
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthetic glycosidases for the precise hydrolysis of oligosaccharides and polysaccharides†
Xiaowei
Li
 and
Yan
Zhao
*
and
Yan
Zhao
*
Department of Chemistry, Iowa State University, Ames, Iowa 50011-3111, USA. E-mail: zhaoy@iastate.edu
First published on 5th November 2020
Abstract
Glycosidases are an important class of enzymes for performing the selective hydrolysis of glycans. Although glycans can be hydrolyzed in principle by acidic water, hydrolysis with high selectivity using nonenzymatic catalysts is an unachieved goal. Molecular imprinting in cross-linked micelles afforded water-soluble polymeric nanoparticles with a sugar-binding boroxole in the imprinted site. Post-modification installed an acidic group near the oxygen of the targeted glycosidic bond, with the acidity and distance of the acid varied systematically. The resulting synthetic glycosidase hydrolyzed oligosaccharides and polysaccharides in a highly controlled fashion simply in hot water. These catalysts not only broke down amylose with similar selectivities to those of natural enzymes, but they also could be designed to possess selectivity not available with biocatalysts. Substrate selectivity was mainly determined by the sugar residues bound within the active site, including their spatial orientations. Separation of the product was accomplished through in situ dialysis, and the catalysts left behind could be used multiple times with no signs of degradation. This work illustrates a general method to construct synthetic glycosidases from readily available building blocks via self-assembly, covalent capture, and post-modification. In addition, controlled, precise, one-step hydrolysis is an attractive way to prepare complex glycans from naturally available carbohydrate sources.
Introduction
Carbohydrates are the most abundant biomolecules on earth. Cellulose makes up 35–50% of lignocellulosic biomass that is produced at an annual scale of 170–200 billion tons.1,2 Starch, consisting of linear and branched polymers of glucose, is the dominant energy-storage material in plants and the predominant carbohydrate in the human diet. With glycosylation being the most common post-translational modification of proteins, carbohydrates (glycans) mediate important biological events including cell adhesion, bacterial and viral infection, inflammation, and cancer development.3–8To process carbohydrates, most organisms use 1–3% of their genome to encode enzymes for glycosylation and hydrolysis of glycans.9 Many of these enzymes, however, cannot be obtained easily and new catalysts with controlled glycosidic selectivity are in great need for glycomics.3 Moreover, enzymes tend to work only under a narrow set of conditions in aqueous solution. Synthetic catalysts with stronger tolerance to adverse temperatures and solvent conditions are highly desirable for challenging operations such as biomass conversion.10
Chemists have long been interested in creating synthetic glycosidases to hydrolyze glycosides/glycans. In their pioneering work, Bols and co-workers used cyclodextrin to bind p-nitrophenyl β-D-glucopyranoside and acidic groups installed on the macrocycle for hydrolysis.11,12 Striegler et al. developed binuclear copper catalysts to hydrolyze glycosides under basic conditions.13,14 The group of Bandyopadhyay reported azobenzene-3,3′-dicarboxylic acid as a simple glycosidase mimic with photoresponsive properties.15 Although these catalysts only worked on activated glycosides carrying good aryl leaving groups, they proved important design principles with readily available scaffolds. Instead of relying on hydrolysis, Yu and Cowan combined a metal-binding ligand and the sugar-binding domain of odorranalectin (a natural lectin-like peptide) to remove L-fucose selectively through oxidative cleavage.16
The fundamental challenge in building a synthetic glycosidase is two-fold, whether on a purely synthetic platform or a hybrid one as above. First, the catalyst needs to recognize the targeted glycan in an aqueous solution. Molecular recognition of carbohydrates is a long-standing challenge in supramolecular and bioorganic chemistry, due to strong solvation of these molecules in water.17 Also, inversion of a single hydroxyl in a glycan, connecting the monosaccharide building blocks by different hydroxyls, or altering the (α/β) glycosidic linkages could all change the biological properties of a glycan profoundly. Distinguishing these subtle structural changes requires a great level of selectivity in the recognition. Second, catalytic groups need to be installed precisely at the correct glycosidic bond for selective hydrolysis. Although many platforms are available for building artificial enzymes,18,19 this type of precision is still very difficult to achieve for a complex substrate.
In this work, we describe a synthetic glycosidase designed and synthesized rationally through molecular imprinting20–22 within a cross-linked micelle.23 Having an acidic group near the exocyclic oxygen of a particular glycosidic bond while recognizing the adjacent sugar residues with reversible boronate ester and hydrogen bonds, these biomimetic catalysts hydrolyzed oligosaccharides and polysaccharides in a precise manner in 60 °C water, to afford specific glycans in a single step. The catalysts also distinguished sugar building blocks and the glycosidic linkages, especially those bound within the active site. Because synthesis of complex glycans requires extensive protection/deprotection chemistry and is often very challenging, selective hydrolysis of naturally available glycan sources can be a highly attractive alternative.
Results and discussion
Construction of the active site for glycan binding
To build an active site for a glycan, we employed molecular imprinting that can quickly produce binding sites within a cross-linked polymer complementary to the template molecules being used.20–22 The templated polymerization was particularly effective in a nanosized environment such as a surfactant micelle.24Scheme 1 shows the general method of micellar imprinting. The template molecule is first solubilized in the mixed micelle of 1a (or 1b) and 2. These surfactants are functionalized with either terminal alkyne or azide on the headgroup. Micellization brings these functional groups into close proximity, and they react readily by Cu(I)-catalyzed cycloaddition to cross-link the surface of the micelle. The mixed micelle also contains divinylbenzene (DVB) and 2,2-dimethoxy-2-phenylaceto-phenone (DMPA, a photoinitiator). UV irradiation triggers free-radical polymerization/cross-linking in the core of the surface-cross-linked micelle, among DVB and the methacrylate of the surfactants around the template molecule. The doubly cross-linked micelle is then decorated with a layer of hydrophilic ligand (3) by a second round of click reaction. Precipitation into acetone and washing with organic solvents removes the template and other impurities to yield water-soluble molecularly imprinted nanoparticles (MINPs) with template-complementary binding sites.23
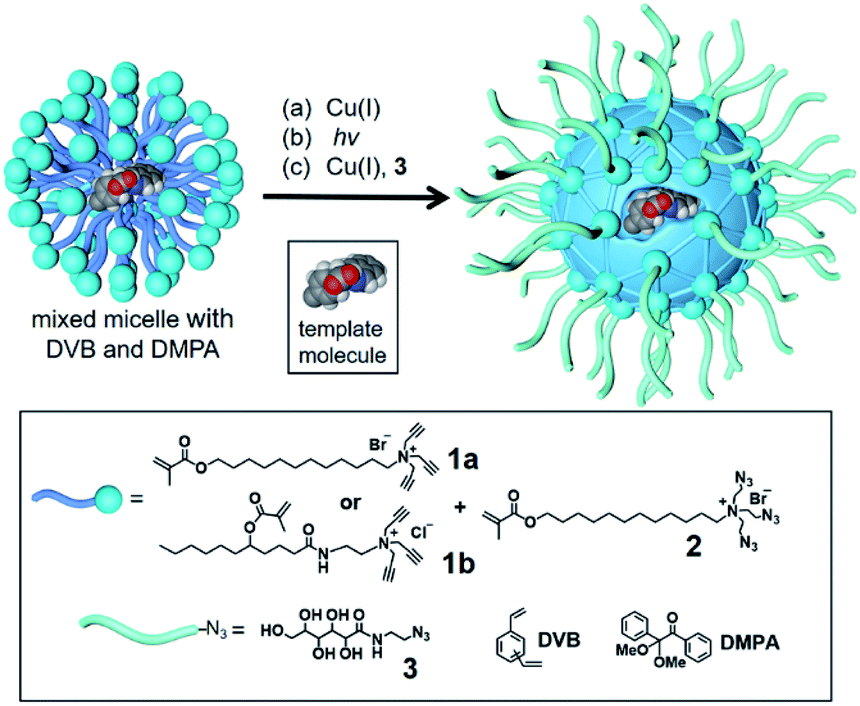 | ||
| Scheme 1 The preparation of molecularly imprinted nanoparticles (MINPs) through templated polymerization in micelles. | ||
Boronic acid25–31 and benzoboroxole derivatives32–35 are known to bind specific 1,2- or 1,3-diols of sugars through reversible boronate bonds. Vinylbenzoboroxle 4 is particularly useful as a functional monomer (FM) for covalent imprinting36 of sugars.35 Aryl glycosides 5a–c have two important parts—a glycan and an aglycon containing a hydrolyzable imine bond (Scheme 2). These template molecules reacted with FM 4 readily to form amphiphilic, anionic boronate esters such as p-6a, stabilized by the cationic micelle.35 The imine bond in MINP(p-G1) was hydrolyzed in 6 M HCl at 95 °C, and the aldehyde group in the imprinted site of MINP-CHO(p-G1) derivatized through reductive amination with 7a–f in DMF.37 The resulting MINP(p-G1 + 7a–f), i.e., the MINP prepared with template 5a and post-functionalized with amino acid 7a–f, was purified by precipitation into acetone and washing with organic solvents. The MINP is expected to bind the terminal glucose of a glucose-terminated oligo- or polysaccharide at the nonreducing end, with its carboxylic acid near the exocyclic oxygen of the glycosidic bond.
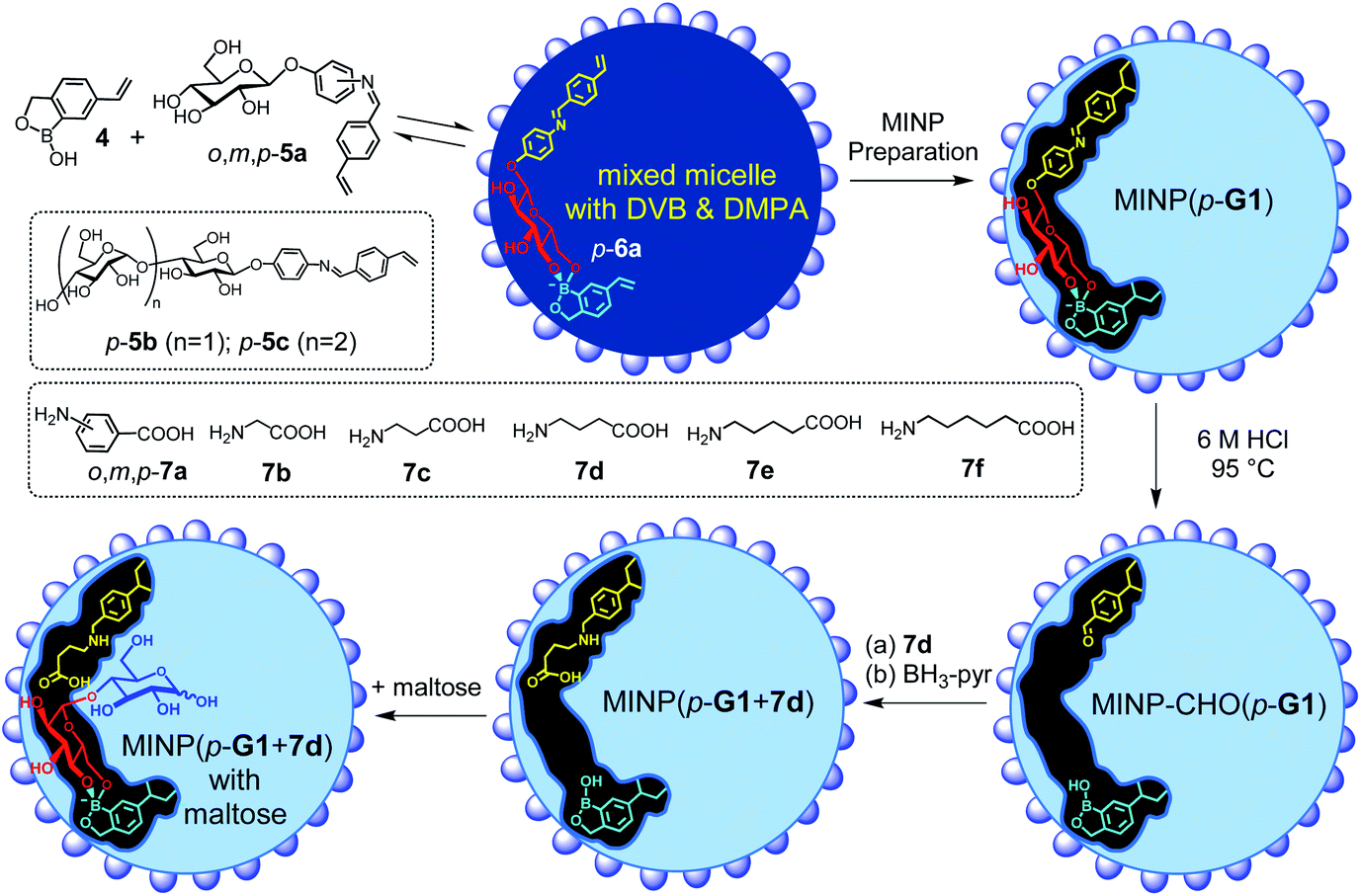 | ||
| Scheme 2 The preparation of the artificial glycosidase MINP(p-G1 + 7d) and its binding of maltose. | ||
The intermediate MINP-CHO(p-G1) indeed was found to bind not only glucose but also maltose and other glucose-terminated oligosaccharides strongly in aqueous buffer (Table 1, entries 1–4). One might consider it strange for a binding site imprinted from a monosaccharide template to bind a larger oligosaccharide, as several works of ours indicate that fitting a smaller guest in a larger imprinted pocket affords a reduced binding, but fitting a larger guest in a small pocket is impossible.35,38,39 However, the amphiphilicity of the template-FM complex (p-6a) demands it stay near the surface of the micelle, a feature helpful to not only the removal of the template after imprinting but also anchoring the binding site near the surface of the micelle. In this way, a glucose-terminated oligosaccharide could use its terminal glucose to interact with the boroxole in the MINP binding pocket, with the rest of the structure residing in water, as shown in Scheme 2. Ability to bind a longer sugar is a prerequisite to the catalysis. Had a longer sugar been excluded, the acid-functionalized MINP(p-G1) would not be able to recognize its substrate (G2 and above). Table 1 also shows that the binding of MINP-CHO(p-G1) weakened steadily with an increase in the chain length of the sugar guest. We attributed the trend to the 1,4-α-glycosidic linkage that creates a significant curvature to the oligosaccharide backbone.40 The longer sugar, with its folded backbone, likely experienced some steric repulsion with the MINP receptor, which was covered with a layer of hydrophilic ligands and averaged ∼5 nm in size according to dynamic light scattering (Fig. S8–10†) and transmission electron microscopy (Fig. S4†).
| Entry | MINP | Oligosaccharide | K a (103 M−1) | ΔG (kcal mol−1) | ΔH (kcal mol−1) | TΔS (kcal mol−1) | N |
|---|---|---|---|---|---|---|---|
a The FM/template ratio in MINP synthesis was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The cross-linkable surfactants were a 3:2 mixture of 1b and 2. The titrations were performed in 10 mM HEPES buffer at pH 7.4 at 298 K.
b
N is the average number of binding sites per nanoparticle measured via ITC curve fitting.
c Nonimprinted nanoparticles (NINPs) were prepared with the same amount of FM 4 as all the MINPs but without any template.
d Binding was extremely weak; because the binding constant was estimated from ITC, −ΔG and N are not listed. :1. The cross-linkable surfactants were a 3:2 mixture of 1b and 2. The titrations were performed in 10 mM HEPES buffer at pH 7.4 at 298 K.
b
N is the average number of binding sites per nanoparticle measured via ITC curve fitting.
c Nonimprinted nanoparticles (NINPs) were prepared with the same amount of FM 4 as all the MINPs but without any template.
d Binding was extremely weak; because the binding constant was estimated from ITC, −ΔG and N are not listed.
|
|||||||
| 1 | MINP-CHO(p-G1) | Glucose (G1) | 8.85 ± 0.68 | −5.38 | −3.10 ± 0.15 | 2.28 | 1.03 ± 0.03 |
| 2 | MINP-CHO(p-G1) | Maltose (G2) | 6.69 ± 0.40 | −5.22 | −2.53 ± 0.12 | 2.69 | 1.26 ± 0.04 |
| 3 | MINP-CHO(p-G1) | Maltotriose (G3) | 5.72 ± 0.29 | −5.13 | −8.00 ± 0.68 | −2.87 | 0.95 ± 0.07 |
| 4 | MINP-CHO(p-G1) | Maltohexaose (G6) | 1.56 ± 0.35 | −4.35 | −3.49 ± 2.83 | 0.86 | 0.83 ± 0.61 |
| 5 | MINP-CHO(p-G2) | Glucose (G1) | 12.9 ± 1.1 | −5.62 | −0.97 ± 0.04 | 4.65 | 1.03 ± 0.03 |
| 6 | MINP-CHO(p-G2) | Maltose (G2) | 27.20 ± 6.47 | −6.05 | −1.33 ± 0.11 | 4.72 | 1.22 ± 0.08 |
| 7 | MINP-CHO(p-G2) | Maltotriose (G3) | 11.30 ± 1.52 | −5.53 | −1.57 ± 0.10 | 3.96 | 1.01 ± 0.04 |
| 8 | MINP-CHO(p-G3) | Glucose (G1) | 7.02 ± 0.43 | −5.24 | −2.44 ± 0.07 | 2.80 | 1.10 ± 0.02 |
| 9 | MINP-CHO(p-G3) | Maltose (G2) | 11.10 ± 0.90 | −5.51 | −3.77 ± 0.20 | 1.74 | 0.95 ± 0.04 |
| 10 | MINP-CHO(p-G3) | Maltotriose (G3) | 35.70 ± 2.98 | −6.21 | −13.4 ± 0.6 | −7.15 | 1.14 ± 0.03 |
| 11 | NINPc | Glucose (G1) | <0.05 d | —d | —d | —d | —d |
Templates p-5b and p-5c were similar to p-5a, except that their glycan was maltose and maltotriose, respectively. Similar hydrolysis of the MINPs in 6 M HCl afforded MINP-CHO(p-G2) and MINP-CHO(p-G2). All three MINP-CHO's could bind glucose (G1), maltose (G2), and maltotriose (G3). Notably, among the three sugar guests, the MINP always bound its templating sugar most strongly (Table 1), consistent with successful imprinting. The nonimprinted nanoparticles (NINPs) showed very weak binding, not measurable by ITC. The imprinting factor, defined as the MINP/NINP binding ratio, was >170 for glucose.
It is good that the binding of MINP-CHO(p-G1) for glucose in aqueous buffer (Ka = 8.85 × 103 M−1) already approached those for monosaccharides by natural lectins (Ka = 103 to 104 M−1).5,41 The strong binding suggests that the polyhydroxylated surface ligand 3 could not fold back to interact with the boroxole group in the imprinted site (to interfere with the guest binding). On the other hand, the stronger binding of MINP-CHO(p-G1) for glucose than the longer sugars is a potential problem in catalytic hydrolysis, as glucose is the expected product from the hydrolysis and product inhibition would be inevitable (vide infra).
Installation of acidic functionality for catalytic hydrolysis
In the initial stage of catalysis, we used maltose (G2) as a model oligosaccharide and studied its hydrolysis by the acid-functionalized MINP(5a + 7a–f). It is extremely encouraging that these MINPs could hydrolyze maltose simply in hot water, without any additives (Table 2). Control experiments (entries 14–16) indicated that neither the nanoparticle itself (i.e., NINP) nor the amino acid used for post-modification (e.g., 7d) was able to catalyze the hydrolysis.| Entry | Catalyst | Surfactant | Yield (%) |
|---|---|---|---|
| a Reactions were performed in duplicate with 0.2 mM maltose and 20 μM MINP in 1.0 mL of water. Yields were determined via LC-MS using calibration curves generated from authentic samples (Fig. S32). b NINP is a nonimprinted nanoparticle prepared without any template or post-modification. | |||
| 1 | MINP(o-G1 + o-7a) | 1a + 2 | 18 ± 2 |
| 2 | MINP(m-G1 + m-7a) | 1a + 2 | 26 ± 4 |
| 3 | MINP(p-G1 + p-7a) | 1a + 2 | 32 ± 4 |
| 4 | MINP(p-G1 + o-7a) | 1a + 2 | 28 ± 4 |
| 5 | MINP(p-G1 + m-7a) | 1a + 2 | 11 ± 2 |
| 6 | MINP(p-G1 + p-7a) | 1b + 2 | 54 ± 7 |
| 7 | MINP(p-G1 + 7b) | 1b + 2 | 17 ± 3 |
| 8 | MINP(p-G1 + 7c) | 1b + 2 | 70 ± 4 |
| 9 | MINP(p-G1 + 7d) | 1b + 2 | 70 ± 8 |
| 10 | MINP(p-G1 + 7e) | 1b + 2 | 31 ± 4 |
| 11 | MINP(p-G1 + 7f) | 1b + 2 | 13 ± 4 |
| 12 | MINP(p-G1 + 7g) | 1b + 2 | 76 ± 4 |
| 13 | MINP(p-G1 + 7h) | 1b + 2 | 82 ± 6 |
| 14 | 7d | — | <1 |
| 15 | NINPb + 7d | 1b + 2 | 0 |
| 16 | None | — | 0 |
To optimize the catalytic design, we varied the position of the imine bond on the phenyl ring in the template. By using the ortho, meta, or para derivative, we could change the position of the to-be-installed acid group relative to the glycan.
Our initial hypothesis was that the ortho template might allow the acid group to be particularly close to the exocyclic glycosidic oxygen of the glycan substrate to be bound (Scheme 2).
Hydrolysis of maltose, however, increased steadily from the ortho-to the meta- and then to the para-derived MINP, from 18 to 32% yield (Table 2, entries 1–3). Molecular imprinting and guest-binding in MINP have a strong driving force in water from hydrophobic interactions.23 During imprinting, the free hydroxyls of o-, m-, or p-6a need to stay close to the surface of the micelle (to be solvated by water) but the aglycon and the aryl group of the boroxole prefer to stay inside the micelle due to their hydrophobicity. It is possible that these requirements were best met in the para derivative, given the geometrical constraints set by the nanodimensioned micelle.
When the ortho and meta amino acid were used in the post-modification of MINP(p-G1), hydrolysis of maltose became less efficient (Table 2, entries 4 & 5). The reductive amination protocol was established previously37 and imine formation was found to correlate with the binding of the amine in the imprinted site.42 The lower yields in entries 4 & 5 could come from a less complete post-modification (due to a mismatch in the dimensions of the reactants with the imprinted sites) and/or poor positioning of the acid in the active site.
Surfactant 1b equips the MINP with a layer of hydrogen-bonding amides near the surface, within the hydrophobic core of the micelle.43 Hydrogen bonds are weakened by competition from water in aqueous solution. They are known, however, to be much stronger inside the hydrophobic microenvironment of a micelle.35,44 Indeed, switching the surfactant from 1a to 1b in the MINP preparation increased the yield of maltose hydrolysis from 32% to 54%, suggesting that hydrogen bonds played important roles in the binding of the substrate.
We also evaluated linear amino acids 7b–f in the post-modification, reasoning that flexibility of the acidic group might be beneficial to the catalysis. The hypothesis was confirmed by the catalysis. Although MINP(p-G1 + 7b) was very inactive, MINP(p-G1 + 7c/d) gave a much higher yield (70%) in maltose hydrolysis. Accurate positioning of the acidic group was clearly key to the hydrolysis, as too short or too long a spacer in the amino acid diminished the yield (Table 2, entries 7, 10, and 11).
Note that, although the MINP active site contained both an acid and a basic amino group, the acid remained active. Similar situations are frequently found in enzyme active sites, where acidic and basic groups co-exist but do not “self-destruct” through intramolecular acid–base reaction, because the resulting ionic species are poorly solvated and thus unstable in a hydrophobic microenvironment.45–47
Precise hydrolysis of oligosaccharides
With the structure of the template and the distance between the acid and glycan optimized, we prepared a series of other MINPs using p-5b–c as the templates, following similar procedures illustrated in Scheme 2. Since the corresponding MINPs were expected to bind glucose, maltose, and maltotriose, respectively, our goal was to hydrolyze maltohexaose (G6) and even a glucose-based polymer in a controlled fashion (Scheme 3).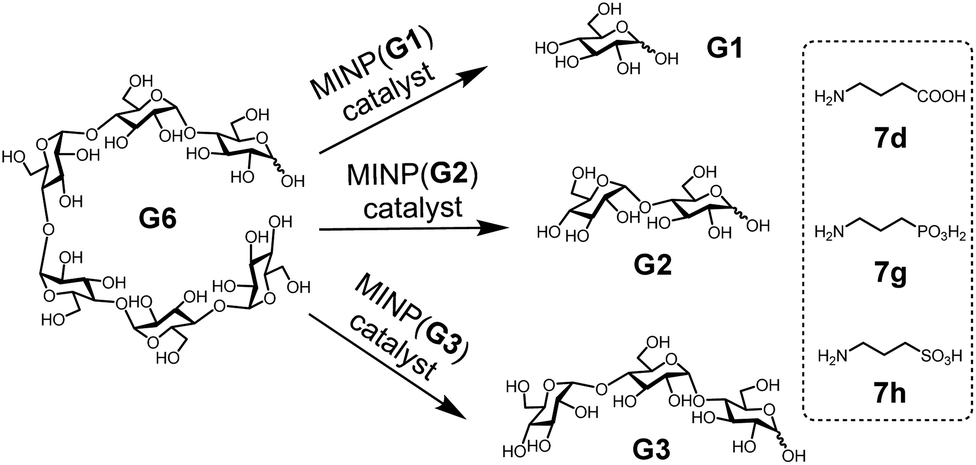 | ||
| Scheme 3 The selective hydrolysis of maltohexaose (G6) by MINPs. | ||
Table 3 shows the hydrolysis of G6 in water under our standard conditions (60 °C for 24 h). We also studied the hydrolysis in buffer at pH 5.5–7.5 and found that the reaction yield in water was close to the highest yield obtained at pH 6 (Table S2†).
| Entry | Catalyst | Yield G1 (μM) | Yield G2 (μM) | Yield G3 (μM) |
|---|---|---|---|---|
| a MINPs were prepared with surfactants 1b and 2. Reactions were performed with 0.1 mM maltohexaose (G6) and 20 μM MINP in 1.0 mL of water. Yields were determined via LC-MS using calibration curves generated from authentic samples (Fig. S32). | ||||
| 1 | MINP(p-G1 + 7d) | 106 ± 18 | 24 ± 6 | 31 ± 8 |
| 2 | MINP(p-G1 + 7g) | 199 ± 22 | 17 ± 6 | 21 ± 4 |
| 3 | MINP(p-G1 + 7h) | 253 ± 41 | 12 ± 4 | 42 ± 8 |
| 4 | MINP(p-G2 + 7d) | 11 ± 2 | 117 ± 11 | 14 ± 4 |
| 5 | MINP(p-G2 + 7g) | 10 ± 2 | 127 ± 14 | 9 ± 2 |
| 6 | MINP(p-G2 + 7h) | 6 ± 2 | 144 ± 17 | 7 ± 2 |
| 7 | MINP(p-G3 + 7d) | 7 ± 2 | 8 ± 2 | 64 ± 11 |
| 8 | MINP(p-G3 + 7g) | 7 ± 2 | 10 ± 4 | 74 ± 10 |
| 9 | MINP(p-G3 + 7h) | 14 ± 6 | 17 ± 5 | 98 ± 17 |
In this set of experiments, we varied the acidity of the acid catalyst, using 7d, 7g, and 7h (Scheme 3), respectively, in the post-modification. To our delight, the yield of the intended products—i.e., glucose (G1) from MINP(G1), maltose (G2) from MINP(G2), and maltotriose (G3) from MINP(G3)—correlated positively with the increased acidity. Meanwhile, the yields of the unintended hydrolytic products were quite random (and low), suggesting that these products probably came from uncontrolled hydrolysis. Positive correlation between the acidity and the hydrolysis was also observed for maltose in Table 2 (entries 9, 12, and 13).
Fig. 1a shows the full characterization of the hydrolyzed products by MINP(p-G1 + 7h), including the starting material (G6) and all the possible hydrolyzed fragments (G1–G5). The theoretical yield of glucose was 600 μM from 100 μM G6. The yield of glucose increased from 45% (24 h at 60 °C) to 77% (48 h at 90 °C) and finally to 86% (96 h at 90 °C). Most importantly, when the three MINPs were used in the hydrolysis, the dominant product was always the intended one, in a yield of 77%, 82%, and 88% for glucose, maltose, and maltotriose, respectively (Fig. 1b).
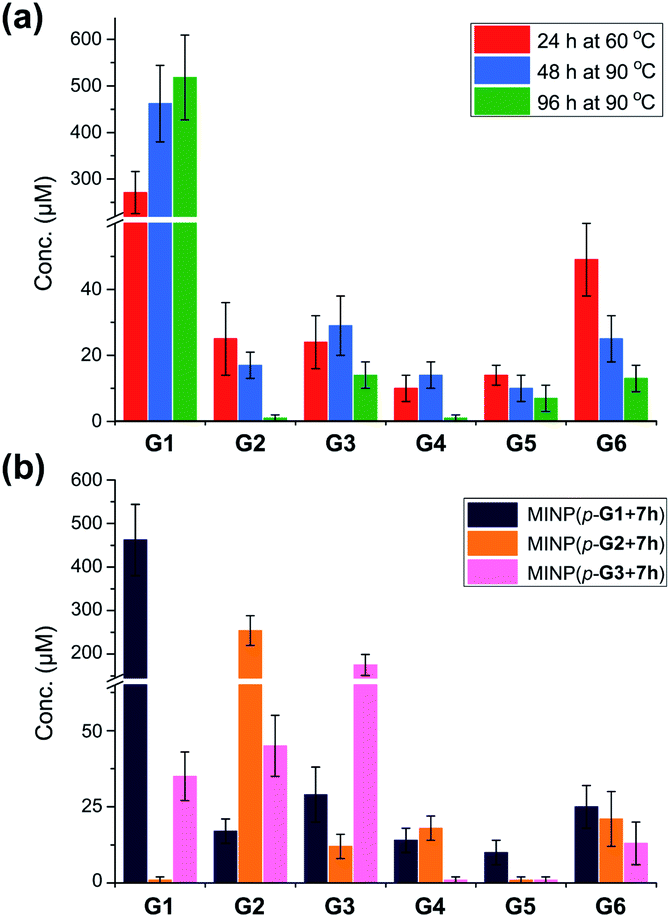 | ||
| Fig. 1 (a) Product distributions for the hydrolysis of 100 μM maltohexaose (G6) under different conditions using 20 μM MINP(p-G1 + 7h) in 10 mM MES buffer (pH 6). (b) Product distributions for the hydrolysis of 0.1 mM maltohexaose (G6) using different MINPs (20 μM) in 10 mM MES buffer (pH 6) at 90 °C for 48 h. See Table S4† for the exact numbers. | ||
Another interesting observation was the noticeable “absence” of intermediate products (G2–G5) when G6 was hydrolyzed by MINP(p-G1).48 Whether at moderate (∼50%) or higher (>80%) conversion, these intermediate products were insignificant in comparison to G1. These results were in line with the observation that the binding of MINP-CHO(p-G1) decreased with increasing chain length of the sugar (i.e., glucose > maltose > maltotriose > maltohexaose, Table 1). Since hydrolysis requires the binding of the sugar by the MINP, the shorter fragments, with a stronger binding for the MINP catalyst, would be hydrolyzed preferentially over the starting material. Essentially, once the hydrolysis starts on a long sugar, it prefers to go all the way to break down the sugar (although not necessarily of the same molecule).
An inevitable result from the stronger binding of the shorter sugars, unfortunately, was product inhibition. Fig. 1a shows that, even at 90 °C for 96 h, MINP(p-G1 + 7h) could not hydrolyze maltohexaose completely. To better understand this behavior, we first measured the Michaelis–Menten parameters for all three MINPs (Table 4). For MINP(p-G1 + 7h), we also performed the study with both maltose and maltohexaose as the substrate.
| Entry | MINP | Substrate | V max (μM min−1) | K m (μM) | k cat (×10−3 min−1) | k cat/Km (M−1 min−1) |
|---|---|---|---|---|---|---|
| a Reaction rates were measured in water at 60 °C, based on the disappearance of the reactant. [MINP] = 20 μM. | ||||||
| 1 | MINP(p-G1 + 7h) | Maltose (G2) | 0.37 ± 0.01 | 336 ± 25 | 18.26 | 54.1 |
| 2 | MINP(p-G1 + 7h) | Maltohexaose (G6) | 0.17 ± 0.01 | 691 ± 90 | 8.70 | 12.6 |
| 3 | MINP(p-G2 + 7h) | Maltohexaose (G6) | 0.19 ± 0.01 | 541 ± 39 | 9.36 | 16.5 |
| 4 | MINP(p-G3 + 7h) | Maltohexaose (G6) | 0.20 ± 0.01 | 474 ± 24 | 9.92 | 19.5 |
The Km value of MINP(p-G1 + 7h) for maltose was about half of that for maltohexaose, indicating that the catalyst bound maltose more strongly. Interestingly, the catalytic turnover (kcat) for maltose was also double that for maltohexaose. The catalytic efficiency (kcat/Km) of the MINP for maltose was thus more than 4 times higher than that for maltohexaose. These results supported our above explanation for the “absence” of intermediate products in the hydrolysis of G6 by MINP(p-G1 + 7h).
For the hydrolysis of G6, the three MINP catalysts showed similar kcat but stronger binding for the substrate as the active site was designed to bind a longer sugar—i.e., Km decreased in the order of MINP(p-G1 + 7h) > MINP(p-G2 + 7h) > MINP(p-G3 + 7h). The trend was similar to what was observed in the ITC-determined binding constants for the corresponding sugars (Table 1, entries 1, 6, and 10). Both should be derived from a larger binding interface of a longer sugar with its complementary imprinted binding site. Not only could the substrate form more hydrogen bonds with the amides of (cross-linked) 1b in the MINP, but more water molecules in the active site would also be released during binding.49
We then performed the Michaelis–Menten study for the hydrolysis of maltose by MINP(p-G1 + 7h), with different amounts of glucose added to the reaction mixture. The inhibition constant (Ki) was found to be ∼68 μM (Fig. S27 and S28†), which was 5–10 times smaller than the Km value for maltose and maltohexaose. Thus, strong product inhibition indeed was present.
Fortunately, with MINP being much larger than the sugars and the staring material (G6) also larger than the desired products (G1, G2, and G3), we could overcome product inhibition simply by performing the hydrolysis inside a dialysis membrane that was permeable to the desired product but impermeable to the starting material and the catalyst. In this way, the starting material and the MINP catalyst would stay inside the membrane during hydrolysis, and the product would escape into the bulk solution. This simple change in reaction setup then could turn the adversity into an advantage because product inhibition would no longer be a problem when the concentration of the product inside the membrane was diluted ∼40 times under our dialysis condition. Not only that, the product would be isolated in situ from the starting material and the catalyst, greatly simplifying the purification of the product and reuse of the catalyst (vide infra).
To test this hypothesis, we chose dialysis tubing with a MW-cutoff (MWCO) of 500 Da, which should let G1 (MW 180) and G2 (MW 342) easily escape but might have difficulty with G3 (MW 504). Indeed, hydrolysis of maltohexaose into glucose, maltose, and even maltotriose all improved significantly with the catalysis performed inside the dialysis membrane. The improvements can be seen by comparing the solid and dashed lines in Fig. 2a–c. At the end of 24 h, the yields of the desired product went from 43% to 87% for glucose (G1), 39% to 89% for maltose (G2), and 49% to 72% for maltotriose (G3). The stronger benefits of dialysis on G1 and G2 over G3 supported our experimental hypothesis, since G3 (MW 504) was very close to the MWCO of the membrane.
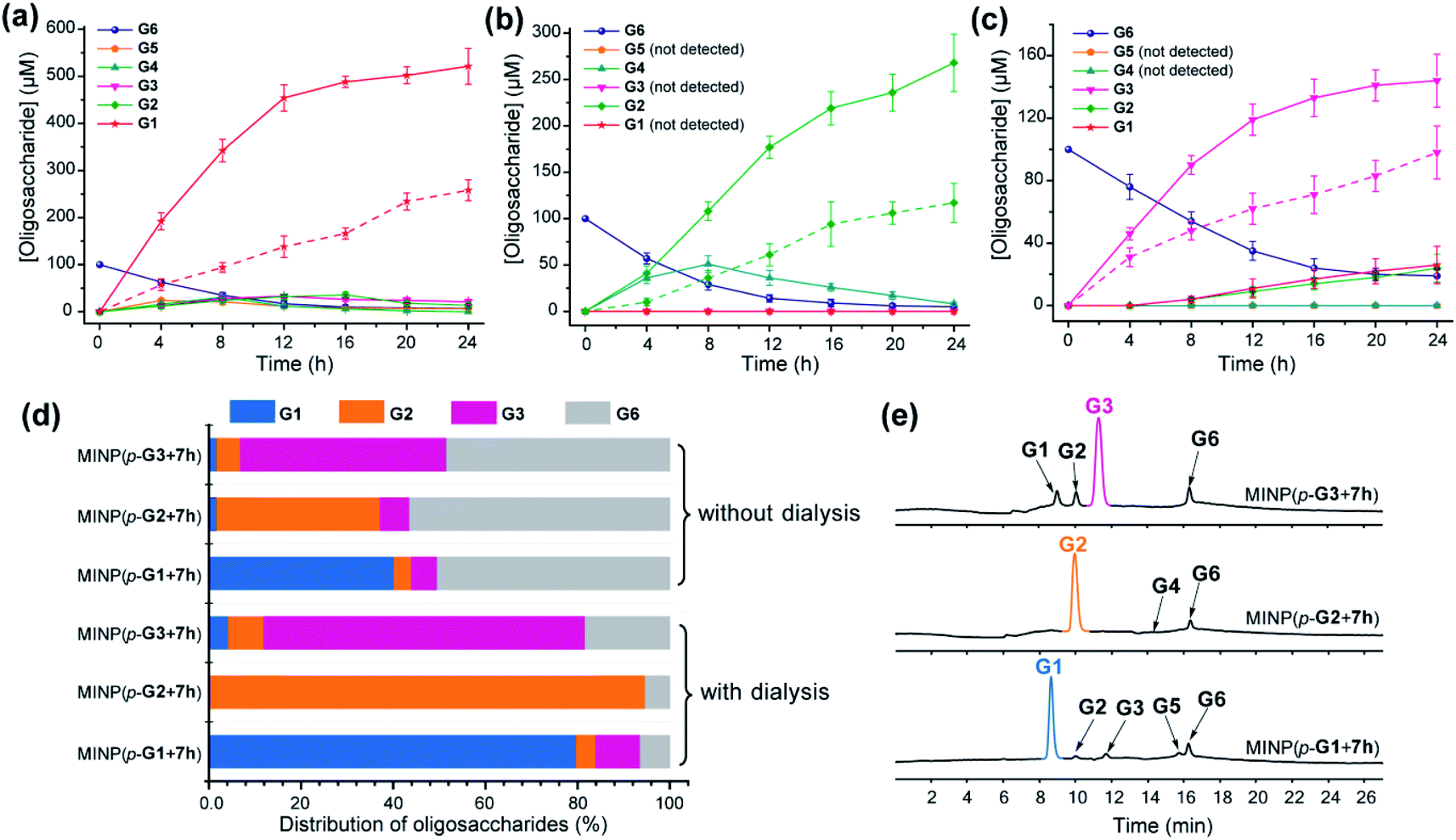 | ||
| Fig. 2 Production distributions in G6 hydrolysis catalyzed by (a) MINP(p-G1 + 7h), (b) MINP(p-G2 + 7h), and (c) MINP(p-G3 + 7h) at 60 °C in H2O, with the reaction mixture (1.0 mL) dialyzed against 40 mL of Millipore water using a membrane (MWCO = 500). The points connected by dashed lines represent hydrolysis without dialysis. (d) Comparison of hydrolysis with and without dialysis, showing the amounts of the starting material G6 and G1–G3 products formed with different catalysts. The product distribution was normalized to G6 equivalents by dividing the G1 concentration by 6, G2 by 3, and G3 by 2. (e) Extracted ion chromatograms of the reaction mixtures in G6 hydrolysis catalyzed by different MINP catalysts inside dialysis tubing (MWCO 500) after 24 h at 60 °C in H2O. Yields were determined by LC-MS using calibration curves generated from authentic samples (Fig. S32†). [Maltohexaose] = 100 μM. [MINP] = 20 μM. | ||
Fig. 2d compares the formation of the desired products with and without dialysis. Fig. 2e shows the LC-MS analyses of the reaction mixtures with the three MINP catalysts. The data indicate that the desired sugar could always be produced as the major product, with the yield increased substantially with dialysis.
Controlled hydrolysis of polysaccharides and recyclability of the catalysts
A rich source of polysaccharides is found in nature. Their precise cleavage based on our selective one-step hydrolysis can be a convenient and economical way to produce glycans that otherwise require complex multistep synthesis and extensive protective/deprotective chemistry.5 Gratifyingly, not only could these MINP glycosidases hydrolyze maltohexaose in a highly controlled fashion, but they could also hydrolyze amylose, a polysaccharide of glucose connected by the same 1,4-α-glycosidic linkage, with equally good selectivity (Fig. 3a). The hydrolysis once again happened inside the dialysis membrane, with the polysaccharide and MINP catalysts trapped inside and the product released into the bulk solution.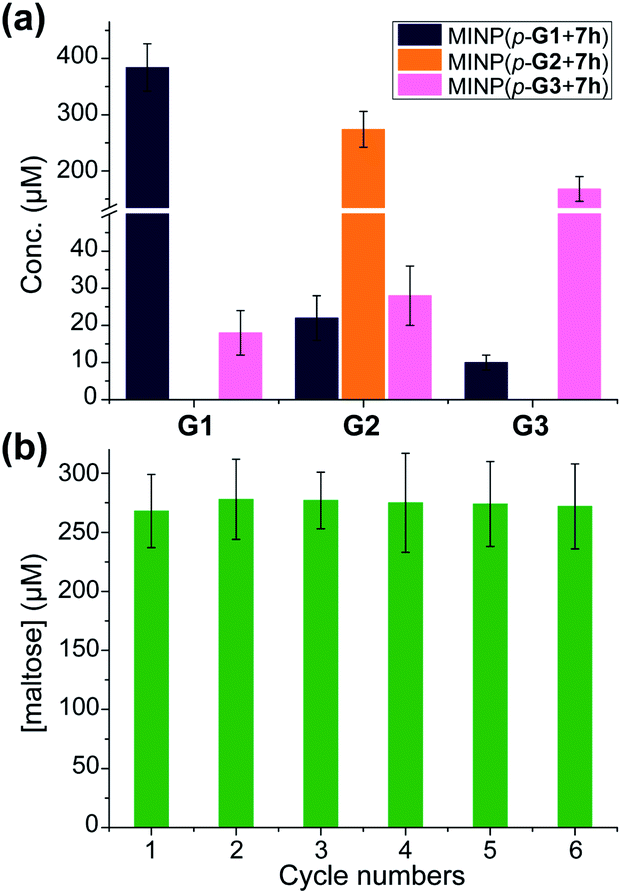 | ||
| Fig. 3 (a) The product distribution (G1, G2, and G3) in the hydrolysis of amylose using the MINP catalysts after 24 h at 60 °C in H2O, with the reaction mixture (1.0 mL) dialyzed against 40 mL of deionized water using a membrane (MWCO = 500). [Amylose] = 1 mg mL−1, [MINP] = 20 μM. (b) The recyclability of MINP(p-G2 + 7h) for maltohexaose hydrolysis. [Maltohexaose] = 100 μM. [MINP] = 20 μM. | ||
Another benefit of performing the hydrolysis inside a dialysis membrane was the facile recycling of the catalyst. As highly cross-linked polymeric nanoparticles, our MINP-based artificial glycosidase could be reused many times without any concerns regarding loss of activity when maltohexaose was repeatedly added into the dialysis tubing that contained MINP(p-G2 + 7h) (Fig. 3b).
Substrate selectivity
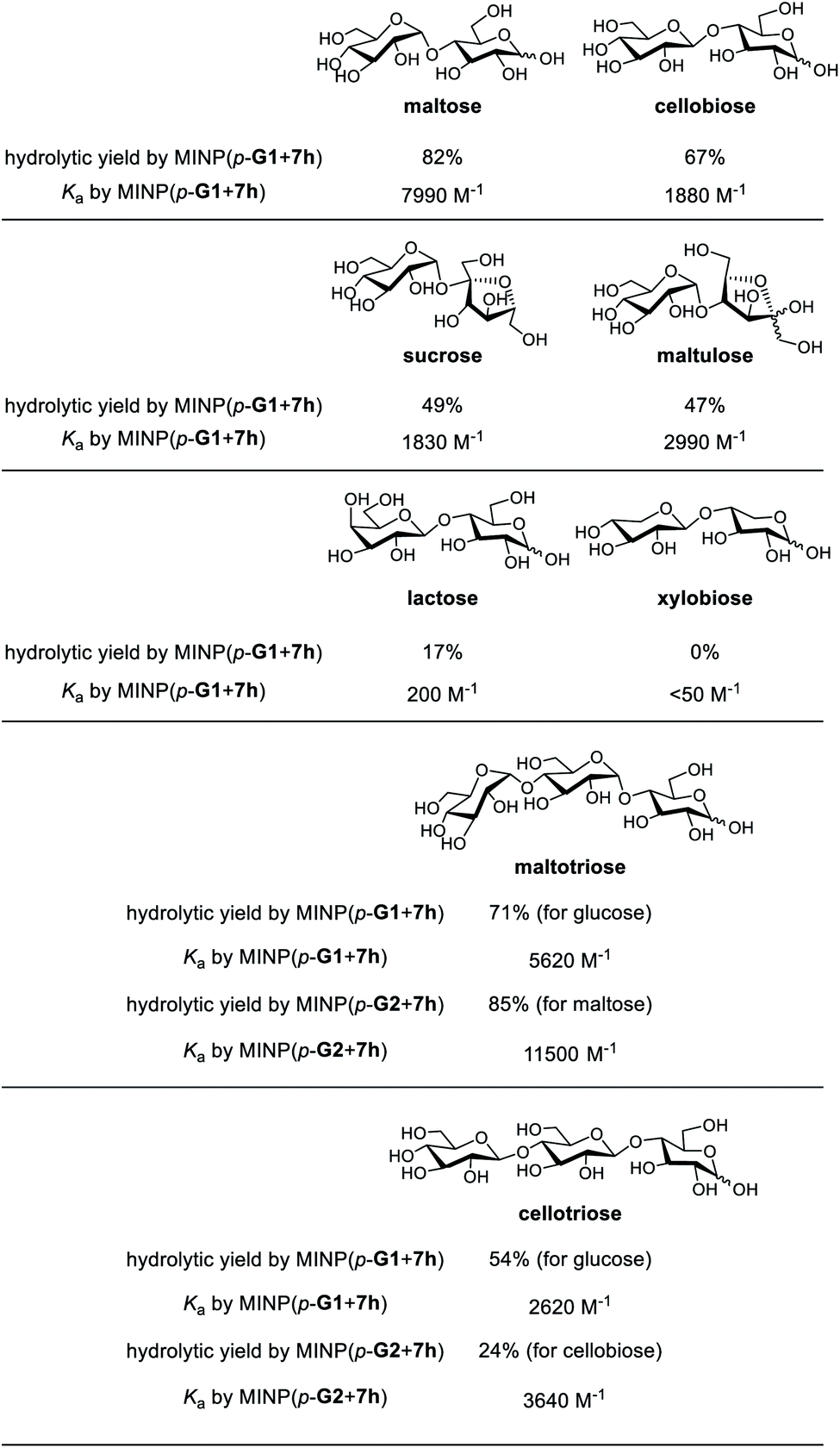
Substrate selectivity is one of the most important performance criteria for a synthetic glycosidase, since different building blocks, connection sites, and spatial orientations of the glycosidic linkage can influence the property of a glycan profoundly. Table 5 shows the hydrolysis of a number of oligosaccharides by our MINP catalysts. The yields were for hydrolysis at 60 °C after 24 h and the binding constants were for the same MINP determined by ITC at 25 °C.| a The hydrolysis experiments were performed at 60 °C in water for 24 h, with [oligosaccharide] = 0.2 mM and [MINP] = 20 μM. Yields were determined by LC-MS using calibration curves generated from authentic samples (Fig. S32). Isothermal titration calorimetry (ITC) data are reported in Table S1. |
|---|
|
Consistent with the binding-derived catalysis, there was an overall correlation between the hydrolytic yields and the Ka values. For example, among the disaccharides, maltose gave the best yield with MINP(p-G1 + 7h) and its binding was also the strongest. Xylobiose was completely inactive and its binding was also the weakest. For the sugars with intermediate binding constants (cellobiose, sucrose, and maltulose), the correlation was weak.
Another conclusion from the hydrolyses was the importance of the boronate ester formation to the substrate selectivity. MINP(p-G1 + 7h) was designed to bind the terminal glucose of a suitable oligo- or polysaccharide at the non-reducing end (Scheme 2). Thus, it was not a surprise that cellobiose, sucrose, and maltulose could all be hydrolyzed by this MINP. The reducing sugar residue on these molecules is expected to reside in water, outside the active site. For the same reason, MINP(p-G1 + 7h) should NOT be particularly selective for the reducing sugar, whether in its chemical structure or spatial orientation. Meanwhile, MINP(p-G1 + 7h) should be much more selective toward the sugar at the nonreducing end, especially if the hydroxyl involved in boronate formation is altered. Lactose, with a galactose at the non-reducing end, indeed gave a very poor hydrolytic yield and binding constant, because the C4 hydroxyl was involved in boronate formation (Scheme 2). It is quite impressive that inversion of a single hydroxyl decreased the yield of hydrolysis from 67% for cellobiose to 17% for lactose. Xylobiose is missing the hydroxymethyl from cellobiose. Its inactivity indicates that the C6 hydroxyl was also essential to the binding.
For a monosaccharide-derived catalyst such as MINP(p-G1 + 7h), its only selectivity was in the terminal sugar at the non-reducing end and the α/β selectivity was low. For a disaccharide-derived catalyst, the situation was different because the α/β linkage between the first two sugar residues would affect the binding of the substrate.
MINP(p-G1+7h) can hydrolyze maltotriose and cellotriose into glucose. Table 5 shows that the yield of glucose was 71% and 54% from the two trisaccharides, respectively. The α/β selectivity (1.3:1) was slightly higher than that observed in maltose/cellobiose (1.2:1), possibly because two hydrolyses were needed to hydrolyze the trisaccharides, but only one for the disaccharides, which magnified the α/β selectivity. When MINP(p-G2 + 7h) was used, however, the yield for the desired (disaccharide) product was 85% from maltotriose and only 24% from cellotriose. This was because the imprinted site was designed to bind maltose in this catalyst (Scheme 2). Thus, the β glycosidic bond in between the first and second sugar from the nonreducing end of cellotriose would weaken the binding of this substrate.
Conclusions
Micellar imprinting provided a rational method for constructing robust synthetic glycosidases from readily synthesized small-molecule templates. The natural glucan 1,4-alpha-glucosidase removes one glucose residue at a time from the nonreducing end of amylose,50 and beta-amylase removes two glucose residues (i.e., maltose) at a time.51 Our synthetic glycosidases not only duplicated the selectivities of these enzymes but also had selectivity not available from natural biocatalysts—i.e., the selective formation of maltotriose from maltohexaose or amylose. Substrate selectivity was mainly determined by the sugar residues bound within the active site, including their spatial orientations. As cross-linked polymeric nanoparticles, the MINPs tolerate high temperature,23,42 organic solvents,42 and extreme pH,37 outperforming natural enzymes completely in these aspects.Importantly, the design of our synthetic glycosidase is general, using molecular imprinting to create a glycan-specific active site, followed by post-modification to install an acidic group right next to the glycosidic bond to be cleaved. Similar designs should be applicable to complex glycans.52 The total synthesis of carbohydrates is often extremely challenging.5 Selective one-step hydrolysis using a rationally designed synthetic glycosidase potentially can be a powerful method to produce complex glycans from precursor oligosaccharides or polysaccharides that are either naturally available or prepared through enzymatic synthesis. The facile separation of the products by dialysis demonstrated in this work, the excellent reusability of the MINP catalysts, and the simplicity of the hydrolysis requiring only hot water are attractive features for such purposes, and can open up new avenues in glycoscience and technology.
Experimental section
Typical Procedure for the Synthesis of MINP Catalysts35
A solution of 6-vinylbenzoxaborole (4) in methanol (0.0004 mmol) was added to imine template p-5a in methanol (0.0004 mmol) in a vial containing methanol (5 mL). After the mixture was stirred for 6 h at room temperature, the methanol was removed in vacuo to afford the final sugar-boronate templates p-6a. A micellar solution of compound 1a or 1b (0.03 mmol), compound 2 (0.02 mmol), divinylbenzene (DVB, 2.8 μL, 0.02 mmol), and 2,2-dimethoxy-2-phenylacetophenone (DMPA, 10 μL of a 12.8 mg mL−1 solution in DMSO, 0.0005 mmol) in H2O (2.0 mL) was added to the sugar-boronate complex. The mixture was subjected to ultrasonication for 10–15 min until the mixture become clear. Then, CuCl2 (10 μL of a 6.7 mg mL−1 solution in H2O, 0.0005 mmol) and sodium ascorbate (10 μL of a 99 mg mL−1 solution in H2O, 0.005 mmol) were added to the mixture. After the reaction mixture was stirred slowly at room temperature for 12 h, the reaction mixture was sealed with a rubber stopper, degassed with N2 three times and purged with nitrogen for 15 min, and irradiated in a Rayonet reactor for 12 h. Compound 3 (10.6 mg, 0.04 mmol), CuCl2 (10 μL of a 6.7 mg mL−1 solution in H2O, 0.0005 mmol), and sodium ascorbate (10 μL of a 99 mg mL−1 solution in H2O, 0.005 mmol) were added. The progress of the reaction was monitored by 1H NMR spectroscopy and dynamic light scattering (DLS). After being stirred for another 6 h at room temperature, the reaction mixture was poured into acetone (8 mL). The precipitate collected by centrifugation was washed with a mixture of acetone/water (5 mL/1 mL) three times, followed by acetone (5 mL) two times before being dried in air to afford the MINP(p-G1). The solid was then rinsed with acetone (5 mL) two times and dried in air to afford the final MINPs. Typical yields were >80%.MINP(p-G1) obtained above was dissolved in 6 N HCl aqueous solution (2 mL) and the solution was stirred at 95 °C for 2 h. After being cooled down to room temperature, the reaction mixture was poured into acetone (8 mL). The precipitate collected by centrifugation was washed with a mixture of acetone/water/CH3OH (5 mL/1 mL/1 mL) three times, and acetone (5 mL) twice, and dried in air. The resulting MINP-CHO(p-G1) (5 mg, 0.0001 mmol) was dissolved in dry DMF (0.5 mL), followed by the addition of 7h (10 μL of 0.1 M stock solution in DMSO, 0.001 mmol). After the reaction mixture was stirred at 50 °C for 6 h, borane–pyridine complex (10 μL of 0.1 M stock solution in dry DMF, 0.001 mmol) was added. The mixture was stirred at 50 °C overnight. After being cooled down to room temperature, the DMF solution was poured into acetone (8 mL). The precipitate collected by centrifugation was washed with acetone/methanol (5 mL/1 mL), methanol/HCl (5 mL/0.1 mL, 1 M) three times, and acetone (5 mL) twice, and dried in air to afford the final MINP(p-G1 + 7h).
Hydrolysis of Oligosaccharides and Polysaccharides
Hydrolysis experiments without dialysis were carried out as follows. In general, a 200 μL aliquot of a 100 μM MINP stock solution in Millipore water was diluted by water or a 10 mM MES buffer (pH 6.0) to 990 μL and sonicated for 0.5 min. To this solution, a 10 μL aliquot of a 10/20 mM oligosaccharide stock solution was added. The reaction mixture was allowed to react in a Benchmark heating block at 60 or 90 °C for the indicated time. The reaction mixture was centrifuged (20000 RPM for 10 min) to remove the MINP catalyst before LC-MS analysis using calibration curves generated from authentic samples (Fig. S32†). Hydrolysis experiments with dialysis were carried out as follows. In general, a 200 μL aliquot of a 100 μM MINP catalyst in Millipore water was diluted with Millipore water to 990 μL and sonicated for 0.5 min, and then the solution was added to a dialysis tubing (MWCO 500), followed by the addition of a 10 μL aliquot of a 10 mM maltohexaose stock solution (or 1 mg of amylose). The reaction mixture was dialyzed against 40 mL of Millipore water at 60 °C. The hydrolysis was monitored by LC-MS analysis of the external solution using calibration curves generated from authentic samples (Fig. S32†).
Conflicts of interest
Iowa State University Research Foundation has filed a patent application relating to the technology.Acknowledgements
We thank NSF (CHE-1708526) and NIGMS (R01GM138427) for financial support of this research.Notes and references
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS.
- Z. Zhang, J. Song and B. Han, Chem. Rev., 2017, 117, 6834–6880 CrossRef CAS.
- N. R. C. (U.S.). Transforming glycoscience: a roadmap for the future, National Academies Press, Washington, D.C., 2012 Search PubMed.
- C. R. Bertozzi and L. L. Kiessling, Science, 2001, 291, 2357–2364 CrossRef CAS.
- J. P. Kamerling and G.-J. Boons, Comprehensive glycoscience: from chemistry to systems biology, Elsevier, Amsterdam ; Boston, 1st edn, 2007 Search PubMed.
- D. Solís, N. V. Bovin, A. P. Davis, J. Jiménez-Barbero, A. Romero, R. Roy, K. Smetana and H.-J. Gabius, Biochim. Biophys. Acta, Gen. Subj., 2015, 1850, 186–235 CrossRef.
- K. K. Palaniappan and C. R. Bertozzi, Chem. Rev., 2016, 116, 14277–14306 CrossRef CAS.
- D. H. Dube and C. R. Bertozzi, Nat. Rev. Drug Discov., 2005, 4, 477–488 CrossRef CAS.
- G. J. Davies, T. M. Gloster and B. Henrissat, Curr. Opin. Struct. Biol., 2005, 15, 637–645 CrossRef CAS.
- R. M. Wahlström and A. Suurnäkki, Green Chem., 2015, 17, 694–714 RSC.
- C. Rousseau, N. Nielsen and M. Bols, Tetrahedron Lett., 2004, 45, 8709–8711 CrossRef CAS.
- F. Ortega-Caballero, C. Rousseau, B. Christensen, T. E. Petersen and M. Bols, J. Am. Chem. Soc., 2005, 127, 3238–3239 CrossRef CAS.
- S. Striegler, J. D. Barnett and N. A. Dunaway, ACS Catal., 2012, 2, 50–55 CrossRef CAS.
- B. Sharma and S. Striegler, Biomacromolecules, 2018, 19, 1164–1174 CrossRef CAS.
- M. Samanta, V. S. R. Krishna and S. Bandyopadhyay, Chem. Commun., 2014, 50, 10577–10579 RSC.
- Z. Yu and J. A. Cowan, Angew. Chem., Int. Ed., 2017, 56, 2763–2766 CrossRef CAS.
- A. P. Davis and T. D. James, Carbohydrate Receptors, Wiley-VCH, Weinheim, 2005 Search PubMed.
- R. Breslow, Artificial enzymes, Wiley-VCH, Weinheim, 2005 Search PubMed.
- M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1734–1787 RSC.
- G. Wulff, Chem. Rev., 2002, 102, 1–28 CrossRef CAS.
- K. Haupt and K. Mosbach, Chem. Rev., 2000, 100, 2495–2504 CrossRef CAS.
- L. Ye and K. Mosbach, Chem. Mater., 2008, 20, 859–868 CrossRef CAS.
- J. K. Awino and Y. Zhao, J. Am. Chem. Soc., 2013, 135, 12552–12555 CrossRef CAS.
- K. Chen and Y. Zhao, Org. Biomol. Chem., 2019, 17, 8611–8617 RSC.
- T. D. James, M. D. Phillips and S. Shinkai, Boronic acids in saccharide recognition, RSC Publishing, Cambridge, 2006 Search PubMed.
- K. T. Kim, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, J. Am. Chem. Soc., 2009, 131, 13908–13909 CrossRef CAS.
- A. Pal, M. Bérubé and D. G. Hall, Angew. Chem., Int. Ed., 2010, 49, 1492–1495 CrossRef CAS.
- X. Wu, Z. Li, X.-X. Chen, J. S. Fossey, T. D. James and Y.-B. Jiang, Chem. Soc. Rev., 2013, 42, 8032–8048 RSC.
- S. D. Bull, M. G. Davidson, J. M. H. Van den Elsen, J. S. Fossey, A. T. A. Jenkins, Y. B. Jiang, Y. Kubo, F. Marken, K. Sakurai, J. Z. Zhao and T. D. James, Acc. Chem. Res., 2013, 46, 312–326 CrossRef CAS.
- G. Wulff and W. Vesper, J. Chromatogr., 1978, 167, 171–186 CrossRef CAS.
- J. K. Awino, R. W. Gunasekara and Y. Zhao, J. Am. Chem. Soc., 2016, 138, 9759–9762 CrossRef CAS.
- H. Kim, Y. J. Kang, S. Kang and K. T. Kim, J. Am. Chem. Soc., 2012, 134, 4030–4033 CrossRef CAS.
- M. Dowlut and D. G. Hall, J. Am. Chem. Soc., 2006, 128, 4226–4227 CrossRef CAS.
- M. Bérubé, M. Dowlut and D. G. Hall, J. Org. Chem., 2008, 73, 6471–6479 CrossRef.
- R. W. Gunasekara and Y. Zhao, J. Am. Chem. Soc., 2017, 139, 829–835 CrossRef CAS.
- G. Wulff, Angew. Chem., Int. Ed. Engl., 1995, 34, 1812–1832 CrossRef CAS.
- X. Xing and Y. Zhao, New J. Chem., 2018, 42, 9377–9380 RSC.
- J. K. Awino and Y. Zhao, Org. Biomol. Chem., 2017, 15, 4851–4858 RSC.
- J. K. Awino, R. W. Gunasekara and Y. Zhao, J. Am. Chem. Soc., 2017, 139, 2188–2191 CrossRef CAS.
- The curved α-1,4-glycosidic linkage also explains the facile formation of cyclic oligomers (i.e., cyclodextrins) from glucose.
- B. Wang and G.-J. Boons, Carbohydrate recognition: biological problems, methods, and applications, Wiley, Hoboken, N.J., 2011 Search PubMed.
- X. Xing and Y. Zhao, Org. Biomol. Chem., 2018, 16, 2855–2859 RSC.
- M. D. Arifuzzaman and Y. Zhao, J. Org. Chem., 2016, 81, 7518–7526 CrossRef CAS.
- J. S. Nowick, J. S. Chen and G. Noronha, J. Am. Chem. Soc., 1993, 115, 7636–7644 CrossRef CAS.
- F. H. Westheimer, Tetrahedron, 1995, 51, 3–20 CrossRef CAS.
- D. Matulis and V. A. Bloomfield, Biophys. Chem., 2001, 93, 37–51 CrossRef CAS.
- G. Chadha and Y. Zhao, Chem. Commun., 2014, 50, 2718–2720 RSC.
- For MINP(p-G2), significant amounts of G4 were observed in the early part of reaction but disappeared at higher conversions (Fig. 2b).
- R. U. Lemieux, Acc. Chem. Res., 1996, 29, 373–380 CrossRef CAS.
- D. French and D. W. Knapp, J. Biol. Chem., 1950, 187, 463–471 CAS.
- P. Bernfeld, in Methods in Enzymology, Academic Press, 1955, vol. 1, pp. 149–158 Search PubMed.
- M. Zangiabadi and Y. Zhao, Nano Lett., 2020, 20, 5106–5110 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc05338d |
| This journal is © The Royal Society of Chemistry 2021 |