
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHexakis-adducts of [60]fullerene as molecular scaffolds of polynuclear spin-crossover molecules†
Mario
Palacios-Corella‡
a,
Javier
Ramos-Soriano‡
b,
Manuel
Souto
 ac,
Duarte
Ananias
c,
Joaquín
Calbo
a,
Enrique
Ortí
*a,
Beatriz M.
Illescas
b,
Miguel
Clemente-León
*a,
Nazario
Martín
*bd and
Eugenio
Coronado
*a
ac,
Duarte
Ananias
c,
Joaquín
Calbo
a,
Enrique
Ortí
*a,
Beatriz M.
Illescas
b,
Miguel
Clemente-León
*a,
Nazario
Martín
*bd and
Eugenio
Coronado
*a
aInstituto de Ciencia Molecular (ICMol), Universidad de Valencia, Catedrático José Beltrán 2, 46980 Paterna, Spain. E-mail: miguel.clemente@uv.es; eugenio.coronado@uv.es; Enrique.Orti@uv.es
bDepartamento de Química Orgánica, Facultad de Ciencias Químicas, Universidad Complutense, 28040 Madrid, Spain
cCICECO-Aveiro Institute of Materials, Department of Chemistry, Universidade de Aveiro, Campus Universitário de Santiago, 3810-193 Aveiro, Portugal
dIMDEA-Nanoscience, C/Faraday 9, Campus de Cantoblanco, 28049 Madrid, Spain. E-mail: nazmar@ucm.es
First published on 16th November 2020
Abstract
A family of hexakis-substituted [60]fullerene adducts endowed with the well-known tridentate 2,6-bis(pyrazol-1-yl)pyridine (bpp) ligand for spin-crossover (SCO) systems has been designed and synthesized. It has been experimentally and theoretically demonstrated that these molecular scaffolds are able to form polynuclear SCO complexes in solution. UV-vis and fluorescence spectroscopy studies have allowed monitoring of the formation of up to six Fe(II)–bpp SCO complexes. In addition, DFT calculations have been performed to model the different complexation environments and simulate their electronic properties. The complexes retain SCO properties in the solid state exhibiting both thermal- and photoinduced spin transitions, as confirmed by temperature-dependent magnetic susceptibility and Raman spectroscopy measurements. The synthesis of these complexes demonstrates that [60]fullerene hexakis-adducts are excellent and versatile platforms to develop polynuclear SCO systems in which a fullerene core is surrounded by a SCO molecular shell.
Introduction
Spincrossover (SCO) complexes constitute one of the most spectacular examples of molecular bistability. These complexes can be reversibly switched between two distinct states (high- and low-spin states) by a variety of external stimuli such as light, temperature, pressure, electric field or analytes.1 Most examples of this kind are provided by Fe(II) complexes, which undergo SCO between the diamagnetic low-spin state (LS, S = 0) and the paramagnetic high-spin state (HS, S = 2).1 A remarkable family of SCO Fe(II) compounds are bis-chelated complexes of the tridentate 2,6-bis(pyrazol-1-yl)pyridine (bpp) ligand and derivatives. Such complexes often present abrupt thermal spin transitions close to room temperature and SCO induced by irradiation at low temperatures in the solid state [Light-Induced Excited Spin State Trapping (LIESST) effect] at relatively high temperatures and long lifetimes.2 The incorporation of these molecular species on a substrate, including a surface or a nanoparticle, is an essential step towards real applications of SCO materials,3 but it still presents severe limitations. Indeed, previous attempts to prepare self-assembled monolayers (SAMs) of [Fe(bpp)2]2+ derivatives on gold4a or metal-oxide surfaces4b have been unsuccessful owing to the too weak bonding to iron(II) of these ligands grafted on surfaces. A most promising result was the preparation of gold nanoparticle arrays decorated with thiol functionalized [Fe(bpp)2]2+ complexes. Still, the thermal spin transition was very gradual and incomplete.5A loosely unexplored possibility is using fullerenes as a carbon-based platform on which SCO complexes are anchored. Since the discovery of fullerene C60, functional fullerene derivatives have attracted a big deal of attention due to their exceptional electrochemical and photophysical properties and the wide range of applications including molecular electronics and biomedical applications.6 In this context, transition metal complexes have been incorporated onto fullerenes for the development of new molecular materials exhibiting additional features and applications7 but, to our knowledge, [60]fullerene has only been used as bulky end-capping group of mononuclear SCO complexes. These complexes were proposed as the first step towards the preparation of single-molecule junctions in which the bis-fullerene derivative acts as a bridge between two graphene electrodes8 or as an anchoring group for nanocarbon materials.9 Very recently, it has also been reported the use of C60 as template for the growth of SCO Fe(II) metal–organic cages.10
Herein, we propose an alternative approach in which [60]fullerene plays the role of a spherical scaffold surrounded by a shell of [Fe(bpp)2]2+ complexes anchored to it. Thus, we have synthesized hexakis-substituted [60]fullerene adducts11 with 12 bpp units arranged with icosahedral symmetry around the fullerene surface (Fig. 1). Owing to the flexibility of the malonate-based spacers, up to six Fe(II)–bpp SCO complexes can be coordinated to the 12 bpp groups. The concentration of bpps on the C60 core is of interest towards the enhancement of interactions between bpp and Fe(II) leading to polynuclear C60-based SCO complexes of enhanced stability and controlled 3D geometry.
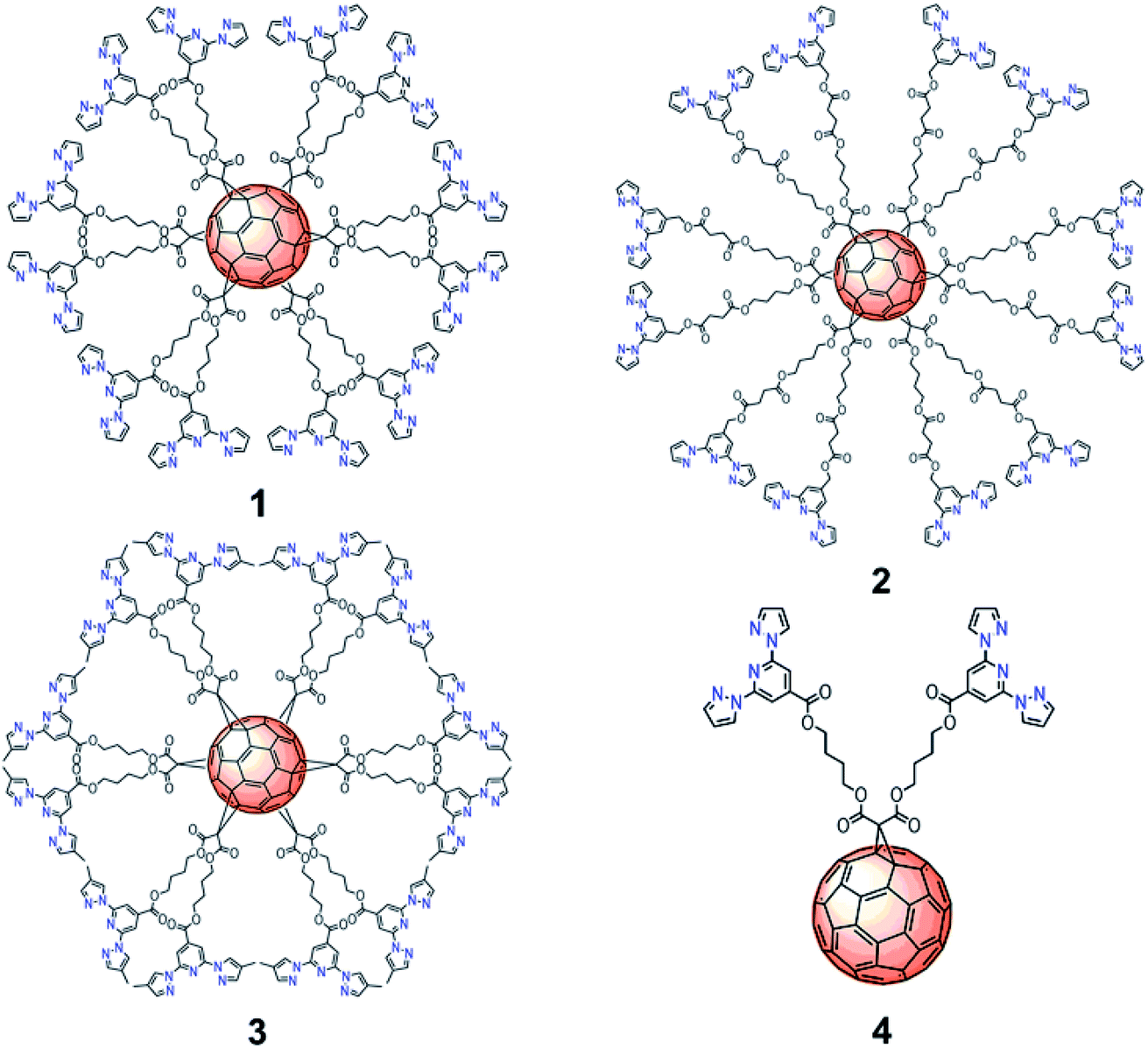 | ||
| Fig. 1 Molecular structures of C60 hexa-adducts 1, 2, 3 and mono-adduct 4 functionalized with bpp ligands. | ||
In order to understand the influence of the spacer and substituents on the behavior of these complexes, we have prepared a set of functionalized C60 derivatives including three hexakis-substituted [60]fullerene adducts with spacers of different length and different bpp substituents (hexakis-adducts 1, 2 and 3) and a model ligand with only two bpp moieties (mono-adduct 4) (Fig. 1). Spectroscopic characterization in solution has been carried out to demonstrate the formation of the polynuclear Fe(II) complexes based on 1, 2 and 3. In addition, DFT calculations have been performed to model the different complexation environments and simulate their electronic properties. Finally, magnetic and Raman studies in the solid state have been used to characterize the SCO properties of these materials with temperature and light as external stimuli. To the best of our knowledge, this is the first time that highly symmetrical 3D hexakis metallo-[60]fullerenes undergoing SCO are reported, opening new avenues towards the preparation and processing of polynuclear SCO scaffolds, which could incorporate additional electronic functionalities.11
Results and discussion
Formation of the Fe(II) complexes in solution
The C60 hexa-adducts 1, 2, 3 and the mono-adduct 4 (Fig. 1) were synthesized following the synthetic strategy depicted in Schemes S1–S4.† Compounds 1–3 were easily prepared by esterification of hexakis-adduct P1 (ref. 11a) with ligands bppCOOH, L4 and L5, respectively, using DCC/DPTS as coupling reagents. After purification using size-exclusion chromatography employing Sephadex, compounds 1–3 were obtained in quantitative yields. Characterization was carried out by standard spectroscopic and analytical techniques (Fig. S1–S3 for 1, S6 and S7 for 2 and S10–S12 for 3†). Owing to the high symmetry of hexakis-adducts 1–3, 1H and 13C NMR were very simple.The assignment of the 1H and 13C NMR spectra was achieved using COSY, HSQC and DEPT NMR spectra. As expected, in the 13C NMR only two signals were observed for the sp2 carbons of the C60 cage (δ ∼ 141 and 146 ppm), thus providing evidence of the highly Th symmetry of these compounds. The absence of the methylene signal bound to the hydroxy group at ∼62.5 ppm (present in compound P1) revealed that the esterification reactions were successfully accomplished.
The synthesis of monoadduct 4 was carried out following a synthetic strategy in three steps, as depicted in Scheme S4.† First, P2 was prepared by Bingel–Hirsch cyclopropanation of malonate L611a to [60]fullerene in the presence of DBU and CBr4 in toluene (see Scheme S4†). Deprotection of the hydroxyl groups by hydrogenation, followed by esterification with bppCOOH using DCC/DPTS afforded 4 in good yield after column chromatography. Compound 4 and its corresponding intermediates were fully characterized by NMR, FTIR spectroscopy and mass spectrometry (Fig. S13–S21†). Thus, the 13C-NMR spectrum reveals the presence of the 15 signals corresponding to the sp2 carbons of the C60 unit, which typically appear for methanofullerenes between 139 and 146 ppm. Furthermore, MS data obtained using MALDI-TOF spectrometry confirmed the presence of the molecular ion peak.
The Fe(II) complexes were obtained by mixing CH2Cl2 solutions of ligands 1, 2, 3 or 4 with CH3CN solutions of Fe(BF4)2·nH2O or Fe(ClO4)2·nH2O (up to 12 equivalents). Rapid color changes from orange to red (1, 3 and 4) and from orange to yellow (2) were observed. The formation of stable solutions after addition of Fe2+ in polar solvents such as CH3CN, in which ligands 1, 2 and 3 are not soluble, is indicative of the presence of discrete, highly charged, polynuclear polar species. The mono-adduct 4 exhibits a different behavior since a fast precipitation of the metal complex is observed upon addition of Fe2+. This could be related to the lower charge of the complex since only one iron ion can be coordinated by 4, in contrast to the six iron ions that can be incorporated by the hexa-adducts, or to the formation of polynuclear or polymeric species. Diluted solutions (∼5 × 10−6 M) of the metal complexes of 1, 2, 3 and 4 were obtained in an excess of CH2Cl2 with a small amount of CH3CN used to dissolve the Fe2+ salts. Such diluted solutions were used for UV-vis and fluorescence spectroscopy studies in order to monitor the formation of the resulting complexes.
Spectroscopic titrations were performed in 2.5 × 10−6 to 7.5 × 10−6 M solutions of ligands 1 and 2 in CH2Cl2 (Fig. 2). The ligands exhibited sharp and intense bpp-centered π–π* and charge-transfer (CT) absorption bands in the UV region at 245, 270 and 335 nm for 1 and at 245, 270 and 306 nm for 2, in agreement with theoretical calculations (see below). The lowest-energy band of 1 is red-shifted, compared to 2 and unsubstituted bpp (λmax = 304 nm), owing to the conjugation of the carboxylic ester group. Addition of aliquots of the Fe2+ salt in CH3CN leads to an intensity decrease of the band at 245 nm and to the appearance of different isosbestic points (Fig. 2). Furthermore, a low-intensity broad band with maximum absorbance at 460 and 430 nm appears after addition of Fe2+ for 1 and 2, respectively, as a result of the formation of the coordination complex. These lowest-energy absorptions are assigned to metal-to-ligand charge transfer (MLCT) bands in agreement with previous results reported for Fe(II)–bpp complexes12,13 and theoretical calculations (see below). Saturation of the MLCT bands was reached upon adding 6 equivalents of Fe2+ to solutions of 1 and 2 (Fig. 2). Titration of model bpp ligands of 1 and 2 functionalized with an ethyl ester (bppCOOEt) or a hydroxymethyl group (bppCH2OH), respectively, showed similar trends for the π–π* UV bands (Fig. S22†), thus confirming that the changes in the absorption spectra are induced by the coordination to Fe2+. Furthermore, the MLCT bands of the model complexes appear at similar wavelengths to those observed for 1 and 2 and those reported for complexes formed by bpp ligands functionalized with a pyrene group through ester or methyl ether groups.14
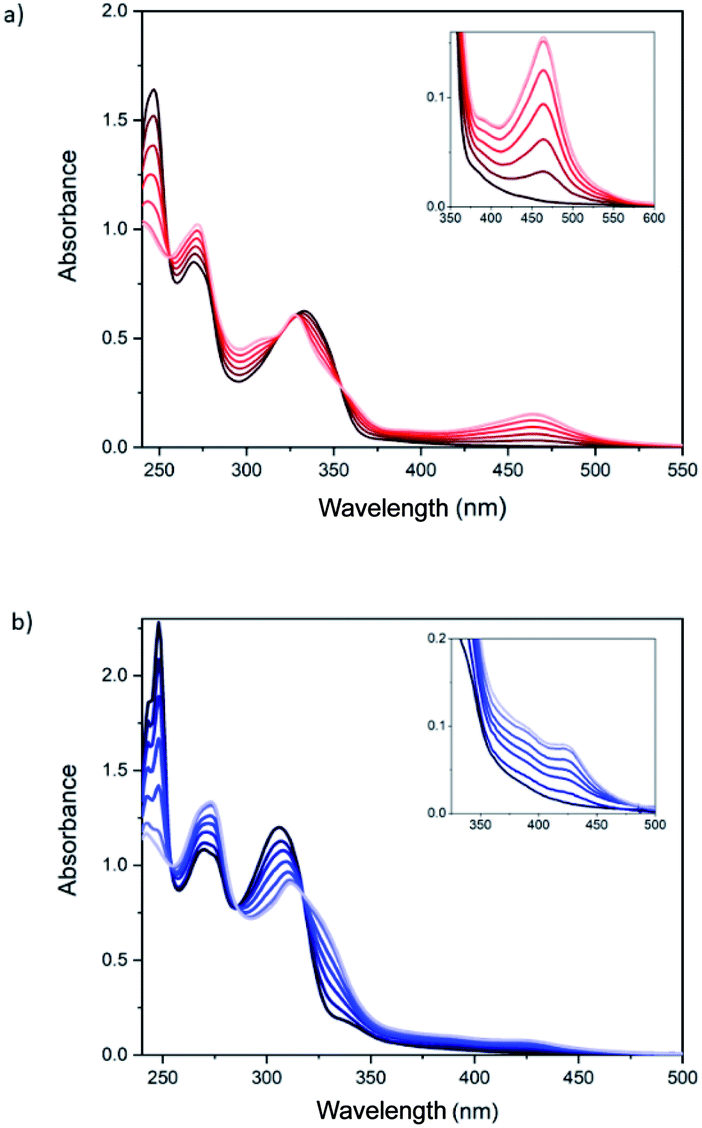 | ||
| Fig. 2 UV-vis spectra of (a) 1 and (b) 2 solutions (5 × 10−6 M, in black) and after addition of up to six equivalents of Fe(ClO4)2. Color scale with increasing transparency represents increasing amount of Fe(ClO4)2 in steps of 1 equivalent. | ||
It is worth to note that the wavelength of the MLCT bands is related to the different spin state (HS or LS) of the complex. Indeed, UV-vis spectra of pure LS bpp compounds present MLCT bands centered around λmax = 420 nm (εmax = 4–6 × 103 M−1 cm−1), whereas MLCT bands attributed to HS complexes appear not well-resolved at lower wavelengths.15 This behavior is consistent with a LS state for 1 and a mixture of LS and HS states in the case of 2 after addition of 6 equivalents of Fe2+. The LS MLCT bands for 1 and bppCOOEt complexes are shifted to higher wavelengths with respect to those of 2 or bppCH2OH complexes, as expected for carboxylic ester derivatives due to enhanced conjugation. The increase of the HS fraction in complex 2 with respect to those of complex 1 is also consistent with that observed in solution for Fe(II)–bpp complexes with similar substituents. Indeed, Evans method indicates that the Fe(II) complex of the hydroxymethyl derivative of bpp is around 90% HS,16 whereas that of the carboxylic ester derivative is around 50% HS in CD3CN.17 Finally, molar extinction coefficient values (εmax) of the MLCT bands for complexes 1 and 2 support a concentration of Fe(II) complexes in the range of that obtained with more simple model compounds assuming that all bpp ligands belonging to 1 and 2 are coordinated (Fig. S23†). The εmax value measured for complex 1 at 460 nm divided by six (due to the six coordinated iron atoms) is ∼5800 M−1 cm−1, which is consistent with that registered for other pure LS Fe(II)–bpp complexes in solution (5600 M−1 cm−1).18 Otherwise, the εmax values recorded for complex 2 at 385 and 420 nm (around 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1 in both cases) in comparison with εmax values reported in the literature for pure LS and HS complexes is indicative of a ∼75% HS.17 The presence of polynuclear species of 1 and 2 coordinated to Fe2+ is also confirmed by mass spectroscopy of the precipitates obtained in acetonitrile solutions with an excess of Fe2+ (see 5.1 section in the ESI†). Theoretical calculations (see below) indicate that the intramolecular complexation of Fe(II) is favored in both 1 and 2 with respect to an intermolecular complex formation. These calculations and the similar spectroscopic titrations of 1 and 2 together with the absence of precipitates and the solubility in MeCN, mentioned above, support the presence of discrete polynuclear species rather than oligomeric or polymeric ones formed by coordination to Fe(II) by bpp ligands from different molecules.
000 M−1 cm−1 in both cases) in comparison with εmax values reported in the literature for pure LS and HS complexes is indicative of a ∼75% HS.17 The presence of polynuclear species of 1 and 2 coordinated to Fe2+ is also confirmed by mass spectroscopy of the precipitates obtained in acetonitrile solutions with an excess of Fe2+ (see 5.1 section in the ESI†). Theoretical calculations (see below) indicate that the intramolecular complexation of Fe(II) is favored in both 1 and 2 with respect to an intermolecular complex formation. These calculations and the similar spectroscopic titrations of 1 and 2 together with the absence of precipitates and the solubility in MeCN, mentioned above, support the presence of discrete polynuclear species rather than oligomeric or polymeric ones formed by coordination to Fe(II) by bpp ligands from different molecules.
To get more insight into the optical properties of the Fe(II) complexes formed by 1 and 2 in solution, fluorescence measurements were carried out by using the same solutions and by excitation at the isosbestic points (320 nm for 1 and 285 nm for 2). Luminescence of hexa-adducts 1 and 2 in CH2Cl2 showed broad emission bands (Fig. 3) with maxima at 380 nm (1) and 345 nm (2), which are similar to those shown by the bpp ligand (335 nm in DMF),19 bppCOOEt (370 nm in CH2Cl2) and bppCH2OH (340 nm in CH2Cl2) (Fig. S24†). These bands correspond to ligand fluorescence as confirmed by a small Stokes shift and a short excited-state lifetime (see Fig. S25†).20 Ligand luminescence is completely quenched after addition of 6 equivalents of Fe2+, confirming the 6:1 metal:ligand ratio for 1 and 2 (Fig. 3). This complete quenching is maintained after correcting inner-filter effects taking into account that a small part of the emitted light from the ligand is re-absorbed by the complex, especially in the case of 2 (Fig. S26†).21 Interestingly, this process was found to be reversible and the emission of 1 was recovered after adding 1,10-phenanthroline (phen), which is a well-known bidentate ligand, leading to [Fe(phen)3]2+ (Fig. S27†). A similar behavior was observed in UV-vis and fluorescence spectra with titrations, regardless of using Fe(ClO4)2 or Fe(BF4)2 (Fig. S23†). This indicates that the different ion-pair formation with these two anions has an almost negligible effect on the formation and the spin state of the resulting Fe(II) complexes. The fluorescence of the model systems bppCOOEt and bppCH2OH shows an almost complete quenching of the emission after addition of 1 equivalent of Fe2+ (see Fig. S24†). These results confirm that complete coordination of bpp ligands 1 and 2 to Fe2+ quenches the emission from bpp, in contrast to what has been observed for indazolylpyridine ligands.15 In this context, partial fluorescence quenching of similar derivatives based on the tridentate ligand 2,6-bis(pyrazol-3-yl)pyridine when coordinated to different metals (Co2+, Cu2+, Cd2+, Pb2+ and Zn2+) in solution has been reported.22
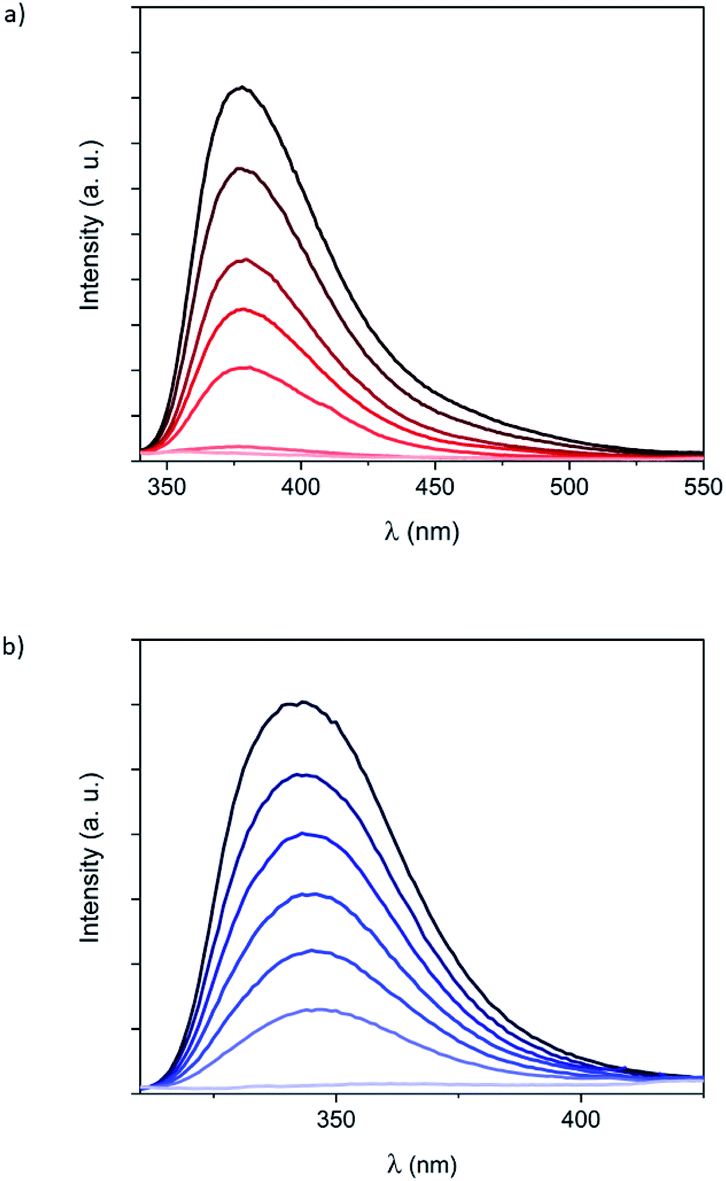 | ||
| Fig. 3 Fluorescence spectra of (a) 1 and (b) 2 solutions (5 × 10−6 M, in black) and after addition of up to six equivalents of Fe(ClO4)2. Color scale with increasing transparency represents increasing amount of Fe(ClO4)2 in steps of 1 equivalent. Excitation wavelength is 320 nm for 1 and 285 nm for 2. | ||
It is worth to note that maximum quenching of the emission bands for 1 and 2 is reached upon addition of the expected six equivalents of Fe2+ (Fig. 3). However, in the case of model systems bppCOOEt and bppCH2OH, at comparable ligand concentrations, a slight excess of Fe2+ (0.75 equivalents) is required instead of the expected 0.5 equivalents (Fig. S24†). This observation suggests that confinement of six Fe(II) complexes around the C60 hexa-adducts 1 and 2 enhances the affinity of bpp towards Fe2+ and decreases the reversibility with respect to that of less confined bppCOOEt or bppCH2OH ligands.
The methylated derivative 3 shows an optical behavior upon addition of Fe2+, both in the UV-Vis and emission spectra, very similar to 1, confirming that the methyl substituent attached to the pyrazolyl rings also favors the LS state (see Fig. S28†).16,17 The absorption and emission spectra of the monoadduct 4, which presents only one malonate branch and two bpp ligands, show a more complex behavior (Fig. S29 and S30†). Due to the smaller number of bpp ligands, the absorption bands in the UV-vis spectrum at 260 (shoulder), 330 and 426 nm (shoulder), characteristic of [60]fullerene mono-adducts,8,9 are more clearly distinguished than in 1. After addition of Fe2+, a MLCT band centered at 460 nm is observed, suggesting a LS state, similar to that observed for 1. Maximum absorbance of the MLCT band is reached after addition of around 1.5 and 0.75 Fe2+ equivalents for 1.5 × 10−5 and 4.5 × 10−5 M solutions, respectively (Fig. S29 and S30†), instead of the expected 1 Fe2+ equivalent. The emission spectrum of the ligand presents a shoulder at lower wavelengths and is not completely quenched after addition of an excess of Fe2+ for the more diluted solution. This suggests a different behavior to that of the hexakis-adducts 1, 2 and 3, which could be due to the formation of mixtures of intramolecular and intermolecular complexes. Indeed, theoretical calculations suggest that intramolecular Fe(II) complexes with two bpp ligands from the same malonate branch—the only ones which could be formed in 4—would present a very distorted geometry (see below).
In an attempt to detect the spincrossover behavior in solution, variable-temperature UV-vis spectra in the range 298 to 283 K of a 7.5 × 10−6 M solution of 2 with six equivalents of Fe2+ were recorded. We choose this compound since is the one with the highest fraction of HS Fe(II) at room temperature. A slight increase in the relative intensity of the band with maximum at 420 nm with respect to the shoulder at 385 nm is observed when the temperature is decreased from 298 to 283 K (Fig. S32†). This is consistent with an increase of the LS fraction at decreasing temperature, which is a clear evidence of a spin transition in solution. Still, lower temperatures should be required to complete this preliminary study. Note that 1H NMR measurements (Evans method) could not be used to evaluate the HS/LS fractions in solution due to the low concentrations available in CH2Cl2 of these species. DOSY and Evans NMR experiments require higher concentrations, typically 1 mM (i.e., a hundred times higher than the available concentration).
Density functional theory (DFT) studies
Theoretical calculations have been performed in a multi-level approach to shed light into the complexation and electronic properties of the Fe(II)-based complexes. First, several Fe(II) environments were modelled using a mono- or a di-malonate derivative bearing short bppCOOR arms as in ligands 1 and 4, or long bppCH2OR arms as in 2. The situations modelled include complex formation between the two bpp arms of a malonate branch (A), between bpp arms of two malonate branches (B) and between bpp arms of vicinal C60 molecules (C) (Fig. 4a and S33†). Note that configuration B cannot occur in the mono-branched adduct 4. Complexes of hexa-adducts 1 and 2 were also calculated, and their structures are displayed in Fig. 4b. All the structures were fully optimized at the cost-effective GFN2-xTB level of theory including representative BF4− counterions for electroneutrality. Binding energies for complex formation were calculated at the same theory level (see the ESI for full computational details†) and are summarized in Table 1.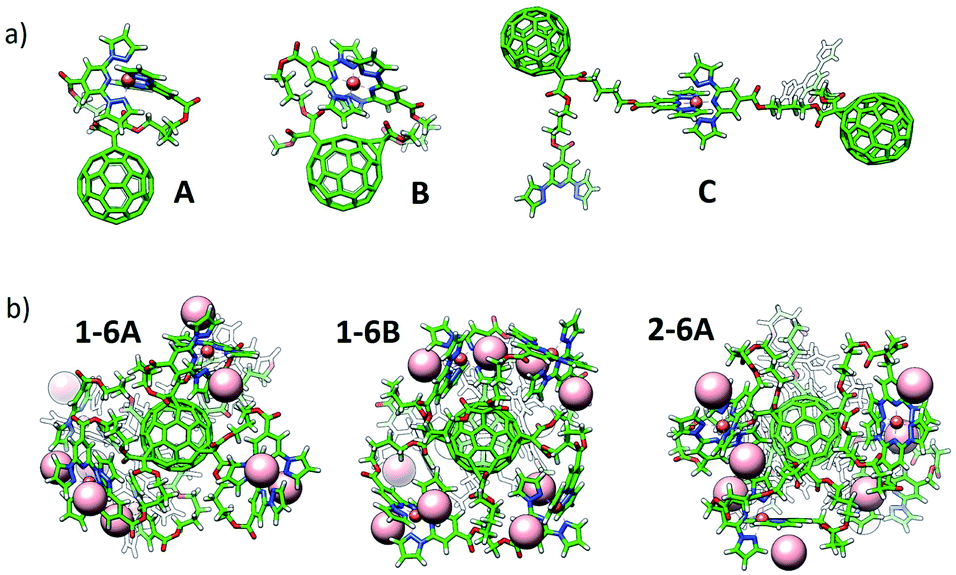 | ||
| Fig. 4 Minimum-energy geometry calculated for (a) representative configurations A, B and C of Fe(II)-complexes with bppCOOR ligands, and (b) hexakis Fe(II)-complexes of hexa-adducts 1 (1-6A and 1-6B) and 2 (2-6A). BF4− counterions are omitted in (a) for clarity (see Fig. S33†), and are represented with light pink spheres in (b). | ||
| Structure | Ligand bppCOOR | Ligand bppCH2OR |
|---|---|---|
| A | −7.53 | −42.95 |
| B | −15.39 | −38.63 |
| C | −19.59 | −23.48 |
| 6A | −14.61 | −178.91 |
| 6B | −68.08 | — |
Theoretical calculations indicate that the alkyl chain in bppCOOR ligands is too short to efficiently coordinate to Fe2+ within a unique malonate branch (A), as evidenced by a faulty octahedral coordination (Fig. S33 and S34a†) and a predicted binding energy (ΔEbind) as small as −7.53 kcal mol−1 (Table 1). However, iron complexation by bppCOOR ligands of two branches (B) is relatively more effective (ΔEbind = −15.39 kcal mol−1), approaching the stability of the lowest-energy configuration C (intermolecular complexation), which is predicted with a ΔEbind = −19.59 kcal mol−1. The significantly lower stability predicted for the complexation involving the two arms of the same malonate branch supports the incomplete Fe(II) complexation experimentally recorded for mono-adduct 4.
On the other hand, the longer chains of the bppCH2OR arms present in 2 provide enough ligand flexibility for an efficient octahedral complexation (Fig. S33 and S34b†), showing very similar ΔEbind values for configurations A and B (ca. −40 kcal mol−1; see Table 1). In this case, complexation between different molecules (C, −23.48 kcal mol−1) is significantly unfavoured compared to intramolecular complexation due to the absence of intra-ligand noncovalent interactions (Fig. S33†).
Fe(II) complexes of hexa-adducts 1 and 2 bearing short-chain bppCOOR and long-chain bppCH2OR ligands, respectively, were also modelled, and their minimum-energy optimized geometries are shown in Fig. 4b. For 1, intra-branch (1-6A) and inter-branch (1-6B) complexations were considered. Theoretical calculations indicate that configuration A is unlikely to be formed for hexakis metallo-fullerene complexes of 1 because a low total binding energy of only −14.61 kcal mol−1 (−2.44 kcal mol−1 per complex formation) is predicted. In contrast, complexation between bpp ligands on different malonate branches (B) is more stable, with a ΔEbind per complex of −11.35 kcal mol−1 (slightly lower than that calculated for the monoadduct, −15.39 kcal mol−1). The long malonate chains of bppCH2OR in 2 allow a significantly more efficient complexation within the six branches, and the hexakis complex is predicted with a very large binding energy of −178.91 kcal mol−1 (−29.81 kcal mol−1 per complex). Note that in all optimized structures, the BF4− counterions remain close to the iron complex to compensate its +2 charge (Fig. 4b and S33†).
Electronic structure calculations were performed on model Fe(II) complexes bearing bpp (C0), bppCOOEt (C1) and bppCH2OCOCH3 (C2) ligands (see Fig. S35†). The geometry and stability of LS vs. HS configurations is assessed by using the density functional theory (DFT) framework at the B3LYP/def2-SVP/LANL2DZ(Fe) level. The minimum-energy structures computed for low and high-spin states of complexes C0–2 are displayed in Fig. S36†, and show coordination distances of around 0.2 Å longer in the case of the HS configuration, as expected. Theoretical calculations predict a very small energy difference between the two spin states (<0.07 eV) for all the complexes, being the LS configuration slightly more stable than the HS upon inclusion of solvent effects (Table S1†). Calculations indicate that the HS state is closer in energy to the LS state for the bppCH2OR-based complex C2 than for C1 bearing the bppCOOEt ligand by ca. 0.02 eV. This nicely supports the experimental observation of a higher percentage of HS in the Fe(II) complex of 2 compared to 1.
Theoretical absorption spectra were calculated for the simplified complexes C1 and C2 and their respective bppCOOEt (L1) and bppCH2OCOCH3 (L2) ligands to fully assign the experimental UV-Vis bands recorded for the hexa-adducts 1 and 2, and the absorption changes upon Fe(II) complexation. The lowest-lying electronic excited states of L1/L2 and C1/C2 (Fig. S35†) were computed at the B3LYP*/def2-SVP/LANL2DZ(Fe) level, and the simulated absorption spectra are displayed in Fig. 5a and S37,† respectively (see the ESI for computational details†). The three bands experimentally recorded for 1 at 335, 270 and 245 nm (Fig. 2a) are closely reproduced by the calculations of ligand L1 with maximum absorption wavelengths at 331, 264 and 248 nm (Fig. 5a), which are described by transitions to the S1, S7 and S12 singlet excited states, respectively. The lowest-lying state S1 is predicted with a significant charge-transfer (CT) character, since it implies an electron density transfer from the electron-rich bpp moiety to the electron-withdrawing ester group, whereas S7 and S12 are bpp-centered π–π* states (Fig. S38†). Upon Fe(II) complexation, several low-lying excitations arise above 350 nm for both LS and HS species (Fig. S37a†). Assuming that Fe(II) is in the LS configuration, as experimentally suggested, the titration simulation of L1 to form C1 predicts the appearance of a low-energy intense transition at 400 nm (Fig. 5b), originating from a MLCT excitation (Fig. S37a and S39†), which can be assigned to the growing band experimentally observed at 460 nm (Fig. 2a). In good agreement with the experimental spectra, the three bands corresponding to the ligand are also calculated for C1, with the same nature but small wavelength shifts and oscillator strength differences (Fig. 5b).
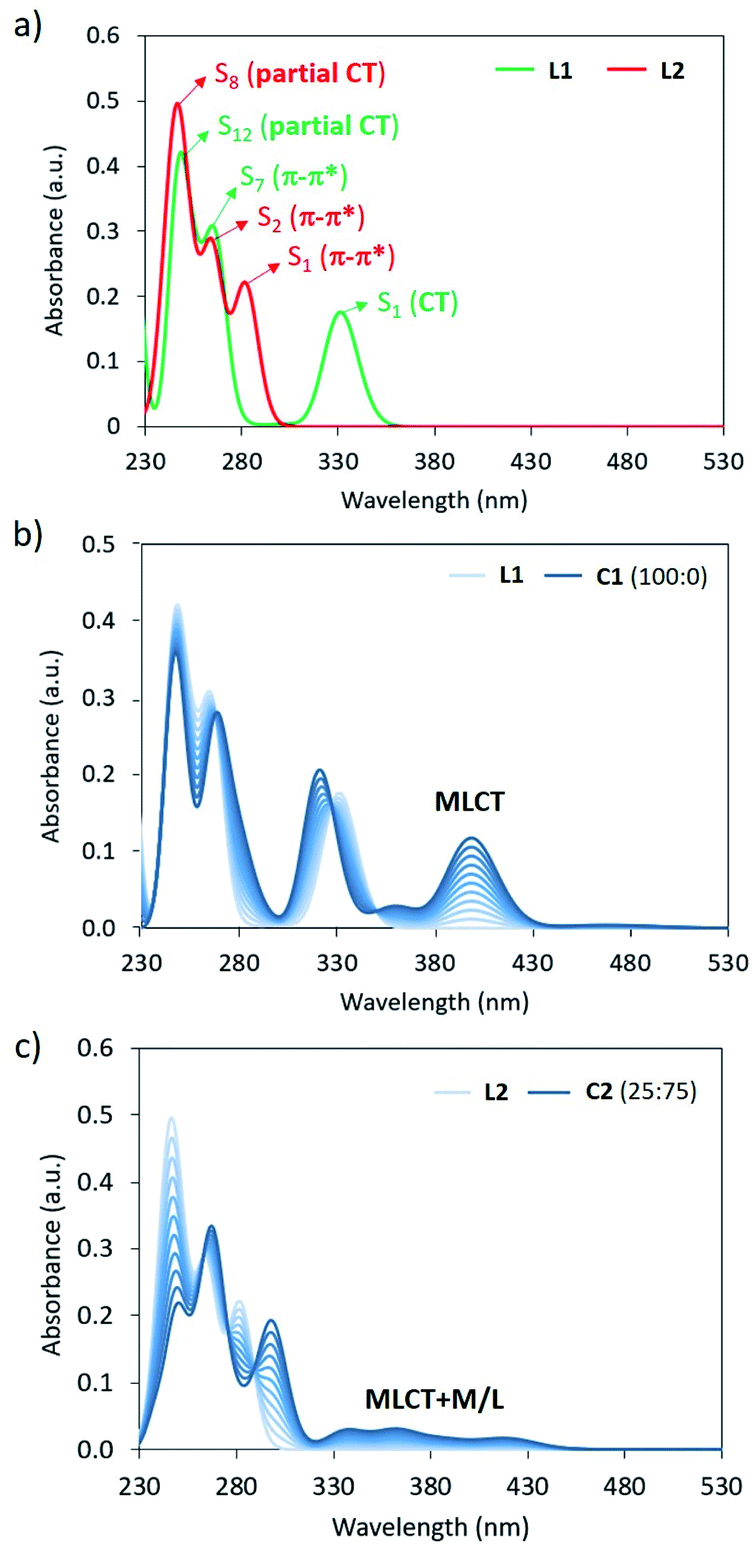 | ||
| Fig. 5 (a) Theoretical absorption spectra calculated for L1 and L2 at the B3LYP*/def2-SVP/LANL2DZ(Fe) level including solvent effects. (b) and (c) Theoretical titration simulations of the absorption spectrum of L1 and L2 upon Fe(II) complexation considering a ratio of 100:0 LS:HS for C1, and of 25:75 LS:HS for C2. | ||
Moving to ligand L2, theoretical calculations predict intense S0 → S1, S2 and S8 excitations at 282, 264 and 245 nm with oscillator strengths (f) of 0.217, 0.256 and 0.261, respectively (Fig. 5a). These electronic transitions nicely correlate with the experimental bands of derivative 2 recorded at 306, 270 and 245 nm (Fig. 2b). In this case, S1 presents a π–π* character with negligible CT character due to the low electron activity of the attached –CH2OR group (Fig. S41†). This explains the significant blue-shift of the lowest-lying absorption band predicted and recorded for 2 compared to 1 (Fig. 2). Similar to that described for L1, several low-lying excitations are calculated upon Fe(II) complexation (Fig. S37†). Considering a LS:HS ratio of 25:75, the titration simulation of L2 to form C2 predict a long and weak absorption between 330 and 430 nm (Fig. 5c), which correlates with the experimental tail recorded above 330 nm for the Fe(II) complex of 2 (Fig. 2b), and is ascribed to mixed metal/ligand (M/L) and MLCT excitations of both LS and HS states (Fig. S37b, S42 and S43†). High-energy absorptions of L2 are predicted similarly in C2 (Fig. 5c), with wavelength and intensity differences that match the experimental evolution of the UV-Vis spectrum of 2 upon complexation (Fig. 2b).
The origin of the emission quenching upon complexation has also been analyzed by means of quantum chemical calculations. The relative energy position of the ground state and the lowest-lying singlet (S1) and triplet (T1) excited states was calculated at the time-dependent TD-(U)B3LYP*/def2-SVP/LANL2DZ(Fe) level for isolated ligands L0–2 and for complexes C0–2 (see the ESI for details†). Theoretical calculations predict a large energy gap between the lowest-lying singlet/triplet excited states and the ground state for ligands L0–L2, with values between 2.5 and 4.4 eV in all cases (Table S2†). Interestingly, excited-state geometry relaxation allows prediction of the emission fluorescence energy, which is calculated at 374 (3.32) and 333 nm (3.72 eV) for L1 and L2, respectively, in good accord with the experimental values of 380 and 345 nm for 1 and 2. Upon Fe(II)-complexation, several singlet and triplet transitions are predicted in the low-energy range. In the LS state, the singlet S1 state is vertically calculated at ∼2.1 eV above the ground state for the three complexes C0–2, whereas the triplet T1 is computed at ∼1.0 eV (Table S2†). Excited-state relaxation from the Frank–Condon geometry leads to an energy difference of ∼1.5 eV between S0 and S1 at the S1 minimum-energy geometry, whereas T1 is computed to be more stable than S0 at the T1 potential well in all complexes (Table S2†). In the HS configuration, the lowest-lying quintuplet excited state Q1 is at 0.21 eV in the Frank–Condon region, and goes below Q0 upon excited-state geometry relaxation (Q0–Q1 energy crossing). Theoretical calculations therefore predict that complexes C0–2 present low-lying excited states (triplet for LS and quintuplet for HS) that enhance non-radiative relaxation paths and quench photoluminescence processes.
Studies in the solid state
The Fe(II) complexes of 1 and 2, hereafter named 5 and 6, respectively, were isolated in the solid state by fast precipitation after addition of diethyl ether to the CH3CN/CH2Cl2 (∼7.5/1 ratio) solutions containing 1 or 2 (6.4 × 10−4 M) and 12 equivalents of Fe2+ to achieve a complete coordination. Amorphous materials were obtained in both cases, which were characterized by elemental analysis, infrared (IR) and X-ray photoelectron spectroscopy (XPS) and thermogravimetric analysis (TGA) (see Fig. S44–S47 and Section 5.1 in the ESI†). These techniques support the presence of six iron per fullerene unit in the solid state samples. Attempts to obtain crystalline materials by slowing precipitation were unsuccessful. Similar results were obtained using 6 equivalents of Fe2+.The electrospray ionization mass spectrometry (ESI-MS) of 5 (obtained from the Fe(ClO4)2 salt) and 6 (obtained from the Fe(BF4)2 salt) were performed by dissolving these solids in a mixture of dimethylformamide and methanol or CH3CN. The spectra showed the presence of several species with one molecule of 1 or 2 and a variable number of Fe2+ ions (from one to six) plus counteranions (see Section 5.1†). Although this technique does not give an exact view of the composition of the precipitate since it requires re-dissolution and measurement conditions in which the compound could decompose, the presence of fragments with m/z values corresponding to 1 and 2 plus six Fe2+ suggests that the hexanuclear species are present in the solid samples.
Magnetic measurements of 5 and 6 were performed on these solid samples isolated by centrifugation after successive washings with diethyl ether and protected with a small amount of clean diethyl ether or embedded in a grease to keep them solvated, since the magnetic properties of this type of compounds are very sensitive to the presence of solvent molecules.2 Both protection methods gave similar results. In the solvated sample of 5 the LS state is favored, in agreement with the solution data (see above). Thus, magnetic molar susceptibility times temperature (χMT) values are close to zero in the temperature range 5–230 K. At higher temperatures, a gradual increase to reach a value of 8.7 emu K mol−1 at 300 K is observed, which corresponds to a SCO from LS to HS of around 40% of Fe(II) (Fig. 6a) (see Section 5.2 in the ESI for calculation details†). The solvated sample of 6 shows an almost constant χMT value close to 9.5 emu K mol−1 from 50 to 190 K (∼45% of HS fraction) with a gradual increase to reach 22.0 emu K mol−1 (∼100% HS fraction) above 285 K (Fig. 6b). This observation is also in agreement with the results in solution, which indicate that for complexes of 2 the fraction of HS Fe(II) at room temperature is higher than for complexes of 1. Notice that these gradual thermal spin transitions are typical of SCO complexes showing long intermolecular distances and weak intermolecular interactions. Interestingly, the polynuclear nature of our complexes force the metal centers to be in close proximity, thus leading to SCO transitions that are much sharper and more complete (∼40% of Fe(II) centers in 5 upon heating the sample from 230 to 290 K and ∼55% of Fe(II) centers in 6 upon heating the sample from 200 to 280 K) than that observed in the mononuclear Fe(II) complex having fullerenes as end-capping groups. This last compound T shows a very gradual and incomplete spin transition, which extends over a wide temperature range (from 100 to 400 K) in the temperature ranges in which the spin switching occurs in 5 and 6 that corresponds to a spin-crossover of a ∼15%.9
 | ||
| Fig. 6 Temperature dependence of χMT of 5 (a) and 6 (b) measured with a protective layer of clean diethyl ether or a grease to avoid the loss of solvent before (black circles), during (black triangles) and after (red triangles) irradiation with red light at 10 K. | ||
Magnetic measurements after irradiation with red light at 10 K were carried out on solvated samples embedded in a grease. Irradiation with a 633 nm laser at 10 K leads to a drastic increase of the magnetic signal reaching saturation after ca. 1 h. Therefore, both samples exhibit LIESST effect. After switching off the irradiation, the temperature was increased at a scan rate of 0.3 K min−1 (see Fig. 6). The maximum χMT value after irradiation in 5 (6.5 emu K mol−1) corresponds approximately to a photo-induced LS to HS transition of two of the six LS Fe(II) centers (Fig. 6).
Finally, these magnetic and photomagnetic effects were observed to be less pronounced in the same samples of 5 and 6 measured without protecting them with the washing solvent (diethyl ether) or grease to avoid the loss of solvent molecules. These desolvated samples showed higher HS fractions at low temperatures than the solvated ones (see Section 5.6 in the ESI and Fig. 7). These differences are frequent in the solvates of [Fe(bpp)2]2+ complexes and their derivatives. In fact, very often, partial or complete desolvation of SCO compounds causes the loss of the spin transition. The use of a solvent or grease covering layer is then a typical procedure to protect the sample,24 avoiding this undesired process. Indeed, elemental analyses of 5 and 6 in contact with air are consistent with the presence of water molecules (see Section 5.6 in the ESI†). This indicates that the volatile solvent molecules that could be found in the structure of 5 and 6 (dichloromethane, acetonitrile or diethyl ether) are lost and replaced by water molecules from air upon filtering.
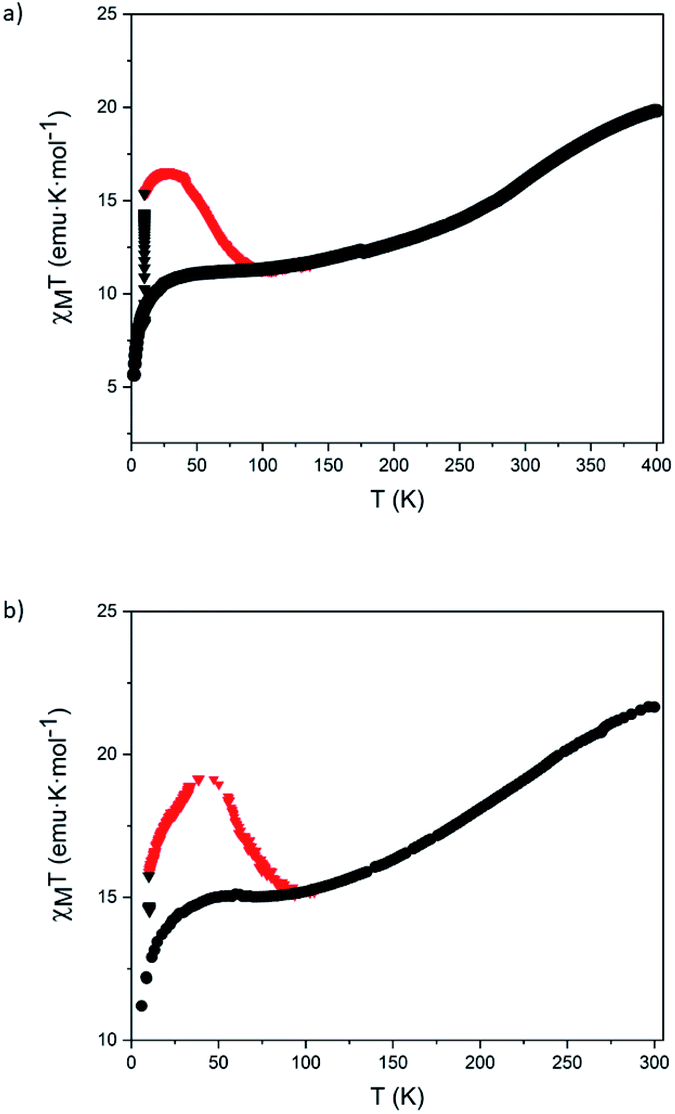 | ||
| Fig. 7 Temperature dependence of χMT of filtered samples of 5 (a) and 6 (b) before (black circles), during (black triangles) and after (red triangles) irradiation with red light at 10 K. | ||
Raman μ-spectroscopy also confirmed the incomplete thermal SCO in desolvated samples of 5 and 6 (Fig. S48†). Focusing on the pyridine vibration located in the range of 1000−1100 cm−1, we observed that at 298 K the most intense peak for the two samples appears at ∼1015 cm−1 accompanied by a low intense peak at ∼1040 cm−1 for 5. At 88 K, the relative intensity of this peak increases for 5, and a new peak at the same position (∼1040 cm−1) appears in the spectrum of 6. Interestingly, the initial spectrum of 6 is recovered after heating again at 298 K confirming the reversibility of the SCO process. The changes in intensity of the peaks at 1040 cm−1 (LS) and 1012 cm−1 (HS) are in agreement with those observed for [FeII(bpp)2]2+ complexes in the literature.23 This suggests that the coordination environment of Fe(II) is very similar in 5 and 6 and close to that encountered for Fe(II) bpp complexes. In addition, the changes in intensity are consistent with the magnetic properties that indicate that the HS fraction decreases from 80 to 50% in 5 and from 100 to 70% in 6 in the same range of temperatures (see Section 5.6 in the ESI and Fig. S49†). On the other hand, Raman spectrum of 1 was also recorded for comparative purposes (Fig. S49†). The Raman spectra were also theoretically simulated and confirm the assignment of the Raman-active vibrational modes that allow differentiation between HS vs. LS configurations (see Section 5.7 in the ESI and Fig. S50†).
Conclusions
In this work, we have designed and synthesized different hexakis-substituted [60]fullerene adducts of the well-known bpp ligand. Spectroscopic characterization in solution and theoretical calculations have demonstrated that they are able to form polynuclear spin-crossover Fe(II) complexes. The variety of behaviors exhibited by the different fullerene adducts indicates that the spin state of Fe(II) in these species can be tuned by a proper choice of the bpp substituents. Indeed, some of these polynuclear complexes show SCO properties induced by temperature and light as external stimuli, with spin transitions that, although gradual, are much sharper than those reported in mononuclear complexes based on fullerene. These results demonstrate that [60]fullerene hexa-adducts are excellent and versatile platforms to develop new spin-switching materials.In these magnetic molecules, [60]fullerene has been used as a molecular carbon substrate on which 12 peripheral tridentate ligands have been covalently connected through the use of flexible spacers adopting a well-defined 3D spatial arrangement and allowing to organize up to six [Fe(bpp)2]2+ SCO complexes on this substrate. This hybrid nanostructure can provide an ideal molecular platform to investigate hybrid nanostructures formed by SCO complexes anchored on flat 2D substrates such as gold or HOPG. In fact, the preparation of SAMs based on monolayers of these SCO [Fe(bpp)2]2+ complexes has so far been elusive. The various attempts have failed owing to the lability of these Fe(II) complexes,4 a problem that could be overcome using the ligands developed in the present work as they can be designed so as to exhibit much higher binding energies than those reported in the parent [Fe(bpp)2]2+complex.
Finally, in this work we have demonstrated the incorporation of multiple bpp ligands to a central fullerene scaffold. This feature opens the possibility to process these systems from solution, something that was not possible to achieve when [60]fullerene is functionalized with one bpp ligand.8,9 The resulting polynuclear SCO molecules, conveniently protected to avoid a loss of solvent, may be of interest in molecular spintronics since a modulation of their spin properties by electrical or light stimuli may be possible, as it has recently been demonstrated for single SCO molecules5 and nanoparticles.25
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the EU (ERC Advanced Grant MOL-2D 788222, FET-OPEN COSMICS 766726 and COST action MOLSPIN CA15128), the Spanish MCIU (MAT-2017-89993-R, PGC2018-099568-B-I00 and Unidad de Excelencia María de Maeztu CEX2019-000919-M), the Spanish MINECO (CTQ2017-84327-P, CTQ2017-83531-R), the Generalitat Valenciana (PROMETEO program and iDiFEDER/2018/061) and the Ministry of Education and Science of Russian Federation (Agreement No. 14.W03.31.0001) is gratefully acknowledged. M. S. and D. A. thank the project CICECO-Aveiro Institute of Materials (UIDB/50011/2020 & UIDP/50011/2020). We thank J. M. Martínez-Agudo and G. Agustí for magnetic measurements and Prof. M. A. Halcrow for helpful discussions.Notes and references
- See for general reviews: (a) Spin Crossover in Transition Metal Compounds, Topics in Current Chemistry, ed. P. Gütlich and H.A. Goodwin, Springer Verlag, Berlin-Heidelberg-New York, 2004, vol. 233–235 Search PubMed; (b) Spin-Crossover Materials: Properties and Applications, ed. M. A. Halcrow, John Wiley & Sons, Chichester, UK, 2013 Search PubMed.
- (a) M. A. Halcrow, Coord. Chem. Rev., 2005, 249, 2880–2908 CrossRef CAS; (b) M. A. Halcrow, Coord. Chem. Rev., 2009, 253, 2493–2514 CrossRef CAS; (c) J. Olguín and S. Brooker, Coord. Chem. Rev., 2011, 255, 203–240 CrossRef; (d) M. A. Halcrow, New J. Chem., 2014, 38, 1868–1882 RSC; (e) L. J. Kershaw Cook, R. Mohammed, G. Sherborne, T. D. Roberts, S. Alvarez and M. A. Halcrow, Coord. Chem. Rev., 2015, 289–290, 2–12 CrossRef CAS.
- (a) R. Torres-Cavanillas, R. Sanchis-Gual, J. Dugay, M. Coronado-Puchau, M. Giménez-Marqués and E. Coronado, Adv. Mater., 2019, 31, 190039 Search PubMed; (b) A. Enriquez-Cabrera, A. Rapakousiou, M. Piedrahita Bello, G. Molnár, L. Salmon and A. Bousseksou, Coord. Chem. Rev., 2020, 419, 213396 CrossRef CAS; (c) E. Coronado, Nat. Rev. Mater., 2020, 5, 87 CrossRef.
- (a) L. Pukenas, F. Benn, E. Lovell, A. Santoro, L. J. Kershaw Cook, M. A. Halcrow and S. D. Evans, J. Mater. Chem. C, 2015, 3, 7890–7896 RSC; (b) V. García-López, M. Palacios-Corella, V. Gironés-Pérez, C. Bartual-Murgui, J. A. Real, E. Pellegrin, J. Herrero-Martín, G. Aromí, M. Clemente-León and E. Coronado, Inorg. Chem., 2019, 58, 12199–12208 CrossRef.
- (a) T. G. Gopakumar, F. Matino, H. Naggert, A. Bannwarth, F. Tuczek and R. Berndt, Angew. Chem., Int. Ed., 2012, 51, 6262–6266 CrossRef CAS; (b) E. J. Devid, P. N. Martinho, M. V. Kamalakar, I. Salitros, Ú. Prendergast, J. F. Dayen, V. Meded, T. Lemma, R. González-Prieto, F. Evers, T. E. Keyes, M. Ruben, B. Doudin and S. Jan van der Molen, ACS Nano, 2015, 9, 4496–4507 CrossRef CASK. Bairagi, O. Iasco, A. Bellec, A. Kartsev, D. Li, J. Lagoute, C. Chacon, Y. Girard, S. Rousset, F. Miserque, Y. J. A. Dappe Smogunov, C. Barreteau, M. L. Boillot, T. Mallah and V. Repain, Nat Commun., 2016, 7, 12212 CrossRef CAS.
- (a) H. W. Kroto, J. R. Heath, S. C. O'Brien, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162–163 CrossRef CAS; (b) F. Giacalone and N. Martín, Chem. Rev., 2006, 106, 5136–5190 CrossRef CAS; (c) N. Martín, Chem. Commun., 2006, 2093–2104 RSC; (d) B. M. Illescas, J. Rojo, R. Delgado and N. Martín, J. Am. Chem. Soc., 2017, 139, 6018–6025 CrossRef CAS.
- W. Yan, C. Réthoré, S. Menning, G. Brenner-Weiß, T. Muller, P. Pierrat and S. Bräse, Chem.–Eur. J., 2016, 22, 11522–11526 CrossRef CAS.
- E. Nuin, W. Bauer and A. Hirsch, Eur. J. Org. Chem., 2017, 2017, 790–798 CrossRef CAS.
- K. Senthil Kumar, I. Salitros, N. Suryadevara, E. Moreno-Pineda and M. Ruben, Eur. J. Inorg. Chem., 2018, 2018, 5091–5097 CrossRef.
- W. K. Hang, H. X. Zhang, Y. Wang, W. Liu, X. Yan, T. Li and Z. G. Gu, Chem. Commun., 2018, 54, 12646–12649 RSC.
- (a) J. Ramos-Soriano, J. J. Reina, A. Pérez-Sánchez, B. Illescas, J. Rojo and N. Martín, Chem. Commun., 2016, 52, 10544–10546 RSC; (b) A. Muñoz, D. Sigwalt, B. M. Illescas, J. Luczkowiak, L. Rodríguez-Pérez, I. Nierengarten, M. Holler, J. S. Remy, K. Buffet, S. P. Vincent, J. Rojo, R. Delgado, J. F. Nierengarten and N. Martín, Nat. Chem., 2016, 8, 50–57 CrossRef; (c) J.-F. Nierengarten, Chem. Commun., 2017, 53, 11855–11868 RSC; (d) J. Ramos-Soriano, J. J. Reina, B. M. Illescas, J. Rojo and N. Martín, J. Org. Chem., 2018, 83, 1727–1736 CrossRef CAS; (e) J. Ramos-Soriano, J. J. Reina, B. M. Illescas, N. de la Cruz, L. Rodríguez-Pérez, F. Lasala, J. Rojo, R. Delgado and N. Martín, J. Am. Chem. Soc., 2019, 141, 15403–15412 CrossRef CAS.
- J. M. Holland, J. A. McAllister, C. A. Kilner, M. Thornton-Pett, A. J. Bridgeman and M. A. Halcrow, J. Chem. Soc., Dalton Trans., 2002, 2002, 548–554 RSC.
- Y. Hasegawa, K. Takahashi, S. Kume and H. Nishihara, Chem. Commun., 2011, 47, 6846–6848 RSC.
- K. Senthil Kumar, I. Salitros, E. Moreno-Pineda and M. Ruben, Dalton Trans., 2017, 46, 9765–9768 RSC.
- A. Santoro, L. J. Kershaw Cook, R. Kulmaczewski, S. A. Barrett, O. Cespedes and M. A. Halcrow, Inorg. Chem., 2015, 54, 682–693 CrossRef CAS.
- M. A. Halcrow, I. Capel Berdiell, C. M. Pask and R. Kulmaczewski, Inorg. Chem., 2019, 58, 9811–9821 CrossRef CAS.
- L. J. Kershaw Cook, R. Kulmaczewski, R. Mohammed, S. Dudley, S. A. Barrett, M. A. Little, R. J. Deeth and M. A. Halcrow, Angew. Chem., Int. Ed., 2016, 13, 4327–4331 CrossRef.
- J. M Holland, S. A. Barrett, C. A. Kilner and M. A. Halcrow, Inorg. Chem. Commun., 2002, 5, 328–332 CrossRef.
- Y. Jiao, J. Zhu, Y. Guo, W. He and Z. Guo, J. Mater. Chem. C, 2017, 5, 5214–5222 RSC.
- J. M. Stanley, X. Zhu, X. Yang and B. J. Holliday, Inorg. Chem., 2010, 49, 2035–2037 CrossRef CAS.
- A. Credi and L. Prodi, Spectrochim. Acta, Part A, 1998, 54, 159–170 CrossRef.
- (a) Y. Pi, H. Liu and D. J. Wang, Adv. Mater. Res., 2014, 906, 96–100 Search PubMed; (b) H. Liu, F. X. Li, Y. Pi, D. J. Wang, Y. J. Hu and J. Zheng, Spectrochim. Acta, Part A, 2015, 145, 588–593 CrossRef CAS.
- M. Cavallini, I. Bergenti, S. Milita, J. C. Kengne, D. Gentili, G. Ruani, I. Salitros, V. Meded and M. Ruben, Langmuir, 2011, 27, 4076–4081 CrossRef CAS.
- G. Chastanet, C. Desplanches, C. Baldé, P. Rosa, M. Marchivie and P. Guionneau, Chem. Sq., 2018, 2, 2 Search PubMed.
- J. Dugay, M. Giménez-Marqués, T. Kozlova, H. W. Zandbergen, E. Coronado and H. S. van der Zant, Adv. Mater., 2015, 27, 1288–1293 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, spectroscopic characterization, theoretical studies and studies in the solid state. Fig. S1–S51. See DOI: 10.1039/d0sc05875k |
| ‡ M. P.-C and J. R.-S contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |