
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReversible carbon–boron bond formation at platinum centers through σ-BH complexes†
Pablo
Ríos
 a,
Rocío
Martín-de la Calle
a,
Pietro
Vidossich
b,
Francisco José
Fernández-de-Córdova
a,
Agustí
Lledós
*c and
Salvador
Conejero
*a
a,
Rocío
Martín-de la Calle
a,
Pietro
Vidossich
b,
Francisco José
Fernández-de-Córdova
a,
Agustí
Lledós
*c and
Salvador
Conejero
*a
aInstituto de Investigaciones Químicas (IIQ), Departamento de Química Inorgánica, CSIC and Universidad de Sevilla, Centro de Innovación en Química Avanzada (ORFEO-CINQA), C/ Américo Vespucio 49, Sevilla, 41092, Spain. E-mail: sconejero@iiq.csic.es
bMolecular Modeling & Drug Discovery Laboratory, Istituto Italiano di Tecnologia, Via Morego 30, 16163 Genoa, Italy
cDepartament de Química, Universitat Autònoma de Barcelona, Edifici Cn, Cerdanyola del Vallès, 08193, Spain. E-mail: agusti@klingon.uab.es
First published on 10th November 2020
Abstract
A reversible carbon–boron bond formation has been observed in the reaction of the coordinatively unsaturated, cyclometalated, Pt(II) complex [Pt(ItBuiPr′)(ItBuiPr)][BArF], 1, with tricoordinated boranes HBR2. X-ray diffraction studies provided structural snapshots of the sequence of reactions involved in the process. At low temperature, we observed the initial formation of the unprecedented σ-BH complexes [Pt(HBR2)(ItBuiPr′)(ItBuiPr)][BArF], one of which has been isolated. From −15 to +10 °C, the σ-BH species undergo a carbon–boron coupling process leading to the platinum hydride derivative [Pt(H)(ItBuiPr–BR2)(ItBuiPr)][BArF], 4. Surprisingly, these compounds are thermally unstable undergoing carbon–boron bond cleavage at room temperature that results in the 14-electron Pt(II) boryl species [Pt(BR2)(ItBuiPr)2][BArF], 2. This unusual reaction process has been corroborated by computational methods, which indicate that the carbon–boron coupling products 4 are formed under kinetic control whereas the platinum boryl species 2, arising from competitive C–H bond coupling, are thermodynamically more stable. These findings provide valuable information about the factors governing productive carbon–boron coupling reactions at transition metal centers.
1. Introduction
The formation of C–B bonds, mediated by transition metal complexes, has become an important tool for the synthesis of organoboron compounds,1 an important family of organic molecules in synthetic organic chemistry and in materials science.2 From the mechanistic point of view, there have been several reports that point to the participation of σ-BH (σ-borane) complexes as key intermediates in the formation of the carbon–boron bonds in metal-alkyl or -aryl compounds. For example, Hartwig et al. have shown, by means of DFT calculations, that σ-BH complexes of rhodium and tungsten are transient species in the borylation of alkanes, where the driving force is the formation of the C–B bonds (Scheme 1).3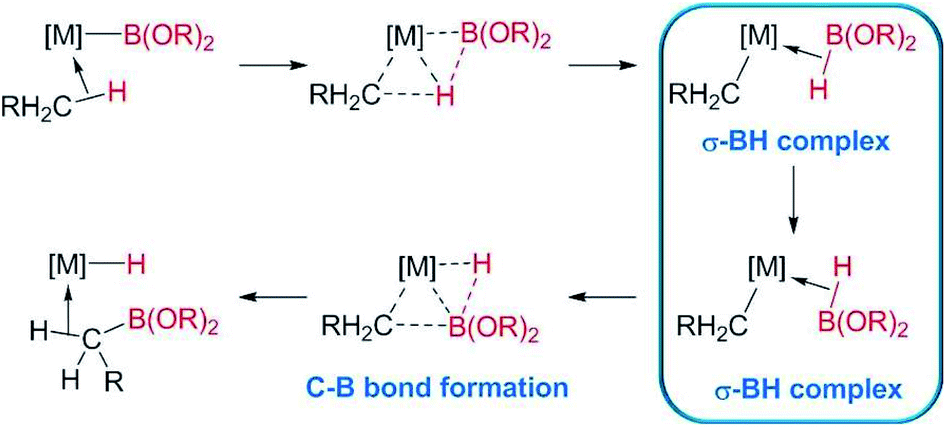 | ||
| Scheme 1 σ-BH complexes proposed as intermediates in the borylation of C–H bonds of alkanes reported by Hartwig et al.3 | ||
Although most of this type of C–H borylation processes have been carried out using rhodium and iridium systems,4 there have been a few reports on the utilization of platinum complexes for the formation of C–B bonds, either in stoichiometric reactions or in catalytic processes.5 Turculet and Stradiotto reported the dehydrogenative coupling of B–H/C–H bonds at a Pt(II) center,5a whereas Chatani, Tobisu et al. suggested the participation of σ-BH complexes in the platinum-catalyzed borylation of arenes.5b,c However, the interaction of tricoordinated hydroboranes with platinum centers leading to σ-BH complexes has proven elusive.6 In addition, examples of transition metal σ-borane alkyl complexes [M(R)(HBR2)] are extremely rare, and those that have been isolated did not show further reactivity involving the formation of carbon–boron bonds.7 Isolation of this type of compounds might offer an invaluable opportunity for determining the factors that can lead to competitive carbon–boron or carbon–hydrogen coupling products.
In this article, we report the spectroscopic observation and isolation of platinum σ-BH complexes that undergo either a carbon–boron coupling process or formation of platinum-boryl complexes as a function of temperature. The whole sequence of events underlying these processes has been followed by NMR and X-ray diffraction studies and investigated by DFT calculations.
2. Results and discussion
2.1 Synthesis of Pt(II) boryl complexes
In previous studies we have reported that some low-coordinate cationic platinum(II) complexes can interact with tetra-coordinated boranes, L·BH3 (L = pyridine, amine), leading to stable adducts or to catalytic boron–nitrogen coupling products (amine–borane dehydrocoupling).8 In this context we have examined the reactivity of these platinum systems with tri-coordinated boranes derived from dioxa- and diaza-boroles. We have chosen cationic platinum(II) complex [Pt(ItBuiPr′)(ItBuiPr)][BArF], 1, as starting material (where ItBuiPr is 1-tert-butyl-3-isopropylimidazol-2-ylidene, and ItBuiPr′ its cyclometalated form through the tert-butyl substituent), since this system has been successfully used for the isolation of related σ-SiH derivatives.9 When complex 1 was treated with either pinacol- (HBpin, a) or catechol–borane (HBcat, b), or 1,3,2-benzodiazaborolane (HBdab, c) in CH2Cl2 at 25 °C, boryl complexes 2a–c were formed quantitatively according to NMR spectroscopy (Scheme 2). The reaction proceeds smoothly but reaction times vary considerably from dioxaboroles to the diazaborole, from 10 min to 15 h. The 1H NMR spectra of these species reveal a highly symmetric environment of the NHC ligands, indicating their chemical equivalence.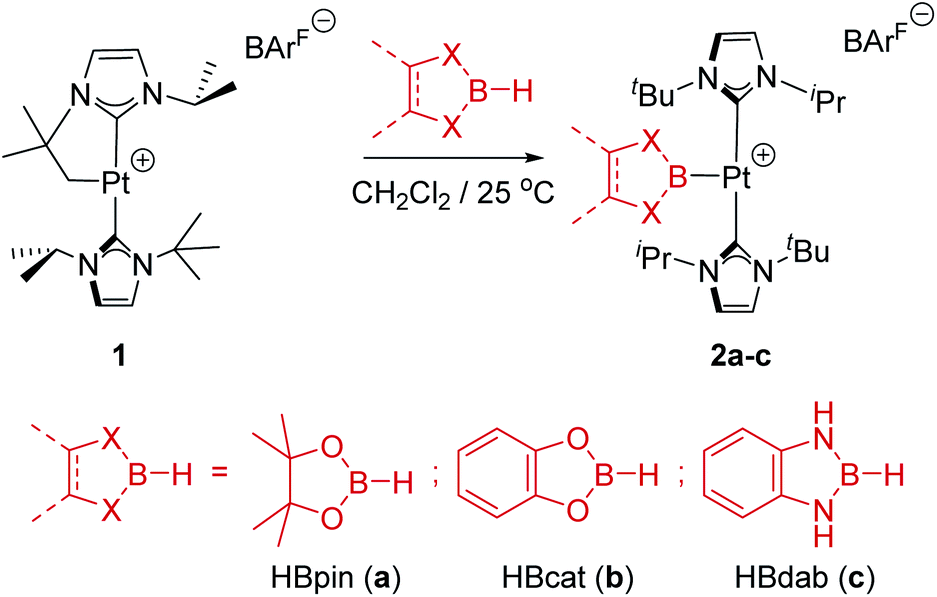 | ||
| Scheme 2 Reaction of complex 1 with hydroboranes. | ||
The 11B{1H} NMR spectra exhibit two sets of signals, one for the Pt–B fragment and the other for the BArF anion. The former resonate at δ 12.7, 14.7 and 8.8 for 2a–c, respectively, showing coupling to 195Pt in the range 1390–1700 Hz (see ESI†). Crystals of 2c suitable for X-ray diffraction studies were grown by slow diffusion of CH2Cl2 solutions into pentane. Fig. 1 shows an ORTEP-type view of the cation. The platinum atom is surrounded by only three ligands, with a formal vacant site trans to the boryl moiety. Very weak, at best, agostic interactions are present in the molecule (the closest C to Pt is 3.1 Å away).10 The Pt–B distance of 1.988(6) Å is similar to that reported by Braunschweig in related systems of the type trans-[(Cy3P)2Pt{B(X)X′}].11
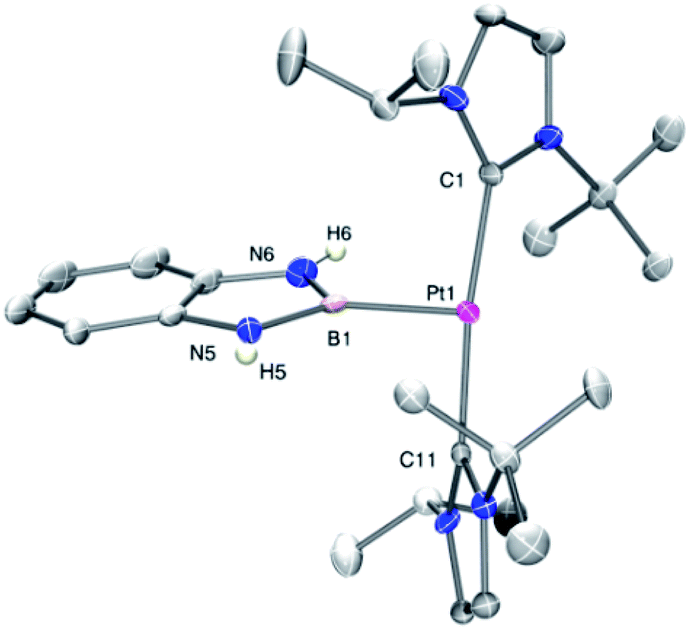 | ||
| Fig. 1 ORTEP representation of the cation of complex 2c. Hydrogen atoms (except those of the NH moieties) have been omitted for clarity. Selected bond distances (Å) and angles (°): Pt1–B1, 1.988(6); Pt1–C1, 2.015(5); Pt1–C11, 2.016(5); B1–N6, 1.424(7); B1–N5, 1.433(7); C1–Pt1–C11, 173.5(2); B1–Pt1–C1, 92.9(2); B1–Pt1–C11, 93.6(2). | ||
2.2 Low-temperature reactivity studies: characterization and isolation of σ-BH Pt(II) complexes
In order to look for intermediates in the reaction process, and in particular to detect σ-BH complexes, low temperature NMR experiments have been carried out. The reaction of 1 with a slight excess of pinacolborane in an NMR tube in CD2Cl2 at −30 °C led to a clean reaction with formation of a new species that, according to multinuclear NMR, is the σ-BH complex 3a (Scheme 3). The 1H NMR is characterized by a broad signal in the hydride region at −3.93 ppm, showing coupling to 195Pt (1JH,Pt = 357 Hz). This signal becomes narrower upon 11B decoupling (see Fig. S11 in the ESI†) and it has been assigned to the bridging Pt–H–B proton. The 11B{1H} spectrum shows, besides the signal for the BArF anion and some excess of HBpin, a very broad signal at 21.8 ppm.12 A similar behavior has been observed in the reaction of 1 with HBcat leading to 3b (1H NMR: −3.28 ppm, 1JH,Pt = 327 Hz; 11B{1H} NMR: 21.3 ppm) and with HBdab producing 3c (1H NMR: −2.32 ppm, 1JH,Pt = 339 Hz; 11B{1H} NMR: 18.6 ppm). All these sigma complexes are stable at temperatures ranging from −15 to +10 °C, with complex 3c being the most stable. In line with these findings, DFT calculations estimate favorable ΔG of formation at −30 °C of −2.3, −0.6 and −3.1 kcal mol−1 for 3a, 3b and 3c, respectively (see Fig. S33 in the ESI†).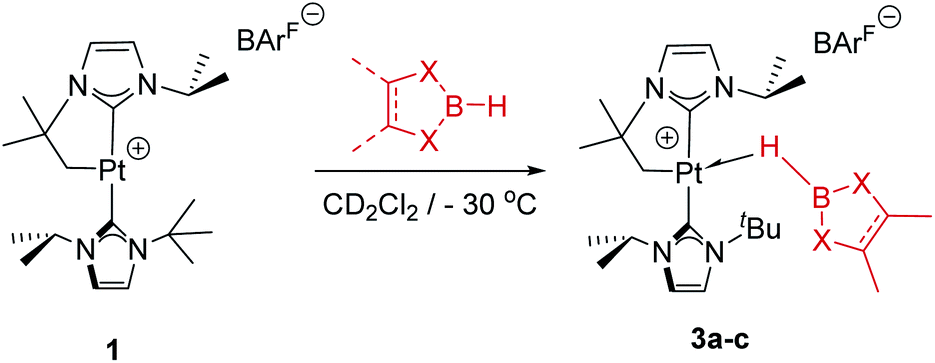 | ||
| Scheme 3 Formation of σ-BH complexes 3a–c at low T. | ||
The increased thermal stability of complex 3c has allowed us to obtain crystals suitable for X-ray diffraction analysis by slow diffusion of CH2Cl2 solution into pentane at −25 °C (Fig. 2). The platinum center is found in a nearly planar environment, with two NHCs and one –CH2– unit occupying three coordination sites and the B–H moiety the fourth. The angle defined by atoms C13–Pt1–H1 and Pt1–H1–B1 are 168.5(1)° and 105(2)°, respectively, very close to those observed in the related σ-SiH derivative [Pt(HSiEt3)(ItBuiPr′)(ItBuiPr)][BArF] (173.1(1)° and 103(2)°).9 The Pt1–H1 and H1–B1 bond distances are 1.69(4) Å and 1.19(3) Å, respectively. The latter is slightly elongated with respect to the B–H bond of boranes13 and comparable to related σ-BH primary aminoborane complexes.14 The Pt1⋯B1 distance of 2.31 Å is considerably larger than the sum of covalent radii (2.2 Å), suggesting that, if any, the interaction between these two atoms is weak.15 In fact, topological analysis of the electron density and its laplacian (see ESI†) revealed the presence of bond critical points (BCPs) between Pt and H, and H and B, yet no BCP between Pt and B was observed.16 This fact suggests that the coordination mode is better described as η1-BH instead of the common η2-BH mode reported for other transition metals.6,13,17 Bonding interactions were further analyzed in terms of localized orbitals.18 The centroid of the localized orbital corresponding to the B–H bond (XBH, Fig. 3) is close to the H center, in agreement with a marked hydride character of this H atom, and is displaced from the B–H axis towards Pt, indicative of a perturbed sigma bond. At the same time, one Pt centroid (XPt) is slightly moved away (0.431 Å) from the nucleus, compared to the other Pt centroids (0.345–0.398 Å), and pointing to B. We have previously characterized similar interactions in cationic Pt(II) σ-silane complexes, which adopt a rare η1-SiH coordination mode.9
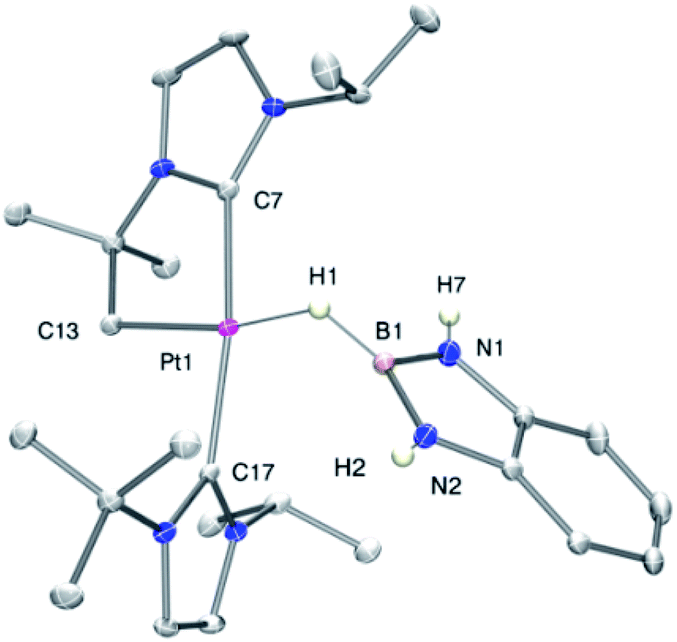 | ||
| Fig. 2 Molecular structure of the cationic component as determined by X-ray diffraction of complex 3c (ellipsoids at 30% probability). Selected bond distances (Å) and angles (°): Pt1–H1, 1.69(4); H1–B1, 1.19(3), Pt1–C13, 2.054(2); Pt1–H1–B1, 105(2); C13–Pt1–H1, 168.5(1). | ||
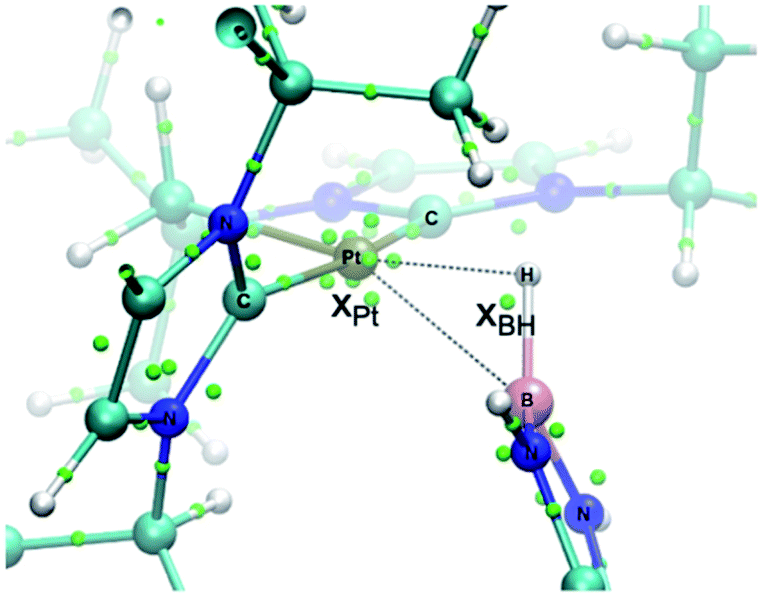 | ||
| Fig. 3 Localized orbital analysis of 3c. The molecular structure is overlaid with the centroids of the localized orbitals (small green dots). Dotted lines highlight the Pt–B and Pt–H distances. | ||
As in the previously characterized η1-SiH platinum complexes,9 the coordination of the σ-bonded ligand is very flexible. We computed the energetic cost of deforming the Pt–H–B angle in 3c (see Table S4 and Fig. S40†). Starting from the energy minimum at 101.6°, closing the angle to 78° or opening it to 154° requires less than 6 kcal mol−1. More acute angles (68°) lead to borane dissociation instead of B–H bond cleavage, which contrasts with what was observed between 1 and hydrosilanes, for which closing the Pt–H–Si angle led ultimately to the Si–H oxidative addition.9 Importantly, QTAIM analysis did not reveal any BCP between Pt and B in structures with acute Pt–H–B angles (Fig. S38 and S39†).
2.3 Thermal evolution of σ-BH Pt(II) complexes: reversible C–B coupling en route to boryl complexes
As mentioned before complexes 3a–c are only marginally thermally stable, and upon warming at temperatures above −15 °C (3a), 0 °C (3b) or +10 °C (3c) undergo a rearrangement that involves the cleavage of the B–H bond and the formation of a new C–B bond leading to complexes 4a–c (Scheme 4). The reaction is particularly clean in the case of complex 3a, for which a progressive formation of 4a is observed before it undergoes further rearrangement (see below). When complex 3a is allowed to stand at −5 °C for ca. 70 min it undergoes B–H bond cleavage of the borane leading to hydride species 4a, arising from a C–B coupling process with the Pt–CH2 unit. In the 1H NMR spectrum the signal of the BH proton of complex 3a progressively disappears, whereas a new narrow signal in the hydride region at −27.11 ppm simultaneously appears, showing a large coupling constant to 195Pt of 2167 Hz. This value is smaller than that observed for complex [Pt(H)(ItBuiPr)2][BArF] (1JH,Pt = 2496 Hz).19 In addition, the diastereotopic Pt–CH2 protons in 3a merge into a singlet resonance at 2.44 ppm in 4a,20 that correlates in the 1H, 13C-HSQC spectrum with a very broad resonance at 26.8 ppm. The 11B{1H} NMR spectrum shows a very broad signal at 32.9 ppm. All these data support the platinum-hydride structure shown in Scheme 4, but definite evidence came from X-ray diffraction studies of complex 4a obtained at −20 °C. Fig. 4 shows the cationic fragment of complex 4a. The hydride ligand has been localized in the difference Fourier map. The structure confirms the formation of a C–B bond, as anticipated by NMR analysis. Moreover, one of the oxygen atoms of the pinacol moiety is interacting with the platinum center (Pt–O bond distance, 2.247(3) Å), trans to the hydride ligand, which explains the reduction of the 1JH,Pt coupling constant of the hydride compared to that in [Pt(H)(ItBuiPr)2][BArF].19,21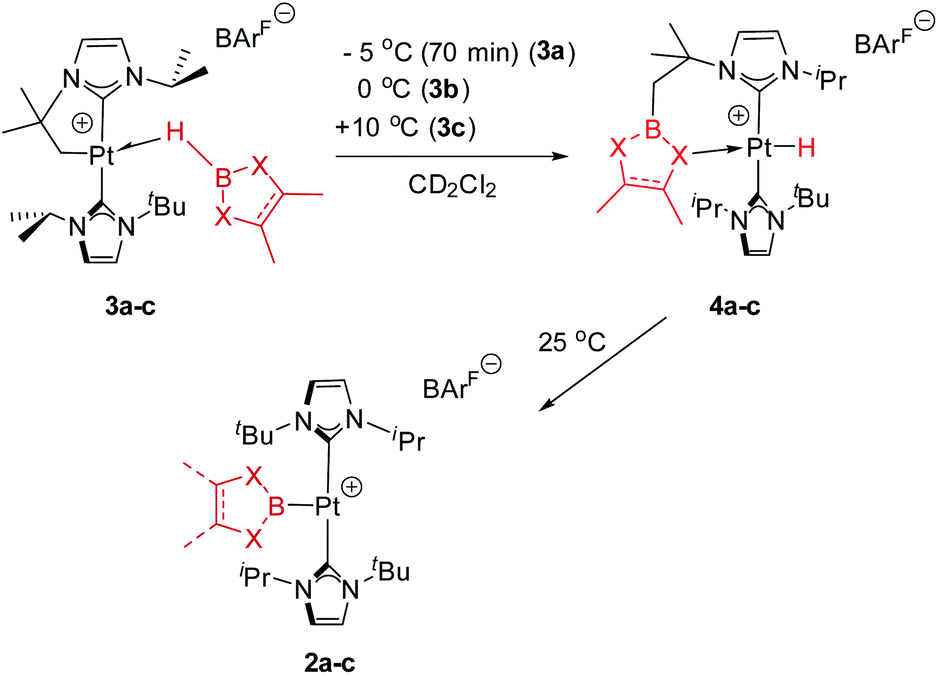 | ||
| Scheme 4 Rearrangement of complexes 3a–c. | ||
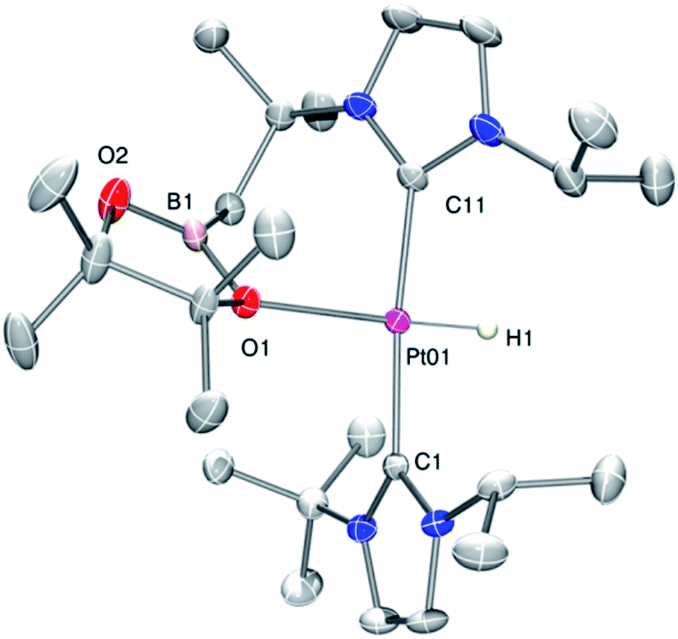 | ||
| Fig. 4 Molecular structure of the cationic component as determined by X-ray diffraction of complex 4a (ellipsoids at 30% probability). | ||
With respect to complexes 3b and 3c, a similar rearrangement is observed, although in a different range of temperatures. However, as the temperature increases, mixtures of these compounds together with intermediates 4b, c and the final boryl complexes 2b, c coexist, hampering full characterization of intermediates 4b, c (see Fig. S25–S28 in the ESI†). Despite this, the signals observed in the hydride region of the 1H NMR spectra are consistent with formation of these transient species (4b: −24.7 ppm, 1JH,Pt = 2459 Hz at 5 °C; 4c: −21.06; 1JH,Pt = 1905 Hz at 20 °C), as those in the 11B{1H} NMR (4b: 33.4 ppm; 4c: 29.6 ppm). It's worth noting the variations in the magnitude of the 1JH,Pt coupling constants of these Pt–H hydrides. In 4b the value of 2459 Hz is higher than that of complex 4a, and close to the hydride derivative [Pt(H)(ItBuiPr)2] [BArF], whereas a smaller value of 1906 Hz is observed in 4c. Since the 1JH,Pt values are sensitive to the presence and nature of ligands trans to the hydride, the interaction of the oxygen atom with the platinum center in 4b is weaker than in 4a whereas, expectedly, the nitrogen atom has a stronger interaction with the platinum atom in 4c.21 This has important implications in the subsequent reactivity. All complexes 4 evolve at 25 °C into boryl species 2, but the time required is slightly different between 4a and 4b, and markedly different for 4c. The fastest is 4b, requiring 10 min to be fully converted into 2b, while 4a rearrange in ca. 35 min. On the other hand, 4c undergoes the C–B bond cleavage after 15 h. Thus, it appears that the stronger the interaction of the X (X = O, N) atom of the boryl fragment with the platinum center in complexes 4, the longer it takes to transform 4 into the boryl species 2. Therefore, we reasoned that de-coordination of the X atom to open a vacant site at the platinum center is required for the cleavage of the C–B bond leading to the boryl species 2. In order to prove this, intermediate 4a was reacted at −5 °C with tBuNC (Scheme 5), leading to the 16-electron Pt(II) complex 5. This compound is very stable, and no cleavage of the C–B bond is observed at 25 °C in solution, therefore corroborating the need for the vacant site to promote the rearrangement of hydride complexes 4 into boryl species 2.
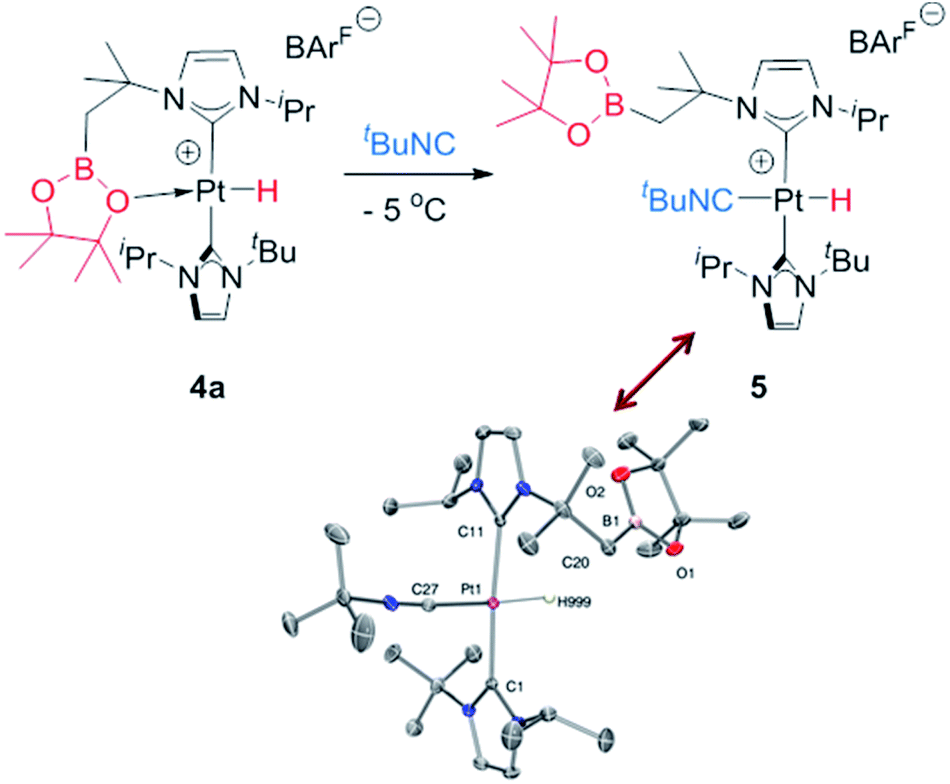 | ||
| Scheme 5 Synthesis of complex 5 (top) and molecular structure of its cationic component (bottom). | ||
2.4 Mechanistic analysis of C–B and C–H bond formation
The observed reactivity of the 1 + HBR2 systems summarized in Scheme 4 suggests a sequential mechanism in which first C–B and Pt–H bonds are formed (compounds 4a–c) and then cleaved22 leading to C–H and Pt–B bonds (compounds 2a–c). To unravel atomic and electronic rearrangements taking place in these systems, we carried out DFT calculations (M06 functional) in dichloromethane solvent (SMD continuum model) using species 1 and HBpin (a). Details of the calculations are given in the ESI.† Contrary to expectations, however, we found that it is not possible to directly interconvert 4a into 2a. All attempts made to find such a pathway resulted in high energy species (see High Energy Transition States section in ESI†). The solution to this issue came by recognizing the analogy with the borylation of alkanes (Scheme 1).3 Considering the σ-BH complex (here 3a) the system can evolve towards the formation of a C–B bond (complex 4a) or with the formation of a C–H bond (complex 2a). Moreover, when raising the temperature, 4a transforms into 2a (Scheme 6).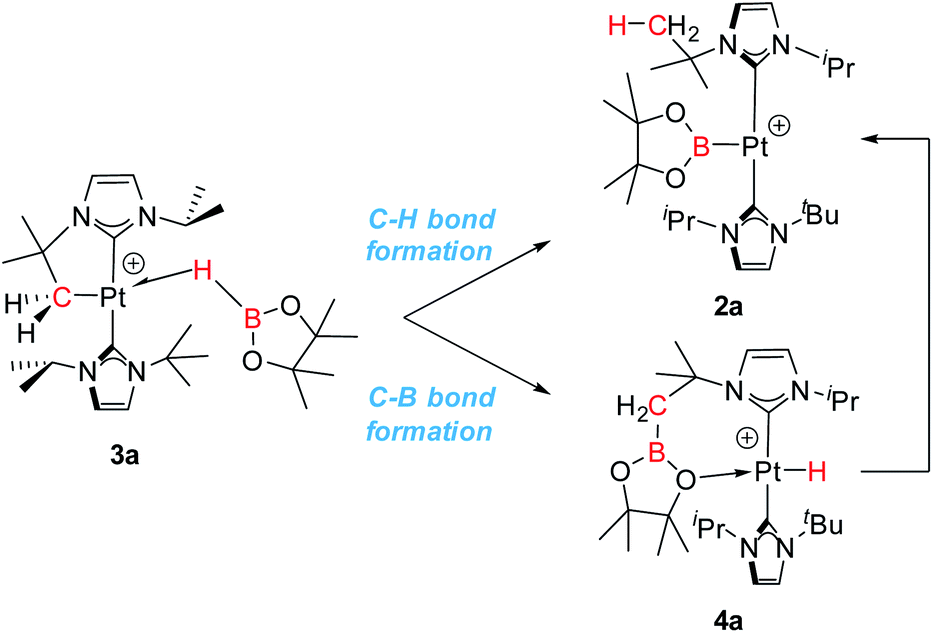 | ||
| Scheme 6 Different reactivity pathways considered in DFT calculations. | ||
We started the exploration from σ-BH complex 3a. However, with the trans-NHC arrangement in 3a, all transition states for transferring a H atom to the cyclometalated methylene fragment and forging a Pt–B bond, yielding complex 2a (C–H bond formation), or transferring the Bpin and forming a Pt–H bond, yielding 4a (C–B bond formation), are high in energy (34.0 and 28.5 kcal mol−1 above the reagents, respectively; see Fig. S48 and S49 in the ESI†) and inconsistent with the experimental observations. We thus searched for alternative routes.
Pt(NHC)2 fragments, even with bulky NHC ligands, are known to be rather flexible around the C–Pt–C angle.23 In previous works we have shown that trans–cis isomerizations of Pt(NHC)2 fragments can occur in their reactions with H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 24 and silanes.19 Thus, we investigated mechanisms containing species with a cis geometry of the NHC ligands. Starting from complex 1, trans–cis isomerization requires overcoming a barrier of 12.8 kcal mol−1 and yields species 1-cis, 11.1 kcal mol−1 above the origin (see Fig. S50 in the ESI†) presumably due to the increased steric bulk between the isopropyl chain of the cyclometalated NHC ligand and the other NHC fragment. Coordination of HBpin to 1-cis gives a σ-BH complex, for which we found two possible orientations of the HBpin ligand relative to the Pt(NHC)2 fragment (3a-cis and 3a-cis′, Fig. 5). Both display a η1-BH coordination mode, with geometrical parameters of the Pt–H–B fragment resembling those of 3a. In 3a-cis (7.8 kcal mol−1 above 1 + HBpin), the B atom is closer to the methylene unit than the H atom (C⋯B(H)pin = 2.84 Å vs. C⋯H(Bpin) = 3.05 Å) with a Pt–H–B angle of 100.5°. On the contrary, in 3a-cis′ (10.0 kcal mol−1) HBpin places its H atom closer to the methylene unit than the Bpin fragment (C⋯H(Bpin) = 2.57 Å vs. C⋯B(H)pin = 3.54 Å) with a Pt–H–B angle of 109.2°.
24 and silanes.19 Thus, we investigated mechanisms containing species with a cis geometry of the NHC ligands. Starting from complex 1, trans–cis isomerization requires overcoming a barrier of 12.8 kcal mol−1 and yields species 1-cis, 11.1 kcal mol−1 above the origin (see Fig. S50 in the ESI†) presumably due to the increased steric bulk between the isopropyl chain of the cyclometalated NHC ligand and the other NHC fragment. Coordination of HBpin to 1-cis gives a σ-BH complex, for which we found two possible orientations of the HBpin ligand relative to the Pt(NHC)2 fragment (3a-cis and 3a-cis′, Fig. 5). Both display a η1-BH coordination mode, with geometrical parameters of the Pt–H–B fragment resembling those of 3a. In 3a-cis (7.8 kcal mol−1 above 1 + HBpin), the B atom is closer to the methylene unit than the H atom (C⋯B(H)pin = 2.84 Å vs. C⋯H(Bpin) = 3.05 Å) with a Pt–H–B angle of 100.5°. On the contrary, in 3a-cis′ (10.0 kcal mol−1) HBpin places its H atom closer to the methylene unit than the Bpin fragment (C⋯H(Bpin) = 2.57 Å vs. C⋯B(H)pin = 3.54 Å) with a Pt–H–B angle of 109.2°.
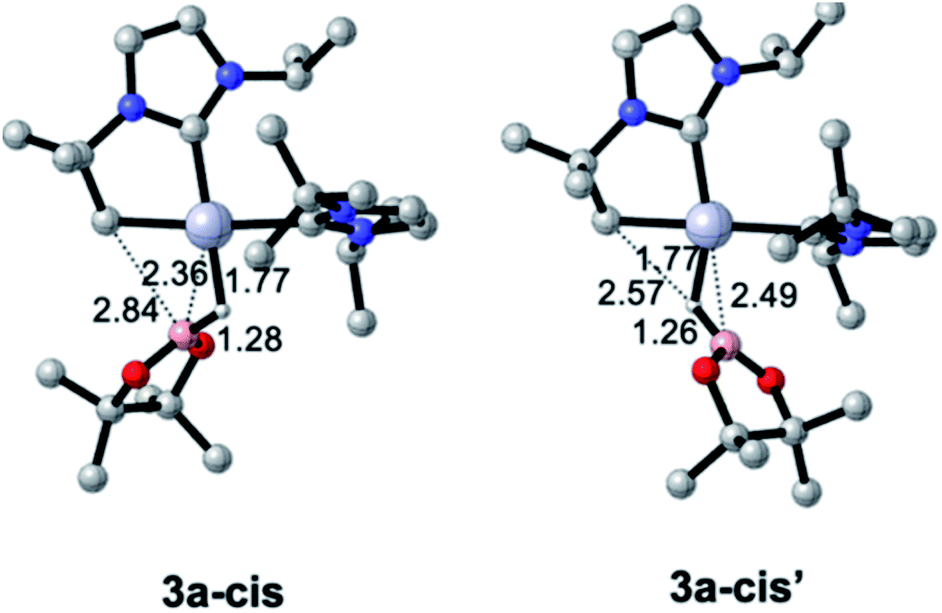 | ||
| Fig. 5 Optimized geometries of σ-BH complexes with a cis-arrangement of NHC ligands. | ||
The computed Gibbs energy profile assuming a cis arrangement of the NHC ligands is depicted in Fig. 6. 3a-cis is the starting point of the C–B bond coupling pathway (Fig. 6, green trace). From this point, the B–H bond is cleaved through TS1 (15.6 kcal mol−1) in a process where the boryl group is transferred to the methylene fragment (C–B bond formation) at the same time that a Pt–H bond is created. The resulting hydride species Int1 (7.5 kcal mol−1) features an agostic interaction through one of the C–H bonds from the CH2 group (H–Pt = 1.89 Å, C–H–Pt = 107.5°)10 that stabilizes the vacant position of the complex. Further stabilization is observed when one of the oxygen groups from Bpin coordinates to platinum as in 4a-cis (2.0 kcal mol−1), due to its higher electron donation ability. This exchange of ligands takes place through TS2 (15.4 kcal mol−1). Once 4a-cis is formed, cis to trans isomerization through TS3 (13.6 kcal mol−1) yields experimentally observed complex 4a, which exhibits both NHC ligands in a trans geometry. During isomerization, coordination of the Bpin fragment through oxygen is lost, giving species Int2, which consists of a cationic Pt(II) hydride complex analogous to [Pt(H)(ItBu2)2]+, previously calculated by our group (the main difference being the presence of Bpin in one of the carbene ligands and isopropyl instead of tert-butyl substituents).25 Finally, coordination of one oxygen of the Bpin fragment to the metal yields the C–B coupling product 4a, at −6.8 kcal mol−1.
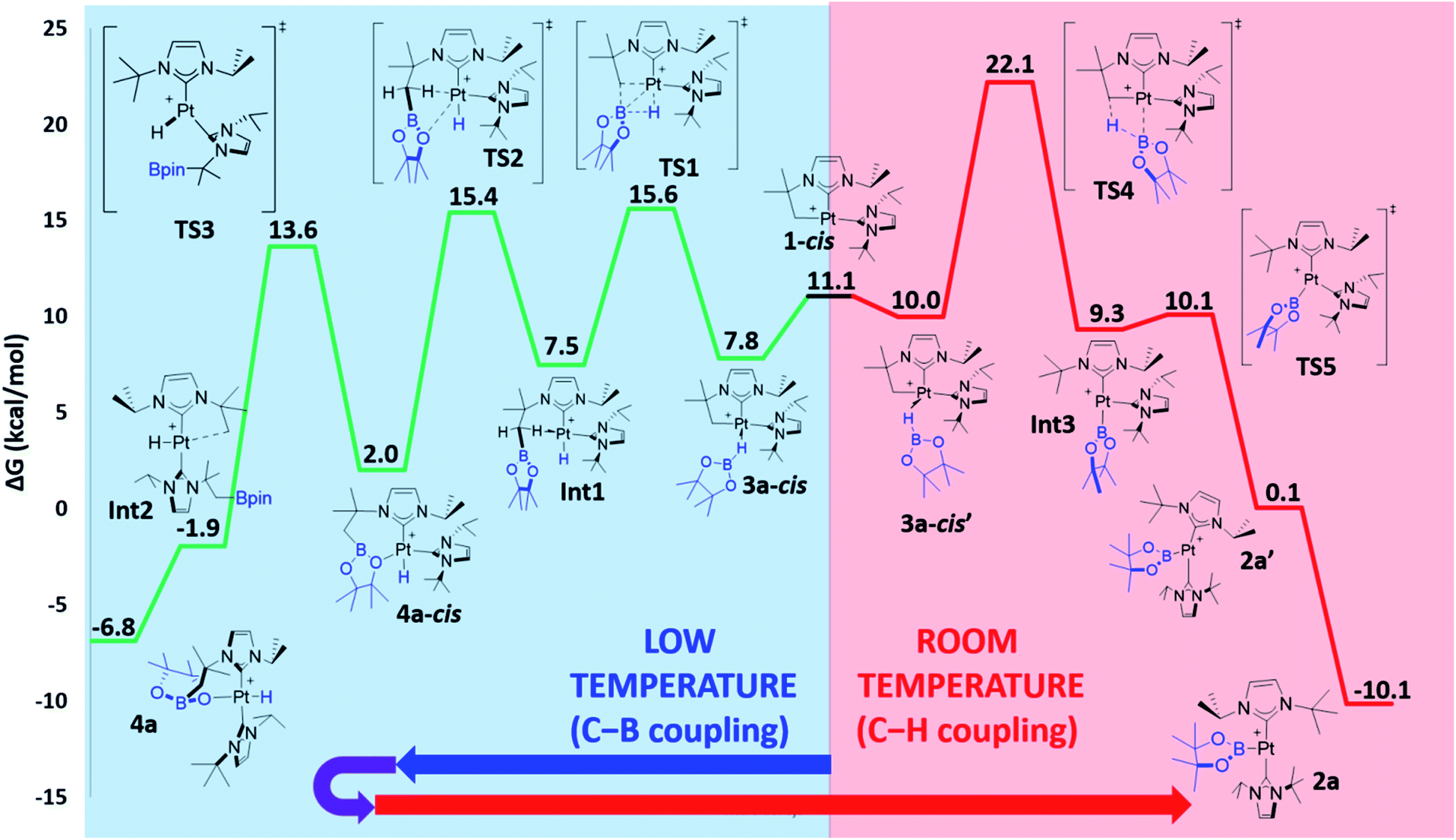 | ||
| Fig. 6 Gibbs energy profile in dichloromethane for the C–B (green trace) and C–H (red trace) bond formation pathways (NHCs in cis) upon reaction between 1 and HBpin. Gibbs energies at 298 K in kcal mol−1. The Gibbs energy of 1 + HBpin has been taken as zero-energy. | ||
3a-cis′ has the right orientation of the HBpin ligand to start C–H bond coupling pathway (Fig. 6, red trace). From here, the H atom is transferred to the C atom at the same time than the Pt–B bond is formed. This step takes place through TS4 (22.1 kcal mol−1), after which the corresponding boryl complex Int3 is formed. Right after, isomerization viaTS5 (10.1 kcal mol−1) gives the experimentally observed boryl derivative 2a, which is the thermodynamically most stable compound of the whole energy profile.
From the Gibbs energy profile in Fig. 6 it can be inferred that, at low temperature, the product entailing crossing the lowest barrier will be formed (kinetic control). This is the hydride species 4a, arising from a C–B coupling process with the Pt–CH2 unit, which is formed from 3a-cis after crossing a barrier of only 7.8 kcal mol−1 (TS1). However, going back from 4a to 3a-cis requires an activation of only 22.4 kcal mol−1, which can be provided raising enough the temperature. At 25 °C transition state TS4 (22.1 kcal mol−1) can be overcome and the reaction is driven by the formation of the most stable product, 2a (thermodynamic control). The boryl complex 2a is formed via a C–H coupling process. The rationalization for the experimental observations combines the C–B (Fig. 6, green trace) and C–H (Fig. 6, red trace) bond formation pathways: at low temperatures, C–B bond formation takes place through the aforementioned described mechanism, yielding 4a. Then, increasing the temperature to 25 °C allows the system to undergo the reverse pathway, reaching 3a-cis and, after a conformational change, 3a-cis′, which initiates the C–H coupling pathway leading to the most stable product 2a. The different energetic barriers observed from 3a-cis and 3a-cis′ leading to C–B or C–H coupling, respectively, might have (at least in part) a steric origin. The orientation of the pinacol substituents in 3a-cis′ is likely inducing some steric repulsions with the spectator NHC ligand in cis to it, and this might account for the different Pt⋯B bond distances (0.13 Å) observed in both 3a-cis and 3a-cis′. Reducing the steric effects on the borane might reduce the energy differences. In fact, as stated before, in the reaction carried out with HBcat, complexes 4b and 2b coexist in a certain range of temperatures, suggesting that, in this case, formation of C–B and C–H bonds have very similar energetic barriers.
2.5 Electronic analysis of C–B and C–H coupling processes
Exploration of the potential energy surface of 1 + HBpin system has allowed disclosing the mechanism for the formation of C–B and C–H coupling products and their interconversion. Calculations have shown that rupture of B–H and H2C–Pt bonds and formation of either C–B and Pt–H or C–H and Pt–B bonds entail similar energy requirements. To reveal these bonding rearrangements we applied a localized orbital approach, which turned highly useful to analyze other organometallic reactions.18 In this approach Kohn–Sham molecular orbitals are transformed into maximally localized orbitals (LMOs) and the centroids of these LMOs are computed for selected structures along the reaction path. The movement of the LMOs centroids along the reaction describes the electronic rearrangements taking place in the bond-breaking and bond-forming processes. In this way an arrow-pushing description of the C–B and C–H coupling processes can be obtained from DFT calculations. Recently, we applied this approach to analyze C–Si bond coupling reactions.19Fig. 7 shows the relevant LMOs together with their centroids in three representative structures of the key chemical step of each process. The corresponding arrow-pushing schemes are also depicted in Fig. 7. The same LMOs are involved in both processes, one describing the H2C–Pt bond and another corresponding to the B–H bond. Similarly to 3c, in the initial structures 3a-cis and 3a-cis′ the centroid of the B–H bond is displaced and very close to the H center, pointing to the hydride character of this atom. In the formation of the C–H bond the centroid of the C–Pt bond moves from the C–Pt axis to the C–H axis, while the centroid of the B–H bond moves from the B–H axis to the B–Pt axis. This corresponds to a sigma-CAM type reaction,26 in which, formally, a proton is being transferred to the methylene fragment and a boryl moiety is being formed. Conversely, in the formation of the C–B bond the centroid of the C–Pt bond moves from the C–Pt axis to the B–C axis, while the centroid of the B–H bond moves from the B–H axis to the H–Pt axis (again, a sigma-CAM rearrangement). Formally, a borinium27 is being transferred to the methylene fragment and a metal-hydride is being formed. Therefore, the possibility to generate both boryl and borinium species from a platinum σ-BH complex is the underlying reason that allows both C–B and C–H bond coupling processes to take place in this system.
 | ||
| Fig. 7 C–H (top) and C–B (bottom) bond forming events from the analysis of the movements of the centroids of the localized orbitals, represented by small purple dots. Isosurfaces of the localized orbitals involved in the bond-breaking/bond-forming processes are also shown. For each process the deduced arrow-pushing scheme is also shown. | ||
3. Conclusion
The low-electron count Pt(II) complex [Pt(ItBuiPr′)(ItBuiPr)][BArF], 1, reacts with tri-coordinated boranes in a set of reactions that start with formation of σ-BH complexes, 3, in which the interaction of the B–H fragment with the cationic platinum center adopts a rare η1-BH coordination mode. These species evolve, as a function of temperature, by either formation of hydride complexes [Pt(H)(ItBuiPr′-BR2)(ItBuiPr)][BArF], 4, or platinum boryl derivatives [Pt(BR2)(ItBuiPr)2][BArF], 2. We were able to detect species 3, 4 and 2 by means of low temperature NMR spectroscopy and, in some cases, by X-ray diffraction, providing snapshots of this unusual reactivity. DFT-based analyses support σ-complex assisted metathesis mechanisms (σ-CAM),26 preceded by trans to cis isomerization of the NHC ligands. These calculations indicate that carbon-boron coupling products are energetically more accessible and that the competitive process leading to C–H bond coupling is more energy demanding. However, as the temperature increases, the thermodynamic stability of the final products determines the formation of the C–H bond coupling products. Steric effects between the substituents at the boryl fragment and the spectator NHC ligand in complexes 3a-cis and 3a-cis′ might be behind the different energetic barriers. These results contribute to the mechanistic understanding of the factors that can lead to productive borylation and advance the design of new effective catalytic systems for such reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support (FEDER contribution) from the MINECO (Projects CTQ2016-76267-P, CTQ2017-87889-P, PID2019-109312GB-I00 and RED2018-102387-T), the CSIC (AEPP_ CTQ2016-76267-P) and the use of computational facilities of the Supercomputing Center of Galicia (CESGA) is gratefully acknowledged. P. R. thanks the Junta de Andalucía for a research grant.Notes and references
- (a) J. V. Obligacion and P. J. Chirik, Nat. Rev. Chem., 2018, 2, 15–34 CrossRef CAS; (b) M. Oestreich, E. Hartmann and M. Mewald, Chem. Rev., 2013, 113, 402–441 CrossRef CAS; (c) I. Beletskaya and C. Moberg, Chem. Rev., 2006, 106, 2320–2354 CrossRef CAS; (d) T. Ishiyamaa and N. Miyaura, Chem. Rec., 2004, 3, 271–280 CrossRef; (e) K. Burgess and M. J. Ohlmeyer, Chem. Rev., 1991, 91, 1179–1191 CrossRef CAS; (f) T. B. Marder and N. C. Norman, Topics in Catal, ed. W. Leitner and D. G. Blackmond, Baltzer Science Publishers, Amsterdam, 1998, vol. 5, pp. 63−73 Search PubMed.
- (a) Synthesis and Applications of Organoboron Compounds, in Topics in Organometallic Chemistry, ed. E. Fernández and A. Whiting, Springer, Heidelberg, 2015 Search PubMed; (b) Boronic acids: preparation and applications in organic synthesis, medicine and materials, ed. D. G. Hall, Wiley-VCH, Weinheim, 2nd edn, 2011, vol. 1 and 2 Search PubMed.
- (a) J. F. Hartwig, K. S. Cook, M. Hapke, C. D. Incarvito, Y. Fan, C. E. Webster and M. B. Hall, J. Am. Chem. Soc., 2005, 127, 2538–2552 CrossRef CAS; (b) C. E. Webster, Y. Fan, M. B. Hall, D. Kunz and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 858–859 CrossRef CAS.
- (a) A. Ros, R. Fernández and J. M. Lassaletta, Chem. Soc. Rev., 2014, 43, 3229–3243 RSC; (b) J. F. Hartwig, Acc. Chem. Res., 2012, 45, 864–872 CrossRef CAS; (c) J. F. Hartwig, Chem. Soc. Rev., 2011, 40, 1992–2002 RSC; (d) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS; (e) T. Ishiyama and N. Miyaura, J. Organomet. Chem., 2003, 680, 3–11 CrossRef CAS.
- (a) C. M. Kelly, J. T. Fuller III, C. M. Macaulay, R. McDonald, M. J. Ferguson, S. M. Bischof, O. L. Sydora, D. H. Ess, M. Stradiotto and L. Turculet, Angew. Chem., Int. Ed., 2017, 56, 6312–6316 CrossRef CAS; (b) T. Furukawa, M. Tobisu and N. Chatani, Bull. Chem. Soc. Jpn., 2017, 90, 332–342 CrossRef CAS; (c) T. Furukawa, M. Tobisu and N. Chatani, J. Am. Chem. Soc., 2015, 137, 12211–12214 CrossRef CAS.
- (a) M. A. Esteruelas, A. M. López and M. Oliván, Chem. Rev., 2016, 116, 8770–8847 CrossRef CAS; (b) I. M. Riddlestone, J. A. B. Abdalla and S. Aldridge, Adv. Organomet. Chem., 2015, 63, 1–38 CrossRef CAS; (c) G. Alcaraz, M. Grellier and S. Sabo-Etienne, Acc. Chem. Res., 2009, 42, 1640–1649 CrossRef CAS; (d) Z. Lin, Transition Metal σ-borane complexes, in Contemporary metal boron chemistry I: borylenes, boryls, borane σ-complexes and borohydrides, ed. T. B. Marder and Z. Lin, Springer, 2008, vol. 130, pp. 123−148 Search PubMed.
- (a) A. Cassen, Y. Gloaguen, L. Vendier, C. Duhayon, A. Poblador-Bahamonde, C. Raynaud, E. Clot, G. Alcaraz and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2014, 53, 7569–7573 CrossRef CAS; (b) C. Y. Tang, A. L. Thompson and S. Aldridge, J. Am. Chem. Soc., 2010, 132, 10578–10591 CrossRef CAS.
- (a) M. Roselló-Merino, R. J. Rama, J. Díez and S. Conejero, Chem. Commun., 2016, 52, 8389–8392 RSC; (b) M. Roselló-Merino, J. López-Serrano and S. Conejero, J. Am. Chem. Soc., 2013, 135, 10910–10913 CrossRef.
- P. Ríos, H. Fouilloux, P. Vidossich, J. Díez, A. Lledós and S. Conejero, Angew. Chem., Int. Ed., 2018, 57, 3217–3221 CrossRef.
- M. Brookhart, M. L. H. Green and G. Parkin, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6908–6914 CrossRef CAS.
- (a) H. Braunschweig, K. Radacki and K. Uttinger, Chem.–Eur. J., 2008, 14, 7858–7866 CrossRef CAS; (b) H. Braunschweig, P. Brenner, A. Müller, K. Radacki, D. Rais and K. Uttinger, Chem.–Eur. J., 2007, 13, 7171–7176 CrossRef CAS; (c) H. Braunschweig, K. Radacki, D. Rais and D. Scheschkewitz, Angew. Chem., Int. Ed., 2005, 44, 5651–5654 CrossRef CAS.
- G. Alcaraz and S. Sabo-Etienne, Coord. Chem. Rev., 2008, 252, 2395–2409 CrossRef CAS.
- (a) J. C. Babón, M. A. Esteruelas, I. Fernández, A. M. López and E. Oñate, Inorg. Chem., 2018, 57, 4482–4491 CrossRef; (b) M. A. Esteruelas, I. Fernández, C. García-Yebra, J. Martín and E. Oñate, Organometallics, 2017, 36, 2298–2307 CrossRef CAS; (c) K. K. Pandey, Inorg. Chem. Commun., 2008, 11, 288–292 CrossRef CAS.
- (a) A. Kumar, N. A. Beattie, S. D. Pike, S. A. Macgregor and A. S. Weller, Angew. Chem., Int. Ed., 2016, 55, 6651–6656 CrossRef CAS; (b) M. C. MacInnis, R. McDonald, M. J. Ferguson, S. Tobisch and L. Turculet, J. Am. Chem. Soc., 2011, 133, 13622–13633 CrossRef CAS; (c) G. Alcaraz, L. Vendier, E. Clot and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2010, 49, 918–920 CrossRef CAS; (d) C. Y. Tang, A. L. Thompson and S. Aldridge, Angew. Chem., Int. Ed., 2010, 49, 921–925 CrossRef CAS.
- K. K. Pandey, J. Mol. Struct.: THEOCHEM, 2008, 855, 18–26 CrossRef CAS.
- Different functionals and basis sets have been employed in these calculations, and no BCP between B and Pt has been observed in any case (see ESI† for more details).
- (a) H. Braunschweig, K. Kraft, T. Kupfer and E. Siedler, Z. Anorg. Allg. Chem., 2010, 636, 2565–2567 CrossRef CAS; (b) T. J. Hebden, M. C. Denney, V. Pons, P. M. B. Piccoli, T. F. Koetzle, A. J. Schultz, W. Kaminsky, K. I. Goldberg and D. M. Heinekey, J. Am. Chem. Soc., 2008, 130, 10812–10820 CrossRef CAS; (c) M. G. Crestani, M. Muñoz-Hernández, A. Arévalo, A. Acosta-Ramírez and J. J. García, J. Am. Chem. Soc., 2005, 127, 18066–18073 CrossRef CAS; (d) V. Montiel-Palma, M. Lumbierres, B. Donnadieu, S. Sabo-Etienne and B. Chaudret, J. Am. Chem. Soc., 2002, 124, 5624–5625 CrossRef CAS; (e) S. Schlecht and J. F. Hartwig, J. Am. Chem. Soc., 2000, 122, 9435–9443 CrossRef CAS; (f) J. F. Hartwig, C. N. Muhoro and X. He, J. Am. Chem. Soc., 1996, 118, 10936–10937 CrossRef CAS.
- G. Sciortino, A. Lledós and P. Vidossich, Dalton Trans., 2019, 48, 15740–15752 RSC.
- P. Ríos, H. Fouilloux, J. Díez, P. Vidossich, A. Lledós and S. Conejero, Chem.–Eur. J., 2019, 25, 11346–11355 Search PubMed.
- Likely, a dynamic low energy barrier process (4.9 kcal mol-1 from 4a to Int2; see Figure 6) involving de-coordination/coordination of the O atom from platinum is responsible for the NMR equivalence of these CH2 protons.
- M. Ortuño, S. Conejero and A. Lledós, Beilstein J. Org. Chem., 2013, 9, 1352–1382 CrossRef.
- For some examples of reversible C–B bond formation processes in organoboron compounds see: (a) Z.-C. He, S. K. Mellerup, L. Liu, X. Wang, C. Dao and S. Wang, Angew. Chem., Int. Ed., 2019, 58, 6683–6687 CrossRef CAS; (b) S. K. Mellerup, C. Li, X. Wang and S. Wang, J. Org. Chem., 2018, 83, 11970–11977 CrossRef CAS; (c) A. F. Eichhorn, S. Fuchs, M. Flock, T. B. Marder and U. Radius, Angew. Chem., Int. Ed., 2017, 56, 10209–10213 CrossRef CAS; (d) B. E. Cowie and D. J. H. Emslie, Organometallics, 2015, 34, 2737–2746 CrossRef CAS; (e) D.-T. Yang, S. K. Mellerup, X. Wang, J.-S. Lu and S. Wang, Angew. Chem., Int. Ed., 2015, 56, 5498–5501 CrossRef.
- G. C. Fortman, N. M. Scott, A. Linden, E. D. Stevens, R. Dorta and S. P. Nolan, Chem. Commun., 2010, 46, 1050–1052 RSC.
- O. Rivada-Wheelaghan, M. Rosellό-Merino, M. A. Ortuño, P. Vidossich, E. Gutiérrez-Puebla, A. Lledόs and S. Conejero, Inorg. Chem., 2014, 53, 4257–4268 CrossRef CAS.
- M. A. Ortuño, P. Vidossich, S. Conejero and A. Lledós, Angew. Chem., Int. Ed., 2014, 53, 14158–14161 CrossRef.
- R. N. Perutz and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2007, 46, 2578–2592 CrossRef CAS.
- (a) D. Franz and S. Inoue, Chem.–Eur. J., 2019, 25, 2898–2926 CrossRef CAS; (b) W. E. Piers, S. C. Bourke and K. D. Conroy, Angew. Chem., Int. Ed., 2005, 44, 5016–5036 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, spectroscopic, computational data (PDF). X-ray crystallographic data (CIF). Cartesian coordinates of the selected DFT optimized structures (XYZ). CCDC 2033931–2033934. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc05522k |
| This journal is © The Royal Society of Chemistry 2021 |