
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntegrating hydrogen production with anodic selective oxidation of sulfides over a CoFe layered double hydroxide electrode†
Lina
Ma‡
 a,
Hua
Zhou‡
b,
Ming
Xu
a,
Peipei
Hao
a,
Xianggui
Kong
a and
Haohong
Duan
*b
a,
Hua
Zhou‡
b,
Ming
Xu
a,
Peipei
Hao
a,
Xianggui
Kong
a and
Haohong
Duan
*b
aState Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing 100029, China
bDepartment of Chemistry, Tsinghua University, Beijing 100084, China. E-mail: hhduan@mail.tsinghua.edu.cn
First published on 11th November 2020
Abstract
Replacing the sluggish oxygen evolution reaction (OER) with oxidation reactions for the synthesis of complex pharmaceutical molecules coupled with enhanced hydrogen evolution reaction (HER) is highly attractive, but it is rarely explored. Here, we report an electrochemical protocol for selective oxidation of sulfides to sulfoxides over a CoFe layered double hydroxide (CoFe-LDH) anode in an aqueous-MeCN electrolyte, coupled with 2-fold promoted cathodic H2 productivity. This protocol displays high activity (85–96% yields), catalyst stability (10 cycles), and generality (12 examples) in selective sulfide oxidation. We demonstrate its applicability in the synthesis of four important pharmaceutical related sulfoxide compounds with scalability (up to 1.79 g). X-ray spectroscopy investigations reveal that the CoFe-LDH material evolved into amorphous CoFe-oxyhydroxide under catalytic conditions. This work may pave the way towards sustainable organic synthesis of valuable pharmaceuticals coupled with H2 production.
Introduction
Electrochemical water splitting is considered to be a promising hydrogen production approach to deal with the increasing global energy demand and environmental problems associated with fossil fuel utilization.1 Water electrolysis involves two half-reactions, including H2 and O2 evolution reactions (HER and OER).2 The overall reaction rate is often restricted by the anodic OER because of its more sluggish kinetics.2c As a result, much higher overpotential is needed for the OER to match the H2 production rate, thereby undermining the overall energy conversion efficiency.3 Although advanced non-noble-metal OER electrocatalysts have been developed, accomplishing an OER efficiency comparable with that of the HER still remains a challenge.4 In addition, the economic value of O2 is far inferior to that of H2. Recently, electrooxidation of low-cost organic agents has emerged as an alternative strategy to replace the OER, achieving lower overpotential for total water splitting and at the same time producing high-value chemicals.5 Electrooxidation has been exploited for converting biomass-derived platform chemicals, e.g., ethanol, glycerol, and 5-hydroxymethyfurfural (HMF).6 Despite the advanced concept of co-production of valuable chemicals and H2, only a handful of simple molecules were studied.6f,g The anodic synthesis of functional and complex organic molecules, especially pharmaceuticals with medicinal significance, has rarely been explored.In the family of pharmaceuticals, organosulfur compounds play essential roles in many biological processes linked with human disease therapy.7 For example, sulforaphane offers efficient chemoprotection against various cancers (e.g. prostate, lung, breast, and colon cancer).7g Direct oxidation of their sulfide precursors is a straightforward and atom-economical synthetic method, and thus has been widely applied for the synthesis of sulfoxides.8 However, current synthetic methods often require homogeneous catalysts (e.g. Mn- and Fe-based catalysts) and strong oxidizing agents such as H2O2.9 Additional oxidants such as peroxy acids, prevalent iodine reagents or oxone are also needed.10 In 2012, He and co-workers reported a catalyst-free protocol for the selective oxidation of sulfides with an inorganic oxidant oxone in ethanol.11 However, the utilization of oxone may result in purification issues and environmental contamination. More importantly, over-oxidation of sulfoxide to sulfone or co-oxidation of other functional groups in the sulfoxide molecule would occur and generate medically useless products. Recently, Xu and co-workers reported an efficient heterogeneous polyoxovanadate catalyst for sulfide oxidation; nevertheless the reaction was assisted by a strong oxidant (tert-butylhydroperoxide (TBHP)) at elevated temperatures.12 Jiang's group reported a photocatalysis strategy to achieve sulfide selective oxidation driven by visible light under mild conditions, but the use of homogeneous catalysts (UO2(OAc)2·2H2O) would make separation difficult.12 Therefore, the selective oxidation of sulfides using heterogeneous catalysts under ambient conditions remains challenging.
Electrochemical oxidation by using heterogeneous electrocatalysts shows promise to achieve selective sulfide oxidation under mild conditions while avoiding separation issues. Recently, an environmentally benign electrochemical oxidation of sulfides was reported by Laudadio and colleagues, who evaluated the electrocatalysis with continuous reactors that shows great promise for practical applications.13 Non-noble metal-based layered double hydroxides (LDHs), a class of inorganic layered materials with unique 2D structures in which di- and tri-valence cations are dispersed within the layers at the atomic level,14 have emerged as efficient electrocatalysts for the OER15 and anodic organic agents for electrooxidation, for example converting HMF to value-added 2,5-furandicarboxylic acid (FDCA).16 However, there is no report of developing LDHs as the electrocatalyst for sulfide selective oxidation with the goal of synthesis of sulfoxide-containing pharmaceuticals.
In this regard, we report an electrochemical approach for selective oxidation of sulfides to replace the OER over CoFe layered double hydroxides supported on carbon cloth (CoFe-LDH/CC) as the anode in an aqueous-MeCN electrolyte, coupled with 2-fold promoted cathodic H2 productivity (Fig. 1a). A variety of aryl, heteroaryl and alkyl sulfides at the CoFe-LDH/CC anode can be selectively converted into the corresponding sulfoxides with 85–96% yields under ambient conditions (Fig. 1b). Importantly, this method was successfully extended to the synthesis of complex pharmaceutical compounds with sulfoxide moieties from their sulfide precursors in good yields, including ricobendazole (78%), omeprazole (70%), sulindac (63%) and amino acid methionine (89%). Moreover, the catalyst CoFe-LDH/CC was used in the gram-scale synthesis of diphenyl sulfoxide with 83% yield (up to 1.63 g) and amino acid methionine with 86% yield (up to 1.79 g). Preliminary investigations suggest that the in situ formed amorphous metal oxyhydroxide acts as the active species for selective oxidation of sulfides to sulfoxides via a radical process.
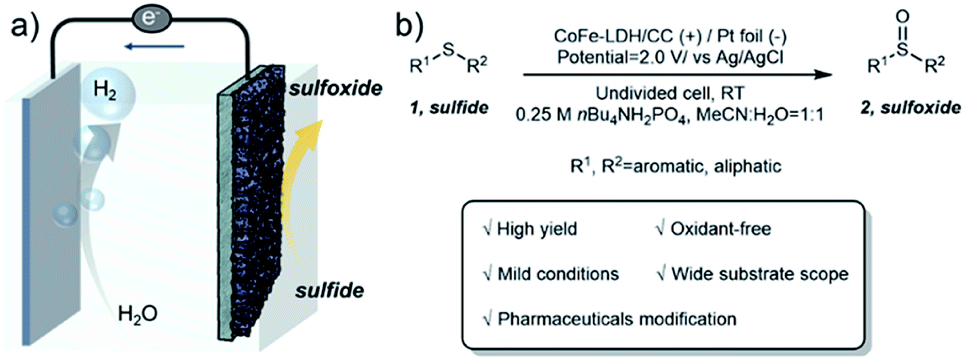 | ||
| Fig. 1 (a) Electrocatalytic oxidation of sulfides 1 coupled with the HER. (b) Oxidation of sulfides 1 to form sulfoxides 2. | ||
Results and discussion
Initially, CoFe-LDH/CC was fabricated by a facile electrodeposition method (see the ESI† for details).17 As illustrated in Fig. 2a, Co and Fe nitrate precursors were converted to CoFe hydroxide by electrolysis with the formation of a LDH nanoarray grown on a carbon cloth cathode. As shown in the scanning electron microscope (SEM) and transmission electron microscope (TEM) images (Fig. 2b and c, ESI, Fig. S1a†), the cross-linked LDH nanosheets are homogeneously grown on CC with an average thickness of 9.4 nm (ESI, Fig. S1b and c†). Meanwhile, the atomic Co/Fe ratio is 3.01 as determined by inductively coupled plasma-atomic emission spectrometry (ICP-AES, ESI, Table S1†), which is consistent with the stoichiometry of the introduced values. The energy dispersive spectrometry (EDS) mapping technique reveals the uniform dispersion of Co and Fe species in the nanosheet (Fig. 2d). A high-resolution TEM image (HRTEM, Fig. 2e) shows that the interplanar spacing is approximately 0.26 nm, which is assigned to the (012) plane of the CoFe-LDH phase.18 The characteristic (003), (006), (012), and (110) planes of the LDH structure were also observed in the selected area electron diffraction (SAED) pattern of the nanosheet (Fig. 2f). Consistently, these diffractions were observed in the X-ray diffraction (XRD) pattern of the CoFe-LDH/CC (Fig. 2g). | ||
| Fig. 2 (a) Schematic illustration of the preparation of the CoFe-LDH nanoarray supported on carbon cloth. (b) SEM, (c) HAADF-STEM, (d) EDS mapping, (e) high-resolution TEM, (f) SAED pattern, and (g) XRD pattern of the CoFe-LDH/CC material. | ||
The selective electrooxidation of diphenyl sulfide (1a) was chosen as a model reaction to evaluate the activity of CoFe-LDH/CC and to establish the optimal reaction conditions. In the reaction, diphenyl sulfoxide (2a) was the desired product, and diphenyl sulfone (3a) was the over-oxidized byproduct. To investigate the effect of the applied voltage, the potential was increased from 2.0 to 3.5 V vs. Ag/AgCl, resulting in the yield of over-oxidized byproduct 3a increasing from trace to 30%. This result suggests that the relatively low potential was a key factor for inhibiting the formation of over-oxidized byproducts, in agreement with the previous literature.13 After extensive optimization of the electrolyte, solvent, cathode and atmosphere (ESI, Table S2†), we found that upon using MeCN/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) as the mixed solvent and nBu4NH2PO4 as the electrolyte, the desired product 2a was obtained in 85% yield and 85% faradaic efficiency (FE) in 2 h at room temperature (entry 1 of Table 1), which represents a high performance compared to existing homo- and heterogeneous catalysts (ESI, Table S3†). Specifically, H2O was initially tested as the solvent for its wide use in the OER and anodic oxidations, but moderate conversion and yield of 2a were obtained, accompanied by overoxidation (entry 2, Table 1). Replacing it by MeCN or other organic solvents showed even lower activities (entry 3 in Table 1, entries 1–4 in the ESI, Table S2†). We found that using a mixed solvent of MeCN and H2O could furnish significantly higher efficacy, which is likely due to MeCN showing higher conductivity and capacity to dissolve the electrolyte, substrate, and reactants compared with other employed solvents, which may lead to catalytic enhancement (entries 5–6 in the ESI, Table S2†).19 The use of electrolytes other than nBu4NH2PO4 showed lower activity (entries 7–9 in the ESI, Table S2†), which is due to the promotion effect of nBu4NH2PO4 for the oxidation reaction.12b In addition, the yield of 2a didn't show obvious change when the reaction atmosphere was varied from air to N2 (entry 4 in Table 1), indicating that H2O in the electrolyte could likely be the source of oxygen in 2a. It should be noted that under our reaction conditions in air, it remains possible that oxygen in the product can be from the air. A blank experiment using CC as the anode showed much lower conversion for sulfide oxidation, suggesting CoFe-LDH/CC was essential for catalytic activity of sulfide oxidation (entry 5 in Table 1). Finally, no reaction occurred in the absence of electric current, demonstrating the nature of electrocatalysis (entry 6 in Table 1). Pt ions were not observed in the electrolyte after electrooxidation by using an Inductive Coupled Plasma Emission Spectrometer (ICP), indicating that Pt was not leached into the electrolyte. According to the conversion of 1a and total yields of products 2a (or with 3a), there remained other byproducts while their structures can't be recognized as no other signals were observed by crude NMR analysis.
:1 v/v) as the mixed solvent and nBu4NH2PO4 as the electrolyte, the desired product 2a was obtained in 85% yield and 85% faradaic efficiency (FE) in 2 h at room temperature (entry 1 of Table 1), which represents a high performance compared to existing homo- and heterogeneous catalysts (ESI, Table S3†). Specifically, H2O was initially tested as the solvent for its wide use in the OER and anodic oxidations, but moderate conversion and yield of 2a were obtained, accompanied by overoxidation (entry 2, Table 1). Replacing it by MeCN or other organic solvents showed even lower activities (entry 3 in Table 1, entries 1–4 in the ESI, Table S2†). We found that using a mixed solvent of MeCN and H2O could furnish significantly higher efficacy, which is likely due to MeCN showing higher conductivity and capacity to dissolve the electrolyte, substrate, and reactants compared with other employed solvents, which may lead to catalytic enhancement (entries 5–6 in the ESI, Table S2†).19 The use of electrolytes other than nBu4NH2PO4 showed lower activity (entries 7–9 in the ESI, Table S2†), which is due to the promotion effect of nBu4NH2PO4 for the oxidation reaction.12b In addition, the yield of 2a didn't show obvious change when the reaction atmosphere was varied from air to N2 (entry 4 in Table 1), indicating that H2O in the electrolyte could likely be the source of oxygen in 2a. It should be noted that under our reaction conditions in air, it remains possible that oxygen in the product can be from the air. A blank experiment using CC as the anode showed much lower conversion for sulfide oxidation, suggesting CoFe-LDH/CC was essential for catalytic activity of sulfide oxidation (entry 5 in Table 1). Finally, no reaction occurred in the absence of electric current, demonstrating the nature of electrocatalysis (entry 6 in Table 1). Pt ions were not observed in the electrolyte after electrooxidation by using an Inductive Coupled Plasma Emission Spectrometer (ICP), indicating that Pt was not leached into the electrolyte. According to the conversion of 1a and total yields of products 2a (or with 3a), there remained other byproducts while their structures can't be recognized as no other signals were observed by crude NMR analysis.
|
|
||||
|---|---|---|---|---|
| Entry | Deviation from standard conditions | Conv.a (%) | Yield 2aa (%) | Yield 3aa (%) |
|
a Conversion and yields were determined by 1H NMR using CH2Br2 as an internal standard. nd = not detected. The isolated yields are given in parentheses. Standard reaction conditions and variations on the electrochemical oxidation of diphenyl sulfide: 1a (0.5 mmol), nBu4NH2PO4 (0.5 mmol), MeCN/H2O (2.0 mL, 1:1 v/v), RT, 2 h, CoFe-LDH anode (working area: 1 cm2), Pt cathode, applied potential: 2.0 V vs. Ag/AgCl (passing charge of about 96C), under air, unless otherwise noted.
|
||||
| 1 | None | >99 | 85(81) | Trace |
| 2 | H2O as solvent | 39 | 35(33) | 2 |
| 3 | MeCN as solvent | 12 | Trace | nd |
| 4 | N2 atmosphere | >99 | 81(81) | Trace |
| 5 | CC as the anode | 39 | 11(10) | Trace |
| 6 | No electric current | <5 | nd | nd |

In the following catalytic studies, the catalytic reactions were examined over the as-synthesized CoFe-LDH/CC under the optimized conditions. Fig. 3a shows the linear sweep voltammetry (LSV) curves in the presence and absence of 1a over the CoFe-LDH/CC anode. After adding 1a, the current density significantly increases, and the required potential for gaining current density of 5 mA cm−2 dramatically decreased from 1.90 V (without 1a) to 1.39 V (with 1a). The desired product 2a increased with the time, and to our delight, the overoxidized product (sulfone 3a) was rarely observed (<5% yield, Fig. 3b). More importantly, 2a can be obtained on a gram scale with 83% yield (up to 1.63 g) (for details, see Experimental procedures in the ESI†). We then assembled a two-electrode setup in which CoFe-LDH/CC was used as the anode for 1a oxidation and Pt as the cathode for the HER. Impressively, it shows two-fold higher activity for H2 production compared with the one without 1a that follows the OER process (Fig. 3c), indicative of enhanced energy efficiency. To a certain extent, the organic solvent MeCN has some slight effect on the faradaic efficiency of H2 (90%), lower than the faradaic efficiency of 96% in aqueous electrolyte. After ten-cycle runs, no apparent decrease of activity was observed (Fig. 3d), indicative of promising catalytic stability. The structural stability of the used CoFe-LDH/CC anode was also demonstrated (ESI, Fig. S2†).
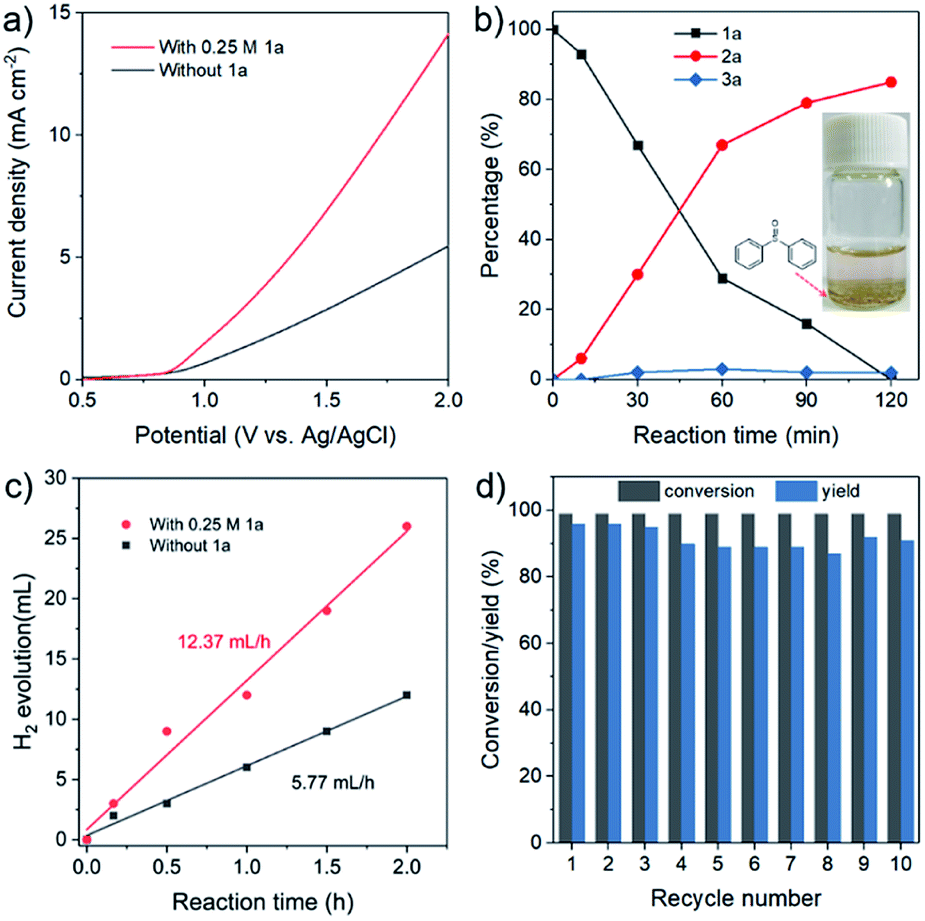 | ||
| Fig. 3 (a) LSV curves of the CoFe-LDH/CC anode at a scan rate of 50 mV s−1 in MeCN/H2O (2.0 mL, 1:1 v/v) with and without 0.5 mmol 1a. (b) Tracking experiments of the sulfoxidation procedure. (c) H2 production with and without 1a. (a–c) CoFe-LDH/CC anode and Pt cathode. (d) Cycle-dependent conversions for (methylsulfinyl)benzene. | ||
Subsequently, the generality of the electrocatalytic selective oxidation using CoFe-LDH/CC was studied by using 12 examples (Fig. 4). In general, the sulfides and their derivatives with functional groups including –H, –F, –Cl and –Br worked very well to produce sulfoxides (2a–2g) under the optimized conditions. The reaction efficiency was not affected by steric hindrance (2d, 2f and 2g). A multi-substituted sulfoxide (2h) was also produced efficiently. Aliphatic and benzyl groups could be tolerated, and target products 2i and 2j were furnished in 91% and 86% yields, respectively. Notably, nitrogen-containing hetero-aromatics can be selectively oxidized to form the corresponding sulfoxide (2k) without N-oxide formation. Finally, alkyl sulfoxide (2l) was well tolerated under our reaction conditions, given that it is often sensitive to oxygenation conditions.13
 | ||
| Fig. 4 Substrate scope for the formation of sulfoxides. Yields were determined by 1H NMR using CH2Br2 as an internal standard. The isolated yields were given in parentheses. | ||
The late-stage oxygenation of complex molecules to construct sulfoxides is often accompanied by the existence of various functional groups, and maintaining these functional groups is very important but remains challenging. By using our selective method, we expect that complex pharmaceuticals can be precisely modified at the last stage, in which sulfide is oxidized to sulfoxide while the side reactions on other functional groups are avoided. As a proof-of-concept, we focused on four important drug molecules with sulfoxide moieties (Fig. 5). Specifically, the corresponding sulfides were synthesized first by the reported procedures (for Experimental procedures see the ESI†), which was followed by electrochemical oxidation to the targeted sulfoxides by using CoFe-LDH/CC as the anode under the optimized conditions. The catalytic results show that, ricobendazole20 (2m), an effective drug for a potential anticancer agent, was obtained by selective oxidation in 78% yield. Omeprazole20 (2n), a well-known drug for treating gastroesophageal reflux, was also obtained in 70% yield. Sulindac20 (2o), another anticancer reagent, was generated with 63% yield even in the presence of oxygen-sensitive sequential conjugated alkenes. Amino acid methionine (2p),20 a biologically relevant compound, could be efficiently synthesized in good yield. Moreover, this electrochemical protocol could be used in the gram-scale synthesis of sulfoxides (2p) with 1.79 g and high yield of 86% was still obtained. Therefore, this protocol potentially enables access to electrochemical synthesis of valuable pharmaceutical related sulfoxides without external oxidants. In addition, the CoFe-LDH/CC electrocatalyst potentially overcomes disadvantages, such as catalyst reusability, which are often encountered by using homogeneous catalysts in conventional methods.
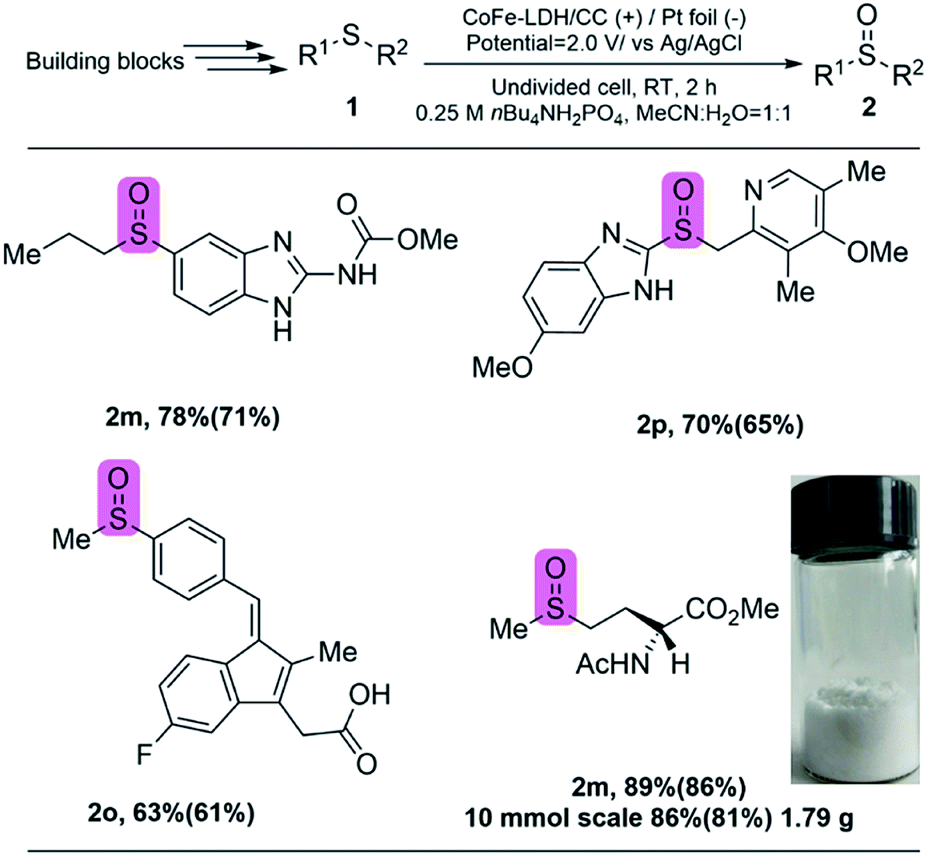 | ||
| Fig. 5 Synthesis of existing pharmaceuticals: showcasing the applicability of this protocol by integrating with current procedures. Yields were determined by 1H NMR using CH2Br2 as an internal standard. The isolated yields are given in parentheses. 2m, ricobendazole. 2n, omeprazole. 2o, sulindac. 2p, amino acid methionine. The sulfoxide moieties in the pharmaceuticals are highlighted in pink color. | ||
In order to understand the reaction mechanism, a set of control experiments were carried out (Fig. 6). When the standard reaction was performed in the presence of 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) or butylated hydroxytoluene (BHT) as the radical scavengers,21 the reaction was completely inhibited (Fig. 6a). According to a study on oxygenation of sulfides,12b,21 the sulfide radical (Ph2S˙) or persulfoxide radical (Ph2SOO˙) was generally present in the process. The EPR experiments were designed to detect radical intermediates by adding the radical trapping agent DMPO (5,5-dimethyl-1-pyrroline N-oxide). No obvious signals could be observed in the absence of 1a (Fig. 6b, black line). Under the standard conditions, a typical signal of the DMPO-radical adduct was determined by the EPR during reaction (Fig. 6b, red line). We speculated that it might be a sulfide radical (Ph2S˙) or persulfoxide radical (Ph2SOO˙) generated in the reaction with the DMPO adduct.22 These findings suggest that the electrooxidation reaction over CoFe-LDH/CC may involve a radical process. To confirm the source of the oxygen atom in the formed product, oxidation of 1a was carried out in MeCN–H218O electrolyte, and the GC mass spectrum analysis shows that one 18O atom was introduced into 2a (Fig. 6c). This result suggests that water could likely supply oxygen for the sulfoxide oxidation under our reaction conditions.
 | ||
| Fig. 6 (a) Radical trapping experiments, (b) EPR spectra of the DMPO-adduct under different conditions, (c) mass spectra of 2a from isotope labelling experiments with H2O or H218O, respectively. | ||
To gain an insight into the active species in this catalytic system, the structural evolution of the CoFe-LDH/CC anode was in-depth investigated using XRD, TEM, SAED, X-ray photoelectron spectroscopy (XPS), and X-ray absorption fine structure spectroscopy (XAFS).23 As shown in Fig. 7a, the XRD pattern of the used CoFe-LDH/CC (1st recycled CoFe-LDH/CC) shows only two broad diffraction peaks at 24.5° and 44.0°, which are attributed to carbon cloth, indicating that CoFe-LDH undergoes dramatic reconstruction to form an amorphous structure under reaction conditions. Consistently, the SAED further confirmed the amorphous nature of the used CoFe-LDH (ESI, Fig. S3†).
 | ||
| Fig. 7 (a) XRD patterns, (b) Co 2p XPS spectra, and (c) O1s XPS spectra of fresh and 1st recycled CoFe-LDH/CC. (d) Normalized Co K-edge XANES spectra and (e and f) Fourier-transform Co K-edge EXAFS spectra of reference samples, fresh and used CoFe-LDH/CC. | ||
We then investigated the structure of CoFe-LDH before and after reaction using XPS and XAFS techniques. As shown in Fig. 7b, the XPS signal of Co2p3/2 in the starting material shows a typical satellite peak at 787.7 eV and a peak at 783.0 eV attributed to CoII in LDHs. After the reaction, the intensity of the peak of Co2+ decreased with the appearance of a new peak at 780.1 eV which could be attributed to Co3+, suggesting the oxidation of Co2+ to Co3+ in the electrooxidation process.24 Concomitantly, the deprotonation of hydroxyl on CoFe-LDH occurred, giving rise to a peak at 529.1 eV in the O1s XPS spectrum of the used CoFe-LDH that can be attributed to the typical M-O-M species in metal oxyhydroxides (Fig. 7c).24,25 Meanwhile, the Fe2p XPS spectra reveal a stable FeIII state in the fresh and the used CoFe-LDH (ESI, Fig. S4†). In addition, X-ray absorption near-edge structure (XANES) analysis in Fig. 7d shows that the Co K edge profile of the used CoFe-LDH/CC moved to a higher energy and next to that of the crystalline CoFe-oxyhydroxide reference (c-CoFeOOH, obtained from CoFe-LDH/CC by anodization in 1 M KOH, ESI, Fig. S5†), indicating that some CoII ions in starting CoFe-LDH/CC were oxidized to CoIII during the reaction, which is consistent with the XPS data. In addition, the extended XAFS spectra reveal that the length of the first coordination shell C–O (2.03 Å) and the second coordination shell Co–Co(Fe) (3.13 Å) in the initial CoFe-LDH was decreased to 1.88 Å and 2.83 Å, respectively (Fig. 7e), which are identical to those of c-CoFeOOH, reflecting its metal oxyhydroxide nature. This structural transformation during electrooxidation was induced by oxidation of CoII ions to CoIII.26 Notably, the intensities of both Co–O and Co–Co(Fe) in the used CoFe-LDH are lower than those of fresh CoFe-LDH and c-CoFeOOH in the spectra of wavelet transformed EXAFS (ESI, Fig. S6†), owing to its amorphous structure. Moreover, the EXAFS analysis show that the as-formed oxyhydroxide structure was stable after four cycle reactions (Fig. 7f). We can conclude that the starting CoFe-LDH/CC material evolved into the corresponding amorphous oxyhydroxide during electrooxidation as the real active species. In addition, SCN−, often adopted to poison metal sites,27 was used as an indicator for active sites. As shown in Fig. S7,† a remarkable decrease of 2a yield was observed by adding SCN−, thus indicating that the metals in CoFe-LDH contribute to the catalytic activity. As an electrochemical reaction takes place at the interface between the catalyst and electrolyte, we evaluated the stability of CoFe-LDH for 7 h using chronoamperometry (CA) at 2.0 V versus the Ag/AgCl electrode (Fig. S8†). A current densityof ∼11 mA cm−2 was retained, indicating the stability of the catalyst over long reaction times.
On the basis of our results and the previous literature,6d,28 a reaction mechanism of this catalytic system is proposed in Fig. 8. In the neutral aqueous-organic electrolyte (MeCN as the organic phase), water molecules undergo Volmer and Heyrovsky reactions to produce H2 and hydroxide ions (OH−) over the Pt cathode under work potential bias. Meanwhile, the starting CoFe-LDH was oxidized and evolved into amorphous CoFe-oxyhydroxide via the deprotonation process with the assistance of OH− very fast in a few minutes (<5 minutes), and it may complete as soon as the reaction starts, as demonstrated by the anode color change and XRD and XPS results (Fig. S9†), ensuring the maintenance of its activity during the reaction time course. We synthesized reference materials (Co(OH)2/CC and Fe(OH)3/CC) and compared their catalytic activity with that of CoFe-LDH/CC. As shown in Fig. S10a,† Co(OH)2/CC showed much higher activity than Fe(OH)3/CC, suggesting that the Co(OH)2-derived structure is the main active phase. The CoFe-LDH/CC exhibits the highest performance among the three catalysts, indicating that a synergistic effect exists between Co and Fe in this reaction. The Co2p3/2 XPS spectra of the used CoFe-LDH/CC, the derived CoFeOOH structure, exhibits a 0.43 eV shift to higher binding energy compared with that of the used Co(OH)2/CC (Fig. S10b†), indicating the modulation of the local electronic structure of Co cations in the presence of Fe species. Because of the modulated electronic structure, the redox behavior of Co cations could probably be manipulated by Fe incorporation, leading to the active phases in sulfide electrooxidation with higher activity, which is in agreement with recent reports.29 This amorphous oxyhydroxide might act as the active phase for selective sulfide oxidation using water as the oxygen source.
 | ||
| Fig. 8 Proposed reaction mechanism. | ||
Conclusions
In summary, we demonstrate an efficient electrochemical method for selective electrooxidation of sulfides with promoted HER over a CoFe-LDH/CC electrode. A variety of sulfide model compounds were selectively electrooxidized to sulfoxides in high yields (85–96%) at the anode under ambient conditions, showing high catalytic performance compared to the existing homo- and heterogeneous catalysts. Importantly, this method can be applied to the synthesis of four complex pharmaceuticals with sulfoxide moieties from their sulfide precursors in good yields, including ricobendazole (78%), omeprazole (70%), sulindac (63%) and amino acid methionine (89%). Moreover, CoFe-LDH/CC was used in the gram-scale synthesis of diphenyl sulfoxide with 83% yield (up to 1.63 g) and amino acid methionine with 86% yield (up to 1.79 g). In addition, the CoFe-LDH/CC electrocatalyst could be reused, maintaining the yield for more than ten cycles. Mechanistic studies indicate that the reaction pathway proceeds through a radical process, and the in situ formed CoFe-oxyhydroxide may serve as the active species for the sulfide oxidation. The efficiency of this reaction system may pave the way for the electrochemical synthesis of valuable organic molecules by using heterogeneous catalysts without external oxidants under ambient conditions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was financially supported by the National Natural Science Foundation of China (21978147, 21935001), and the China Postdoctoral Science Foundation (2019M660421). We greatly thank Profs. Duan Xue and Yadong Li for long-term support.Notes and references
- (a) H. Jin, C. Guo, X. Liu, J. Liu, A. Vasileff, Y. Jiao, Y. Zheng and S. Z. Qiao, Chem. Rev., 2018, 118, 6337–6408 CrossRef CAS; (b) S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS; (c) Y. Yang, H. L. Fei, G. D. Ruan and J. M. Tour, Adv. Mater., 2015, 27, 3175–3180 CrossRef CAS; (d) Y. Yu, Y. Shi and B. Zhang, Acc. Chem. Res., 2018, 51, 1711–1721 CrossRef CAS; (e) J. X. Feng, S. Y. Tong, Y. X. Tong and G. R. Li, J. Am. Chem. Soc., 2018, 140, 5118–5126 CrossRef CAS; (f) X. H. Gao, H. X. Zhang, Q. G. Li, X. G. Yu, Z. L. Hong, X. W. Zhang, C. D. Liang and Z. Lin, Angew. Chem., Int. Ed., 2016, 55, 6290–6294 CrossRef CAS; (g) C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS; (h) Y. Wang, D. Yan, S. E. Hankari, Y. Zou and S. Wang, Adv. Sci., 2018, 5, 1800064 CrossRef.
- (a) Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC; (b) W. J. Zhou, J. Jia, J. Lu, L. J. Yang, D. M. Hou, G. Q. Li and S. W. Chen, Nano Energy, 2016, 28, 29–43 CrossRef CAS; (c) J. Luo, J. H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N. G. Park, S. D. Tilley, H. J. Fan and M. Grätzel, Science, 2014, 345, 1593–1596 CrossRef CAS; (d) M. Gong, W. Zhou, M. C. Tsai, J. Zhou, M. Guan, M. C. Lin, B. Zhang, Y. Hu, D. Y. Wang and J. Yang, Nat. Commun., 2014, 5, 4695–4700 CrossRef CAS; (e) H. Yin, S. Zhao, K. Zhao, A. Muqsit, H. Tang, L. Chang, H. Zhao, Y. Gao and Z. Tang, Nat. Commun., 2015, 6, 6430–6437 CrossRef CAS; (f) M. W. Kanan and D. G. Nocera, Nature, 2008, 321, 1072–1075 CAS; (g) P. Zhou, Y. Wang, C. Xie, C. Chen, H. Liu, R. Chen, J. Huo and S. Wang, Chem. Commun., 2013, 53, 11778–11781 RSC.
- (a) F. Song, L. Bai, A. Moysiadou, S. Lee, C. Hu, L. Liardet and X. Hu, J. Am. Chem. Soc., 2018, 140, 7748–7759 CrossRef CAS; (b) D. S. He, D. P. He, J. Wang, Y. Lin, P. Q. Yin, X. Hong, Y. Wu and Y. D. Li, J. Am. Chem. Soc., 2016, 138, 1494–1497 CrossRef CAS; (c) H. Sun, X. Xu, Z. Yan, X. Chen, F. Cheng, P. S. Weiss and J. Chen, Chem. Mater., 2017, 29, 8539–8547 CrossRef CAS; (d) P. Zhang, X. Sheng, X. Chen, Z. Fang, J. Jiang, M. Wang, F. Li, L. Fan, Y. Ren, B. Zhang, B. Timmer, M. Ahlquist and L. Sun, Angew. Chem., Int. Ed., 2019, 58, 9155–9159 CrossRef CAS; (e) J. Hou, Y. Wu, B. Zhang, S. Cao, Z. Li and L. Sun, Adv. Funct. Mater., 2019, 29, 1908367 Search PubMed.
- (a) T. Binninger, R. Mohamed, A. Patru, K. Waltar, E. Gericke, X. Tuaev, E. Fabbri, P. Levecque, A. Hoell and T. J. Schmidt, Chem. Mater., 2017, 29, 2831–2843 CrossRef CAS; (b) G. Y. Chen, K. A. Kuttiyiel, D. Su, M. Li, C. H. Wang, D. Buceta, C. Y. Du, Y. Z. Gao, G. P. Yin, K. Sasaki, M. B. Vukmirovic and R. R. Adzic, Chem. Mater., 2016, 28, 5274–5281 CrossRef CAS; (c) K. Xu, P. Z. Chen, X. L. Li, Y. Tong, H. Ding, X. J. Wu, W. S. Chu, Z. M. Peng, C. Z. Wu and Y. Xie, J. Am. Chem. Soc., 2015, 137, 4119–4125 CrossRef CAS; (d) P. W. Cai, J. H. Huang, J. X. Chen and Z. H. Wen, Angew. Chem., Int. Ed., 2017, 56, 4858–4861 CrossRef CAS; (e) X. Zou, Y. Liu, G. D. Li, Y. Y. Wu, D. P. Liu, W. Li, H. W. Li, D. J. Wang, Y. Zhang and X. X. Zou, Adv. Mater., 2017, 29, 1700404 CrossRef; (f) T. T. Liu, D. N. Liu, F. L. Qu, D. X. Wang, L. Zhang, R. X. Ge, S. Hao, Y. J. Ma, G. Du and A. M. Asiri, Adv. Mater., 2017, 7, 1700020 Search PubMed; (g) T. T. Liu, X. Ma, D. N. Liu, S. Hao, G. Du, Y. J. Ma, A. M. Asiri, X. P. Sun and L. Chen, ACS Catal., 2017, 7, 98–102 CrossRef CAS.
- (a) B. You, X. Liu, N. Jiang and Y. Sun, J. Am. Chem. Soc., 2016, 138, 13639–13646 CrossRef CAS; (b) H. G. Cha and K.-S. Choi, Nat. Chem., 2015, 7, 328–333 CrossRef CAS; (c) Y. Zhou, Y. Gao, X. Zhong, W. Jiang, Y. Liang, P. Niu, M. Li, G. Zhuang, X. Li and J. Wang, Adv. Funct. Mater., 2019, 29, 1807651 CrossRef; (d) S. Li, X. Sun, Z. Yao, X. Zhong, Y. Cao, Y. Liang, Z. Wei, S. Deng, G. Zhuang, X. Li and J. Wang, Adv. Funct. Mater., 2019, 42, 1904780 CrossRef; (e) Q. Zhang, Y. Cao, Y. Yan, B. Yuan, H. Zheng, Y. Gu, X. Zhong and J. Wang, J. Mater. Chem. A, 2020, 8, 2336–2342 RSC.
- (a) X. Jin, M. Zhao, C. Zeng, W. Yan, Z. Song, P. S. Thapa, B. Subramaniam and R. V. Chaudhari, ACS Catal., 2016, 6, 4576–4583 CrossRef CAS; (b) J. S. Luterbacher, J. M. Rand, D. M. Alonso, J. Han, J. T. Youngquist, C. T. Maravelias, B. F. Pfleger and J. A. Dumesic, Science, 2014, 343, 277–280 CrossRef CAS; (c) Y. Huang, X. Chong, C. Liu, Y. Liang and B. Zhang, Angew. Chem. Int., Ed., 2018, 57, 13163–13166 CrossRef CAS; (d) C. Huang, Y. Huang, C. Liu, Y. Yu and B. Zhang, Angew. Chem. Int., Ed., 2019, 58, 12014–12017 CrossRef CAS; (e) A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS; (f) Y. Tong, P. Chen, M. Zhang, T. Zhou, L. Zhang, W. Chu, C. Wu and Y. Xie, ACS Catal., 2017, 8, 1–7 Search PubMed; (g) J. Zhang, H. Wang, Y. Tian, Y. Yan, Q. Xue, T. He, H. Liu, C. Wang, Y. Chen and B. Y. Xia, Angew. Chem., Int. Ed., 2018, 57, 7649–7653 CrossRef CAS.
- (a) S. Otocka, M. Kwiatkowska, L. Madalińska and P. Kiełbasiński, Chem. Rev., 2017, 117, 4147–4181 CrossRef CAS; (b) I. Fernandez and N. Khiar, Chem. Rev., 2003, 103, 3651–3706 CrossRef CAS; (c) B. M. Trost and M. Rao, Angew. Chem., Int. Ed., 2015, 54, 5026–5043 CrossRef CAS; (d) J. A. Ellman, T. D. Owens and T. P. Tang, Acc. Chem. Res., 2002, 35, 984–995 CrossRef CAS; (e) D. Kaldre, I. Klose and N. Maulide, Science, 2018, 361, 664–667 CrossRef CAS; (f) R. R. Merchant, J. T. Edwards, T. Qin, M. M. Kruszyk, C. Bi, G. Che, D.-H. Bao, W. Qiao, L. Sun, M. R. Collins, O. O. Fadeyi, G. M. Gallego, J. J. Mousseau, P. Nuhant and P. S. Baran, Science, 2018, 360, 75–80 CrossRef CAS; (g) H. Yehuda, S. Khatib, I. Sussan, R. Musa, J. Vaya and S. Tamir, Biofactors, 2009, 35, 295–305 CrossRef CAS.
- (a) Y. Li, M. Wang and X. Jiang, ACS Catal., 2017, 7, 7587–7592 CrossRef CAS; (b) J. Legros, J. R. Dehli and C. Bolm, Adv. Synth. Catal., 2005, 347, 19–31 CrossRef CAS.
- (a) R. Trivedi and P. Lalitha, Synth. Commun., 2006, 36, 3777–3782 CrossRef CAS; (b) M. Matteucci, G. Bhalay and M. Bradley, Org. Lett., 2003, 5, 235–237 CrossRef CAS; (c) F. Hosseinpoor and H. Golchoubian, Tetrahedron Lett., 2006, 47, 5195–5197 CrossRef CAS; (d) M. D. A. Maria, P. Mastrorilli and C. F. Nobile, J. Mol. Catal. A: Chem., 1996, 108, 57–62 CrossRef; (e) J. Legros and C. Bolm, Angew. Chem., Int. Ed., 2004, 43, 4225–4228 CrossRef CAS; (f) J. Legros and C. Bolm, Angew. Chem., Int. Ed., 2003, 42, 5487–5489 CrossRef CAS; (g) H. Egami and T. Katsuki, J. Am. Chem. Soc., 2007, 129, 8940–8941 CrossRef CAS; (h) B. Li, A.-H. Liu, L.-N. He, Z.-Z. Yang, J. Gao and K.-H. Chen, Green Chem., 2012, 14, 130–135 RSC.
- (a) B. Yu, A.-H. Liu, L.-N. He, B. Li, Z.-F. Diao and Y.-N. Li, Green Chem., 2012, 14, 957–962 RSC; (b) E. S. Priestley, D. L. Cheney, I. DeLucca, A. Wei, J. M. Luettgen, A. R. Rendina, P. C. Wong and R. R. Wexler, J. Med. Chem., 2015, 58, 6225–6236 CrossRef CAS; (c) D. P. de Sousa, C. Wegeberg, M. S. Vad, S. Mørup, C. Frandsen, W. A. Donald and C. J. McKenzie, Chem.–Eur. J., 2016, 22, 3810–3820 CrossRef CAS; (d) M. V. Gómez, R. Caballero, E. Vázquez, A. Moreno, A. de la Hoz and A. Díaz-Ortiz, Green Chem., 2007, 9, 331–336 RSC; (e) Y. Liu, A. J. Howarth, J. T. Hupp and O. K. Farha, Angew. Chem., Int. Ed., 2015, 54, 9001–9005 CrossRef CAS.
- B. Yu, A.-H. Liu, L.-N. He, B. Li, Z.-F. Diao and Y.-N. Li, Green Chem., 2012, 14, 957–962 RSC.
- (a) J.-P. Cao, Y.-S. Xue, N.-F. Li, J.-J. Gong, R.-K. Kang and Y. Xu, J. Am. Chem. Soc., 2019, 141, 19487–19497 CrossRef CAS; (b) Y. Li, S. A.-e.-A. Rizvi, D. Hu, D. Sun, A. Gao, Y. Zhou, J. Li and X. Jiang, Angew. Chem., Int. Ed., 2019, 58, 13499–13506 CrossRef CAS.
- G. Laudadio, N. J. W. Straathof, M. D. Lanting, B. Knoops, V. Hessel and T. Noël, Green Chem., 2017, 19, 4061–4066 RSC.
- (a) L. Zhou, M. F. Shao, C. Zhang, J. W. Zhao, S. He, D. M. Rao, M. Wei, D. G. Evans and X. Duan, Adv. Mater., 2017, 29, 1604080 CrossRef; (b) X. Long, G. X. Li, Z. L. Wang, H. Y. Zhu, T. Zhang, S. Xiao, W. Y. Guo and S. H. Yang, J. Am. Chem. Soc., 2015, 137, 11900–11903 CrossRef CAS; (c) C. Zhang, M. F. Shao, L. Zhou, Z. H. Li, K. M. Xiao and M. Wei, ACS Appl. Mater. Interfaces, 2016, 8, 33697–33703 CrossRef CAS; (d) Q. Wang and D. O'Hare, Chem. Rev., 2012, 112, 4124–4155 CrossRef CAS; (e) M. Shao, F. Ning, J. Zhao, M. Wei, D. G. Evans and X. Duan, J. Am. Chem. Soc., 2012, 134, 1071–1077 CrossRef CAS; (f) Y. Zhao, B. Li, Q. Wang, W. Gao, C. J. Wang, M. Wei, D. G. Evans, X. Duan and D. O'Hare, Chem. Sci., 2014, 5, 951–958 RSC; (g) L. Yin, S. Li, X. Liu and T. Yan, Sci. China Mater., 2019, 62, 1537–1555 CrossRef CAS.
- (a) F. Song and X. Hu, Nat. Commun., 2014, 5, 4477–4485 CrossRef CAS; (b) X. Zou, A. Goswami and T. Asefa, J. Am. Chem. Soc., 2013, 135, 17242–17245 CrossRef CAS; (c) B. Hunter, J. Blakemore, M. Deimund, H. Gray and J. Winkler, J. Am. Chem. Soc., 2014, 136, 13118–13121 CrossRef CAS; (d) F. Li, J. Du, X. Li, J. Shen, Y. Wang, Y. Zhu and L. Sun, Adv. Energy Mater., 2018, 8, 1702598 CrossRef; (e) X. Wu, Y. Zhao, T. Xing, P. Zhang, F. Li, H. Lee, F. Li and L. Sun, ChemSusChem, 2018, 11, 1761–1767 CrossRef CAS; (f) L. Huang, Z. He, J. Guo, S. Pei, H. Shao and J. Wang, Nano Res., 2020, 13, 246–254 CrossRef CAS; (g) H. Yang, H. Wang, Y. Zhang and Q. Wang, Sci. China Mater., 2019, 62, 681–689 CrossRef CAS.
- (a) H. Zhang, H. Li, B. Akram and X. Wang, Nano Res., 2019, 12, 1327–1331 CrossRef CAS; (b) B. You, N. Jiang, X. Liu and Y. Sun, Angew. Chem., Int. Ed., 2016, 55, 9913–9917 CrossRef CAS; (c) B. You, X. Liu, N. Jiang and Y. Sun, J. Am. Chem. Soc., 2016, 138, 13639–13646 CrossRef CAS.
- J. Zhao, Z. Lu, M. Shao, D. Yan, M. Wei, D. G. Evans and X. Duan, RSC Adv., 2013, 3, 1045–1049 RSC.
- R. Yang, Y. Zhou, Y. Xing, D. Li, D. Jiang, M. Chen and S. Yuan, ACS Appl. Energy Mater., 2018, 1, 1200–1209 CrossRef.
- C. Kingston, M. D. Palkowitz, Y. Takahira, J. C. Vantourout, B. K. Peters, Y. Kawamata and P. S. Baran, Acc. Chem. Res., 2020, 53, 72–83 CrossRef CAS.
- For detailed references. See the ESI. Section 2.†.
- Z. Cheng, P. Sun, A. Tang, W. Jin and C. Liu, Org. Lett., 2019, 22, 8925–8929 CrossRef.
- (a) R. I. Sayler, B. M. Hunter, W. Fu, H. B. Gray and R. D. Britt, J. Am. Chem. Soc., 2020, 142, 1838–1845 CrossRef CAS; (b) Z. Guan, S. Zhu, S. Wang, H. Wang, X. Zhong, F. Bu, H. Cong and A. Lei, Angew. Chem., Int. Ed., DOI:10.1002/anie.202011329.
- (a) X. Lu, L. Gu, J. Wang, J. Wu, P.-Q. Liao and G.-R. Li, Adv. Mater., 2017, 29, 1604437 CrossRef; (b) J. A. Koza, C. M. Hull, Y.-C. Liu and J. A. Switzer, Chem. Mater., 2013, 25, 1922–1926 CrossRef CAS; (c) M. Ludvigsson, J. Lindgren and J. Tegenfeldt, J. Mater. Chem., 2001, 11, 1269–1276 RSC; (d) T. Pauporte, L. Mendoza, M. Cassir, M. C. Bernard and J. Chivot, J. Electrochem. Soc., 2005, 152, 49–53 CrossRef.
- (a) Z.-F. Huang, J. Song, Y. Du, S. Xi, S. Dou, J. M. V. Nsanzimana, C. Wang, Z. J. Xu and X. Wang, Nat. Energy, 2019, 4, 328–338 Search PubMed; (b) Z. Li, H. Duan, M. Shao, J. Li, D. O'Hare, M. Wei and Z. L. Wang, Chem, 2018, 4, 1–12 CrossRef.
- (a) Z. Li, K. Liu, K. Fan, Y. Yang, M. Shao, M. Wei and X. Duan, Angew. Chem., Int. Ed., 2019, 58, 3962–3965 CrossRef CAS; (b) F. Dionigi, Z. Zeng and I. Sinev, et al. , Nat. Commun., 2020, 11, 2522 CrossRef CAS.
- (a) J. Huang, J. Chen, T. Yao, J. He, S. Jiang, Z. Sun, Q. Liu, W. Cheng, F. Hu, Y. Jiang, Z. Pan and S. Wei, Angew. Chem., Int. Ed., 2015, 54, 8722–8727 CrossRef CAS; (b) A. Bergmann, T. E. Jones, E. Martinez Moreno, D. Teschner, P. Chernev, M. Gliech, T. Reier, H. Dau and P. Strasser, Nat. Catal., 2018, 1, 711–719 CrossRef CAS.
- (a) E. Zhang, T. Wang, K. Yu, J. Liu, W. Chen, A. Li, H. Rong, R. Lin, S. Ji, X. Zheng, Y. Wang, L. Zheng, C. Chen, D. Wang, J. Zhang and Y. Li, J. Am. Chem. Soc., 2019, 141, 16569–16573 CrossRef CAS; (b) X. Li, W. Bi, M. Chen, Y. Sun, H. Ju, W. Yan, J. Zhu, X. Wu, W. Chu, C. Wu and Y. Xie, J. Am. Chem. Soc., 2017, 139, 14889–14892 CrossRef CAS.
- C. Huang, Y. Huang, C. Liu, Y. Yu and B. Zhang, Angew. Chem., Int. Ed., 2019, 58, 12014–12017 CrossRef CAS.
- X. Han, C. Yu, S. Zhou, C. Zhao, H. Huang, J. Yang, Z. Liu, C. Zhao and J. Qiu, Adv. Energy Mater., 2017, 7, 1602148 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc05499b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |