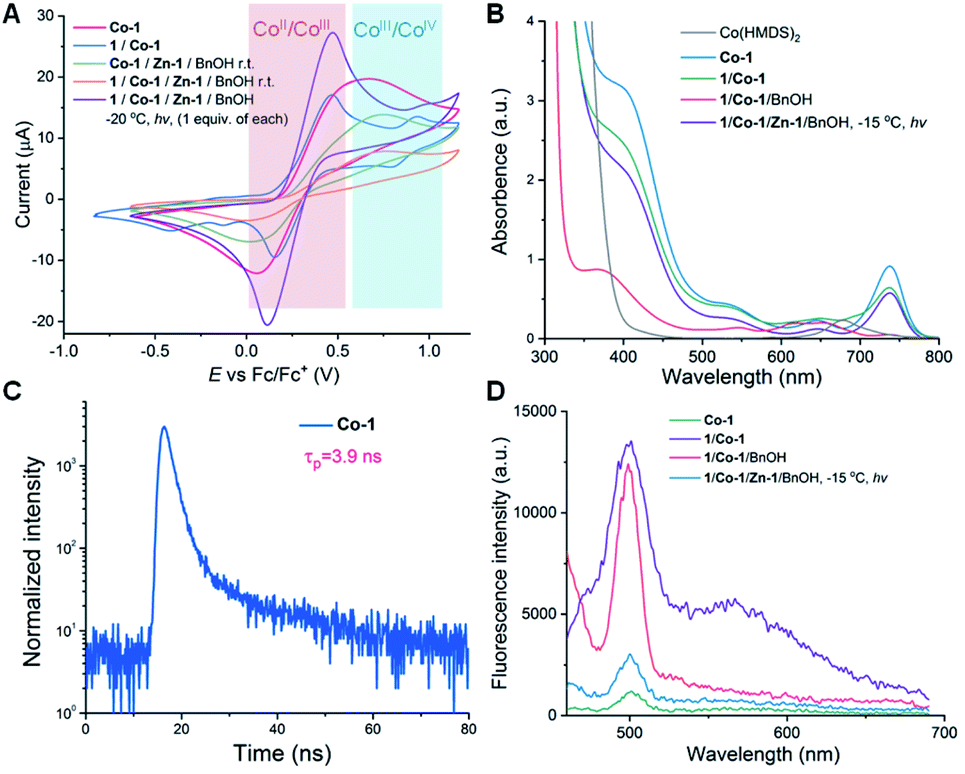 | ||
| Fig. 2 Electrochemical and photophysical studies of the photoredox decarboxylation reactions mediated by Co-1. (A) Cyclic voltammograms of Co-1 and the ring-opening reaction mixtures (1 equiv. of each) at various conditions. Scan rate: 100 mV s−1. Solvent: 0.1 M tetrabutylammonium hexafluorophosphate in THF. Initial scanning direction: zero to positive. (B) UV/vis absorption spectra of Co-1 and the reaction mixtures (1 equiv. of each). [Co] = 2.0 mM in THF in all samples. (C) Photoluminescence decay of Co-1 recorded in deaerated THF with excitation at 350 nm. (D) Emission spectra of Co-1 and the reaction mixtures (1 equiv. of each) with the excitation at 435 nm. [Co] = 2.0 mM in THF in all samples. | ||
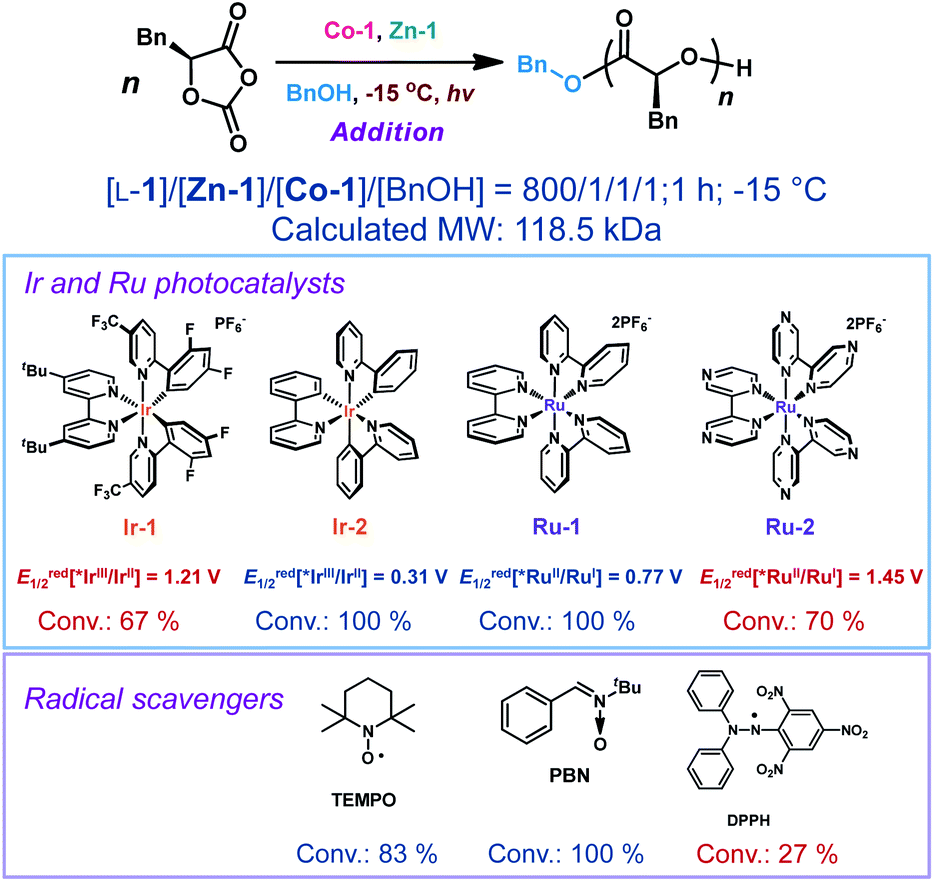 | ||
| Scheme 4 Effects of the addition of precious-metal photocatalysts and radical scavengers into the photoredox ring-opening polymerization of L-1.a aAbbreviations: conv., monomer conversion; TEMPO, 2,2,6,6-tetramethylpiperidine-N-oxyl; PBN, N-tert-butyl-α-phenylnitrone; DPPH, 2,2-diphenyl-1-picrylhydrazyl. For all polymerization reactions, [Co-1] = [Zn-1] = [BnOH]. Ered1/2 values for photocatalysts are based on ref. 62 Detailed polymerization data in Table S5.† | ||
The involvement of radical intermediates in Co/Zn-mediated photoredox ROP was confirmed by the use of radical scavengers. We found that nitroxide-based radical scavengers, such as 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO), and N-tert-butyl-α-phenylnitrone, were ineffective at inhibiting the polymerization (Scheme 4; Table S5,† entries 6 and 7), different from that in Ni/Ir-mediated photoredox reaction.22 This may be due to TEMPO's inability to trap radicals in certain organometallic reactions.73,74 However, another powerful radical scavenger, 2,2-diphenyl-1-picrylhydrazyl (DPPH), effectively disrupted chain propagation (entry 8), indicating the possible formation of the photoexcited radical species.
We then found that the absorption spectrum of Co-1 exhibited a maximum around 398 nm that did not correspond to the ligand (bpy, Fig. 2B). Upon photoexcitation at 298 K, a photoluminescence band with a lifetime of 3.9 ns was observed (Fig. 2C), which is longer than many first-row transition metal complexes.57,75 This band likely corresponds to a metal-to-ligand charge-transfer excited state,76 a possibility that was supported by density functional theory calculations: the lowest unoccupied molecular orbital was located exclusively on the π-system of the bpy ligand in Co-1, whereas the highest occupied molecular orbital (HOMO), HOMO−1, HOMO−3 and HOMO−4, were dominated by contributions from the Co dyz, dxz, dxy, and dx2–y2 orbitals (Fig. S22†).
Additionally, L-1/Co-1 exhibited a metal-to-ligand charge-transfer absorption band (Fig. 2B). Notably, the time-dependent density functional theory computed absorption spectra of both Co-1 and L-1/Co-1 are in good agreement with our experimental spectra (Fig. S23†). We also noticed that the intensity of the emission band at 499 nm was 11.6 times that of Co-1; and a broad peak with a maxima at 567 nm was also only found in L-1/Co-1 (excitation wavelength, 435 nm; Fig. 2D). This suggested that a transient photoactive cobaltacycle adduct formed after oxidative insertion of Co-1 into L-1. Attempts to isolate such photoactive CoIII species were unsuccessful. Because no CoII/CoI and minimal CoIV/CoIII (1.308 V versus SCE) redox couples were observed in the mixture of Co-1, Zn-1, BnOH and L-1 (1 equiv. of each) under irradiation with light at −20 °C (Fig. 2A), the photoexcited CoIII complex (II in Scheme 5) likely functioned as a viable photo-oxidant. Calculations based on the photophysical and electrochemical results indicated that such a CoIII intermediate had an estimated Ered1/2(*CoIII/CoII) of 2.878 V (Table S6†). Given the oxidation potential of the amino acid carboxylate (Ered1/2 = 0.832 V vs. SCE for N-(carbobenzyloxy)-L-phenylalanine; Table S6†) and literature reports,77,78 the photoexcited CoIII-mediated decarboxylation process was thermodynamically feasible.77
 | ||
| Scheme 5 Proposed Co/Zn-mediated photoredox ring-opening polymerization of O-carboxyanhydrides. | ||
Next we explored the state of the Co complex after photoredox decarboxylation. Comparison of cyclic voltammograms (Fig. 2A), the absorption and fluorescence spectra (Fig. 2B and D), and magnetic moments of Co-1 alone (4.78 μB) with those of the L-1/Co-1/Zn-1/BnOH mixture (1 equiv. of each) irradiated at −15 °C (4.94 μB) indicates that a CoII complex was likely to have been regenerated after the decarboxylation. Note that the magnetic moment of a L-1/Co-1 mixture decreased substantially from 4.78 to 4.16 μB, owing in part to the formation of a CoIII adduct, which is assumed to have a lower magnetic moment than CoII.79 On this basis, we hypothesize that the reduced CoII species and alkoxy radical III are likely generated via the successive loss of CO2 (Scheme 5). We note that considering the low-intensity absorption of the L-1/Co-1/BnOH mixture (Fig. 2B), it is less likely that a photoexcited Co–alkoxide species was involved in the decarboxylation.
Regioselective Co/Zn-mediated ring-opening of OCAs
We then investigated whether CoII could oxidatively add to the OCA in a regioselective manner (Scheme 6). We initially attempted to use 13C NMR spectroscopy to study the reaction of L-1 mediated by Co-1, Zn-1, and BnOH (1 equiv. of each), but the presence of the paramagnetic Co-1 complex resulted in less informative NMR spectra. We found that the use of 13C-labeled L-1 allowed us to conveniently monitor the reaction intermediates in 13C NMR, because broad peaks can be avoided when using 13C-labeled compounds in 13C NMR to decrease nuclear relaxation rates.80 To differentiate the insertion site of CoII complex in OCA, we prepared [13C2]-L-1 and [13C5]-L-1 (Fig. S25†). The peak for ester carbonyl carbon at 169 ppm in the spectrum of the photoredox reaction mixture obtained using [13C5]-L-1 at −20 °C suggested that Co-1 probably inserted regioselectively into the O1–C5 bond of L-1 (Fig. 3), in a manner similar to that observed for ROP of N-carboxyanhydrides81 and our previously reported Ni/Zn/Ir-mediated photoredox ROP.22,31 Only a small carbonate peak at 160 ppm was observed in the 13C NMR spectra obtained when [13C2]-L-1 was used in the photoredox reaction at −20 °C (Fig. 3, orange line), indicating that efficient photoredox decarboxylation occurred after the oxidative insertion. In contrast, in the absence of light at −20 °C, the same mixture showed multiple peaks at 163–160 ppm in the 13C NMR spectrum (Fig. 3, green line), suggesting that decarboxylation was inefficient without light irradiation. Note that mixing Co-1 with BnOH in the absence of L-1 led to the formation of a precipitate, which rules out the possibility that a Co–alkoxide inserted into L-1. | ||
| Scheme 6 Plausible oxidative insertion reactions between O-carboxyanhydrides and Co/Zn complexes. | ||
 | ||
| Fig. 3 13C NMR studies of oxidative addition reactions between L-1 and Co/Zn complexes. To improve the 13C NMR spectra qualities (due to the paramagnetic Co complexes) and determine the regioselectivity, [13C2]-1 and [13C5]-1 were used to study the ring-opening reaction (600 MHz, THF-d8). The red area highlighted in spectra indicates 13C2(O) carbonate peaks. [L-1]/[Co-1]/[Zn-1]/[BnOH] = 1/1/1/1. | ||
Additionally, the ESI-MS spectrum obtained for the reaction mixture of L-1/Co-1/Zn-1/BnOH at room temperature rather than at −15 °C exhibited two sets of peaks, suggesting that side reactions occurred at room temperature (Fig. S26a†). We initially speculated that decarbonylation at room temperature occurred, similar to the case of Ni/Zn/Ir-mediated ROP of OCA. However, Fourier transform IR spectroscopy indicated no obvious Co–carbonyl peaks (∼1940 cm−1) were observed in the mixtures of L-1/Co-1 (1/1) and L-1/Co-1/Zn-1/BnOH (1 equiv. of each), even in the presence of (PPh3)3RhCl, an extremely effective CO scavenger (Fig. S27†).82 We thus hypothesized that the alkoxy radical species, which was generated following decarboxylation, could mediate a chain-scission side reaction at room temperature (Scheme 7). The alkoxy radical-mediated β-scission-like reaction has been recently reported in Ce and Mn mediated photoredox reactions.53,83–85 It is also known that free primary alcohols are good O-nucleophiles and prone to 2e− oxidations that generate carboxylic acids or aldehydes under oxidative conditions.52,53 We noticed that the absence of Zn complex seemed to not affect this side reaction (Fig. S26b†). This radical-mediated side reaction can be completely suppressed in the cold temperature (Fig. S26c†), which may explain the temperature's influence on the chain propagation and MWs shown in Fig. 1A.
 | ||
| Scheme 7 Plausible radical-induced chain scission side reactions at room temperature. | ||
On the basis of our results, we propose the following mechanism (Scheme 5). First, regioselective oxidative addition of CoII complex to OCA leads to a transient CoIII intermediate I, which can be photoexcited to produce a strong photo-oxidant II. Next, the photoexcited cobaltacycle undergoes decarboxylation, which is thermodynamically feasible based on photophysical and electrochemical studies, to generate alkoxyl radical species III and reduced CoII species. Although it remains unclear how a reactive alkoxy radical mediates chain-scission side reactions at room temperature,86,87 at this point, we believe that this radical species should be rapidly intercepted by the Zn complex to generate a reactive Zn–alkoxide terminus at low temperature, thereby enabling the regeneration of the CoII catalyst.
Discovery of Co/Hf-mediated photoredox polymerization of OCAs
Based on our success of using Co/Zn complexes for photoredox ROP of OCAs, it was conceivable that other metal alkoxides may replace Zn–alkoxides to promote chain propagation in the polymerization. We then began our investigation by screening the reactivity of different metal alkoxides, e.g., Zr, Hf, Y, and in situ prepared Mg–alkoxides in the photoredox polymerization of L-1 in the presence of Co complexes at −15 °C (Table S7†). We found that the use of Co-2 (Scheme 8), a CoII complex with a less bulky pyridine ligand than that of Co-1, together with hafnium isopropoxide showed decent polymerization activities (entry 2 versus 1), yet with little control over the MWs when adjusting the monomer to catalyst feeding ratios (entries 2–4). Notably, the substitute of Co-2 with (bpy)Ni/Ir-1 resulted in the diminished reactivity (entry 5); and hafnium isopropoxide itself was unable to initiate polymerization (entry 6). Other metal alkoxides (Zr, Y and Mg) were essentially unreactive towards L-1 (entries 7–9). The addition of macrodentate complex to Hf alkoxide that forms Hf-1 (Scheme 8) enhanced the reactivity with an increased Mn value of 91.0 kDa, and a narrow Ð of 1.02 (entries 10–11). However, such strategy did not work for Zr complexes (entry 12). Our results agree with the recent studies about the reactivities of metal alkoxides or silylamides towards ROP of OCAs by other groups,88,89 which may be related to the decarboxylation capability of metal complexes.90 To this end, the identification of the active Hf complexes allows us to carry out stereoselective photoredox ROPs to synthesize different stereoregular polymers (vide infra). | ||
| Scheme 8 Co/Hf- or Co/Zn-mediated photoredox ring-opening polymerization. | ||
Stereoselective photoredox ring-opening polymerization of OCAs
We next explored whether our methods could be applied to stereoselective photoredox ROP of racemic OCAs. Similar to our studies in eROP, we found that the ligand of the Co complex markedly affected the polymerization stereoselectivity: under the optimized photoredox conditions at −15 °C, the use of Zn-2 and Co-1 ([L-1]/[D-1]/[Co-1]/[Zn-2]/[BnOH] = 150/150/1/1/1; Scheme 9) afforded a stereoblock (sb) copolymer poly(sb-1) with a Mn of 67.0 kDa, a narrow Ð of 1.05, and a high Pm of 0.96 (Pm, probability of meso dyad formation; Table S8, entry 1; Fig. S30a†). In contrast, Co-2/Zn-3 initiated controlled polymerization of rac-1 ([L-1]/[D-1]/[Co-2]/[Zn-3]/[BnOH] = 100/100/1/1/1) and afforded syndiotactic (sd) copolymer poly(sd-1) with a Pr (probability of racemic dyad formation) of 0.88 (Mn = 57.7 kDa, Ð = 1.14; entry 2; Fig. S30b†). Note that when Zn-2 was replaced with Zn-1, polymerization was not efficiently initiated (entries 3–4); when Co-2 was replaced by Co-1, the obtained polymer had a large Ð of 1.28 and a decreased Pr value (entry 5; Fig. S30c†); Zn-2 or Zn-3 alone was incapable to initiate polymerization of rac-1 (entries 6–7). Either the increase of steric bulky group or introducing electron-withdrawing cyano substituent on the β-diimine ligand of Zn-3 could not improve the syndioselectivity of the polymerization (entries 8–9; Fig. S30d and e†). | ||
| Scheme 9 Stereoselective photoredox ring-opening polymerization of racemic monomer 1 mediated by Co/Zn and Co/Hf complexes to prepare polymers with stereoblock or syndiotactic microstructures.a aAbbreviations: Pm, maximum probability of meso dyad formation; Pr, maximum probability of racemic dyad formation; Tg, glass transition temperature; Tm, melting temperature; sb, stereoblock; sd, syndiotactic. See Table S8† for detailed polymerization data. | ||
Additionally, we found that the combination of Co-2 with Hf-1 (Scheme 9), a reported syndioselective Hf–alkoxide complex for rac-lactide and rac-4 polymerization,88,91 provided syndiotactic copolymers when used for the photoredox ROP of rac-1 with a Pr of 0.89 ([L-1]/[D-1]/[Co-2]/[Hf-1] = 200/200/1/1; Mn = 96.6 kDa, Ð = 1.06; Table S8, entry 10; Fig. S30f†). Changing the [L-1]/[D-1] ratio from 1/1 to 2/1 or 1/2 markedly decreased the Pr value from 0.89 to 0.77 ([Co-2]/[Hf-1] = 1/1, entries 12–13 versus 11), confirming our assigned tetrad peaks. Kinetic studies agreed well with our measured syndioselectivity that kRS ≫ kRR ∼ kSS in both Co-2/Zn-3 and Co-2/Hf-1 catalytic systems (Fig. S31a and b†). The use of Hf-2 with less sterically bulky ligand compared to Hf-1 did not improve the Pr value (entry 15; Fig. S30g†); whereas the sole use of Hf complexes was found to be essentially unreactive towards rac-1 (entries 16–17), different from the literature.88
We next demonstrated that such stereoselective photoredox ROPs could be extended to other OCA monomers (Scheme 10). Notably, the stereoselectivity trends that were observed in ROP of rac-1 varied in other monomers. For example, Co-1/Zn-2 and Co-2/Zn-3 exhibited similar isoselectivity in the photoredox ROPs of rac-2 and rac-4 (Scheme 10A; Table S9, entries 1–2 and 4–5; Fig. S32 and S33†). Intriguingly, the photoredox ROP of rac-4 using Co-2/Hf-1 resulted in stereoblock copolymers, with a Pm of 0.74 ([L-4]/[D-4]/[Co-2]/[Hf-1] = 200/200/1/1; Mn = 32.8 kDa, Ð = 1.04; entry 6; Fig. S33c†), contrast to a syndiotactic poly(sd-4) generated by Hf-1 alone (Pr = 0.82; entry 7; Fig. S33d†). Kinetic studies are consistent with the formation of a stereoblock copolymer: the copolymerization of rac-4 was much slower than the polymerization of either enantiomer separately at the same [4]/[Hf-1] ratio under the same conditions (Fig. S31c†), which excludes the possibility that two isotactic polymers, poly(L-4) and poly(D-4), formed separately.
 | ||
| Scheme 10 Stereoselective photoredox ring-opening polymerization of racemic O-carboxyanhydrides (A) 2 and 4, (B) 3, and (C) 5, which are mediated by Co/Zn and Co/Hf complexes.a aAbbreviations: Pm, maximum probability of meso dyad formation; Pr, maximum probability of racemic dyad formation; Tg, glass transition temperature; Tm, melting temperature; sb, stereoblock; sd, syndiotactic. See Table S9† for detailed polymerization data. | ||
On the other hand, Co-1/Zn-2 provided stereoblock copolymers in the photoredox ROPs of rac-3 and rac-5 (Scheme 10B and C; Table S9,† entries 8 and 11), with Pm values of 0.94 and 0.77, respectively (Fig. S34a and S35a†); whereas Co-2/Zn-3 exhibited moderate syndioselectivity (entries 9 and 12; Fig. S34b and S35b†). However, Co-2/Hf-1 showed syndioselectivity towards rac-3 (entry 10; Fig. S34c†), but could mediate isoselective synthesis of poly(sb-5) with a Pm of 0.97 (entry 13; Fig. S35c†). Furthermore, in the presence of Co-2, the replacement of Hf-1 with Hf-4 (Scheme 10), a dinuclear hafnium complex, resulted in the syndioselective photoredox ROP of rac-5 with a Pr of 0.91 (entry 14; Fig. S35d†). Importantly, we found that Tg values increase as the syndiotacticity Pr values increase in poly(rac-1), higher than their stereoblock and isotactic counterparts (Table S10, entries 1–3; Fig. S36†), similar to the cases in poly(methyl methacrylate)92 and poly(3-hydroxybutyrate).93,94 However, such trend is reserved in the case of poly(5): obtained poly(sb-5) had a Tg of 97 °C, which was similar to that of poly(L-5) and much higher than that of poly(sd-5) (74 °C; Fig. S40;† entries 11–15), similar to that observed in poly(lactide).24,95 Our photoredox ROP thus offers stereoregular polymers with Tg values spanning over ∼100 °C (poly(sb-3) to poly(sb-5), Table S10†). Notably, stereocomplex (sc) of polyesters96 can also lead to polymers exhibiting melting temperatures that are not found in other microstructures (e.g., poly(sc-5), entry 16). To be practically useful in many applications, polymers are expected to have thermal transitions far from room temperature. Specifically, polyesters with Tg values above approximately 90 °C are useful for applications in which a rigid structure must be maintained, such as preventing polyesters deformation in the presence of hot water. The wide range of Tg values also potentially allows for the preparation of multiblock thermoplastic elastomers.97–99
Conclusions
Our work represents an important proof of concept that a catalytic system can mediate controlled ROP by either light (this report) or electricity.32 We have demonstrated that the oxidation status of Co complexes can be modulated by application of light during the polymerization, without the assistance from rare transition metal photocatalysts such as Ru or Ir complexes. Given the growing interest in replacing polyolefins with degradable and recyclable polymers,1 the discovery of Co/Zn catalytic system provides an alternative paradigm to develop stimulus-triggered polymerization strategies in coordination polymerization chemistry.Synergism between metal catalysts has been recently used to improve ROP performances, including using hetrodinuclear catalysts for the ROP of LA,100 and the copolymerization of CO2 and epoxides.101,102 In the case of photoredox ROP of OCAs, combining two metal catalysts not only significantly enhances the reactivity to produce high-MW PAHAs, but also allows for precise control of polymer's tacticity, which can significantly affect PAHA's thermal properties. To our knowledge, most photo- and electrochemically controlled polymerization strategies in polyolefins, however, could not mediate stereoselective polymerization.
Collectively, we believe that this stimulus-triggered controlled polymerization chemistry will prove to be widely applicable in polymer and materials research. Ongoing studies are directed toward the synthesis of new metal initiators to regulate stereoselectivity as well as functionalized polyesters for future production of tough polyesters and thermoplastic elastomers.
Author contributions
Y. Z., Q. F., and R. T. conceived the idea and designed experiments. Y. Z., Q. F., X. W. performed experiments. L. Y. performed density functional theory calculations. A. G. K. performed diffusion-ordered NMR spectroscopy experiments. Y. Z., Q. F., X. W. L. A. M., and R. T. analyzed the data. Y. Z. and R. T. wrote the manuscript.Conflicts of interest
Provisional patents (U.S. Patent Application No: 62/414016 and VTIP No. 19-112) have been filed pertaining to the results presented in this paper. The authors declare no other competing interests.Acknowledgements
This work was supported by start-up funding from Virginia Tech, ACS-Petroleum Research Foundation (57926-DNI-7), and the National Science Foundation (CHE-1807911). We thank Dr N. Murthy Shanaiah for NMR experiments, Mehdi Ashraf-Khorassani for electrospray ionization mass spectrometry studies, Dr Jeffrey Parks for inductively coupled plasma mass spectrometry measurements, Dr Amanda Morris for fluorescence spectra measurements, Dr Guoliang Liu for differential scanning calorimetry, and Dr Webster Santos and Dr John Matson (all from Virginia Tech, Department of Chemistry) for providing anhydrous solvents.References
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed.
- K. Sudesh, H. Abe and Y. Doi, Prog. Polym. Sci., 2000, 25, 1503–1555 CrossRef CAS.
- M. A. Hillmyer and W. B. Tolman, Acc. Chem. Res., 2014, 47, 2390–2396 CrossRef CAS PubMed.
- J.-B. Zhu, E. M. Watson, J. Tang and E. Y.-X. Chen, Science, 2018, 360, 398–403 CrossRef CAS PubMed.
- T. Hayashi, Prog. Polym. Sci., 1994, 19, 663–702 CrossRef CAS.
- B. Martin Vaca and D. Bourissou, ACS Macro Lett., 2015, 4, 792–798 CrossRef CAS.
- Q. Yin, L. Yin, H. Wang and J. Cheng, Acc. Chem. Res., 2015, 48, 1777–1787 CrossRef CAS PubMed.
- R. Tong, Ind. Eng. Chem. Res., 2017, 56, 4207–4219 CrossRef CAS.
- M. Yin and G. L. Baker, Macromolecules, 1999, 32, 7711–7718 CrossRef CAS.
- F. Jing, M. R. Smith and G. L. Baker, Macromolecules, 2007, 40, 9304–9312 CrossRef CAS.
- G. L. Baker, E. B. Vogel and M. R. Smith, Polym. Rev., 2008, 48, 64–84 CrossRef CAS.
- T. L. Simmons and G. L. Baker, Biomacromolecules, 2001, 2, 658–663 CrossRef CAS PubMed.
- Y. Yu, J. Zou and C. Cheng, Polym. Chem., 2014, 5, 5854–5872 RSC.
- R. T. Martin, L. P. Camargo and S. A. Miller, Green Chem., 2014, 16, 1768–1773 RSC.
- S. A. Cairns, A. Schultheiss and M. P. Shaver, Polym. Chem., 2017, 8, 2990–2996 RSC.
- Y. Xu, M. R. Perry, S. A. Cairns and M. P. Shaver, Polym. Chem., 2019, 10, 3048–3054 RSC.
- O. T. du Boullay, E. Marchal, B. Martin-Vaca, F. P. Cossio and D. Bourissou, J. Am. Chem. Soc., 2006, 128, 16442–16443 CrossRef PubMed.
- Y. Zhong and R. Tong, Front. Chem., 2018, 6, 641 CrossRef PubMed.
- R. J. Pounder, D. J. Fox, I. A. Barker, M. J. Bennison and A. P. Dove, Polym. Chem., 2011, 2, 2204–2212 RSC.
- A. Buchard, D. R. Carbery, M. G. Davidson, P. K. Ivanova, B. J. Jeffery, G. I. Kociok-Köhn and J. P. Lowe, Angew. Chem., Int. Ed., 2014, 53, 13858–13861 CrossRef CAS PubMed.
- R. Wang, J. Zhang, Q. Yin, Y. Xu, J. Cheng and R. Tong, Angew. Chem., Int. Ed., 2016, 55, 13010–13014 CrossRef CAS PubMed.
- Q. Feng and R. Tong, J. Am. Chem. Soc., 2017, 139, 6177–6182 CrossRef CAS PubMed.
- Q. Feng, Y. Zhong, L. Xie and R. Tong, Synlett, 2017, 28, 1857–1866 CrossRef CAS.
- M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 RSC.
- J.-F. Lutz, D. Neugebauer and K. Matyjaszewski, J. Am. Chem. Soc., 2003, 125, 6986–6993 CrossRef CAS PubMed.
- B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229–3238 CrossRef CAS PubMed.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed.
- N. Ajellal, J.-F. Carpentier, C. Guillaume, S. M. Guillaume, M. Helou, V. Poirier, Y. Sarazin and A. Trifonov, Dalton Trans., 2010, 39, 8363–8376 RSC.
- C. M. Thomas, Chem. Soc. Rev., 2010, 39, 165–173 RSC.
- S. M. Guillaume, E. Kirillov, Y. Sarazin and J.-F. Carpentier, Chem.–Eur. J., 2015, 21, 7988–8003 CrossRef CAS PubMed.
- Q. Feng, L. Yang, Y. Zhong, D. Guo, G. Liu, L. Xie, W. Huang and R. Tong, Nat. Commun., 2018, 9, 1559 CrossRef PubMed.
- Y. Zhong, Q. Feng, X. Wang, J. Chen, W. Cai and R. Tong, ACS Macro Lett., 2020, 9, 1114–1118 CrossRef CAS.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- B. J. Shields, B. Kudisch, G. D. Scholes and A. G. Doyle, J. Am. Chem. Soc., 2018, 140, 3035–3039 CrossRef CAS PubMed.
- C. B. Larsen and O. S. Wenger, Chem.–Eur. J., 2018, 24, 2039–2058 CrossRef CAS PubMed.
- O. Reiser, Acc. Chem. Res., 2016, 49, 1990–1996 CrossRef CAS PubMed.
- A. Hossain, A. Bhattacharyya and O. Reiser, Science, 2019, 364, eaav9713 CrossRef PubMed.
- Q. M. Kainz, C. D. Matier, A. Bartoszewicz, S. L. Zultanski, J. C. Peters and G. C. Fu, Science, 2016, 351, 681–684 CrossRef CAS PubMed.
- D. M. Flores and V. A. Schmidt, J. Am. Chem. Soc., 2019, 141, 8741–8745 CrossRef CAS PubMed.
- M. E. Weiss, L. M. Kreis, A. Lauber and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 50, 11125–11128 CrossRef CAS PubMed.
- B. D. Ravetz, J. Y. Wang, K. E. Ruhl and T. Rovis, ACS Catal., 2019, 9, 200–204 CrossRef CAS.
- E. Bergamaschi, F. Beltran and C. J. Teskey, Chem.–Eur. J., 2020, 26, 5180–5184 CrossRef CAS PubMed.
- W.-Q. Liu, T. Lei, S. Zhou, X.-L. Yang, J. Li, B. Chen, J. Sivaguru, C.-H. Tung and L.-Z. Wu, J. Am. Chem. Soc., 2019, 141, 13941–13947 CrossRef CAS PubMed.
- C.-H. Lim, M. Kudisch, B. Liu and G. M. Miyake, J. Am. Chem. Soc., 2018, 140, 7667–7673 CrossRef CAS PubMed.
- X. Shen, Y. Li, Z. Wen, S. Cao, X. Hou and L. Gong, Chem. Sci., 2018, 9, 4562–4568 RSC.
- Y. Gao, C. Yang, S. Bai, X. Liu, Q. Wu, J. Wang, C. Jiang and X. Qi, Chem, 2020, 6, 675–688 CAS.
- P. Nuhant, M. S. Oderinde, J. Genovino, A. Juneau, Y. Gagné, C. Allais, G. M. Chinigo, C. Choi, N. W. Sach, L. Bernier, Y. M. Fobian, M. W. Bundesmann, B. Khunte, M. Frenette and O. O. Fadeyi, Angew. Chem., Int. Ed., 2017, 56, 15309–15313 CrossRef CAS PubMed.
- L. Wang, J. M. Lear, S. M. Rafferty, S. C. Fosu and D. A. Nagib, Science, 2018, 362, 225 CrossRef CAS PubMed.
- R.-Z. Liu, J. Li, J. Sun, X.-G. Liu, S. Qu, P. Li and B. Zhang, Angew. Chem., Int. Ed., 2020, 59, 4428–4433 CrossRef CAS PubMed.
- Y. Qiao, Q. Yang and E. J. Schelter, Angew. Chem., Int. Ed., 2018, 57, 10999–11003 CrossRef CAS PubMed.
- A. Hu, J.-J. Guo, H. Pan and Z. Zuo, Science, 2018, 361, 668–672 CrossRef CAS PubMed.
- K. Zhang, L. Chang, Q. An, X. Wang and Z. Zuo, J. Am. Chem. Soc., 2019, 141, 10556–10564 CrossRef CAS PubMed.
- Q. An, Z. Wang, Y. Chen, X. Wang, K. Zhang, H. Pan, W. Liu and Z. Zuo, J. Am. Chem. Soc., 2020, 142, 6216–6226 CrossRef CAS PubMed.
- J.-H. Ye, M. Miao, H. Huang, S.-S. Yan, Z.-B. Yin, W.-J. Zhou and D.-G. Yu, Angew. Chem., Int. Ed., 2017, 56, 15416–15420 CrossRef CAS PubMed.
- X.-J. Wei, I. Abdiaj, C. Sambiagio, C. Li, E. Zysman-Colman, J. Alcázar and T. Noël, Angew. Chem., Int. Ed., 2019, 58, 13030–13034 CrossRef CAS PubMed.
- W.-M. Cheng and R. Shang, ACS Catal., 2020, 10, 9170–9196 CrossRef CAS.
- F. Glaser and O. S. Wenger, Coord. Chem. Rev., 2020, 405, 213129 CrossRef CAS.
- M. C. Carey, S. L. Adelman and J. K. McCusker, Chem. Sci., 2019, 10, 134–144 RSC.
- J. K. McCusker, Science, 2019, 363, 484–488 CrossRef CAS PubMed.
- C. Theunissen, M. A. Ashley and T. Rovis, J. Am. Chem. Soc., 2019, 141, 6791–6796 CrossRef CAS PubMed.
- M. Li, Y. Tao, J. Tang, Y. Wang, X. Zhang, Y. Tao and X. Wang, J. Am. Chem. Soc., 2019, 141, 281–289 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed.
- J. W. Tucker and C. R. J. Stephenson, J. Org. Chem., 2012, 77, 1617–1622 CrossRef CAS PubMed.
- M. Chen, M. Zhong and J. A. Johnson, Chem. Rev., 2016, 116, 10167–10211 CrossRef CAS PubMed.
- J. C. Theriot, C.-H. Lim, H. Yang, M. D. Ryan, C. B. Musgrave and G. M. Miyake, Science, 2016, 352, 1082–1086 CrossRef CAS PubMed.
- S. Dadashi-Silab, S. Doran and Y. Yagci, Chem. Rev., 2016, 116, 10212–10275 CrossRef CAS PubMed.
- N. Corrigan, S. Shanmugam, J. Xu and C. Boyer, Chem. Soc. Rev., 2016, 45, 6165–6212 RSC.
- T. G. Ribelli, D. Konkolewicz, S. Bernhard and K. Matyjaszewski, J. Am. Chem. Soc., 2014, 136, 13303–13312 CrossRef CAS.
- D. Konkolewicz, K. Schröder, J. Buback, S. Bernhard and K. Matyjaszewski, ACS Macro Lett., 2012, 1, 1219–1223 CrossRef CAS.
- M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, R. A. Pascal, G. G. Malliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712–5719 CrossRef CAS.
- M. Haga, E. S. Dodsworth, G. Eryavec, P. Seymour and A. B. P. Lever, Inorg. Chem., 1985, 24, 1901–1906 CrossRef CAS.
- A. C. Albéniz, P. Espinet, R. López-Fernández and A. Sen, J. Am. Chem. Soc., 2002, 124, 11278–11279 CrossRef PubMed.
- J. R. Carney, B. R. Dillon, L. Campbell and S. P. Thomas, Angew. Chem., Int. Ed., 2018, 57, 10620–10624 CrossRef CAS PubMed.
- G. Auböck and M. Chergui, Nat. Chem., 2015, 7, 629–633 CrossRef PubMed.
- D. M. Arias-Rotondo and J. K. McCusker, Chem. Soc. Rev., 2016, 45, 5803–5820 RSC.
- Z. W. Zuo, D. T. Ahneman, L. L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437–440 CrossRef CAS PubMed.
- Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 5257–5260 CrossRef CAS PubMed.
- P. P. Power, Chem. Rev., 2012, 112, 3482–3507 CrossRef CAS PubMed.
- W. Bermel, I. Bertini, I. C. Felli, R. Kümmerle and R. Pierattelli, J. Am. Chem. Soc., 2003, 125, 16423–16429 CrossRef CAS PubMed.
- T. J. Deming and S. A. Curtin, J. Am. Chem. Soc., 2000, 122, 5710–5717 CrossRef CAS.
- K. R. Dunbar and S. C. Haefner, Inorg. Chem., 1992, 31, 3676–3679 CrossRef CAS.
- J.-J. Guo, A. Hu and Z. Zuo, Tetrahedron Lett., 2018, 59, 2103–2111 CrossRef CAS.
- B. D. W. Allen, M. D. Hareram, A. C. Seastram, T. McBride, T. Wirth, D. L. Browne and L. C. Morrill, Org. Lett., 2019, 21, 9241–9246 CrossRef CAS PubMed.
- J. Zhang, Y. Li, F. Zhang, C. Hu and Y. Chen, Angew. Chem., Int. Ed., 2016, 55, 1872–1875 CrossRef CAS PubMed.
- A. Padwa, J. Org. Chem., 2009, 74, 6421–6441 CrossRef CAS PubMed.
- L. Capaldo and D. Ravelli, Chem. Commun., 2019, 55, 3029–3032 RSC.
- Y. Sun, Z. Jia, C. Chen, Y. Cong, X. Mao and J. Wu, J. Am. Chem. Soc., 2017, 139, 10723–10732 CrossRef CAS PubMed.
- Y. Wang and T.-Q. Xu, Macromolecules, 2020, 53, 8829–8836 CrossRef CAS.
- D. H. Gibson, Chem. Rev., 1996, 96, 2063–2096 CrossRef CAS PubMed.
- A. J. Chmura, M. G. Davidson, C. J. Frankis, M. D. Jones and M. D. Lunn, Chem. Commun., 2008, 47, 1293–1295 RSC.
- L. R. Denny, R. F. Boyer and H.-G. Ellas, J. Macromol. Sci., Part B: Phys., 1986, 25, 227–265 CrossRef.
- N. Ajellal, M. Bouyahyi, A. Amgoune, C. M. Thomas, A. Bondon, I. Pillin, Y. Grohens and J.-F. Carpentier, Macromolecules, 2009, 42, 987–993 CrossRef CAS.
- X. Tang and E. Y. X. Chen, Nat. Commun., 2018, 9, 2345 CrossRef PubMed.
- T. M. Ovitt and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 1316–1326 CrossRef CAS PubMed.
- H. Tsuji, Macromol. Biosci., 2005, 5, 569–597 CrossRef CAS PubMed.
- F. S. Bates, M. A. Hillmyer, T. P. Lodge, C. M. Bates, K. T. Delaney and G. H. Fredrickson, Science, 2012, 336, 434–440 CrossRef CAS PubMed.
- G. L. Gregory, G. S. Sulley, L. P. Carrodeguas, T. T. D. Chen, A. Santmarti, N. J. Terrill, K.-Y. Lee and C. K. Williams, Chem. Sci., 2020, 11, 6567–6581 RSC.
- G. S. Sulley, G. L. Gregory, T. T. D. Chen, L. Peña Carrodeguas, G. Trott, A. Santmarti, K.-Y. Lee, N. J. Terrill and C. K. Williams, J. Am. Chem. Soc., 2020, 142, 4367–4378 CrossRef CAS PubMed.
- A. B. Kremer and P. Mehrkhodavandi, Coord. Chem. Rev., 2019, 380, 35–57 CrossRef CAS.
- J. A. Garden, P. K. Saini and C. K. Williams, J. Am. Chem. Soc., 2015, 137, 15078–15081 CrossRef CAS PubMed.
- A. C. Deacy, A. F. R. Kilpatrick, A. Regoutz and C. K. Williams, Nat. Chem., 2020, 12, 372–380 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc05550f |
| This journal is © The Royal Society of Chemistry 2021 |