
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConfined space design by nanoparticle self-assembly
Valentina
Dichiarante
 ,
Claudia
Pigliacelli
*,
Pierangelo
Metrangolo
* and
Francesca
Baldelli Bombelli
,
Claudia
Pigliacelli
*,
Pierangelo
Metrangolo
* and
Francesca
Baldelli Bombelli
Laboratory of Supramolecular and Bio-Nanomaterials (SupraBioNanoLab), Department of Chemistry, Materials, and Chemical Engineering “Giulio Natta”, Politecnico di Milano, Via Luigi Mancinelli 7, 20131 Milan, Italy. E-mail: pierangelo.metrangolo@polimi.it; claudia.pigliacelli@polimi.it
First published on 23rd December 2020
Abstract
Nanoparticle (NP) self-assembly has led to the fabrication of an array of functional nanoscale systems, having diverse architectures and functionalities. In this perspective, we discuss the design and application of NP suprastructures (SPs) characterized by nanoconfined compartments in their self-assembled framework, providing an overview about SP synthetic strategies reported to date and the role of their confined nanocavities in applications in several high-end fields. We also set to give our contribution towards the formation of more advanced nanocompartmentalized SPs able to work in dynamic manners, discussing the opportunities of further advances in NP self-assembly and SP research.
 Valentina Dichiarante | Valentina Dichiarante is a Senior Assistant Professor of Chemistry in the Department of Chemistry, Materials and Chemical Engineering of Politecnico di Milano, where she currently works in the SupraBioNano Lab. She has been a post-doctoral researcher at the Politecnico di Milano (2012–2015), Commissariat à l’Energie Atomique et aux Energies Alternatives CEA-Saclay (France, 2010–2011), and University of Pavia (Photogreen Lab, 2008–2009). She received her PhD in Chemical Sciences in 2008 from the University of Pavia. Her research interests focus on the development of highly fluorinated ligands for nanostructured theranostics and functional materials, and on the synthesis and characterization of supramolecular responsive materials. |
 Claudia Pigliacelli | Claudia Pigliacelli is a Junior Assistant Professor in the Department of Chemistry, Materials, and Chemical Engineering “Giulio Natta” of Politecnico di Milano, where she has been working in the SupraBioNano Lab since 2020. She received her PhD in Pharmacy from the University of East Anglia (2015). She has been a research assistant and post-doc researcher at the Politecnico di Milano (2013–2016), at Aalto University (Finland, 2019), and at the Centro de Investigacions Cientificas Avanzadas (CICA, Spain, 2020). Her research interests include the design of nanoscale systems based on peptides and nanoparticles, and electron microscopy techniques for the characterization of nanomaterials. |
 Pierangelo Metrangolo | Pierangelo Metrangolo has been a full professor at the Politecnico di Milano since 2011, where he is the Executive Deputy Head of the Department of Chemistry, Materials, and Chemical Engineering “Giulio Natta”. He also holds a position as a visiting professor at the Centre of Excellence in Molecular Engineering of Biosynthetic Hybrid Materials of Aalto University, Finland, and at the VTT-Technical Research Centre of Finland. His awards include the 2005 ‘‘G. Ciamician’’ medal of the Division of Organic Chemistry of the Italian Chemical Society, the 2005 Journals Grant Award of the Royal Society of Chemistry, the 2009 IUPAC Young Chemist Award, and the 2019 Fluorous Technologies Award. He is the Chief of the Editorial Board of CrystEngComm (RSC), a Co-Editor of Acta Crystallogr. B (IUCr), and an Editorial Advisory Board member of Cryst. Growth Des. (ACS). He is an author of 290 publications with 17686 citations (H-index: 60). He is currently the Vice President of the Physical and Biophysical Chemistry Division (Div. I) of the IUPAC. |
 Francesca Baldelli Bombelli | Francesca Baldelli Bombelli is an Associate Professor in Chemistry at the Politecnico di Milano. She currently works in the Department of Chemistry, Materials and Chemical Engineering in the SupraBioNanoLab (www.suprabionano.eu). She was the Group Leader at the European Centre of Nanomedicine (www.nanomedicen.eu) in 2013–2015. In 2011–2014, she was a Lecturer in Nanotechnology and Colloid Science at the School of Pharmacy, UEA, Norwich, UK. She was a Post-Doctoral Researcher: 2009–2011 at CBNI, University College of Dublin, Dublin, Ireland; 2006–2009 at the University of Florence; 2004–2006 at Chalmers University, Gothenburg, Sweden. She has got her PhD in Chemical Sciences in 2004 from the University of Florence. Her research interests are focused on the development of supramolecular and nano-scaled functional materials for biomedical applications. |
1. Introduction
Compartmentalization is commonplace in biology, where nanocavities function as selective hosts for ions and molecules, as well as containers for essential biochemical processes, thus enabling spatial and temporal control over biological events.1 In an attempt to emulate Nature's complexity, the rational design of functional nanocavities has long been catching the attention of chemists.2–6 When nanoconfined, molecules show properties and reactivities that cannot be achieved in the dilute solution regime.7 Indeed, isolation from bulk media and restraint in nanometre-sized cavities strongly enhance molecules' ability to transport, accelerate reaction kinetics, and facilitate protein and macromolecule folding, thus resembling the behaviour of biomolecules in biochemical processes.1,8–11A large variety of molecular hosts bearing functional nanocavities have been reported to date, including cyclodextrins, carcerands, cavitands, calixarenes, cryptophanes, and cucurbiturils.12 Moreover, several host–guest systems have shown the ability to tune the photo-optical properties of their guest molecules and facilitate their solubilisation.13,14 In this regard, programmed self-assembly became a key facilitator in the fabrication of innovative compartmentalized nanostructures, making use of non-covalent interactions among distinct molecular building blocks,15,16 and/or exploiting specific templates or stimuli to modulate their self-assembly process.17–19 For example, discrete self-assembled molecular cages have also been reported, in which the restrained compartments were exploited for selective chemical reactions and processes, as well as responsive containers.20–26
More recently, confined space has also been obtained via self-assembly of nanoparticles (NPs).27 NP self-assembly is currently a rising tide in contemporary materials science and efforts done in this topic resulted in a wide variety of NP suprastructures (SPs) with different architectures and properties.28–32 NP packing through self-assembly can be programmed to obtain the formation of confined empty spaces in the resulting SPs, with the formation of new functional cavities for hosting molecules and promoting reactivity and catalytic processes.33–36 Specifically, fine tuning of NP size, shape, and surface functionalization can be used to drive NP self-organization into specific SPs bearing interparticle nano-scale compartments, as schematically represented in Fig. 1. Often, engineering these functional nanocontainers allows combining the ability of SPs to host molecules with innovative and/or collective properties arising from NP proximity.37–40 The intrinsic versatility of compartmentalized SPs is instrumental to several high-end applications, including drug delivery, catalysis, biomimetic processes, and chemical reactions.27,41,42
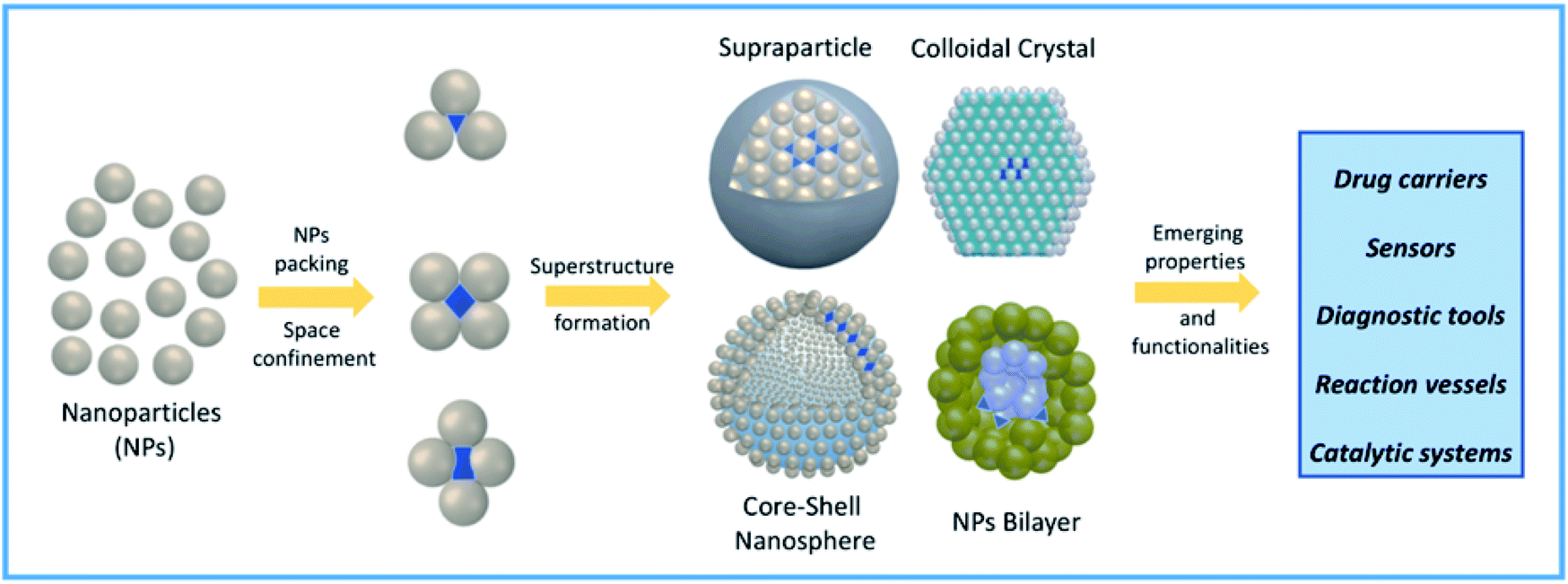 | ||
| Fig. 1 Schematic representation of confined nanocompartment formation in NP-based self-assembled systems and their possible applications in high-end fields. | ||
The concept of nanoconfinement has widely been reviewed in the literature and several contributions provided an excellent overview on the various nanosized molecular systems reported for molecule and reaction confinement.43–45 Here, instead, we aim at giving an outlook on the possible uses of interparticle confined nanocavities in self-assembled NP systems. In particular, we focus on their role as carriers, reaction vessels and catalytic systems. Also, we would like to uncover the possible future developments of nanoconfined SPs, with insights into the unique features offered by these versatile objects and their possible application in not-yet explored fields.
2. Nanoparticle self-assembly and nanocavity design strategies
NP self-assembly refers to the organization of NP building blocks into larger and ordered structures, driven by either direct interparticle specific interactions, or templates, or external stimuli.24,42,4346,47 The arrangement of NPs into assembled nanoscale architectures can involve a wide repertoire of different interactions occurring among chosen building blocks.48 van der Waals forces, electrostatic interactions, biomolecular recognition, protein–protein interactions, metal ligand coordination, and hydrogen bonding (HB) have already been reported as possible non-covalent forces that can take place among nanomaterials and consequently drive the formation of their assemblies and control the spatial distribution of NPs in the final structure (Table 1).49Currently, one of the most exploited non-covalent bonds in driving the self-assembly of NPs is HB, which, thanks to its multi-stimuli responsiveness (pH, temperature, and solvent), allows the formation of reversible and responsive assemblies. For example, Nonappa et al. recently reported cobalt hollow SPs obtained via HB between p-aminobenzoic acid (pABA) stabilized cobalt NPs (CoNPs; Fig. 2). Notably, the formation of these assembled magnetic capsids could be achieved through a one-step, template-free, and reversible procedure, providing an excellent example of potential containers obtained from metal NP assembly.50
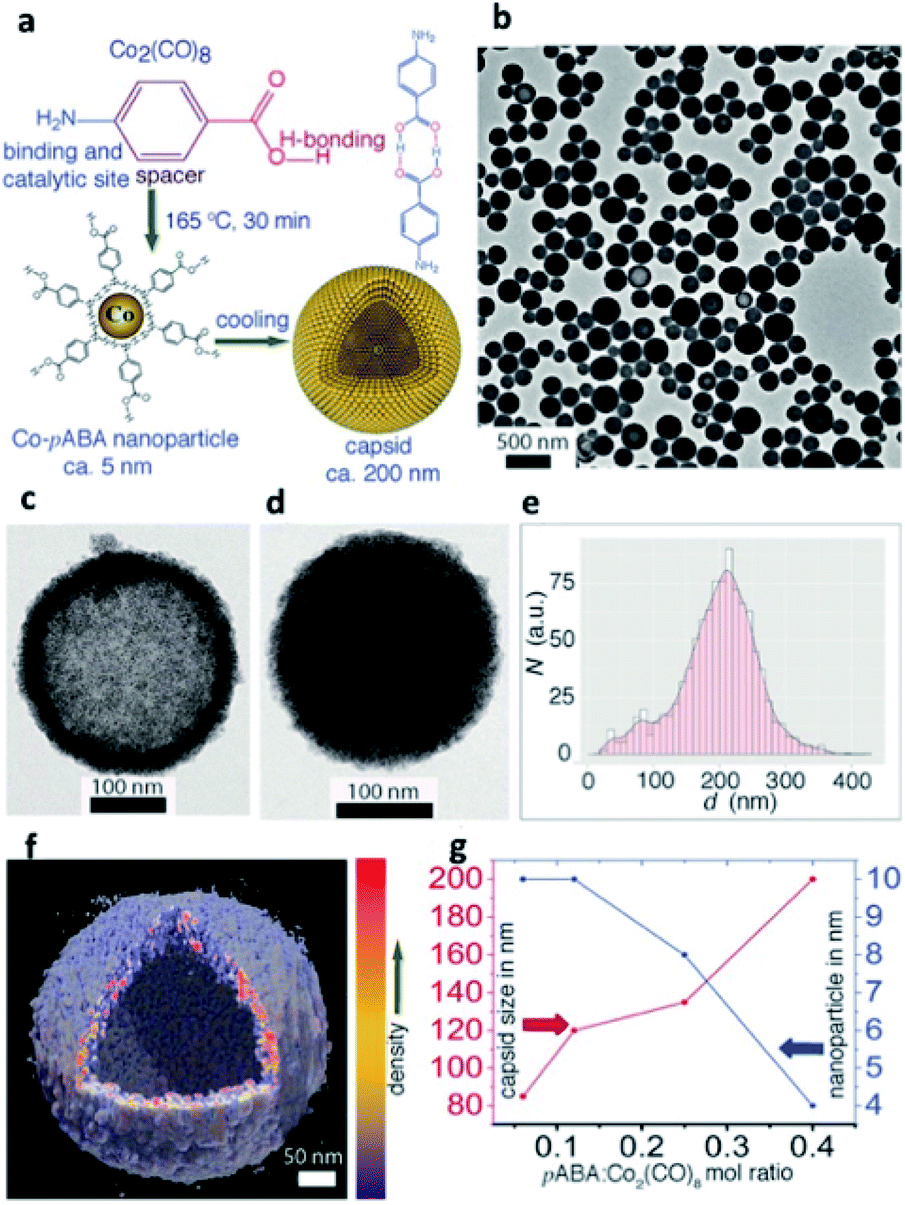 | ||
| Fig. 2 (a) Synthesis of CoNPs using Co2(CO)8 and pABA in 1,2-DCB and their spontaneous self-assembly into shell-like capsids; (b) Transmission Electron Microscopy (TEM) images showing spherical capsids for Co2(CO)8/pABA. Capsids with less (c) and more (d) kinetically trapped pABA in the interior; (e) size distribution of capsids; (f) Electron Tomography (ET) reconstruction resolves clearly the CoNP shell layer from the hollow interior; (g) NP and capsid sizes are tuned by their composition. Reproduced with permission from ref. 50. Copyright 2017 Wiley and Sons. | ||
Together with HB, its counterpart, halogen bonding (XB),51,52 has also shown potential in driving the assembly of gold NPs (AuNPs) into supraspheres and chain-like structures. Indeed, NP functionalization with custom ligands bearing XB donor and/or acceptor moieties enabled to mediate NP assembly via XB, exploiting the unique directionality provided by such an interaction to form chain-like and spheroidal inter-particle assemblies.53,54 Electrostatic forces have also been employed as driving forces for the assembly of polydispersed NPs into monodispersed SPs and a large variety of SP systems have been developed using hydrophobic interactions.55,56 Notably, NPs can also be functionalized with ligands designed to respond to chosen stimuli and trigger their assembly into SPs.57,58
| Nanocavity design strategy | Self-assembly driving interactions or stimuli | Starting building blocks | Potential applications | Ref. |
|---|---|---|---|---|
| Supraparticles | Hydrophobic (or solvophobic) interactions | Au NPs, F–Au NPs, and chiral Au NPs | Drug delivery systems | 70–73, 75, 77, 78 and 84 |
| PEG-S-Au NPs | Uptake of hydrophobic molecules from the bulk dispersing medium | 74 | ||
| Attractive and repulsive competing electrostatic interactions + crosslinking | Cysteine-capped CoO NPs | Cancer treatment | 79 | |
| Short-range attractive and long-range repulsive electrostatic interactions | Penicillin-stabilized Zn NPs | Modular enantioselective catalysts for tyrosine photooxidation | 42 | |
| Halogen bonding (XB) | Au NPs | Chain-like and spheroidal inter-particle suprastructures in solution and on planar surfaces | 53 and 54 | |
| Colloidal crystals | Light-induced self-assembly of photo-responsive surface ligands | Au, Fe3O4, and SiO2 NPs | Dynamic nanoflasks for photochemical reactions | 57, 91 and 92 |
| Oriented attachment | ZnO NPs (nanoporous pyramids) | Photocatalysts for water depollution | 97 | |
| NP bilayers | Template-directed metal growth | Au, Pd, and Pt NPs | Catalysts for solar light-induced dehydrogenation of N-heterocycles | 98 |
| Hollow chain vesicles | van der Waals forces + hydrophobic interactions | BCP–Au NPs | Photoacoustic imaging | 59 |
| Hollow capsids | Hydrogen bonding (HB) | Co NPs + p-aminobenzoic acid | Magnetic reversible containers | 50 |
| Core–shell nanospheres | Spontaneous balanced growth | Pt–CeO2 NPs | Catalysts for CO oxidation and chemical reduction of 4-nitrophenol by ammonia borane | 96 |
An important factor affecting the morphology and structure of SPs is NP surface functionalization, which governs NP–NP interactions and their self-organization. The nature of surface functional groups is not the only parameter driving the NP interaction pattern, as also their grafting density may play a crucial role. Y. Liu and coworkers showed an elegant example of how ligand surface-grafting density can be determinant for the final structures and properties of SP assemblies (Fig. 3).59 In this work, AuNPs, produced with either high or low grafting densities of a block copolymer (BCP), were dispersed in tetrahydrofuran (THF) and assembled upon gradual addition of water by two different self-organization patterns determined by the polymer grafting density. In fact, vesicular SPs having the same overall morphology, but differing in the NP spatial arrangement within the vesicular membranes, were formed. In particular, a high grafting density of NPs yielded a random NP distribution with larger inter-particle distances, while ordered NP chains, having shorter NP–NP distances, were formed by NPs with a lower polymer grafting density. As the cherry on the cake, near infrared (NIR) absorption activation was shown by the former SPs, indicating how NP self-organization at the mesoscale can be transferred to the material as a functional property.
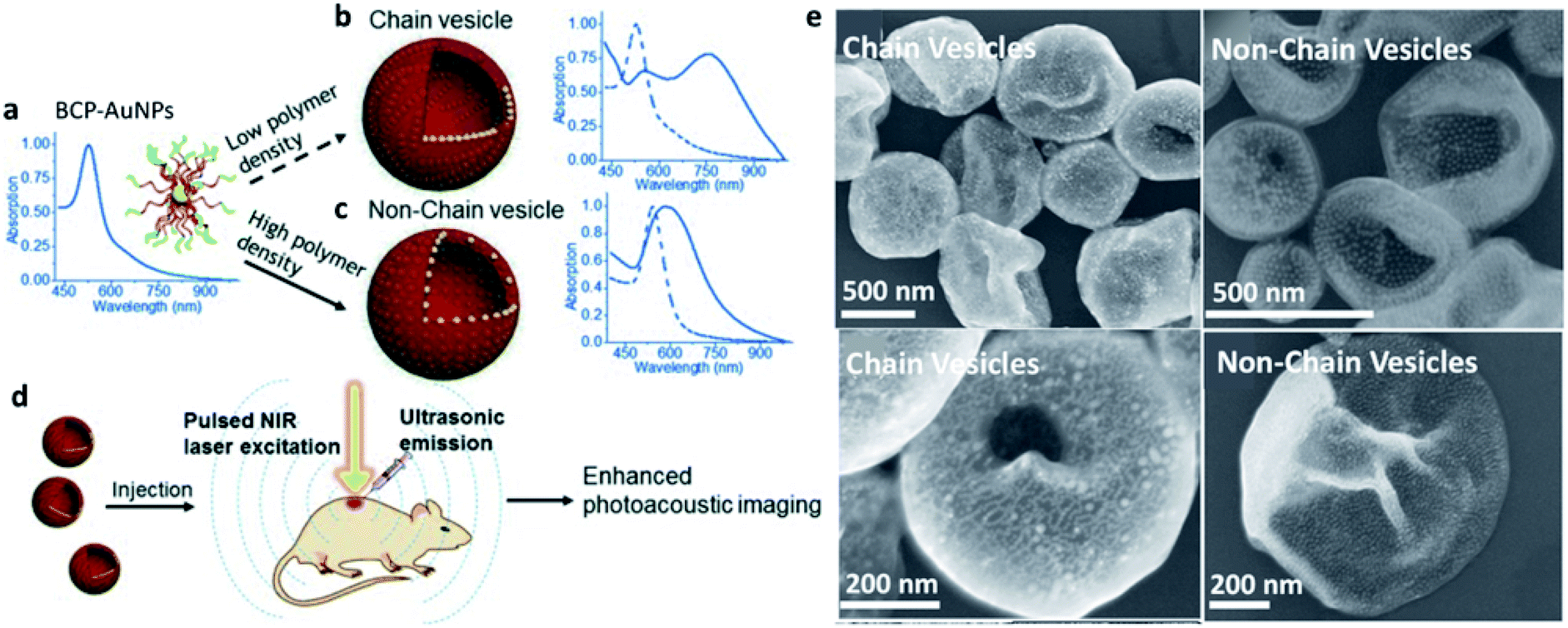 | ||
| Fig. 3 (a–c) Self-assembly of BCP–AuNPs into chain vesicles and non-chain vesicles; (d) enhanced PA imaging with chain vesicles; (e) Scanning Electron Microscopy (SEM) images of chain vesicles (left side) and non-chain vesicles (right side) made from BCP–AuNPs with 13 nm AuNPs as the cores. Reproduced with permission from ref. 59. Copyright 2015 Wiley and Sons. | ||
Controlling NP spatial distribution through non-covalent interactions can be an important tool to build-up ordered NP assemblies endowed with additional collective properties. In nature, fibrillar proteins reach the hierarchical order by finely balancing repulsive and attractive intermolecular interactions through complex biological processes.60 Similarly, NPs can be engineered to experience competing interaction forces (not necessarily directional) in their environment and finally self-assemble forming higher order structures.61,62 In fact, finely tuning these competing forces together with control over NP assembly kinetics often allow for the organization of NP isotropic components into self-limited ordered SPs with low polydispersity and a controlled composition.55,63 Although still emergent, the possible formation of ordered lattices via NP assembly has also been observed.64 Indeed, when orientational, rotational, and translational orders are achieved in the SP system, through a directional self-assembly of the NP surface ligands, SPs can show very high hierarchy and atomic accuracies, at the interparticle and intraparticle levels, resulting in single crystals.65,66
NP self-assembly can also be mediated by a wide variety of templates, including proteins, peptides, DNAs, surfactants, and many more. In this case, the interaction between NPs and NP precursors represents one of the main factors determining the NP spatial distribution and possible formation of inner pockets in the final SP structure.67–69 Overall, the formation of SPs is obtained starting from simple components (NPs, biomolecules, surfactants, etc.) that spontaneously self-aggregate in a solvent creating an “extensive” confined space through a high yield and low-cost process. In the next paragraphs the main applicative fields for these confined compartments are described.
3. Carriers and biomedical systems
In many cases, NP self-assembly is driven by solvophobic interactions, as explained above, creating a confined environment within the SP with opposite physical–chemical properties to those of the dispersing medium. In other words, SPs stabilized in an aqueous environment can be considered as possible carriers of poorly soluble molecules for drug delivery and nanomedical applications.70–73In particular, the formation of SPs offers higher surface areas with respect to analogue ligand-stabilized NPs promoting improved drug encapsulation efficiencies. In some cases, NP assembly also provides an enhanced porosity of the SPs with a possible uptake of molecules from the bulk dispersing medium. Wang et al. showed, for instance, that 200 nm SPs, composed of an array of 4 nm ligand-capped gold NPs, were characterized by an excellent ability not only to encapsulate poorly water-soluble molecules, but also to adsorb them from the dispersing solvent. Their SPs showed an uptake of hydrophobic molecules comparable to that of zeolites/metal–organic frameworks (MOFs) and five orders of magnitude higher than that of individual supramolecular cages/containers (Fig. 4a–d). This enhanced host ability was related to the formation of a closed-packed phase of NPs, in which about 26% of the space within the SP was occupied by voids, for which volume calculations were compatible with the huge amounts of encapsulated drug. In particular, the diffusion of molecular guests from the bulk solution to the inner cavities in the SP was explained through the formation of a percolated network of hydrophobic cavities formed by the interstitial spaces among the NPs. These interstitial spaces consisted of octahedral (Oh) and tetrahedral (Td) voids linked by triangular pores, big enough to guarantee the diffusion of the molecular guests (Fig. 4e–h).74
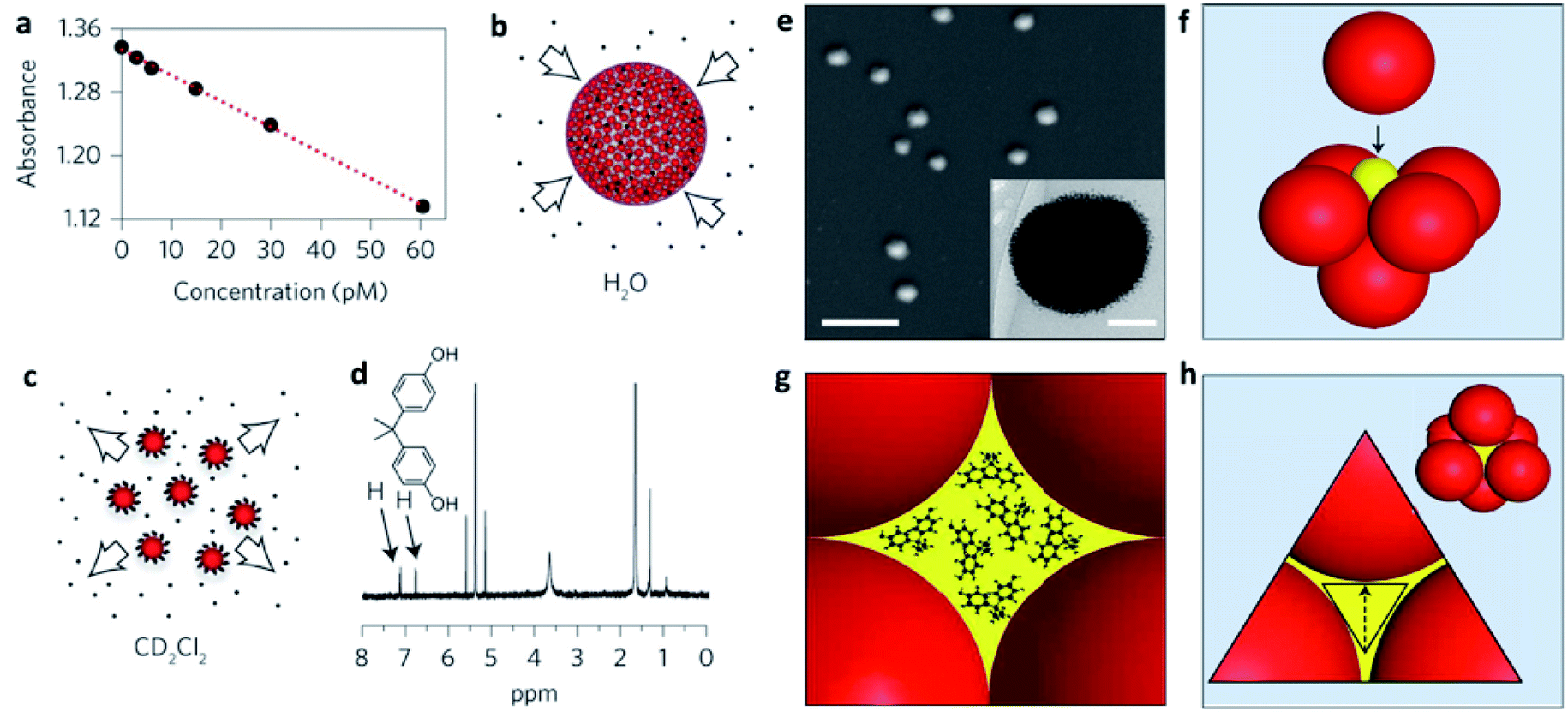 | ||
| Fig. 4 (a) Decrease in the absorbance of the guest bisphenol A (BPA) in water after uptake by increasingly larger concentrations of mercapto-polyethylene glycol (PEG-SH) capped supraspheres; (b) schematic representation of BPA uptake; (c) release of BPA guests upon the dissolution of the supraspheres in CD2Cl2; (d) the BPA guests released into CD2Cl2 were quantified by 1H NMR spectroscopy; (e) SEM image of dried 200 nm diameter PEG-S-capped supraspheres (scale bar, 1 μm). Inset: cryogenic transmission electron microscopy (cryo-TEM) image of a PEG-S-capped suprasphere (scale bar, 50 nm); (f) formation of an octahedral (Oh) hole by closest-packed spheres. For uniform hexanethiolate-capped AuNPs with diameters of 6 nm, 2.5 nm diameter spheres (yellow ball in the figure) fit precisely inside the Oh holes; (g) illustration of an Oh-symmetry hole hosting 1 nm BPA guests; (h) illustration showing a pore (in yellow) and a three-AuNP face of an Oh hole. Reproduced with permission from ref. 74. Copyright 2016 Springer Nature. | ||
Additionally, the versatility in the chemical nature of the surface ligands of the NP components can endow the obtained SPs with an intrinsic chemo-selectivity towards specific guest molecules. An example is shown by our recent work on the development of water-dispersible fluorous SPs, in which the functionalization of the surface of the composing AuNPs with perfluoroalkyl chains allowed the creation of fluorophilic cavities in the SP. These fluorous gold SPs (F-GSPs) were found to more efficiently encapsulate fluorinated drugs (about 30% higher drug loading) with respect to analogue SPs formed by AuNPs capped with fully hydrogenated chains (Fig. 5), affording a valuable carrier for fluoros guest molecules.75,76 In this case, the overall stabilization of the F-GSPs was guaranteed by a protein coating, which hampered a direct contact of the NP interface with the bulk solution as well as the adsorption of guest molecules from the dispersing solvent. In this regard, chemo-selectivity can instead be specifically delivered to the external pores of the SPs, if the NP components are in direct contact with the solvent, through a controlled and selective ligand exchange process at the interface, allowing access of similarly sized akin guests to the internal confined space.77 Although, the presence of a stabilizing biocoating can sometimes be preferred, not only for obtaining a higher colloidal stability, but also because a custom design of the stabilizer can provide SPs with additional features. Engineered recombinant surfactant proteins with exact terminal peptide motifs were, for example, used for obtaining target SPs, while the entrapment of primary Si-SPs in degradable alginate matrices resulted in the formation of long and sustained drug release containers, which can be desired in some applications.78
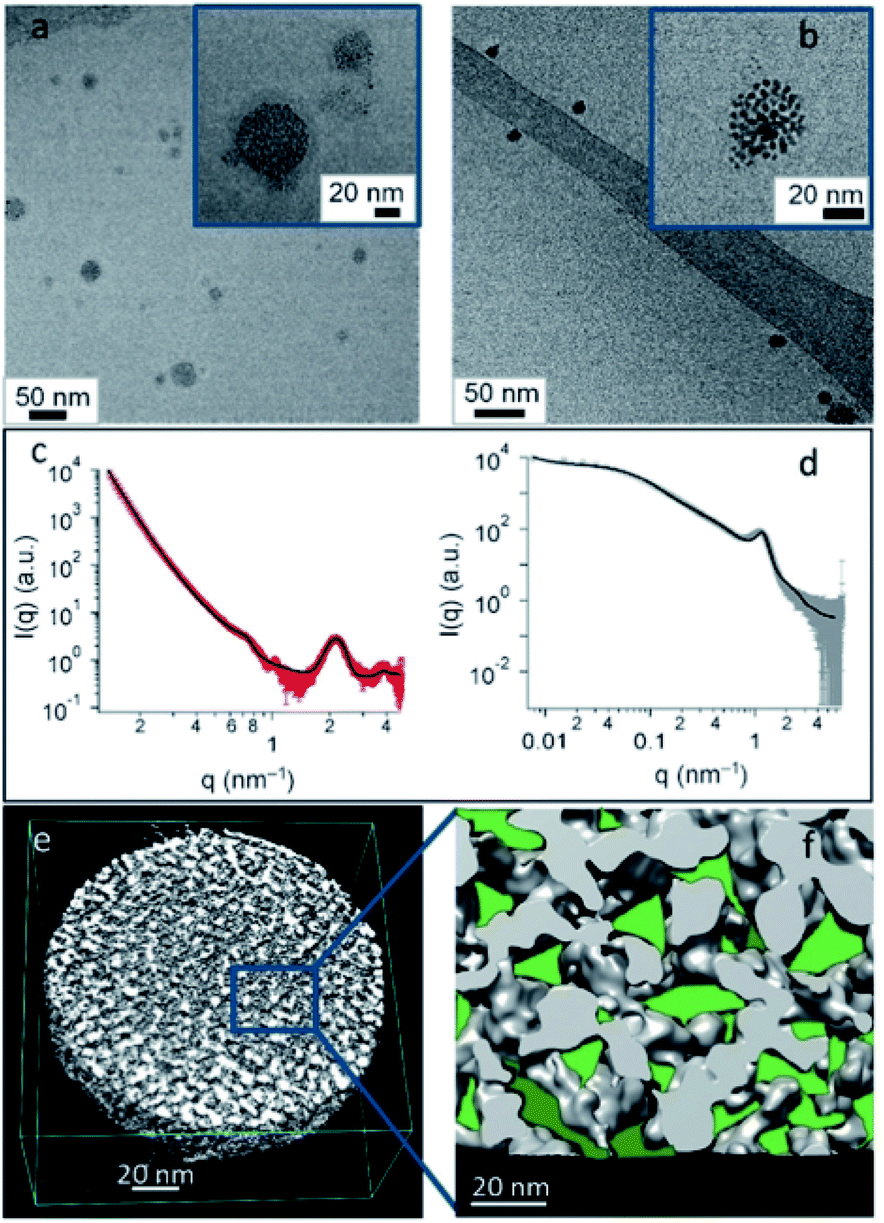 | ||
| Fig. 5 (a and b) Cryo-TEM images of F-GSPs obtained from NCs (a) and NPs (b). (c and d) SAXS pattern and corresponding fit of F-GSPs obtained from NC (c) and NP (d) self-assembly. (e) ET3D reconstruction of an F-GSP obtained from NPs, and (f) an ET 3D enlargement with confined spaces forming fluorinated pockets marked in green. Reproduced with permission from ref. 75. Copyright 2017 Wiley and Sons. | ||
Another interesting system is represented by D-chiral SPs developed by Jaklenec, Langer and coworkers, showing how chirality can be a new tool to tune the interaction of nanosystems with the cellular machinery. In fact, these novel D-chiral SPs, composed of D-cystein capped cobalt-oxide NPs, demonstrated higher resistance to enzymatic digestion and a more enhanced tumour killing efficiency with respect to L-chiral and racemic analogous SPs.79
It is also clear that NP self-assembly, beyond providing a huge space to host molecular guests and enhanced porosity, produces complex SPs with efficient multifunctionality and often responsivity to exogenous (e.g., light, magnetic field, ultrasound, etc.) or endogenous (e.g., pH, enzyme, ionic strength, etc.) stimuli. Typically, the assembly of metal NPs results in collective spectroscopic properties such as superparamagnetism,80 Surface-Enhanced Raman Scattering (SERS),81 NIR absorption, etc.59,82 Hence, it is not surprising that NP assembly has been lately used to obtain effective multiresponsive drug delivery systems (DDSs). Much attention has been given to endow these DDS with a remotely controlled multistage activation, which allows achieving spatial and temporal control upon the trigger of the response.83 Hybrid magneto-plasmonic assemblies were, for instance, obtained mixing magnetic and gold NPs with a free amphiphilic copolymer, resulting in SPs able to host huge drug payloads, which also exhibited strong NIR absorption and an enhanced T2 (transverse relaxation) contrast effect.84 Overall, the co-assembly of multiple types of NPs allowed the formation of stimuli-responsive DDSs, also trackable in vivo by photoacoustic and magnetic resonance imaging, in which drug release was activated by NIR irradiation and release rates were modulated by using a magnetic field. In this case, dual responsivity was entirely exogenous, but there is an increasing trend to build-up multiresponsive DDSs to synergic endogenous/exogenous stimuli.84 For example, photoresponsive gold nanocage assemblies showed both acidic-responsive internalization in cancer cells and light-triggered enhanced tumour accumulation. Additionally, laser-irradiation at the tumour site increased the porosity of the assemblies facilitating drug intracellular liberation providing a mechanism for on-demand drug release.
4. Reaction vessels
The compartmentalization approach to perform a single type of reaction within an enclosed environment, so-called “cavity-directed synthesis”, relies on the use of tailored cavity sizes and geometries to properly pre-organize the starting materials and stabilize the final products. The increase in the local concentration of reactant molecules due to nanoconfinement, together with their orientation in close proximity, may induce specific interactions and associations of substrates, increase the probability of events leading to the reaction, and open up possible new avenues towards unprecedented chemical reactivities and events.85 Indeed, nanoconfinement can effectively change the mechanism of a reaction and generally facilitates the formation of charged or charge-separated species, due to the enhanced proton acceptor or donor abilities of encapsulated solvent molecules. Supramolecular hosts are also known to stabilize reactive species, including high-energy conformations structurally similar to transition states. This usually lowers the activation energy and accelerates the reaction rate, as detailed in the following section. For instance, computational studies on both inter- and intramolecular addition reactions proved that energy barriers are considerably lowered in nanoconfined water.86In some cases, confinement creates an isolated and inert environment around a reactive species, preventing its reaction with external molecules or other stimuli. Ideally, confined spaces could act as nanosized protecting groups that are able to shield isolated regions or functional moieties on the encapsulated molecule from reagents in the bulk.83 Such a protection would be particularly useful when the selective reaction of identical functional groups is needed. The inclusion of linear diterpenoids in a U-shaped conformation within the cavity of a coordination cage formed by six cis end-capped palladium ions and four triazine-cored tridentate pyridine ligands, for example, enabled the site-selective functionalization of the terminal prenyl moiety through conformational fixing and noncovalent protection of internal C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds.87
C bonds.87
Since the initial efforts to build covalent nanoreactors required complex multistep synthesis, several research groups focused on the self-assembly of small molecular components into discrete capsule-like architectures through reversible, non-covalent interactions. Despite looking extremely promising in this sense, supramolecular chemistry has probably not reached its full synthetic potential, yet,88 and only a limited set among the several self-assembled capsules and cages with nanometer-sized cavities reported in the literature was shown to promote chemical reactions. Most of them were based on coordination cages, hydrogen-bonded capsules, protein cages, porous crystals (e.g., zeolites), covalent organic frameworks (COFs), and MOFs.25
An interesting further option might be offered by the aggregation of stimuli-responsive NPs into SPs, whose empty spaces can be used as ‘nanoflasks’.44 SPs obtained from the self-assembly of inorganic NPs should, in principle, exhibit greater chemical stability, mechanical robustness, and membrane-like functionality, compared to other self-assembled compartmentalized systems. On the other hand, a major drawback of these SPs is the difficulty of tailoring the interstices between NPs to control their permeability in terms of selective diffusion and transport of molecules through them.89
To date, the most extensive study in this field is the one performed by Klajn and coworkers.90 First, they focused on surface-confined species and demonstrated that UV irradiation (λ ∼ 365 nm) of ethynylanthracenyl-thiol immobilized on spherical Au NPs gave [4 + 4] dimerization, instead of the well-known selective [4 + 2] Diels–Alder reaction. The [4 + 2] vs. [4 + 4] selectivity could be tuned tailoring the curvature of the underlying NP surface, as well as the flexibility of the linker chains between gold and ethynylanthracene moieties. The large curvature of 2.5 nm Au NPs, for example, prevented the proper orientation for [4 + 4] dimerization, whereas immobilization onto bigger NPs also afforded low yields of the [4 + 4] product.91 In a more recent study, the same group developed gold NPs decorated with mixed monolayers of oligo(ethylene glycol) background ligands and ligands terminating with an azobenzene moiety. The close proximity between the terminal –OH groups of the background ligands and azobenzene's NN bonds promoted the formation of hydrogen-bonded SPs and increased the rate of the back cis–trans isomerization of azobenzenes by about 6000-fold. An additional advantage of this approach was that these NPs became water-soluble by simply co-adsorbing hydrophobic azobenzenes with water-solubilizing ligands.57
Ultraviolet (UV) and visible (vis) light irradiation of NPs functionalized with light-responsive ligands proved to be a functional strategy to reversibly create and destroy confined environments, where trapped molecules can undergo chemical reactions with increased rates and stereoselectivities significantly different from bulk solutions. Azobenzene-functionalized inorganic NPs of different sizes and compositions – including 6 nm gold, 11 nm magnetite (Fe3O4) and 17 nm silica particles – were used as precursors of such dynamic nanoflasks. In contrast to what is usually observed in non-deoxygenated solutions, such SPs were able to exclude oxygen from the confined empty spaces and protect anthracene derivatives against photo-oxidation to anthraquinones, selectively leading to their dimerization (Fig. 6). The stereoselectivity of the confined photo-dimerization was also completely different compared to NP-free solutions. Instead of affording only the thermodynamic anti product, the preorganization of anthracene molecules inside the nanoflasks produced more than 80% of the kinetically favoured syn isomer.92
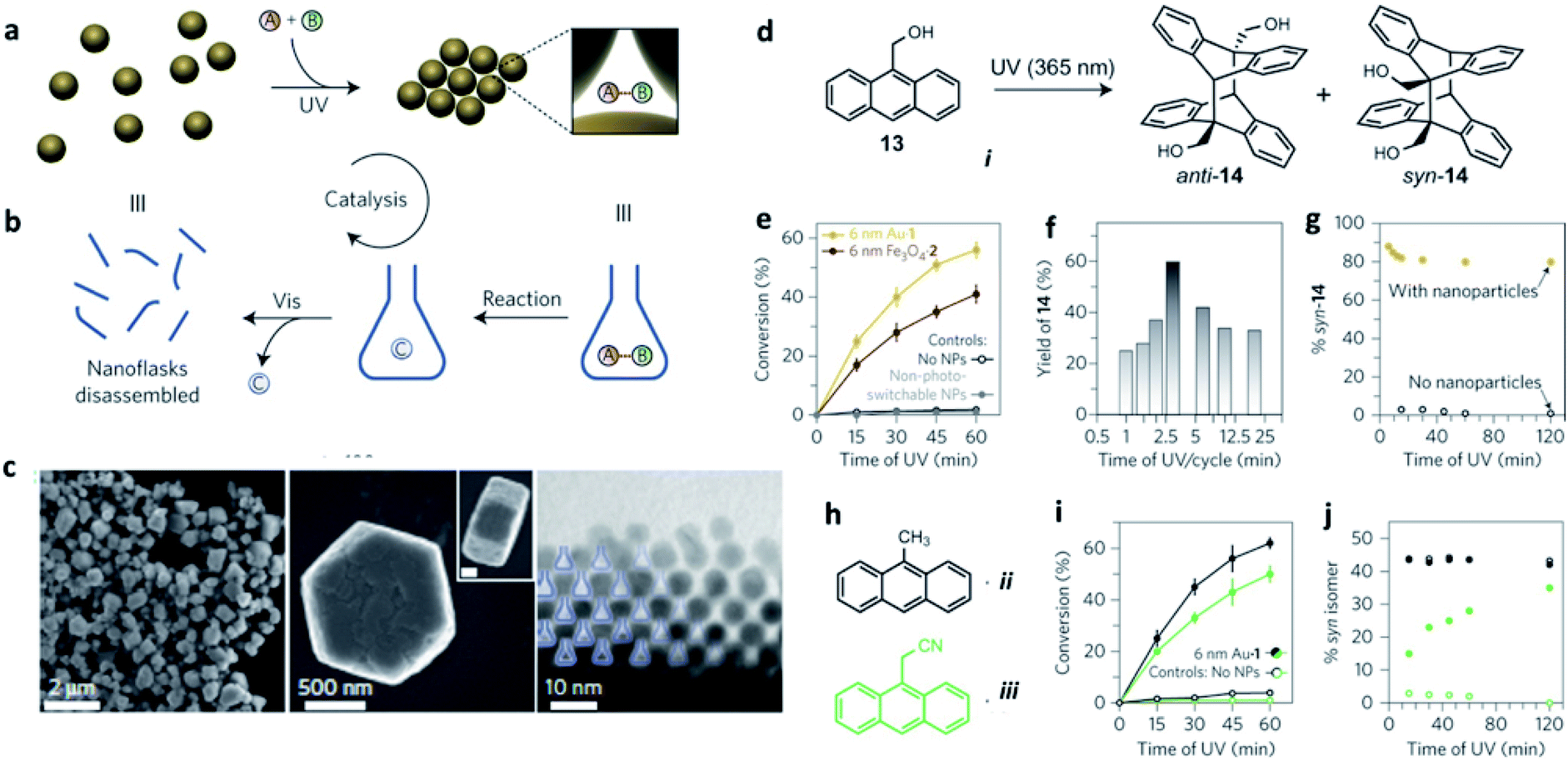 | ||
| Fig. 6 (a and b) Schematic representation of how the reversible formation of confined spaces can accelerate a chemical reaction; (c) electron micrographs (at different magnifications) of colloidal crystals prepared by exposing 6 nm AuNPs to ultraviolet light (scale bar in the inset, 200 nm); (d) schematic representation of the reversible trapping of polar molecules during light-induced self-assembly of photoresponsive NPs. Reaction promotion, example: ultraviolet-induced dimerization of anthracene i; (e) light-accelerated dimerization of anthracene i. Each cycle corresponds to irradiation for 3 min with ultraviolet, followed by 1 min with visible light; (f) effect of the length of ultraviolet exposure per cycle on the conversion of anthracene i to its dimers; (g) stereoselectivity in the dimerization of anthracene i with and without photoswitchable NPs. The composition of isomers was determined from NMR data; (h) Structural formulae of additional anthracenes, ii and iii, the reactivity of which was studied; (i) light-accelerated dimerization of anthracenes ii and iii; (j) stereoselectivity in the dimerization of anthracenes ii and iii with and without photoswitchable nanoparticles. Reproduced with permission from ref. 92. Copyright 2016 Springer Nature. | ||
5. Catalytic systems
Combining the ability of SPs to host reactions in their confined pockets with the catalytic activity of specific NP components, it is possible to engineer efficient catalysts employable in a wide variety of chemical processes. Specifically, catalytic SPs can be obtained from metal, ceramic, and semiconductor NPs, even integrating different constituents into one self-assembled nanostructure, properly designed and optimized for achieving the desired catalytic function and hosting the reaction in their nanocavities.93 Moreover, SPs usually offer high contact areas for interaction with the selected substrates as well as collective excitation and extensive multimodal plasmonic coupling, effects that are further translated into strongly enhanced catalytic activity with respect to single NPs.40,94,95A pioneer example is offered by the work of Li et al. who developed single and multi-component chiral SPs showing enantioselective photocatalysis of tyrosine (Tyr; Fig. 7).42 SPs were formed from the assembly of penicillin (Pen) stabilized ZnNPs, with and without the addition of AuNPs. The obtained SPs showed a diameter of 70–100 nm and were characterized by inner interparticle pores smaller than 2 nm, size that is consistent with the dimensions of reactive centres of many enzymes. Owing to their features, such SPs could efficiently drive the photooxidation of Tyr to its corresponding dimer, diTyr, initiated by illumination at pH 8.0 with UV light at 315–400 nm, which was efficiently absorbed by ZnS and ZnS–Au SPs. Notably, the ZnS SPs carrying L-Pen preferentially catalysed L-Tyr, while SPs made from D-Pen work as catalysts for D-Tyr. Such enantioselectivity was associated with the preferential partitioning of Tyr enantiomers into the SPs.
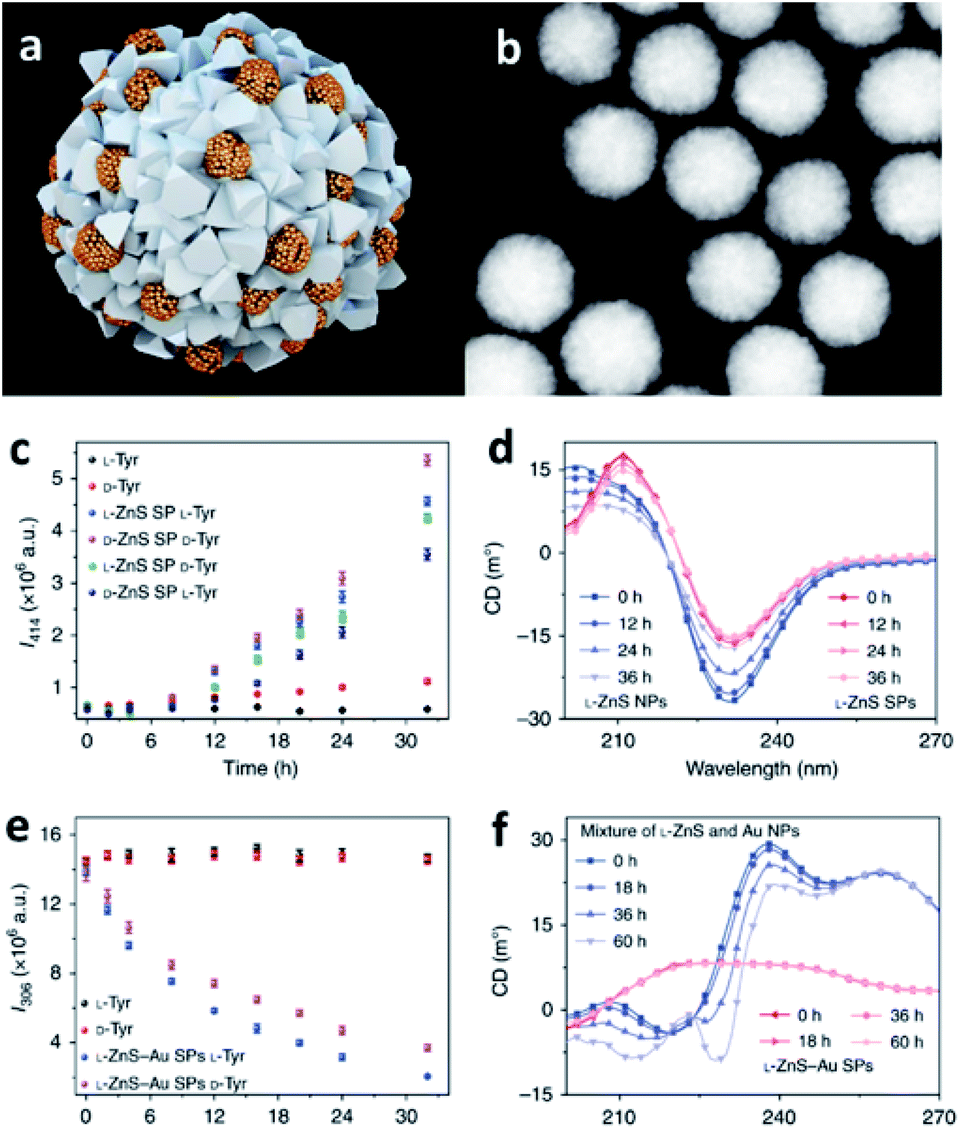 | ||
| Fig. 7 (a) Illustration of multi-component L-ZnS–Au SPs; (b) HAADF-STEM images of multi-component L-ZnS–Au SPs; (c) the dependence of photoluminescence intensity at 414 nm on the time of the photocatalytic reaction with ZnS SPs of different handedness of the catalyst and the substrate; (d) Circular Dichroism (CD) spectra of the mixtures of ZnS NPs and ZnS SPs after UV illumination (115 V, 50/60 Hz, and 0.16 A) for different times; (e) the dependence of photoluminescence intensity at 306 nm on the photocatalytic reaction time with L-ZnS–Au SPs for different Tyr enantiomers; (f) CD spectra of the mixtures of ZnS and Au NPs, ZnS–Au SPs, after UV illumination (115 V, 50/60 Hz, and 0.16 A) for different times. Reproduced with permission from ref. 42. Copyright 2019 Springer Nature. | ||
Another multi-component SP system was devised by Wang et al., who synthesized, via a bottom-up approach, uniform pomegranate-like Pt@CeO2 multicore@shell nanospheres.96 First, ultrasmall Pt and CeO2 NPs were obtained by a redox reaction taking place between Ce(OH)3 and K2PtCl4. Every Pt particle was firmly enclosed by several CeO2 nanoparticles, yielding Pt–CeO2 hybrid structures that spontaneously self-assembled into larger spherical pomegranate-like multicore@shell SPs with a size of about 85 nm. The obtained multicore@shell SPs showed excellent activity and thermal stability when employed as catalysts for CO oxidation. Even after calcination at 600 °C for 5 h in air, they efficiently catalysed 100% conversion of CO into CO2 at a relatively low temperature (145 °C). Pt@CeO2 multicore@shell nanospheres were further supported on reduced graphene oxide (RGO) nanosheets to form Pt@CeO2/RGO nanocomposites that could be employed in the chemical reduction reaction of nitrophenol (NP) by ammonia borane, instead of hazardous H2 or NaBH4. Finally, the Pt@CeO2/RGO system showed a desirable degree of recyclability, further proving the versatility of the developed catalyst.
SPs have also been reported as efficient photocatalysts for the decomposition of organic pollutants in wastewater purification. NPs made of semiconductor materials could hold promise in this field, but their actual usage has been limited by their small size that makes their separation from the products highly challenging. NP assembly in larger SPs can address this challenge while enhancing the photocatalytic activity with respect to single NPs, owing to the generation of a collective response. An excellent example of SPs as photocatalysts for water purification was developed by Liu et al., who designed porous nanopyramids obtained from the oriented self-assembly of 4.7 nm ZnO NPs (Fig. 8).97 The final pyramidal assemblies were characterized by a size of 67 nm and a high surface area (127.7 m2) that, together with uniform nanopores (9.7 nm) and large pore volume fraction (63 v/v%), endowed the system with high absorption capacity for acidic organic molecules. Such a feature was combined with the high photocatalytic efficiency of ZnO SP building blocks, yielding an excellent platform to drive the photodegradation of the organic species absorbed onto the nanopyramid. The developed system provides a versatile tool with high promise in the fields of high-purity separation and purification.
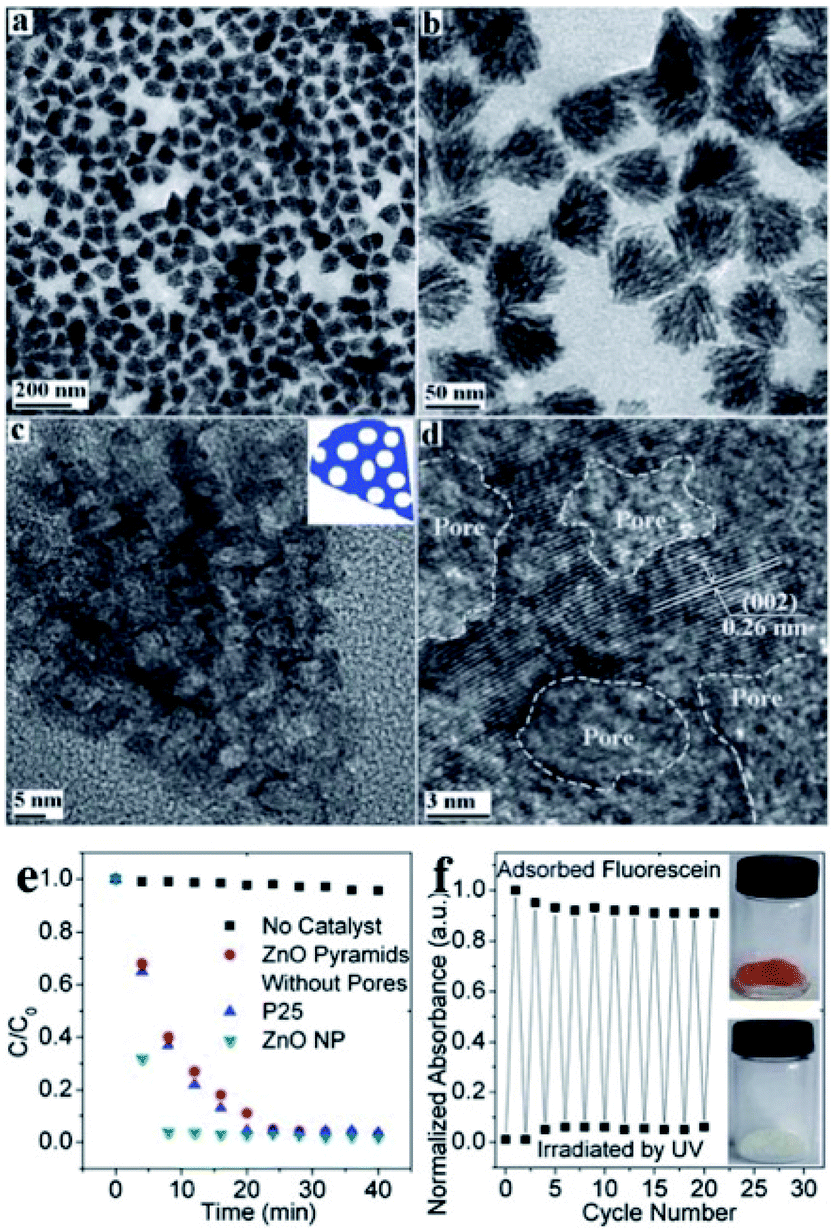 | ||
| Fig. 8 (a–d) TEM images of ZnO nanoporous pyramids with (a) low and (b) high magnification; (c) single ZnO nanoporous pyramid particle, and (d) high resolution TEM image of a ZnO NP; (e) photodegradation of fluorescein by ZnO pyramids without pores and ZnO nanoporous pyramids; (f) adsorption and photodegradation of fluorescein by ZnO nanoporous pyramids repeated more than 20 times, corresponding to the coloration and the decoloration of ZnO nanoporous pyramids, respectively. Reproduced with permission from ref. 97. Copyright 2012 Royal Society of Chemistry. | ||
Another type of SP that has been gaining increasing attention in catalysis is represented by metal NP-based hollow structures. Kumar et al. have recently reported plasmonic-catalytic nanoreactors named “nanocatalosomes (NCat)”, characterized by a metal-bilayer hollow shell-in-shell structure composed of arrays of closely spaced NPs (Fig. 9).98 The interlayer space of NCat presents a large number of plasmonic nanocavities intimately interfaced with the chosen catalytic metal (Au/Pt/Pd). NCat were obtained starting from hollow porous silica nanoshells modified with Au seeds and coated with a 3 nm film of tannic (TA) and iron coordination polymer (TA–Fe). Upon addition of HAuCl4 and hydroquinone, Au seeds grew into larger and closely spaced AuNP units on the outer and inner surfaces of the AuNC@h-SiO2 template, resulting in NCat Au-bilayer structures. Notably, the designed synthetic strategy was also successful with other noble metals, leading to the formation of bilayer structures of Pt and Pd (named Pt-NCat and Pd-NCat). Hence, the synthetic path designed by Kumar et al. offers a valuable tool to prepare metal bilayers endowed with a large number of 3D nanocavities. The sizes and spatial configurations of these nanocavities can be finely tuned adjusting the amounts of metal precursors used in the synthesis, with smaller NPs and larger interparticle pockets obtained when using lower amounts of precursors. The catalytic activity of Pt/Au-NCat was tested in dehydrogenation reactions of N-heterocycles, with no addition of chemicals, using only the solar light as the energy source. 1,2,3,4-Tetrahydroquinoline (THQ) was used as a model substrate in the presence of Pt/Au-NCat and solar light, and the reaction was monitored measuring the formation of the quinoline product by 1H NMR spectroscopy. When Pt/Au-NCat having the highest number of the smallest nanocavities was employed, quinoline started forming within 5 minutes reaching a yield > 99% by 30 min, whereas NCat without Pt and/or with fewer and wider interlayer cavities and plasmonic gaps afforded much lower yields, indicating the important effect of the nanocavities and of the metal components on the catalytic performance.
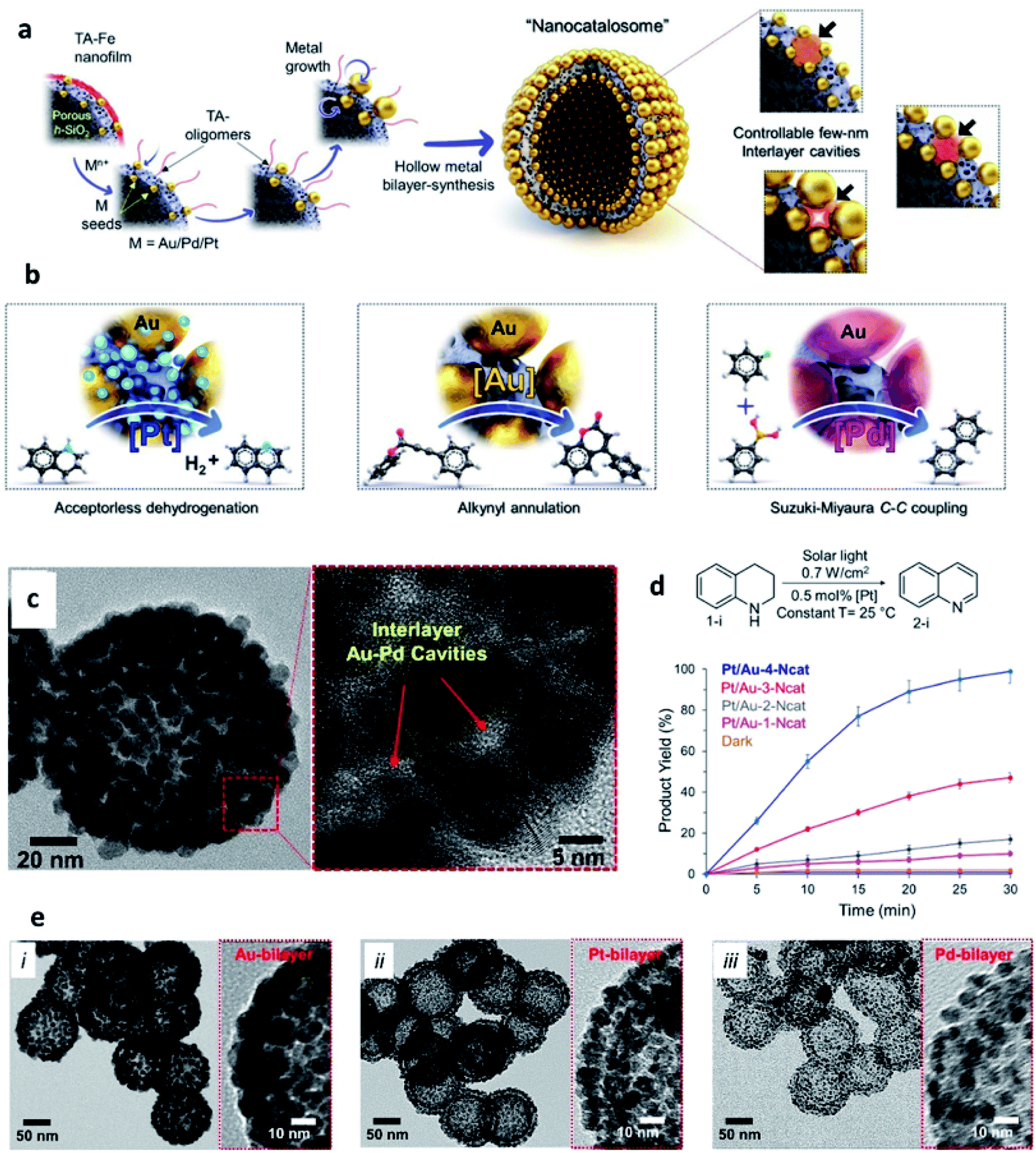 | ||
| Fig. 9 (a) Schematic representation of the synthesis of metallic-bilayer NCat with controllable interlayer cavities of a few nm (right-side), with red shadows depicting change in interlayer cavity-structure; (b) catalytic applications of different NCat for different solar light induced reactions; (c) STEM images of the Pt/Au-1–4-Ncat system; (d) [Pt]-catalysed dehydrogenation of THQ and conversion yields at different reaction times using Pt/Au-1–4-NCat; (e) TEM images of different metal bilayer nanocatalosomes [Au–4-NCat (i), Pt–NCat (ii), and Pd–NCat (iii)]. Reproduced with permission from ref. 98. Copyright 2020 Wiley and Sons. | ||
6. Opportunities and challenges of confined space by nanoparticle self-assembly
The emergence of SP systems endowed with inner cavities inevitably raises the question about possible advances brought about by these recent nano-objects in diverse fields of chemistry and materials science, when compared to the large variety of confined systems, both molecular and self-assembled, previously studied and reported. NPs are unique building blocks for the design of self-organized structures with inner nanocompartments, as the choice of the constituent material, the possibility of tuning the NP size and shape, and functionalizing their surface with specific ligands to ultimately control their assembly offer a plethora of opportunities for the development of SPs bearing custom nanocavities. Such a design strategy, although requiring multiple steps, is highly versatile and NP synthesis mostly involves well-established and quite straightforward procedures.One of the advantages offered by NPs, when compared to molecular building blocks, is their functional nature, as, depending on their constituent material and size, they exhibit exceptional optical and magnetic properties, which are transferred to the obtained SPs and to their inner nanocompartments. For instance, the use of AuNPs as building blocks allows the development of plasmonic SPs with collective and/or innovative properties. Notably, in plasmonic SPs, the optical properties of the system strongly depend on the spatial distribution of the NPs within the self-assembled structure. When the interparticle distance in the SP reaches the nanometer or subnanometer scales, the plasmonic response can be affected by the quantum nature of the electrons, their non-local screening and possibly by electron tunnelling across NP gaps.99 These effects can potentially lead to the formation of diverse plasmonic modes allowed in one single SP, given the different interparticle distance obtained inside and outside SP inner nanocompartments, yielding promising systems for light–matter interactions, plasmonics, resonators and optoelectronic fields.
When compared to individual molecular cages or other hosting systems, confined SPs appear as multi-container assemblies. Indeed, bearing a large number of nanocavities, their interior architecture allows the separation and encapsulation of specific guest molecules, with often higher payloads with respect to molecular hosts. Moreover, host–guest ability is frequently combined with a selective “guest release” mechanism, which can be tuned with chemical or physical stimuli thanks to the specific features of the used NPs as building units of the SPs. Examples previously discussed in this perspective perfectly show the possible exploitation of host–guest chemistry in SPs, using hydrophobic and fluorophobic effects for the encapsulation of high payloads of hydrophobic and fluorinated guests, respectively.74,75 Notably, as already reported for molecular cages, guest uptake and release can also be regulated using post-assembly modifications.100 For instance, it is possible to encapsulate, during the assembly process, molecules able to react with the inner cavity functional groups and trigger the aimed modification to tailor the cavity features and binding properties in situ. Moreover, as shown by Harraq et al., SP post-assembly modification via external stimuli can lead to SP reconfiguration, modulating guest's uptake and SP behaviour, thus allowing a single architecture to perform a variety of functions.101 Finally, SP external surface modification can be employed to tune SP porosity and recognition properties, offering a further possibility to modulate the SP guest ability.101
One of the unmet challenges in SP design is represented by the use of anisotropic NPs as building blocks. Indeed, although several research groups have investigated the self-organization of anisotropic NPs,102 the development of 3D SPs bearing confined nanocavities is still limited and isotropic NPs remain the preferred building blocks for confined SP synthesis. In principle, anisotropy could favor different self-organization modes, with anisotropic NP interaction mimicking more closely directional bonds in molecules, allowing different NP orientations in the assembly pattern. A pioneer example of the 3D assembly of anisotropic NPs and their possible evolution in different architectures was reported by Fava et al., who developed chain-like and 3D spherical assemblies of polystyrene terminated gold nanorods (AuNRs). In detail, by changing the organic/water solvent composition, two competing assembly modes, via end-to-end and side-by-side NR orientation, could be achieved. End-to-end assembly led to the formation of chains in which individual NRs were aligned parallel to the long axis of the chain, while side-by-side assembly resulted in the formation of spheres in which the NRs were aligned parallel to the interface.103 More recently, Nai et al. obtained open-frame cage-like structures from the self-assembly of nanocubes.104 With their asymmetric shapes and consequent inequivalent particle orientations, anisotropic NP-based architectures present both translational and orientational order, posing additional challenges in the development of confined nanocompartments, when compared to their isotropic counterparts. In particular, while isotropic NPs typically self-assemble and crystallize into face-centered cubic or hexagonal close-packed superlattices, anisotropic NPs are capable of affording a wider range of packing symmetries, requiring advanced NP surface chemistry to be tuned and controlled.76 Although still not achieved, it can be envisioned that SPs based on anisotropic NPs could open new opportunities for the development of innovative functional confined compartments and in the further emergence of NP based confined SPs.
7. Future outlook and conclusions
SPs have undoubtedly turned out as key players in nanotechnology progress and, owing to their unique properties, their role in many innovative applications has been steadily consolidating.105 In this perspective, we have been discussing SP design and applications mainly focusing on systems endowed with custom nanocavities, able to enhance SP intrinsic properties or generate new ones. Although several contributions have highlighted, to date, the valuable functionalities of SPs bearing confined nanocavities, these objects are still in their infancy and their potential in practical applications has not fully been explored, yet.Taking increasing inspiration from living species, supramolecular chemistry has lately been focusing on the development of dissipative systems, in which self-organization is governed by kinetic parameters rather than thermodynamic ones, and occurs under out of equilibrium conditions, continuously consuming energy.106 Despite huge research efforts done in this topic, the practical relevance of dissipative systems to date is still quite limited.107 In this context, NPs represent excellent building units for designing dissipative self-assembled systems. In fact, properly functionalizing the NP surface with chemical switches and/or exploiting NP component responsiveness to a certain stimulus, it is possible to control the self-organization process through chemical or physical fuels.108,109 An example of formation of dissipative SPs was recently reported by Grötsch et al., who devised carboxylate-terminated Au and Fe3O4 NPs, whose assembly was regulated by the transient conversion of carboxylate groups to their corresponding N-hydroxysuccinimide (NHS) esters, driven by the hydrolysis of 1-ethyl-3-(3-(dimethylamino)propyl)-carbodiimide (EDC).108 Light has also shown to be a neat fuel to drive dynamic NP self-assembly, as reported by the group of Grzybowski that used photoisomerization of dithiol molecules bound onto NP surfaces to mediate light-induced assembly into ordered, three-dimensional SPs (Fig. 10).110
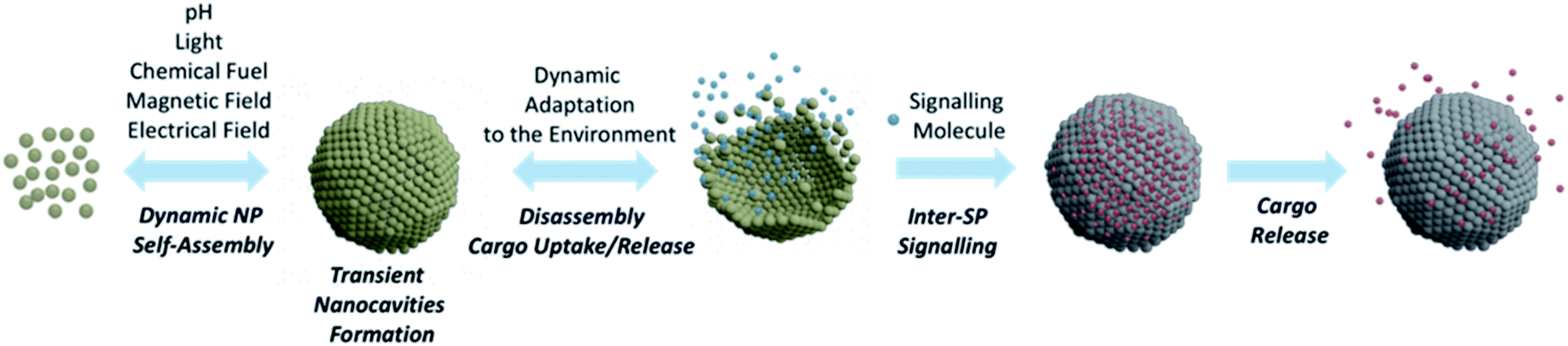 | ||
| Fig. 10 Schematic representation of NP dynamic self-assembly leading to the formation of SP endowed with inner nanocavities. The obtained SP can further adapt dynamically to the surrounding environment affording a conformational rearrangement with possible cargo release/uptake. Furthermore, released cargos can potentially act as signalling molecules for another SP, triggering the release of a second cargo. | ||
The use of chemical and/or physical fuels to manipulate NP assembly may open important avenues towards the fabrication of biologically relevant SPs able to form transient nanocavities within their structure. Indeed, SPs able to actively adapt to the environment by changing their organization, while fuelled by an appropriate energy source, would represent unprecedent progress in nanoscience. Ideally, an active response of SPs to environmental conditions would allow the rearrangement of their conformation to mediate their function, with continuous adjustments to the changes in the surroundings (Fig. 10). Huang et al. have recently highlighted the active responses of SPs as enablers of possible great progress in various applications, such as catalysis, imaging, nanomedicine, and actuators.111 Ohta et al. developed shape-changing SPs by functionalizing differently sized AuNPs with two DNA sequences, one for triggering NP assembly and the other for modulating the SP shape.112 Through a strand displacement mechanism,113 the obtained system could undergo a dynamic conformational change that impacted its optical properties and, importantly, its interaction with cells. Indeed, nanoassemblies' cellular uptake was tuned by the DNA-mediated shape change, which modulated the arrangement of the surface targeting ligands and, thus, the resulting cellular uptake. This system represents a pioneering example of a synthetic nanostructure able to alter its optical and cellular uptake properties by changing its arrangement in response to specific biomolecules, in this case DNA sequences.
We envision that SPs able to produce dynamic responses and work in out of equilibrium conditions might mimic more closely cell organelles, creating specific transient nanocavities for hosting guests and processes, in response to the environment and/or to the applied stimulus/fuel. Such a conformational SP rearrangement would also generate a functional response that would lead to a change in the NP collective properties, a feature that would certainly provide researchers from different fields with unprecedent tools able to fulfil unmet needs. For instance, as shown for micelle-based systems,114 dissipative SP platforms could help overcome the limitations of static DDS, paving the way for more controlled and sustained drug delivery profiles. Dynamic SPs might also enhance the efficacy of self-assembled nanomaterials in the treatment and diagnosis of tumour, actively modulating their shape, size and conformation, in response to the tumour microenvironment, potentially leading to enhanced accumulation and retention, tumour penetration, and/or site-specific cargo release.115 Ideally, by proper functionalization of the SP building blocks, it could also be possible to host different guests in separate compartments of one unique SP or of different SPs, and to modulate their release making use of interacting signals (i.e., connected stimuli or fuels). The possibility of signalling among synthetic SPs would, for instance, allow the encapsulation/release of one substrate to affect the binding of another in a diverse compartment of the same SPs or of a different one (Fig. 10).116,117 Such a cooperativity in nanoscale materials would represent an incredible breakthrough for biomedical applications, as it would lead to new modes of host–guest interactions.118
Beyond the potential applications in the biomedical field, SPs also hold promise in other research fields. We have already discussed the enhanced catalytic activity shown by SPs endowed with nanocavities in comparison to NPs, as well as their potential as reaction vessels. It is foreseeable that SPs with nanocavities might also extend their applications to the field of Li-ion batteries. Ng et al. have recently shown that the nanoconfinement of anodes made of metal oxide (MO) NPs, and their consequent isolation from the electrolyte, could eliminate the primary mechanisms of degradation and coulombic efficiency loss in MO anodes.119 Therefore, NP assembly into SPs might yield self-assembled anodes with improved cycling stability, derived from their 3D nanostructure. For instance, by controlling the geometric configuration of mesoporous iron oxide SPs, Lee et al. could achieve higher cycling stability than random aggregates of iron oxide NPs, thanks to the confinement of the stable solid–electrolyte interphase layer on the outer surface of the SP.120
In summary, with this perspective, we have provided an overview of NP-based supramolecular systems endowed with confined nanocavities and their possible use in several innovative fields. Bearing in mind the multiple opportunities offered by nanoconfinement, in terms of molecular reactivities and chemical process control, and the continuous progress witnessed in SP development, we expect that the research interest towards the study and development of these systems will progressively grow, gaining increasing importance among the scientists of different fields. We therefore envision that NP-based self-assembled systems bearing confined nanocavities will progressively contribute to advances in an array of practical applications and to fulfil unmet technological needs.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors are thankful to the NEWMED project ID: 1175999 (funded by Regione Lombardia POR FESR 2014 2020) and PRIN2017-NiFTy (2017MYBTXC) for funding.References
- B. Worsdorfer, K. J. Woycechowsky and D. Hilvert, Science, 2011, 331, 589–592 CrossRef.
- C. D. Keating, Nat. Chem., 2013, 5, 449–451 CrossRef CAS.
- H. P. M. De Hoog, M. Nallani and N. Tomczak, Soft Matter, 2012, 8, 4552–4561 RSC.
- A. Belluati, I. Craciun, C. E. Meyer, S. Rigo and C. G. Palivan, Curr. Opin. Biotechnol., 2019, 60, 53–62 CrossRef CAS.
- Y. Tu, F. Peng, A. Adawy, Y. Men, L. K. E. A. Abdelmohsen and D. A. Wilson, Chem. Rev., 2016, 116, 2023–2078 CrossRef CAS.
- J.-S. M. Lee and A. I. Cooper, Chem. Rev., 2020, 120, 2171–2214 CrossRef CAS.
- J. Rebek, Acc. Chem. Res., 2009, 42, 1660–1668 CrossRef CAS.
- J. Kang and J. R. Jr, Nature, 1997, 385, 50–52 CrossRef CAS.
- Z. Dong, Q. Luo and J. Liu, Chem. Soc. Rev., 2012, 41, 7890–7908 RSC.
- D. Ajami and J. Rebek, Acc. Chem. Res., 2013, 46, 990–999 CrossRef CAS.
- A. Brinker, G. Pfeifer, M. J. Kerner, D. J. Naylor, F. U. Hartl and M. Hayer-Hartl, Cell, 2001, 107, 223–233 CrossRef CAS.
- Y. Yu, J.-M. Yang and J. Rebek, Chem, 2020, 6, 1265–1274 CAS.
- M. Yamashina, M. M. Sartin, Y. Sei, M. Akita, S. Takeuchi, T. Tahara and M. Yoshizawa, J. Am. Chem. Soc., 2015, 137, 9266–9269 CrossRef CAS.
- K. Ono, J. K. Klosterman, M. Yoshizawa, K. Sekiguchi, T. Tahara and M. Fujita, J. Am. Chem. Soc., 2009, 131, 12526–12527 CrossRef CAS.
- G. M. Whitesides, J. P. Mathias and C. T. Seto, Science, 1991, 254, 1312–1319 CrossRef CAS.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS.
- S. P. R. Kobaku, C. S. Snyder, R. G. Karunakaran, G. Kwon, P. Wong, A. Tuteja and G. Mehta, ACS Macro Lett., 2019, 8, 1491–1497 CrossRef CAS.
- L. Feng, P. Ren, L. Hao, Q. Dong, J. Li, H. Jian, M. Wang, X. Li, A. Wang and S. Bai, Colloids Surf., A, 2019, 573, 22–29 CrossRef CAS.
- A. J. McConnell, C. J. E. Haynes, A. B. Grommet, C. M. Aitchison, J. Guilleme, S. Mikutis and J. R. Nitschke, J. Am. Chem. Soc., 2018, 140, 16952–16956 CrossRef CAS.
- J. Zhao, Y. M. Zhang, H. L. Sun, X. Y. Chang and Y. Liu, Chem.–Eur. J., 2014, 20, 15108–15115 CrossRef CAS.
- M. Petersen, B. Rasmussen, N. N. Andersen, S. P. A. Sauer, M. B. Nielsen, S. R. Beeren and M. Pittelkow, Chem.–Eur. J., 2017, 23, 17010–17016 CrossRef CAS.
- K. Kobayashi and M. Yamanaka, Chem. Soc. Rev., 2015, 44, 449–466 RSC.
- K. Hermann, Y. Ruan, A. M. Hardin, C. M. Hadad and J. D. Badjić, Chem. Soc. Rev., 2015, 44, 500–514 RSC.
- H. Amouri, C. Desmarets and J. Moussa, Chem. Rev., 2012, 112, 2015–2041 CrossRef CAS.
- M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 1909049 CrossRef.
- J. Wankar, N. G. Kotla, S. Gera, S. Rasala, A. Pandit and Y. A. Rochev, Adv. Funct. Mater., 2020, 1909049, 1909049 CrossRef.
- S. Wintzheimer, T. Granath, M. Oppmann, T. Kister, T. Thai, T. Kraus, N. Vogel and K. Mandel, ACS Nano, 2018, 12, 5093–5120 CrossRef CAS.
- N. A. Kotov, Nat. Nanotechnol., 2016, 11, 1002–1003 CrossRef CAS.
- M. Grzelczak, J. Vermant, E. M. Furst and L. M. Liz-Marzán, ACS Nano, 2010, 4, 3591–3605 CrossRef CAS.
- A. K. Boal, F. Ilhan, J. E. Derouchey, T. Thurn-Albrecht, T. P. Russell and V. M. Rotello, Nature, 2000, 404, 746–748 CrossRef CAS.
- J. Shi, Z. Xiao, N. Kamaly and O. C. Farokhzad, Acc. Chem. Res., 2011, 44, 1123–1134 CrossRef CAS.
- C. Pigliacelli, K. B. Sanjeeva, Nonappa, A. Pizzi, A. Gori, F. B. Bombelli and P. Metrangolo, ACS Nano, 2019, 13, 2158–2166 CAS.
- T. Wang, D. LaMontagne, J. Lynch, J. Zhuang and Y. C. Cao, Chem. Soc. Rev., 2013, 2804–2823 RSC.
- J. Guo, W. Yang and C. Wang, Adv. Mater., 2013, 25, 5196–5214 CrossRef CAS.
- C. Chen, C. Nan, D. Wang, Q. Su, H. Duan, X. Liu, L. Zhang, D. Chu, W. Song, Q. Peng and Y. Li, Angew. Chem., Int. Ed., 2011, 50, 3725–3729 CrossRef CAS.
- Z. Wang, C. Schliehe, K. Bian, D. Dale, W. A. Bassett, T. Hanrath, C. Klinke and H. Weller, Nano Lett., 2013, 13, 1303–1311 CrossRef CAS.
- R. Fang, M. Liu and L. Jiang, Adv. Funct. Mater., 2020, 30, 1903351 CrossRef CAS.
- D. Luo, C. Yan and T. Wang, Small, 2015, 11, 5984–6008 CrossRef CAS.
- A. Rao, S. Roy, M. Unnikrishnan, S. S. Bhosale, G. Devatha and P. P. Pillai, Chem. Mater., 2016, 28, 2348–2355 CrossRef CAS.
- J. Wordsworth, T. M. Benedetti, A. Alinezhad, R. D. Tilley, M. A. Edwards, W. Schuhmann and J. J. Gooding, Chem. Sci., 2020, 11, 1233–1240 RSC.
- J.-H. Lee, K.-J. Chen, S.-H. Noh, M. A. Garcia, H. Wang, W.-Y. Lin, H. Jeong, B. J. Kong, D. B. Stout, J. Cheon and H.-R. Tseng, Angew. Chem., 2013, 125, 4480–4484 CrossRef.
- S. Li, J. Liu, N. S. Ramesar, H. Heinz, L. Xu, C. Xu and N. A. Kotov, Nat. Commun., 2019, 10, 4826 CrossRef.
- I. Sinha and P. S. Mukherjee, Inorg. Chem., 2018, 57, 4205–4221 CrossRef CAS.
- A. B. Grommet, M. Feller and R. Klajn, Nat. Nanotechnol., 2020, 15, 256–271 CrossRef CAS.
- W. T. S. Huck, Chem. Commun., 2005, 4143 RSC.
- C. Pigliacelli, R. Sánchez-Fernández, M. D. García, C. Peinador and E. Pazos, Chem. Commun., 2020, 56, 8000–8014 RSC.
- C. Yi, Y. Yang, B. Liu, J. He and Z. Nie, Chem. Soc. Rev., 2020, 49, 465–508 RSC.
- J. Il Park, T. D. Nguyen, G. de Queirós Silveira, J. H. Bahng, S. Srivastava, G. Zhao, K. Sun, P. Zhang, S. C. Glotzer and N. A. Kotov, Nat. Commun., 2014, 5, 3593–3613 CrossRef.
- H. Zhou, J. P. Kim, J. H. Bahng, N. A. Kotov and J. Lee, Adv. Funct. Mater., 2014, 24, 1439–1448 CrossRef CAS.
- Nonappa, J. S. Haataja, J. V. I. Timonen, S. Malola, P. Engelhardt, N. Houbenov, M. Lahtinen, H. Häkkinen and O. Ikkala, Angewandte Chemie International Edition, 2017, 56(23), 6473–6477 CrossRef CAS.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS.
- A. Pizzi, C. Pigliacelli, G. Bergamaschi, A. Gori and P. Metrangolo, Coord. Chem. Rev., 2020, 411, 213242 CrossRef CAS.
- K. Buntara Sanjeeva, C. Pigliacelli, L. Gazzera, V. Dichiarante, F. Baldelli Bombelli and P. Metrangolo, Nanoscale, 2019, 11, 18407–18415 RSC.
- T. Shirman, R. Kaminker, D. Freeman and M. E. van der Boom, ACS Nano, 2011, 5, 6553–6563 CrossRef CAS.
- Y. Xia, T. D. Nguyen, M. Yang, B. Lee, A. Santos, P. Podsiadlo, Z. Tang, S. C. Glotzer and N. A. Kotov, Nat. Nanotechnol., 2011, 6, 580–587 CrossRef CAS.
- A. Sánchez-Iglesias, M. Grzelczak, T. Altantzis, B. Goris, J. Pérez-Juste, S. Bals, G. Van Tendeloo, S. H. Donaldson, B. F. Chmelka, J. N. Israelachvili and L. M. Liz-Marzán, ACS Nano, 2012, 6, 11059–11065 CrossRef.
- Z. Chu, Y. Han, T. Bian, S. De, P. Král and R. Klajn, J. Am. Chem. Soc., 2019, 141, 1949–1960 CrossRef CAS.
- J. Guo, B. L. Tardy, A. J. Christofferson, Y. Dai, J. J. Richardson, W. Zhu, M. Hu, Y. Ju, J. Cui, R. R. Dagastine, I. Yarovsky and F. Caruso, Nat. Nanotechnol., 2016, 11, 1105–1111 CrossRef CAS.
- Y. Liu, J. He, K. Yang, C. Yi, Y. Liu, L. Nie, N. M. Khashab, X. Chen and Z. Nie, Angew. Chem., Int. Ed., 2015, 54, 15809–15812 CrossRef CAS.
- J. K. Mouw, G. Ou and V. M. Weaver, Nat. Rev. Mol. Cell Biol., 2014, 15, 771–785 CrossRef CAS.
- R. Mout, G. Yesilbag Tonga, L. S. Wang, M. Ray, T. Roy and V. M. Rotello, ACS Nano, 2017, 11, 3456–3462 CAS.
- Q. Li, J. C. Russell, T. Y. Luo, X. Roy, N. L. Rosi, Y. Zhu and R. Jin, Nat. Commun., 2018, 9, 1–7 CrossRef.
- E. Piccinini, D. Pallarola, F. Battaglini and O. Azzaroni, Mol. Syst. Des. Eng., 2016, 1, 155–162 CAS.
- M. P. Pileni, Bull. Chem. Soc. Jpn., 2019, 92, 312–329 CrossRef CAS.
- C. Zeng, Y. Chen, K. Kirschbaum, K. J. Lambright and R. Jin, Science, 2016, 354, 1580–1584 CrossRef CAS.
- Q.-Y. Lin, J. A. Mason, Z. Li, W. Zhou, M. N. O'Brien, K. A. Brown, M. R. Jones, S. Butun, B. Lee, V. P. Dravid, K. Aydin and C. A. Mirkin, Science, 2018, 359, 669–672 CrossRef CAS.
- B. Li, N. You, Y. Liang, Q. Zhang, W. Zhang, M. Chen and X. Pang, Energy Environ. Mater., 2019, 2, 38–54 CrossRef CAS.
- D. Aili, K. Enander, L. Baltzer and B. Liedberg, Nano Lett., 2008, 8, 2473–2478 CrossRef CAS.
- B. O. Okesola and A. Mata, Chem. Soc. Rev., 2018, 47, 3721–3736 RSC.
- D. Maiolo, C. Pigliacelli, P. Sánchez Moreno, M. B. Violatto, L. Talamini, I. Tirotta, R. Piccirillo, M. Zucchetti, L. Morosi, R. Frapolli, G. Candiani, P. Bigini, P. Metrangolo and F. Baldelli Bombelli, ACS Nano, 2017, 11, 9413–9423 CrossRef CAS.
- Y. Yu, X. Yang, M. Liu, M. Nishikawa, T. Tei and E. Miyako, ACS Appl. Mater. Interfaces, 2019, 11, 18978–18987 CrossRef CAS.
- Y. Yu, X. Yang, M. Liu, M. Nishikawa, T. Tei and E. Miyako, Nanoscale Adv., 2019, 1, 3406–3412 RSC.
- K. J. M. Bishop, Angew. Chem., Int. Ed., 2016, 55, 1598–1600 CrossRef CAS.
- Y. Wang, O. Zeiri, M. Raula, B. Le Ouay, F. Stellacci and I. a Weinstock, Nat. Nanotechnol., 2016, 12, 170–176 CrossRef.
- C. Pigliacelli, D. Maiolo, Nonappa, J. S. Haataja, H. Amenitsch, C. Michelet, P. Sánchez Moreno, I. Tirotta, P. Metrangolo and F. Baldelli Bombelli, Angew. Chem., Int. Ed., 2017, 56, 16186–16190 CrossRef CAS.
- K. Deng, Z. Luo, L. Tan and Z. Quan, Chem. Soc. Rev., 2020, 49, 6002–6038 RSC.
- S. Chakraborty, C. K. Tiwari, Y. Wang, G. Gan-Or, E. Gadot and I. A. Weinstock, J. Am. Chem. Soc., 2019, 141, 14078–14082 CrossRef CAS.
- Y. Ma, M. Björnmalm, A. K. Wise, C. Cortez-Jugo, E. Revalor, Y. Ju, O. M. Feeney, R. T. Richardson, E. Hanssen, R. K. Shepherd, C. J. H. Porter and F. Caruso, ACS Appl. Mater. Interfaces, 2018, 10, 31019–31031 CrossRef CAS.
- J. Yeom, P. P. G. Guimaraes, H. M. Ahn, B. Jung, Q. Hu, K. McHugh, M. J. Mitchell, C. Yun, R. Langer and A. Jaklenec, Adv. Mater., 2020, 32, 1903878 CrossRef CAS.
- K. B. Vargo, A. Al Zaki, R. Warden-Rothman, A. Tsourkas and D. A. Hammer, Small, 2015, 11, 1409–1413 CrossRef CAS.
- S. Paterson, S. A. Thompson, J. Gracie, A. W. Wark and R. De La Rica, Chem. Sci., 2016, 7, 6232–6237 RSC.
- F. Yang, A. Skripka, M. S. Tabatabaei, S. H. Hong, F. Ren, A. Benayas, J. K. Oh, S. Martel, X. Liu, F. Vetrone and D. Ma, ACS Nano, 2019, 13, 408–420 CrossRef CAS.
- F. Li, Y. Qin, J. Lee, H. Liao, N. Wang, T. P. Davis, R. Qiao and D. Ling, J. Controlled Release, 2020, 322, 566–592 CrossRef CAS.
- Y. Liu, X. Yang, Z. Huang, P. Huang, Y. Zhang, L. Deng, Z. Wang, Z. Zhou, Y. Liu, H. Kalish, N. M. Khachab, X. Chen and Z. Nie, Angew. Chem., Int. Ed., 2016, 55, 15297–15300 CrossRef CAS.
- K. Wang, J. H. Jordan, X. Hu and L. Wang, Angew. Chem., Int. Ed., 2020, 59, 13712–13721 CrossRef CAS.
- D. Muñoz-Santiburcio and D. Marx, Chem. Sci., 2017, 8, 3444–3452 RSC.
- H. Takezawa, T. Kanda, H. Nanjo and M. Fujita, J. Am. Chem. Soc., 2019, 141, 5112–5115 CrossRef CAS.
- D. M. Vriezema, M. C. Aragonès, J. A. A. W. Elemans, J. J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, Chem. Rev., 2005, 105, 1445–1489 CrossRef CAS.
- Z. Xu, G. Xiao, H. Li, Y. Shen, J. Zhang, T. Pan, X. Chen, B. Zheng, J. Wu, S. Li, W. Zhang, W. Huang and F. Huo, Adv. Funct. Mater., 2018, 28, 1–9 Search PubMed.
- A. B. Grommet, L. M. Lee and R. Klajn, Acc. Chem. Res., 2020, 53, 2600–2610 CrossRef CAS.
- T. Zdobinsky, P. S. Maiti and R. Klajn, J. Am. Chem. Soc., 2014, 136, 2711–2714 CrossRef.
- H. Zhao, S. Sen, T. Udayabhaskararao, M. Sawczyk, K. Kučanda, D. Manna, P. K. Kundu, J.-W. Lee, P. Král and R. Klajn, Nat. Nanotechnol., 2016, 11, 82–88 CrossRef CAS.
- F. Giacalone, V. Campisciano, C. Calabrese, V. La Parola, Z. Syrgiannis, M. Prato and M. Gruttadauria, ACS Nano, 2016, 10, 4627–4636 CrossRef CAS.
- K. Hou, J. Han and Z. Tang, ACS Mater. Lett., 2020, 2, 95–106 CrossRef CAS.
- M. Melchionna, P. Fornasiero and M. Prato, APL Mater., 2020, 8, 020905 CrossRef CAS.
- X. Wang, D. Liu, S. Song and H. Zhang, J. Am. Chem. Soc., 2013, 135, 15864–15872 CrossRef CAS.
- Y. Liu, J. Shi, Q. Peng and Y. Li, J. Mater. Chem., 2012, 22, 6539–6541 RSC.
- A. Kumar, N. Kumari, S. Dubbu, S. Kumar, T. Kwon, J. H. Koo, J. Lim, I. Kim, Y. K. Cho, J. Rho and I. S. Lee, Angew. Chem., Int. Ed., 2020, 59, 9460–9469 CrossRef CAS.
- W. Zhu, R. Esteban, A. G. Borisov, J. J. Baumberg, P. Nordlander, H. J. Lezec, J. Aizpurua and K. B. Crozier, Nat. Commun., 2016, 7, 11495–11509 CrossRef CAS.
- C. T. Mcternan, T. K. Ronson and J. R. Nitschke, J. Am. Chem. Soc., 2019, 141, 6837–6842 CrossRef CAS.
- A. Al Harraq, J. G. Lee and B. Bharti, Sci. Adv., 2020, 6, eaba5337 CrossRef CAS.
- A. K. Pearce, T. R. Wilks, M. C. Arno and R. K. O’Reilly, Nat. Rev. Chem., 2021, 5, 21–45 Search PubMed.
- D. Fava, Z. Nie, M. A. Winnik and E. Kumacheva, Adv. Mater., 2008, 20, 4318–4322 CAS.
- J. Nai, B. Y. Guan, L. Yu and X. Wen David Lou, Sci. Adv., 2017, 3, e1700732 CrossRef.
- G. Schmid, Nanoparticles:From Theory to Application, Wiley VCH, 2006 Search PubMed.
- E. Mattia and S. Otto, Nat. Nanotechnol., 2015, 10, 111–119 CrossRef CAS.
- A. Walther, Adv. Mater., 2020, 32, 1–10 CrossRef.
- R. K. Grötsch, C. Wanzke, M. Speckbacher, A. Angı, B. Rieger and J. Boekhoven, J. Am. Chem. Soc., 2019, 141, 9872–9878 CrossRef.
- L. Wang, L. Xu, H. Kuang, C. Xu and N. A. Kotov, Acc. Chem. Res., 2012, 45, 1916–1926 CrossRef CAS.
- R. Klajn, K. J. M. Bishop and B. A. Grzybowski, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10305–10309 CrossRef CAS.
- C. Huang, X. Chen, Z. Xue and T. Wang, Sci. Adv., 2020, 6, eaba1321 CrossRef.
- S. Ohta, D. Glancy and W. C. W. Chan, Science, 2016, 351, 841–845 CrossRef CAS.
- D. Y. Zhang and G. Seelig, Nat. Chem., 2011, 3, 103–113 CrossRef CAS.
- X. D. Guo, L. J. Zhang, Z. M. Wu and Y. Qian, Macromolecules, 2010, 43, 7839–7844 CAS.
- L. E. Low, J. Wu, J. Lee, B. T. Tey, B. H. Goh, J. Gao, F. Li and D. Ling, J. Controlled Release, 2020, 324, 69–103 CrossRef CAS.
- F. J. Rizzuto, L. K. S. von Krbek and J. R. Nitschke, Nat. Rev. Chem., 2019, 3, 204–222 CrossRef.
- C. Giménez, E. Climent, E. Aznar, R. Martánez-Máñez, F. Sancenón, M. D. Marcos, P. Amorós and K. Rurack, Angew. Chem., Int. Ed., 2014, 53, 12629–12633 Search PubMed.
- B. A. Grzybowski, K. Fitzner, J. Paczesny and S. Granick, Chem. Soc. Rev., 2017, 46, 5647–5678 RSC.
- B. Ng, X. Peng, E. Faegh and W. E. Mustain, J. Mater. Chem. A, 2020, 8, 2712–2727 RSC.
- S. H. Lee, S. H. Yu, J. E. Lee, A. Jin, D. J. Lee, N. Lee, H. Jo, K. Shin, T. Y. Ahn, Y. W. Kim, H. Choe, Y. E. Sung and T. Hyeon, Nano Lett., 2013, 13, 4249–4256 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |