
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organic room-temperature phosphorescence from halogen-bonded organic frameworks: hidden electronic effects in rigidified chromophores†
Jiawang
Zhou
 a,
Ljiljana
Stojanović
b,
Andrey A.
Berezin
c,
Tommaso
Battisti
c,
Abigail
Gill
c,
Benson M.
Kariuki
c,
Davide
Bonifazi
cd,
Rachel
Crespo-Otero
*b,
Michael R.
Wasielewski
*a and
Yi-Lin
Wu
*c
a,
Ljiljana
Stojanović
b,
Andrey A.
Berezin
c,
Tommaso
Battisti
c,
Abigail
Gill
c,
Benson M.
Kariuki
c,
Davide
Bonifazi
cd,
Rachel
Crespo-Otero
*b,
Michael R.
Wasielewski
*a and
Yi-Lin
Wu
*c
aDepartment of Chemistry, Institute for Sustainability and Energy at Northwestern, Northwestern University, Evanston, Illinois 60208-3113, USA. E-mail: m-wasielewski@northwestern.edu
bSchool of Biological and Chemical Sciences, Queen Mary University of London, London E1 4NS, UK. E-mail: r.crespo-otero@qmul.ac.uk
cSchool of Chemistry, Cardiff University, Cardiff CF10 3AT, UK. E-mail: WuYL@cardiff.ac.uk
dInstitute of Organic Chemistry, Faculty of Chemistry, University of Vienna, Währinger Str. 38, Vienna 1090, Austria
First published on 5th November 2020
Abstract
Development of purely organic materials displaying room-temperature phosphorescence (RTP) will expand the toolbox of inorganic phosphors for imaging, sensing or display applications. While molecular solids were found to suppress non-radiative energy dissipation and make the RTP process kinetically favourable, such an effect should be enhanced by the presence of multivalent directional non-covalent interactions. Here we report phosphorescence of a series of fast triplet-forming tetraethyl naphthalene-1,4,5,8-tetracarboxylates. Various numbers of bromo substituents were introduced to modulate intermolecular halogen-bonding interactions. Bright RTP with quantum yields up to 20% was observed when the molecule is surrounded by a Br⋯O halogen-bonded network. Spectroscopic and computational analyses revealed that judicious heavy-atom positioning suppresses non-radiative relaxation and enhances intersystem crossing at the same time. The latter effect was found to be facilitated by the orbital angular momentum change, in addition to the conventional heavy-atom effect. Our results suggest the potential of multivalent non-covalent interactions for excited-state conformation and electronic control.
Introduction
Room temperature phosphorescence (RTP) has received increasing interest due to the potential it presents for photonic devices, bio-imaging, anti-counterfeiting, and night-vision applications.1–3 Until recent years, the main sources of RTP luminophores have been inorganic or organometallic complexes, due to the presence of metal atoms being able to promote singlet-to-triplet intersystem crossing (ISC) in the excited states. However, heavy metal complexes or inorganic materials can often be toxic and expensive; through the study of purely organic phosphors, the applications of phosphorescence materials can expand by becoming more biocompatible, cheaper to acquire, and environmentally safer.4,5 While there are many benefits of organic phosphors compared to those containing heavy metals, achieving RTP from purely organic molecules has proven a challenge on account of slow ISC rates and competitive non-radiative processes, in particular.In recent decades, organic phosphorescence has become a more widely explored topic due to the discovery of long-lasting RTP by utilising crystallisation,6–8 aggregation,9,10 halogen bonding,11–14 heavy atoms,15 and carbonyl substituents16–18 to circumvent the aforementioned issues.19–28 Although spin–orbit coupling (SOC) in organic molecules is usually small (on the order of 1 cm−1, cf. 102 to 103 cm−1 for organometallic complexes), the introduction of a carbonyl functionality to aromatic rings often opens up a 1(n–π*) → 3(π–π*) (or 1(π–π*) → 3(n–π*)) channel with SOC ∼100 cm−1.29–33 Such a small increase is sufficient to allow efficient ISC and populate the triplet of, for instance, benzophenone or benzaldehyde with a near-unitary quantum efficiency.34,35 The structure of the as-generated triplet states can be rigidified in the solid state with the aid of non-covalent interactions (e.g. hydrogen and halogen bonds)11–13,20 to suppress non-radiative vibrational relaxation, resulting in nearly quantitative RTP quantum yields in the solid state.19,36
Combining these design principles, the Kim group reported seminal work on efficient RTP luminophores based on 2,5-bis(hexyloxy)-4-bromobenzaldehyde.37 The linear C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯Br halogen-bonding interactions38,39 present in the solid state were suggested to be the major reason to avoid energy dissipation through vibrational motions. The proximity of a fourth-row Br element to the CO group, where the non-bonding electrons originate in the n–π* transition, is believed to enhance SOC as well.40,41 In fact, in a later study by Kim and Dunietz, it was found that moving the Br substituent from the para to the ortho position, closer to the triplet-producing carbonyl functionality, in benzaldehyde increases SOC on the single-molecule level, which enhances both the rates of ISC kISC and phosphorescence kPhos significantly by 5–15 fold.40
O⋯Br halogen-bonding interactions38,39 present in the solid state were suggested to be the major reason to avoid energy dissipation through vibrational motions. The proximity of a fourth-row Br element to the CO group, where the non-bonding electrons originate in the n–π* transition, is believed to enhance SOC as well.40,41 In fact, in a later study by Kim and Dunietz, it was found that moving the Br substituent from the para to the ortho position, closer to the triplet-producing carbonyl functionality, in benzaldehyde increases SOC on the single-molecule level, which enhances both the rates of ISC kISC and phosphorescence kPhos significantly by 5–15 fold.40
Inspired by these findings as well as other successful demonstration of halogen-bond-induced phosphorescence in the solid state, we exploited the naphthalene scaffold, a prototypical building block in organic optoelectronics, to study the effect of the halogen substitution and the role of halogen bonding in mediating triplet formation. Compared to the previously studied bromobenzaldehydes, this system is expected to have less carbonyl-originated n–π* character in the low singlet excited states to drive ISC, thus offering a platform to highlight the halogen effects. Well-developed synthetic methodologies42–45 were used to introduce multiple halogen-bond donors (e.g. Br) and acceptors (e.g. O) in naphthalene to permit multiple non-covalent interactions to occur synergistically, enabling phosphorescence from halogen-bonded frameworks.46
Results and discussion
Naphthalene derivatives with halogen-bond accepting carbonyl functionalities and a various number of halogen-bond donating Br atoms can be prepared readily from 1,4,5,8-naphthalenetetracarboxylic dianhydride (NDA) (Scheme 1).47–49 Bromination of NDA with 1,3-dibromo-5,5-dimethylhydantoin is slow and can produce a mixture of NDA with various numbers of Br substituents.47 However, the application of excess reagents at elevated temperatures for a prolonged reaction time gives tetrabrominated NDA (Br4NDA) as the sole product. Esterification of BrnNDA with ethyl iodide in alkaline ethanol gave a mixture of naphthalene tetracarboxylic ethyl esters, BrnNTE (n = 0, 1, 2, 4; n = 3 can be isolated but it is not discussed here for simplicity).50,51 The individual compounds were isolated by column chromatography on SiO2 and their identity was confirmed by NMR, MS, and single-crystal X-ray crystallography.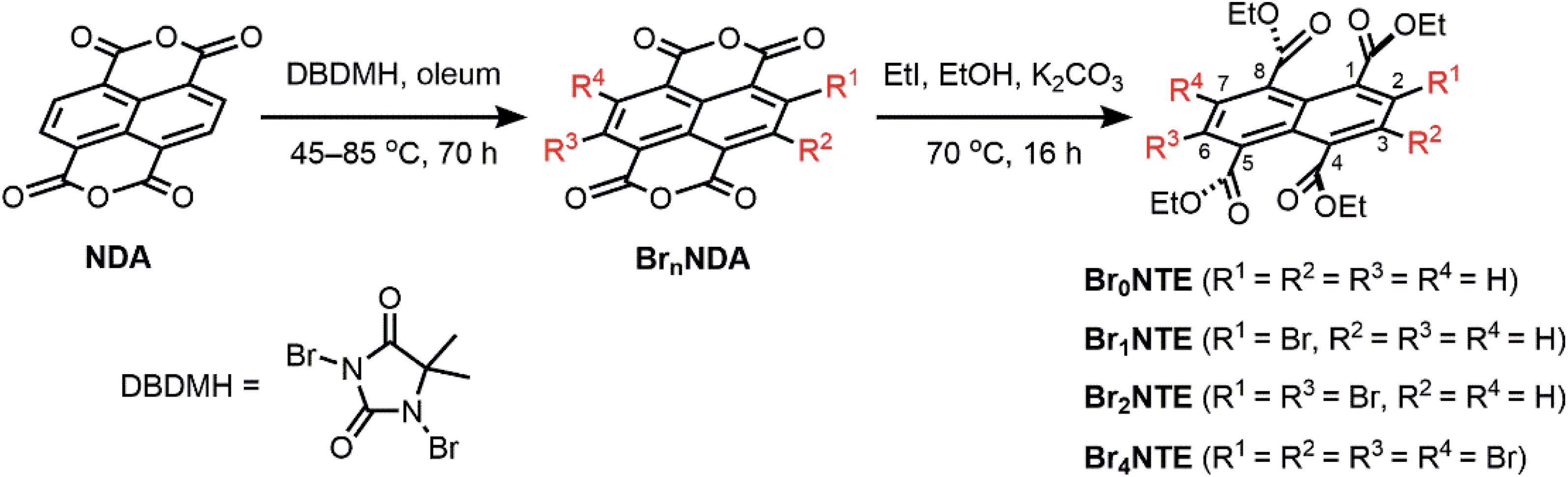 | ||
| Scheme 1 Synthesis of brominated naphthalene tetracarboxylic ethyl ester (BrnNTE). | ||
In the solid state, an extended Br⋯O network can be observed for Br2NTE50 (Fig. 1). For each molecule, each pair of the peri ester groups interacts with a Br atom of a neighbouring molecule to establish bifurcated, slightly asymmetric halogen bonds with d(C–Br⋯OC) = 3.268(2) Å, d(C–Br⋯O–C2H5) = 3.308(2) Å and both θ(C–Br⋯O) ∼150° (i.e. 152.40(9) and 149.21(8)).38 Reciprocally, each Br atom is interacting with two peri ester groups of a nearby molecule of Br2NTE. Being symmetrically substituted with four Br and four ester functionalities, Br4NTE is also embedded in a framework of halogen bonds in the solid state. However, likely due to the steric requirement of large Br atoms, the same arrangement in Br2NTE was not observed. Instead, only two out of the four esters on the 1 and 5 positions form linear C–Br⋯OC short contacts with the Br atoms on the 3 and 7 positions of the neighbouring molecules (values taken from two crystallographically independent molecules: d(Br⋯O) = 3.074(3) and 3.286(3) Å, θ(C–Br⋯O) = 165.5(1) and 168.5(1)°). The remaining two Br atoms on the 2 and 6 positions are engaged in orthogonal (“Type II”)52,53 C–Br⋯Br–C interactions (d(Br⋯Br) = 3.692(1) Å, θ(C–Br⋯Br) = 86.4(1)°).
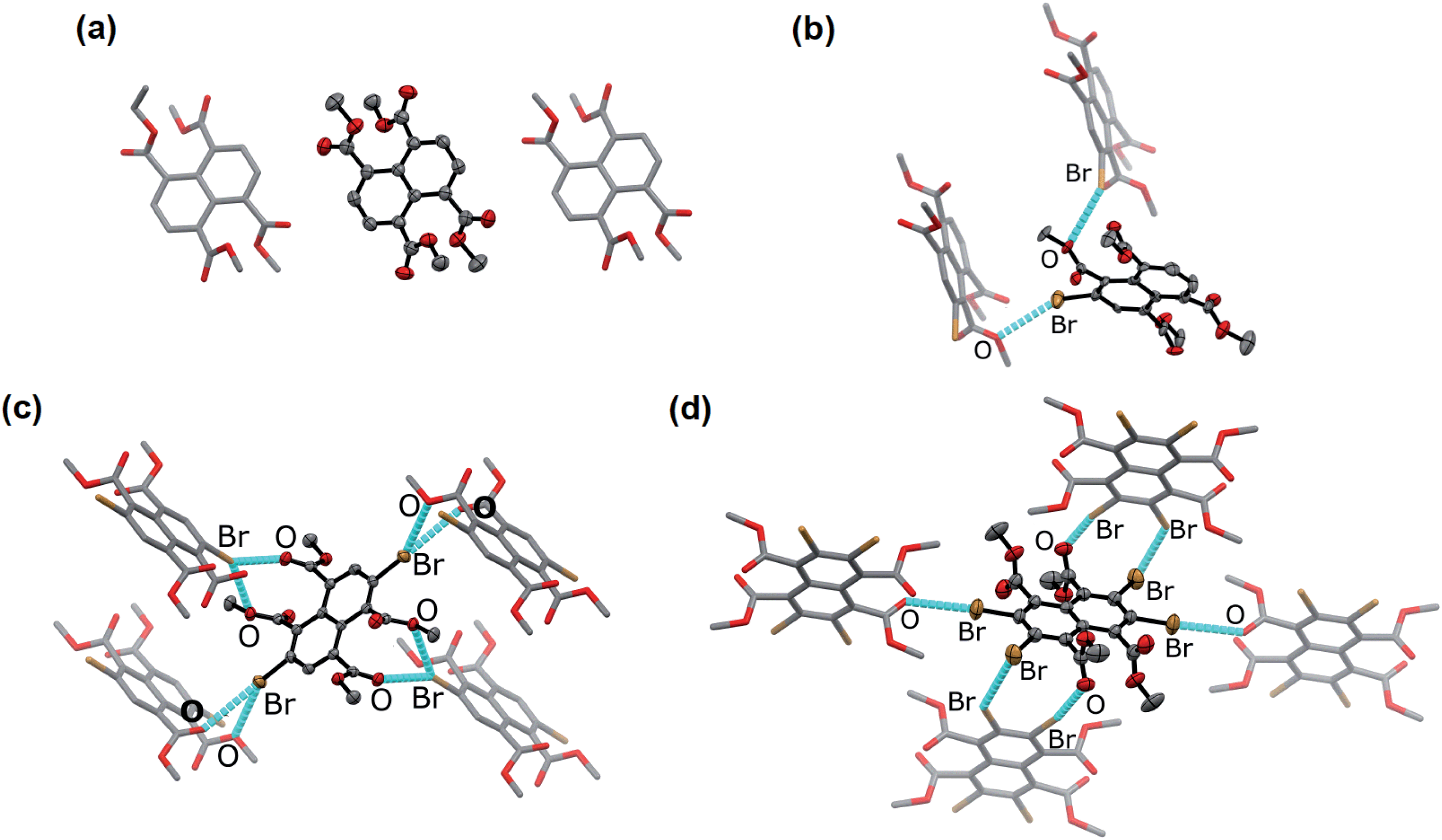 | ||
Fig. 1 Single crystal X-ray molecular structure of (a) Br0NTE (space group P21/n), (b) Br1NTE (P21), (c) Br2NTE (P21/n), and (d) Br4NTE (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) and the close neighbours in crystals. Crystals were obtained by diffusing MeOH vapour into the CHCl3 solutions of BrnNTE. Colour code: C = grey, O = red, Br = brown. For Br1NTE, only the major component of the disorder is shown and discussed. The terminal carbon of the ethyl group and all hydrogen atoms are omitted for clarity. Thermal ellipsoids of the central molecules are shown at the 50% probability level, whereas the neighbouring molecules shown in stick representation. Non-covalent Br⋯O and Br⋯Br interactions are highlighted with cyan dashed lines. ) and the close neighbours in crystals. Crystals were obtained by diffusing MeOH vapour into the CHCl3 solutions of BrnNTE. Colour code: C = grey, O = red, Br = brown. For Br1NTE, only the major component of the disorder is shown and discussed. The terminal carbon of the ethyl group and all hydrogen atoms are omitted for clarity. Thermal ellipsoids of the central molecules are shown at the 50% probability level, whereas the neighbouring molecules shown in stick representation. Non-covalent Br⋯O and Br⋯Br interactions are highlighted with cyan dashed lines. | ||
With only one Br atom per molecule, the ester groups in Br1NTE do not engage in extended halogen-bonded networks. In fact, the shortest d(C–Br⋯O–C2H5) distance is measured to be 3.32(2) Å (θ(C–Br⋯O) = 151.7(9)°), barely shorter than the van der Waals contact distance. At last, no π-stack or short contact between C–H and naphthalene was found in the crystals of Br0NTE.
While naphthalene and its 2-brominated derivatives display electronic absorption <300 nm, ester substitution induces a bathochromic shift of the naphthalene-centred transitions by ca. 50 nm, extending the absorption bands to 350 nm with maxima at ∼300 nm (Fig. 2).54–56 Compared to pristine naphthalene, which has an appreciable fluorescence quantum yield of 23% (40% triplet formation yield),55 no emission was detected from all BrnNTE in deaerated CH2Cl2 up to 0.02 M (near saturation) excited at 330 nm.
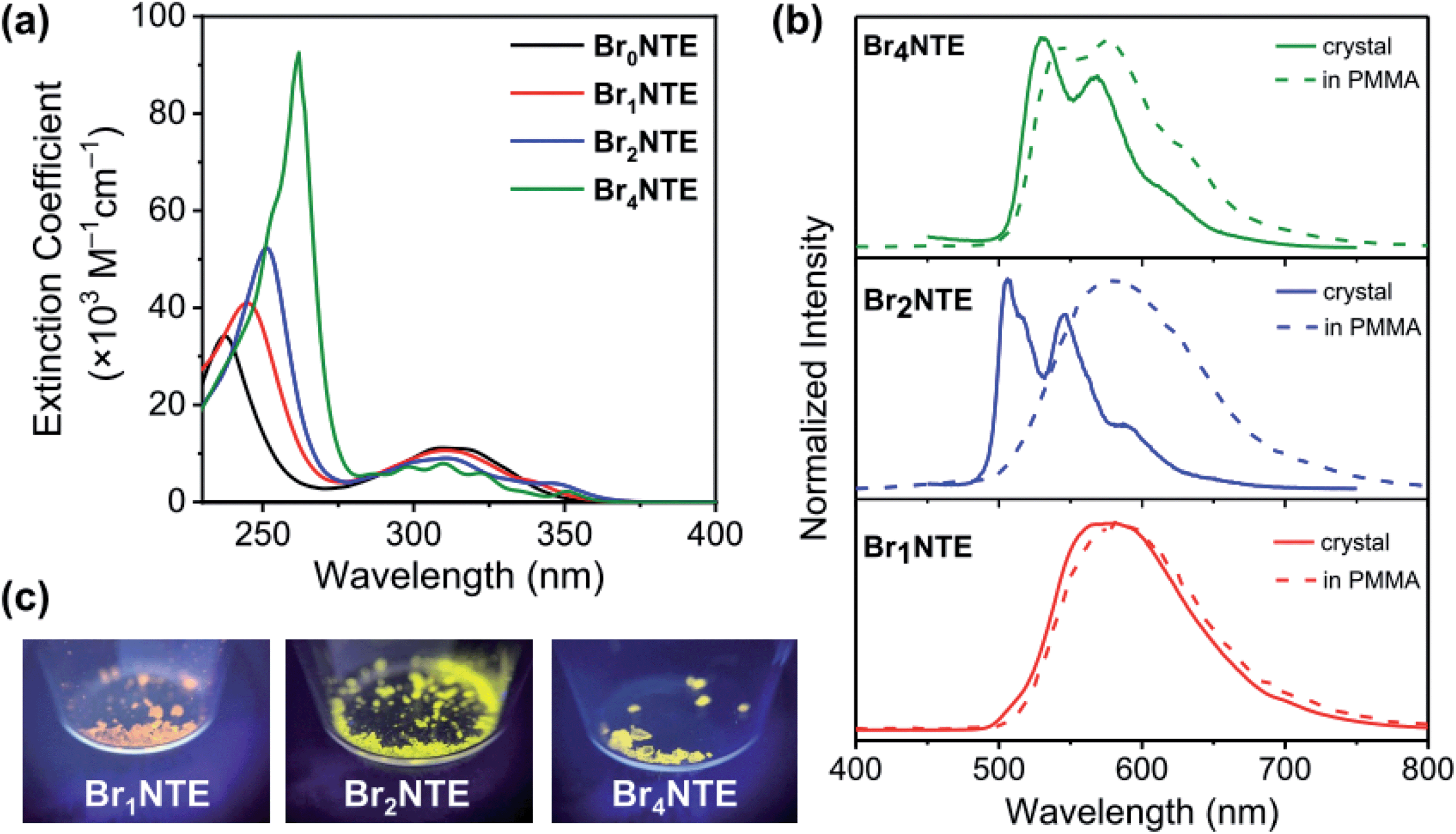 | ||
| Fig. 2 (a) UV-Vis absorption spectra of BrnNTE at (5–8) × 10−5 M in CH2Cl2. (b) Normalised phosphorescence spectra of BrnNTE in the crystalline solid state (solid line) or in PMMA (dashed line). Samples were excited at 300–320 nm. (c) Photographs of solid emission under UV irradiation (365 nm). | ||
Despite the non-radiative energy dissipation in solution, crystalline solids of the brominated molecules display visible phosphorescence in the 500–700 nm region with millisecond lifetimes, whereas non-brominated Br0NTE remains non-emissive (Fig. 2 and Table 1). Powdery crystalline solid samples of BrnNTE, whose powder X-ray diffraction profiles match the pattern based on their single-crystal data, were used in all phosphorescence measurements. Phosphorescence of crystalline Br2NTE and Br4NTE feature clear vibrational progression with a quantum yield of ΦPhos = 19.6% and 9.3%, respectively. Much weaker and structureless emission was observed for Br1NTE (ΦPhos = 1.4%).
| In CH2Cl2 | Crystalline solids | |||
|---|---|---|---|---|
| τ(S1 → Tn)a | τ(T1 → S0)a | τ Phos | Φ Phos , | |
| a From transient absorption measurements. b From (time-resolved) phosphorescence measurements. c The uncertainty is estimated to be 20% of the measured values. d Preceded by the relaxation of hot S1 in (0.9–1.2)±0.3 ps. | ||||
| Br0NTE | 111.7 ± 0.8 psd | 29.1 ± 0.1 μs | n.a. | n.a. |
| Br1NTE | 9.1 ± 0.3 psd | 1.42 ± 0.01 μs | 1.53 ± 0.02 ms | 1.4% |
| Br2NTE | 7.5 ± 0.3 ps | 0.50 ± 0.01 μs | 1.94 ± 0.01 ms | 19.6% |
| Br4NTE | 48.3 ± 0.9 psd | 0.0121 ± 0.0006 μs | 1.11 ± 0.01 ms | 9.3% |
The varying luminescent behaviours suggest that the excited-state dynamics were modulated in a subtle way by Br-specific properties, which is however not directly related to the number of Br atoms in the molecule. It is conceivable that multi-point halogen bonding provides a geometric framework to strengthen the rigidity of BrnNTE in the crystalline state. This effect is especially substantial for Br2NTE where all the peripheral substituents engage in the directional Br⋯O interactions, providing the additional factor to the solid-state effect6,8 of RTP to impede competitive non-radiative relaxation through intramolecular motions. The highest phosphorescence quantum yield was thus observed for the crystalline sample of Br2NTE.
The weaker and non-structured phosphorescence observed for Br1NTE (and Br0NTE) seems to be originated from its looser solid-state packing. If we define the volumetric index Vi as the ratio between the Voronoi volume (VVor)57,58 and the van der Waals volume (VwdW) of a molecule in the crystal, smaller Vi = VVor/VwdW would suggest denser packing. Vi of 1.27–1.30 were found for Br2NTE and Br4NTE embedded in halogen-bonded frameworks, but the values are significantly larger for Br0NTE and Br1NTE (1.36–1.38). The larger free space available to each molecule in the Br0NTE and Br1NTE crystals allows the excited molecules to decay radiatively and non-radiatively on various points of the triplet potential energy surface.
The significance of the inter-BrnNTE Br⋯O interactions is further supported by comparing the phosphorescence of crystalline BrnNTE with that of the dispersed molecules in poly(methyl methacrylate) (PMMA, Mw ∼996 kDa; 2 wt% doping). The rigid polymer matrix is expected to constrain the molecular motion at room temperature but disrupt inter-BrnNTE Br⋯O halogen bonds. The phosphorescence spectra of Br1NTE remained identical in either environment (Fig. 2), indicating that the triplet decay in Br1NTE is largely intrinsic to the monomeric molecule. However, the vibrational progression of Br4NTE, a signature of chromophore rigidity, became less pronounced, and that of Br2NTE completely disappeared and the overall emission profile resembles very well to that of Br1NTE.
Additional support for the efficient population of the triplet excited state was provided by transient absorption measurements. Spectroscopically, all BrnNTE exhibit similar excited-state dynamics: following the initial formation of the singlet excited state, which displays excited-state absorption (ESA) peaking at ca. 490 nm and a broad feature in the near infrared region of 800–1000 nm (Fig. 3 and ESI Section 5†), a new excited-state species with ESA at ca. 480 nm appears with microsecond lifetimes. This long-lived species was assigned to the triplet of each chromophore based on the lifetime and spectral similarity to the triplet–triplet absorption of methyl 1-naphthalate59 and 2-bromonaphthalene.60 Therefore, the decay of the initial state can be ascribed to singlet-to-triplet ISC; time constants on the order of tens of picosecond were observed for this process (Table 1). Compared to the typical fluorescence lifetime (1 ns or longer) of naphthalene derivatives,54,61 the fast ISC process suggests a high triplet forming efficiency. Such efficient ISC on the molecular level is likely due to the combined results of bromo (cf. >90% triplet yield for 2-bromonaphthalene)60 and carbonyl substitution.62 Broadly speaking, the more bromo atoms in a molecule, the faster the S1 → Tn and T1 → S0 processes, in line with the stronger heavy-atom enhanced SOC.63 Unexpectedly, however, the S1 → Tn ISC for Br4NTE is noticeably slower than its less brominated analogues.
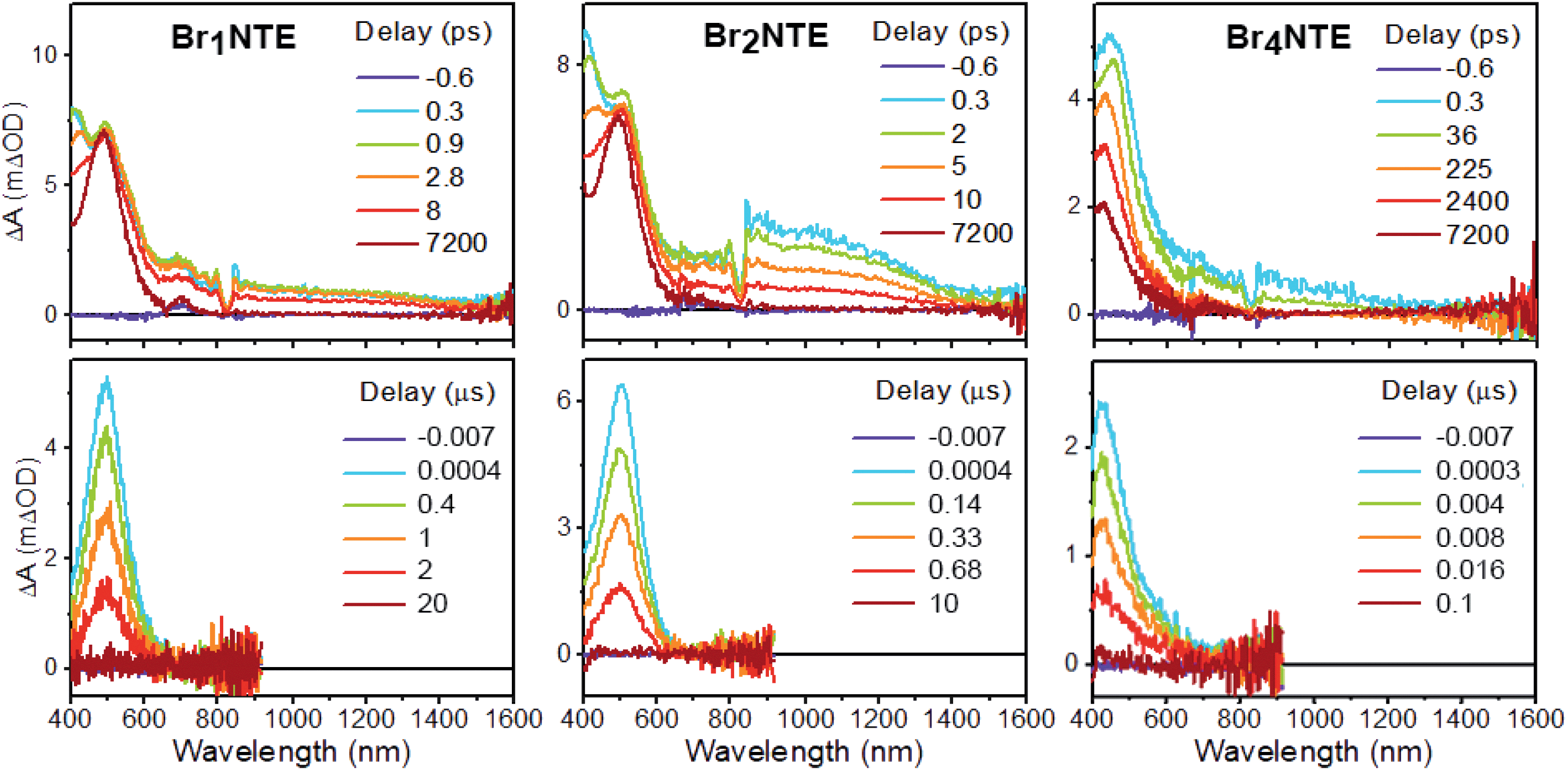 | ||
| Fig. 3 Transient absorption spectra of BrnNTE (n = 1, 2, and 4) in deaerated CH2Cl2 at various pump–probe delay indicated (excitation = 330 nm, see ESI Section 5† for n = 0). | ||
Since the rate of S1 → Tn ISC is largely determined by the energy gap between the singlet and triplet states (ΔEST) and the magnitude of spin–orbit coupling (SOC),64 we evaluated the matrix elements of 〈S1|ĤSO|Tn〉 using the two-layer ONIOM (QM:MM) scheme to simulate the photophysical processes in crystals. The molecular geometry was computed at the level of ωB97X-D/6-31G(d):OPLS-AA, and 〈S1|ĤSO|Tn〉 calculated at the TDA-ωB97X-D/6-311+G(d,p) level of theory based on the ONIOM geometries (Table 2 and ESI Section 7†). The (TD-)DFT calculations were performed using Gaussian 16,65 which was then interfaced with PySOC30 to evaluate the SOC matrix elements. The Tamm–Dancoff approximation (TDA) was exploited to minimise triplet instability.40,66 In all cases, the state energies are not significantly affected by aggregation; thus results from the calculations with one molecule in the QM region are discussed here.
| At S1 geometrya | At T1 geometry | ||||
|---|---|---|---|---|---|
| 〈S1|ĤSO|T1〉 | 〈S1|ĤSO|T2〉 | 〈S1|ĤSO|T3〉 | 〈S1|ĤSO|T4〉 | 〈S0|ĤSO|T1〉 | |
| a States relevant for the intersystem-crossing mechanism are highlighted in bold. | |||||
| Br0NTE | 0.82 | 0.04 | 0.52 | 0.86 | 0.01 |
| Br1NTE | 8.22 | 10.87 | 15.49 | 9.02 | 3.22 |
| Br2NTE | 68.74 | 0.93 | 166.79 | 106.08 | 142.3 |
| Br4NTE | 20.30 | 42.79 | 3.55 | 30.93 | 0.38 |
The ISC process for BrnNTE likely takes place between S1 and the high-lying triplet states. Considering ΔEST alone (<0.5 eV), ISC to T2,3 for Br1NTE, T2–4 for Br2NTE, and T2 for Br4NTE should dominate in the respective molecules, whereas the large energy gap ΔEST > 1.5 eV prevents direct ISC into T1 (see ESI Section 7† for the relative energies). Compared to Br0NTE, incorporating fourth-row Br elements into the naphthalene scaffold increases SOC by 1–2 orders of magnitude. Despite the larger number of Br atoms in the structure, smaller SOC was found for Br4NTE than Br2NTE, in line with the slower triplet formation found experimentally for the former molecule. The 〈S0|ĤSO|T1〉 calculated at the T1 geometry, the key factor determining the rate of phosphorescence, was similarly found to be smaller for Br4NTE than Br2NTE.
A close examination of the electron density of the key states provided hints to the origin of the unexpected drop in SOC for Br4NTE. Fig. 4 shows the electron density difference between the selected excited states and the ground state for Br2NTE (S1 and T3) and for Br4NTE (S1 and T2). These transitions displayed a significant naphthalene-centred π–π* character; the involvement of the Br atoms can be clearly seen and hence the higher SOC in brominated BrnNTE. Comparatively, the carbonyl n–π* contribution, the typical driver for the ISC process in aromatic ketones/aldehydes, appears to be much less substantial. In the case of Br2NTE, the Br-centred transition densities are roughly perpendicular to the naphthalene plane in the S1 state but rotate distinctively in the T3 state, facilitating the orbital angular momentum change for ISC (similar rotation found in T4). In the case of Br4NTE, however, the Br-centred transition densities in S1 and T2 are both perpendicular to the naphthalene plane. The absence of the analogous rotated transition density for Br4NTE is understandable as unfavourable electron repulsion in the region between neighbouring Br atoms would be caused by such a change in density orientation.
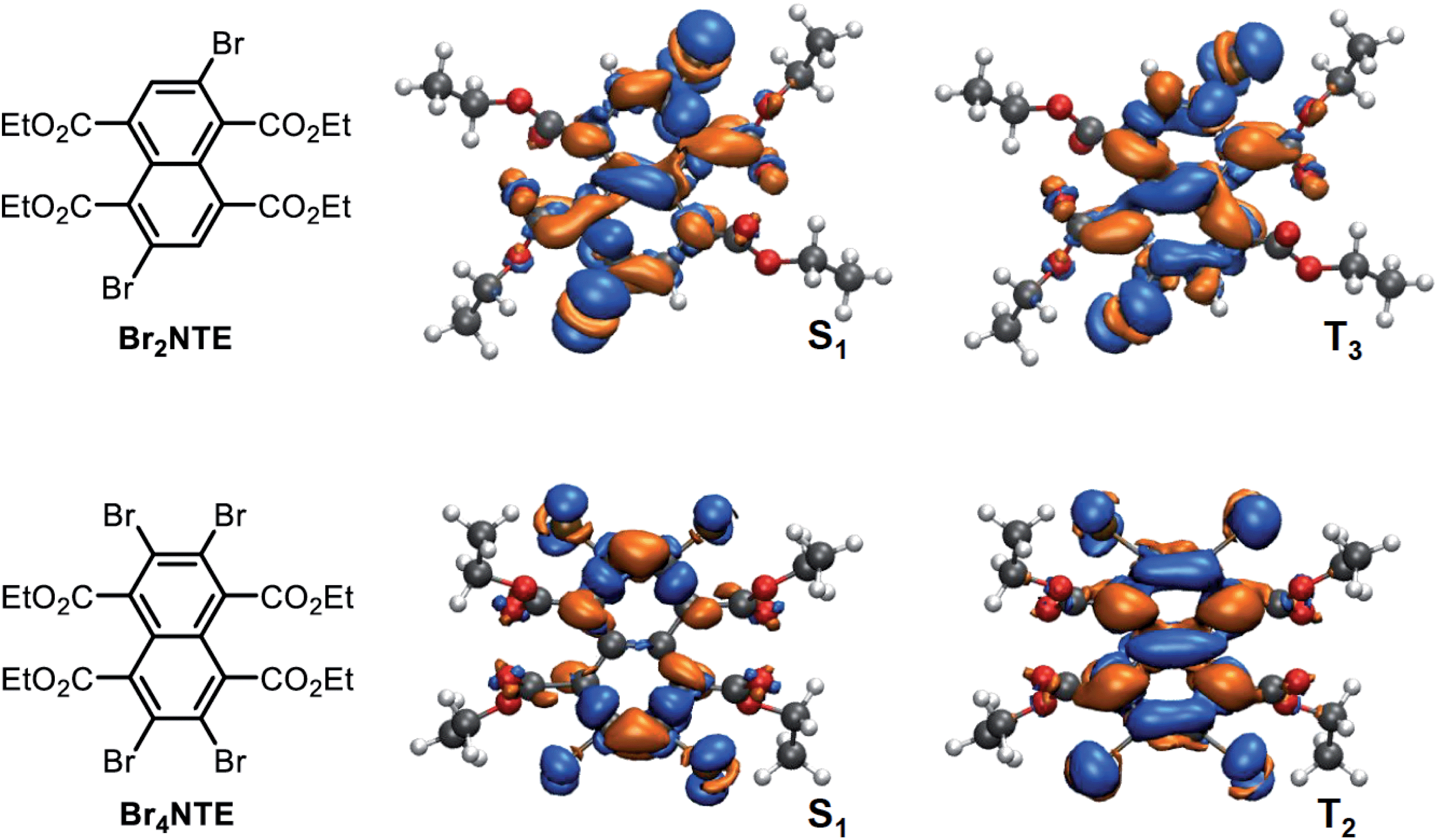 | ||
| Fig. 4 Top-down view of electron density difference plots (0.001 e bohr−3 isovalue) between the selected excited states (S1 or T2/3) and the ground state for Br2NTE (top row) and Br4NTE (bottom row). The molecular orientation is sketched on the left; orange colour represents positive and blue negative values. | ||
Taken together, the judicious heavy-atom positioning in Br2NTE results in the favourable structural and electronic contributions to its efficient RTP. The 2,6-dibromo substitution offers a lock-in mechanism through halogen bonding to inhibit non-radiative relaxation. Furthermore, high SOC and hence efficient ISC are made possible by adding the orbital angular momentum change to the heavy-atom effect in both the triplet-generation (S1 → Tn) and phosphorescence (T1 → S0) processes.
Conclusions
In summary, we have shown that simultaneously incorporating multiple heavy halogens and halogen-bond donor/acceptor pairs in aromatic molecules can enable bright phosphorescence from purely organic materials. The formation of halogen-bonded frameworks in the solid states rigidifies phosphorophores, favouring the radiative decay. However, our results indicate that a fine balance has to be struck in terms of the number and positioning of halogens. Too many large halogen atoms in proximity may prohibit structurally the access of halogen-bond acceptors and electronically the contribution of the non-bonding electrons of halogens for enhancing SOC. The latter effect is especially important to consider in the case of carbonyl-bearing polycyclic aromatic hydrocarbons, such as rylenes and its derivatives in the present study where the S1 state is primarily π–π* in nature. It should be noted that the formation of halogen bonds cannot necessarily be correlated to the increase in ISC and phosphorescence rates; an excited-state analysis will be needed to elucidate the magnitude and origin of SOC when designing organic RTP materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. B. gratefully acknowledges the EU through the MSCA-ITN-ETN (GA No. 722591 – project PHOTOTRAIN) and School of Chemistry at Cardiff University for generous financial support. R. C.-O. acknowledges funding from the EPSRC (EP/R029385/1) and Leverhulme Trust (RPG-2019-122). This research utilised Queen Mary's Apocrita HPC facility. This work was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Award DE-SC0020168 (M. R. W.). The authors thank Professor Kenneth D. M. Harris and Dr Colan E. Hughes (Cardiff) for the help with powder X-ray diffraction.References
- Z. W. Pan, Y. Y. Lu and F. Liu, Nat. Mater., 2012, 11, 58–63 CrossRef CAS.
- T. Maldiney, A. Bessiere, J. Seguin, E. Teston, S. K. Sharma, B. Viana, A. J. J. Bos, P. Dorenbos, M. Bessodes, D. Gourier, D. Scherman and C. Richard, Nat. Mater., 2014, 13, 418–426 CrossRef CAS.
- Y. Li, M. Gecevicius and J. R. Qiu, Chem. Soc. Rev., 2016, 45, 2090–2136 RSC.
- S. M. A. Fateminia, Z. Mao, S. D. Xu, Z. Y. Yang, Z. G. Chi and B. Liu, Angew. Chem., Int. Ed., 2017, 56, 12160–12164 CrossRef CAS.
- G. Q. Zhang, G. M. Palmer, M. Dewhirst and C. L. Fraser, Nat. Mater., 2009, 8, 747–751 CrossRef CAS.
- Y. Gong, L. Zhao, Q. Peng, D. Fan, W. Z. Yuan, Y. Zhang and B. Z. Tang, Chem. Sci., 2015, 6, 4438–4444 RSC.
- R. Yoshii, A. Hirose, K. Tanaka and Y. Chujo, J. Am. Chem. Soc., 2014, 136, 18131–18139 CrossRef CAS.
- W. Z. Yuan, X. Y. Shen, H. Zhao, J. W. Y. Lam, L. Tang, P. Lu, C. L. Wang, Y. Liu, Z. M. Wang, Q. Zheng, J. Z. Sun, Y. G. Ma and B. Z. Tang, J. Phys. Chem. C, 2010, 114, 6090–6099 CrossRef CAS.
- Z. Zhao, X. Zheng, L. Du and Y. Xiong, et al. , Nat. Commun., 2019, 10, 2952 CrossRef.
- G. Bergamini, A. Fermi, C. Botta, U. Giovanella, S. Di Motta, F. Negri, R. Peresutti, M. Gingras and P. Ceroni, J. Mater. Chem. C, 2013, 1, 2717–2724 RSC.
- L. Xiao, Y. Wu, Z. Yu, Z. Xu, J. Li, Y. Liu, J. Yao and H. Fu, Chem.–Eur. J., 2018, 24, 1801–1805 CrossRef CAS.
- S. K. Maity, S. Bera, A. Paikar, A. Pramanik and D. Haldar, Chem. Commun., 2013, 49, 9051–9053 RSC.
- S. Cai, H. Shi, D. Tian, H. Ma, Z. Cheng, Q. Wu, M. Gu, L. Huang, Z. An, Q. Peng and W. Huang, Adv. Funct. Mater., 2018, 28, 1705045 CrossRef.
- Z. Yang, C. Xu, W. Li, Z. Mao, X. Ge, Q. Huang, H. Deng, J. Zhao, F. L. Gu, Y. Zhang and Z. Chi, Angew. Chem., Int. Ed., 2020, 59, 17451–17455 CrossRef CAS.
- A. Kremer, C. Aurisicchio, F. De Leo, B. Ventura, J. Wouters, N. Armaroli, A. Barbieri and D. Bonifazi, Chem.–Eur. J., 2015, 21, 15377–15387 CrossRef CAS.
- J. Xu, A. Takai, Y. Kobayashi and M. Takeuchi, Chem. Commun., 2013, 49, 8447–8449 RSC.
- D. Lee, O. Bolton, B. C. Kim, J. H. Youk, S. Takayama and J. Kim, J. Am. Chem. Soc., 2013, 135, 6325–6329 CrossRef CAS.
- M. Shimizu, A. Kimura and H. Sakaguchi, Eur. J. Org. Chem., 2016, 467–473 CrossRef CAS.
- S. Mukherjee and P. Thilagar, Chem. Commun., 2015, 51, 10988–11003 RSC.
- L. Xiao and H. Fu, Chem.–Eur. J., 2019, 25, 714–723 CrossRef CAS.
- Z. He, W. Zhao, J. W. Y. Lam, Q. Peng, H. Ma, G. Liang, Z. Shuai and B. Z. Tang, Nat. Commun., 2017, 8, 416 CrossRef.
- W. Zhao, Z. He, J. W. Y. Lam, Q. Peng, H. Ma, Z. Shuai, G. Bai, J. Hao and B. Z. Tang, Chem, 2016, 1, 592–602 CAS.
- A. Barbieri, E. Bandini, F. Monti, V. K. Praveen and N. Armaroli, Top. Curr. Chem., 2016, 374, 47 CrossRef.
- M. Hayduk, S. Riebe and J. Voskuhl, Chem.–Eur. J., 2018, 24, 12221–12230 CrossRef CAS.
- M. S. Kwon, D. Lee, S. Seo, J. Jung and J. Kim, Angew. Chem., Int. Ed., 2014, 53, 11177–11181 CrossRef CAS.
- L. Bian, H. Shi, X. Wang, K. Ling, H. Ma, M. Li, Z. Cheng, C. Ma, S. Cai, Q. Wu, N. Gan, X. Xu, Z. An and W. Huang, J. Am. Chem. Soc., 2018, 140, 10734–10739 CrossRef CAS.
- M. Baroncini, G. Bergamini and P. Ceroni, Chem. Commun., 2017, 53, 2081–2093 RSC.
- A. Forni, E. Lucenti, C. Botta and E. Cariati, J. Mater. Chem. C, 2018, 6, 4603–4626 RSC.
- R. Liu, X. Gao, M. Barbatti, J. Jiang and G. Z. Zhang, J. Phys. Chem. Lett., 2019, 10, 1388–1393 CrossRef CAS.
- X. Gao, S. M. Bai, D. Fazzi, T. Niehaus, M. Barbatti and W. Thiel, J. Chem. Theory Comput., 2017, 13, 515–524 CrossRef CAS.
- M. A. El-Sayed, J. Chem. Phys., 1963, 38, 2834–2838 CrossRef CAS.
- F. Dinkelbach, M. Kleinschmidt and C. M. Marian, J. Chem. Theory Comput., 2017, 13, 749–766 CrossRef CAS.
- D. Sasikumar, A. T. John, J. Sunny and M. Hariharan, Chem. Soc. Rev., 2020, 49, 6122–6140 RSC.
- R. H. Compton, K. T. V. Grattan and T. Morrow, J. Photochem., 1980, 14, 61–66 CrossRef CAS.
- T. Itoh, Chem. Phys. Lett., 1988, 151, 166–168 CrossRef CAS.
- S. Hirata, Adv. Opt. Mater., 2017, 5, 1700116 CrossRef.
- O. Bolton, K. Lee, H. J. Kim, K. Y. Lin and J. Kim, Nat. Chem., 2011, 3, 205–210 CrossRef CAS.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CAS.
- P. J. Costa, Phys. Sci. Rev., 2017, 2, 20170136 Search PubMed.
- S. Sarkar, H. P. Hendrickson, D. Lee, F. DeVine, J. Jung, E. Geva, J. Kim and B. D. Dunietz, J. Phys. Chem. C, 2017, 121, 3771–3777 CAS.
- S. J. Ang, T. S. Chwee and M. W. Wong, J. Phys. Chem. C, 2018, 122, 12441–12447 CrossRef CAS.
- M. Al Kobaisi, S. V. Bhosale, K. Latham, A. M. Raynor and S. V. Bhosale, Chem. Rev., 2016, 116, 11685–11796 CrossRef.
- O. Cakmak, J. Chem. Res., 1999, 366–367 RSC.
- A. M. Wagner, A. J. Hickman and M. S. Sanford, J. Am. Chem. Soc., 2013, 135, 15710–15713 CrossRef CAS.
- K. D. Collins, R. Honeker, S. Vasquez-Cespedes, D. T. D. Tang and F. Glorius, Chem. Sci., 2015, 6, 1816–1824 RSC.
- N. Chongboriboon, K. Samakun, T. Inprasit, F. Kielar, W. Dungkaew, L. W. Y. Wong, H. H. Y. Sung, D. B. Ninkovic, S. D. Zaric and K. Chainok, CrystEngComm, 2020, 22, 24–34 RSC.
- A. A. Berezin, A. Sciutto, N. Demitri and D. Bonifazi, Org. Lett., 2015, 17, 1870–1873 CrossRef CAS.
- C. Roger and F. Wurthner, J. Org. Chem., 2007, 72, 8070–8075 CrossRef.
- M. Sasikumar, Y. V. Suseela and T. Govindaraju, Asian J. Org. Chem., 2013, 2, 779–785 CrossRef CAS.
- R. S. K. Kishore, V. Ravikumar, G. Bernardinelli, N. Sakai and S. Matile, J. Org. Chem., 2008, 73, 738–740 CrossRef CAS.
- Y. Ma, X. Zhang, S. Stappert, Z. Yuan, C. Li and K. Müllen, Chem. Commun., 2017, 53, 5310–5313 CAS.
- B. K. Saha, A. Saha and S. A. Rather, Cryst. Growth Des., 2017, 17, 2314–2318 CrossRef CAS.
- B. K. Saha, S. A. Rather and A. Saha, Cryst. Growth Des., 2016, 16, 3059–3062 CrossRef CAS.
- M. Kitamura and H. Baba, Bull. Chem. Soc. Jpn., 1975, 48, 1191–1195 CrossRef CAS.
- A. A. Lamola and G. S. Hammond, J. Chem. Phys., 1965, 43, 2129–2135 CrossRef CAS.
- Y. L. Chow and X. Y. Liu, Can. J. Chem., 1991, 69, 1261–1272 CrossRef CAS.
- F. M. Richards, J. Mol. Biol., 1974, 82, 1–14 CrossRef CAS.
- M. Rivera, M. Dommett, A. Sidat, W. Rahim and R. Crespo-Otero, J. Comput. Chem., 2020, 41, 1045–1058 CrossRef CAS.
- X. Cai, M. Sakamoto, M. Yamaji, M. Fujitsuka and T. Majima, Chem.–Eur. J., 2007, 13, 3143–3149 CrossRef CAS.
- J. C. Scaiano, B. R. Arnold and W. G. Mcgimpsey, J. Phys. Chem., 1994, 98, 5431–5434 CrossRef CAS.
- S. A. Green, D. J. Simpson, G. Zhou, P. S. Ho and N. V. Blough, J. Am. Chem. Soc., 1990, 112, 7337–7346 CrossRef CAS.
- A. I. Privalova, J. P. Morozova, E. R. Kashapova and V. J. Artyukhov, J. Appl. Spectrosc., 2011, 78, 309–317 CrossRef CAS.
- A. Mohan, E. Sebastian, M. Gudem and M. Hariharan, J. Phys. Chem. B, 2020, 124, 6867–6874 CrossRef CAS.
- D. Beljonne, Z. Shuai, G. Pourtois and J. L. Bredas, J. Phys. Chem. A, 2001, 105, 3899–3907 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, et al., Gaussian 16 Rev. C.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- M. J. G. Peach and D. J. Tozer, J. Phys. Chem. A, 2012, 116, 9783–9789 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, additional transient absorption data, synthetic and computational details, and X-ray crystallographic data. CCDC 1949875, 1949880 and 1949883. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc04646a |
| This journal is © The Royal Society of Chemistry 2021 |