
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of structure and solubility of chain transfer agents on the RAFT control of dispersion polymerisation in scCO2†
Ana A. C.
Pacheco
a,
Arnaldo F.
da Silva Filho
a,
Kristoffer
Kortsen
 a,
Magnus W. D.
Hanson-Heine
a,
Vincenzo
Taresco
a,
Jonathan D.
Hirst
a,
Muriel
Lansalot
*b,
Franck
D'Agosto
*b and
Steven M.
Howdle
*a
a,
Magnus W. D.
Hanson-Heine
a,
Vincenzo
Taresco
a,
Jonathan D.
Hirst
a,
Muriel
Lansalot
*b,
Franck
D'Agosto
*b and
Steven M.
Howdle
*a
aUniversity of Nottingham, University Park, Nottingham, England NG7 2RD, UK. E-mail: steve.howdle@nottingham.ac.uk
bUniv. Lyon, Université Claude Bernard Lyon 1, CPE Lyon, CNRS, UMR 5265, Chemistry, Catalysis, Polymers and Processes (C2P2), 43 Bd du 11 Novembre 1918, 69616 Villeurbanne, France. E-mail: franck.dagosto@univ-lyon1.fr; muriel.lansalot@univ-lyon1.fr
First published on 17th November 2020
Abstract
Reversible addition–fragmentation chain transfer (RAFT) dispersion polymerisation of methyl methacrylate (MMA) is performed in supercritical carbon dioxide (scCO2) with 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid (DDMAT) present as chain transfer agent (CTA) and surprisingly shows good control over PMMA molecular weight. Kinetic studies of the polymerisation in scCO2 also confirm these data. By contrast, only poor control of MMA polymerisation is obtained in toluene solution, as would be expected for this CTA which is better suited for acrylates. In this regard, we select a range of CTAs and use them to determine the parameters that must be considered for good control in dispersion polymerisation in scCO2. A thorough investigation of the nucleation stage during the dispersion polymerisation reveals an unexpected “in situ two-stage” mechanism that strongly determines how the CTA works. Finally, using a novel computational solvation model, we identify a correlation between polymerisation control and degree of solubility of the CTAs. All of this ultimately gives rise to a simple, elegant and counterintuitive guideline to select the best CTA for RAFT dispersion polymerisation in scCO2.
Introduction
Supercritical carbon dioxide (scCO2) is a benign solvent (nontoxic, non-flammable, and inert), that has emerged as a potential replacement for organic solvents.1,2 Moreover, scCO2 has an easily attainable critical point (31.0 °C, 73.8 bar), which is advantageous from an energetic perspective. The high solubility of most monomers, and the poor solubility of most polymers in scCO2 makes it an ideal solvent for dispersion polymerisation.3,4 In a dispersion polymerisation all reactants (i.e. monomer, initiator, etc.) are soluble in the continuous phase at the reaction onset. After the polymerisation is initiated and a critical chain length (Jcrit) is achieved, the growing polymer becomes insoluble and the small chains agglomerate and precipitate to form nuclei, which are then captured by stabiliser, leading to a colloidal dispersion.5 These particles are then enlarged by the inward diffusion of the remaining monomer,6 giving a latex with particle diameters spanning from 100 nm to 20 μm.Dispersion polymerisation is reported to be aided by the low viscosity and high diffusivity of scCO2, which can overcome known issues encountered in traditional heterogeneous polymerisations.1,7 Nevertheless, the greatest advantage of conducting dispersion polymerisations in scCO2 is the facile and complete removal of solvent by simple depressurisation, producing a dry, free-flowing powder that requires no further purification (e.g. drying).7 Since the first reported8 successful radical dispersion polymerisations in scCO2, many vinyl monomers have been polymerised in this reaction medium.9–18
In parallel, the advent of reversible-deactivation radical polymerisation (RDRP) has opened up the possibility to exert control over the number-average molecular weight (Mn) and molecular-weight dispersity (Ð), and to access well-defined and complex architectures via a free radical process. There is extensive research on RDRP in scCO2,19 but here we focus on reversible addition–fragmentation chain transfer (RAFT).3 RAFT is a well-established, robust and versatile RDRP technique based on reversible and degenerative transfer.20 The reaction conditions for RAFT polymerisation are very similar to conventional radical polymerisation, with the addition of a chain transfer agent (CTA; Z–C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S)–SR) that can be a dithioester, a trithiocarbonate, a dithiocarbonate (xanthate) or a dithiocarbamate.21,22
S)–SR) that can be a dithioester, a trithiocarbonate, a dithiocarbonate (xanthate) or a dithiocarbamate.21,22
Transposition of RDRP from homogeneous to dispersed systems is not straightforward. As an example, the early attempts to implement RDRP based on reversible termination (using nitroxide mediated polymerisation) for styrene in dispersion in decane23 or alcohols24 showed long polymerisation times, low conversion, poor control and broad particle size distribution. This occurs because in RDRP, a large number of chains remain active and grow slowly at the same time. The slow formation of many low molecular weight chains all growing at the same time is very different from the rapid formation of few chains of high molecular weight in the early stage of a conventional radical polymerisation. This strongly impacts the nucleation stage, and, consequently, both the stability of the resulting particles and the further control of their polymerisation once they have been formed.25,26
This effect was rationalised by Winnik27 in the particular case of RDRP of styrene in dispersion governed by reversible degenerative transfer (RAFT and iodine transfer polymerisation) and conducted in ethanol or mixtures of ethanol and water. To achieve successful control of the polymerisation, the authors showed that the addition of the CTA for RAFT polymerisation must be delayed in order to take advantage of the efficient nucleation taking place in a conventional dispersion polymerisation. The major breakthrough was the understanding of the need to establish the control inside the formed particles quickly after their formation. However, successful RAFT dispersion polymerisations without such issues have been observed in scCO2 and have been reported to show a surprisingly good level of control in just a single step.13,19,28,29
In 2007, dispersion RAFT polymerisation of methyl methacrylate (MMA) in scCO2 was reported using a dithiobenzoate CTA.29 Reasonable control was observed (Ð ∼ 1.5 with good agreement between theoretical and experimental Mn), and the product was obtained at high conversion as a free-flowing powder (1–2 μm spherical particles). Subsequently, a more detailed study on the effects of various CTAs for MMA polymerisation was reported in scCO2.28 Four CTAs carrying a dithiophtalate Z group and a cyanobenzyl R group or dithiobenzoates Z group and stabilized cyanobenzyl, cyanoisoprop-2-yl, or 4-cyano-1-hydroxypent-4-yl R groups were tested. All polymerisations gave fine, free-flowing powder at high conversion (>90%), with ∼1.4 μm spherical particles. Very prolonged induction periods (5–13 h) were observed for the four CTAs; much longer than in bulk/solution.30 Nevertheless, all four CTAs resulted in a linear evolution of Mn with conversion, leading to Mn close to target and low Ð (∼1.20), in accordance with a successful transposition of RAFT polymerisation to scCO2 dispersion polymerisation.
This excellent control across all the CTAs tested can be ascribed to the selection of CTAs carrying strongly stabilized R reinitiating groups giving high chain transfer constant30,31 which are well known to be suited for good RAFT polymerisation of methacrylates.30,32 In addition, the authors explained that good control over dispersion polymerisation in a single step could be attributed to the high mobility of species in the polymer particles that were highly plasticised by the scCO2, thus providing a much reduced viscosity in the particles.28 An additional contributing factor to the control in the reaction is thought to be the reduction of Jcrit, due to the low solvation power of scCO2 for PMMA when compared to other conventional solvents.28 The lower the Jcrit, the smaller would be the CTA effect in delaying nucleation thus leading to better control. However, none of these hypotheses have been proven so far.
Trithiocarbonates are also known to be good CTAs for the control of MMA polymerisation.33 However, the choice of the R group is critical in the case of methacrylates, with the most effective CTA carrying a strongly stabilized R reinitiating group such as a tertiary cyanoalkyl or a cumyl.34 Indeed, DDMAT (structure 1, Fig. 1) which has a tertiary alkyl –R reinitiating group, has been previously reported to be a good CTA for acrylates, but not applicable for methacrylates.35 Indeed, it has been well documented that DDMAT gives essentially no control over polymerisation of methacrylates in solution.34 Nonetheless, initial CTA screening for the preparation of block copolymers from our own work had shown that both DDMAT and the dithiobenzoate 2-cyano-2-propyl benzodithioate (CPDB) could give similar control (low Ð and good agreement between theoretical and experimental Mn) over MMA polymerisation, despite their different transfer constants (Ctr). This initial outcome led us to the successful synthesis of block copolymers based on MMA and 4-vinylpyridine (4VP). As a result, we developed a wide range of block copolymers in scCO2 using PMMA synthesised with DDMAT in RAFT mediated scCO2 dispersion polymerisation as the first block.14,37 The PMMA chain extension was performed at that time without studying in detail the possible mechanism of control. The group further built upon these data to develop fine control of the internal morphology that arises from phase separation for a series of PMMA-based block copolymer microparticles and these were studied via in situ small-angle X-ray scattering (SAXS).36
 | ||
| Fig. 1 Chain transfer agent (CTA) library, with the R group in red and the Z group in blue: (1) DDMAT (2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid); (2) CPAB (4-cyano-4-(phenylcarbonothioylthio)pentanoic acid); (3) CPDT (2-cyano-2-propyl dodecyl trithiocarbonate) and the stabiliser PDMS-MA (methacrylate terminated polydimethylsiloxane). | ||
So why does the DDMAT work well in scCO2? We now report on our attempts to better understand the RAFT polymerisation of MMA in scCO2 dispersion polymerisation, and more broadly to better understand the reaction process and parameters that should be considered for the selection of the best CTAs for successful RAFT or more generally speaking RDRP dispersion polymerisation in scCO2.
Results and discussion
RAFT polymerisation of MMA mediated by DDMAT in solution in toluene and in dispersion in scCO2.DDMAT was used as CTA for the polymerisation of MMA both in toluene solution and in dispersion in scCO2 to assess the control given by this choice of CTA for a methacrylate polymerisation.
The RAFT solution polymerisation in toluene was performed using AIBN as initiator with a CTA/AIBN ratio of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (E1.1, Table 1). The results confirm the inability of DDMAT to finely control MMA polymerisation, leading to PMMA chains with a large molecular-weight dispersity (Ð = 1.60) and a final Mn that does not match the expected theoretical value (Mn = 82.3 kg mol−1vs. Mn,th = 40.1 kg mol−1); as would be expected from the poor reinitiating efficiency of DDMAT for MMA polymerisation.
:1 (E1.1, Table 1). The results confirm the inability of DDMAT to finely control MMA polymerisation, leading to PMMA chains with a large molecular-weight dispersity (Ð = 1.60) and a final Mn that does not match the expected theoretical value (Mn = 82.3 kg mol−1vs. Mn,th = 40.1 kg mol−1); as would be expected from the poor reinitiating efficiency of DDMAT for MMA polymerisation.
| Expt. | CTA | Solvent | Conv.a (%) | M n,th | M n | Ð |
|---|---|---|---|---|---|---|
| a Conversion calculated from 1H NMR. b Theoretical Mn calculated relative to CTA and monomer concentration and given in kg mol−1. c Ð and Mn (in kg mol−1) obtained by THF-SEC with RI detector against PMMA standards. d Results extracted from Kortsen et al.42 See Experimental section for reaction conditions used for toluene solution polymerisation and scCO2 dispersion polymerisation. | ||||||
| E1.1 | 1-DDMAT | Toluene | 66 | 40.1 | 82.3 | 1.60 |
| E1.2 | 1-DDMAT | scCO2 | 99 | 59.4 | 51.1 | 1.20 |
| E2.1 | 2-CPAB | Toluene | 81 | 48.6 | 49.7 | 1.21 |
| E2.2 | 2-CPAB | scCO2 | 97 | 57.9 | 84.9 | 1.49 |
| E3.1 | 3-CPDT | Toluene | 65 | 41.3 | 43.6 | 1.18 |
| E3.2d | 3-CPDT | scCO2 | 98 | 58.6 | 60.0 | 1.20 |
For dispersion polymerisation in scCO2, the temperature and pressure were selected to ensure solubility of the PDMS-MA acting as a stabiliser in the process.37 The CTA/AIBN ratio used in scCO2 (2:1) was lower than in toluene (5:1). The use of a higher concentrations of initiator was established previously13,14,28 because the rate of decomposition of AIBN in scCO2 is 2.5 times slower than in the equivalent reactions in benzene.38 This higher initiator concentration at the start of the reaction ensures reasonable radical generation for initiation of polymer chains.
For clarity, the role of PDMS-MA (Fig. 1) is to stabilise the nuclei formed during the initial stages of the reaction. The rate of consumption of the stabiliser is not fully known, but there is good evidence that it is consumed mainly in the initial stages of the reaction, as the concentration of stabiliser used influences the final PMMA particle size.9,14 Furthermore, as a macromonomer, we expect that some PDMS-MA will co-polymerise with MMA and it has been previously reported that only up to 15% of the stabiliser is covalently bonded to the final product.39,40 The remaining PDMS-MA apparently acts as a stabiliser by anchoring through physical association of the methacrylate terminal group to the PMMA particle surface.
DDMAT presented great control in scCO2 (E1.2), with Ð = 1.20 and Mn (51.1 kg mol−1) close to Mn,th (59.4 kg mol−1). This was in agreement with previous unpublished work, but is an unusual outcome for this choice of CTA. Furthermore, very well-defined spherical particles of 2.11 μm were obtained (Fig. S1 and Table S1†). These results confirm the very good control provided by DDMAT for the polymerisation of MMA in scCO2 and are consistent with the previous results with block copolymers.13,41 Such positive results further hint that the control observed in scCO2 must arise from the mechanisms at play and in the physico-chemistry associated with dispersion polymerisation.
In order to further investigate this point, we made use of a recently developed sampling system42 to try to combine kinetic information with colloidal features of the system during the polymerisation. Previous sampling devices did not allow accurate conversion measurements due to loss of the volatile monomer.28,43 This new sampling system allows both molecular weight and conversion to be efficiently monitored.42
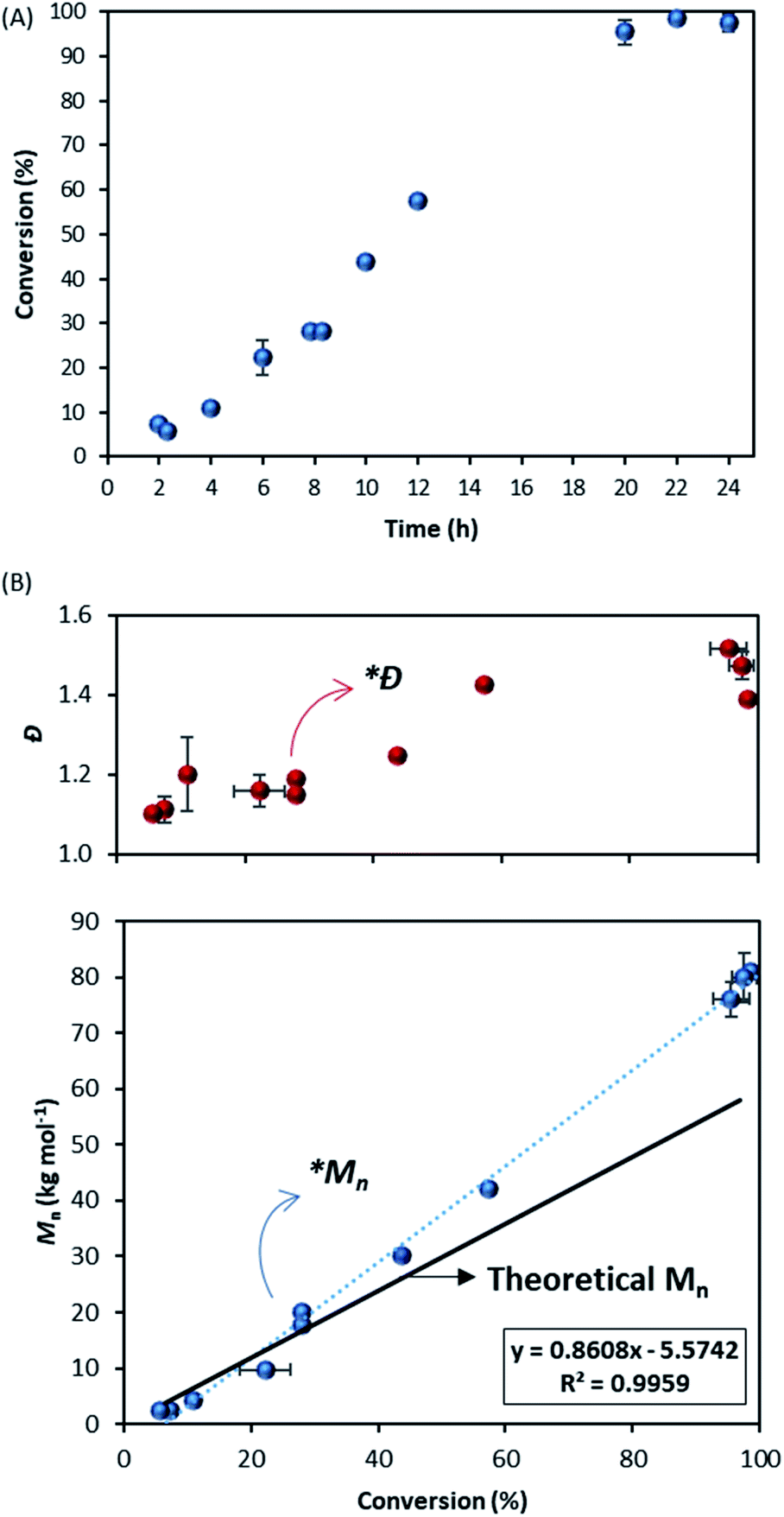
The kinetic study showed an increase of the conversion with time (Fig. 2A) and a linear evolution of Mn with monomer conversion, as expected in RDRP (Fig. 2B and C), although it is fitting the theoretical trend only at higher conversions. A deviation from the theoretical values is clear in the early stage of reaction (0–40% conversion) as noticed in Fig. 2C. The linear trend after 40% conversion (*Mn) is close to the theoretical evolution (black solid line) and it should be noted that dispersity was also consistently low (Ð ∼ 1.30) from this point (*Ð) throughout the reaction (Fig. 2C).
 | ||
| Fig. 2 Dispersion polymerisation of MMA in scCO2 using DDMAT as CTA. (A) Evolution of MMA conversion versus time. (B) Normalised SEC traces showing the molecular weight distributions of the samples withdrawn. (C) Evolution of Mn (blue) and Ð (red) versus conversion; solid trend line is the theoretical Mn and dashed trend line is the linear fitting of experimental data. (Molar ratio DDMAT/AIBN 2:1, 65 °C, 275 bar, 300 rpm stirring rate, 5 wt% of PDMS-MA as stabiliser (based on MMA)). A deviation from expected RDRP behaviour is observed until approximately 40% conversion (*Mn and *Ð). | ||
These results, apart from the early stage deviation, confirm the livingness of MMA dispersion polymerisation in scCO2 with DDMAT. Furthermore, the reaction appears to have a shorter induction time compared to Gregory et al.28 Indeed, they reported induction of up to 12 hours with dithioester CTAs, with no conversion observed before that point. It is important to reiterate that their data were obtained from less reliable kinetic measurements where further precipitation of product in cold hexane could exclude low molecular weight chains and artificially delay the observation of polymerisation onset. Therefore, their induction period was likely shorter.
Although the kinetic results do confirm the very good control obtained with DDMAT, they do not rationalise the surprising behaviour of this CTA in scCO2. In an effort to better understand the process of RAFT dispersion polymerisation in scCO2, we next followed the dispersion polymerisation of MMA visually in a static double window view cell (Fig. S2†) to study the early reaction stage and the onset of nucleation by the appearance of turbidity. Once the Jcrit is achieved, the growing polymer becomes insoluble and nucleation starts, the forming particles causing the once homogeneous system to become turbid.
In the absence of DDMAT, all other conditions remaining the same, a turbid system was observed within the first minute, in agreement with literature observations,44,45 and led to complete obscurity (i.e. no observable light passed through the view cell) within 10 minutes of reaction start (Fig. S3†). Furthermore, our previous kinetic study of conventional radical polymerisation (1 wt% AIBN relative to MMA) in scCO2 has shown conversions of 2.6% at 30 minutes from reaction onset.42 At that time nucleation has already occurred. In fact, Ballauff and Fehrenbacher have previously monitored the early stages (≤300 s) of MMA conventional radical dispersion polymerisation in scCO2via turbidimetry,44,45 and have observed that nucleation started before 0.1% MMA conversion.
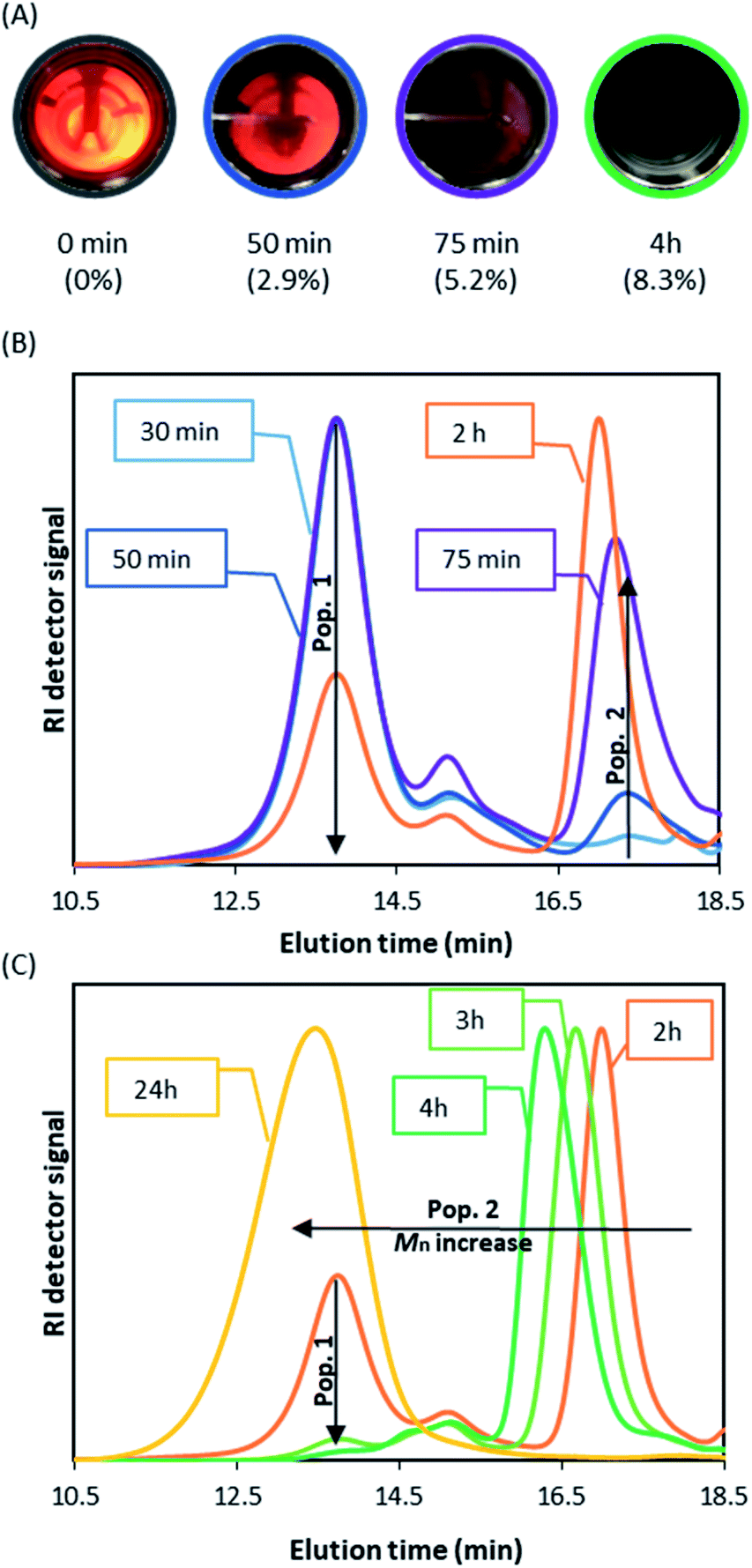
When DDMAT-mediated RAFT dispersion polymerisation was studied, turbidity was first observed 10 minutes after the start of reaction and complete obscurity occurred after circa 75 minutes, at which point conversion was found to be below 4% (Fig. 3A). Therefore, nucleation is very clearly delayed by addition of DDMAT.
 | ||
| Fig. 3 Early stage studies of RDRP dispersion in scCO2 with DDMAT. (A) Photographs of view cell study at different reaction times show the evolution of turbidity in the dispersion polymerisation; conversion is presented in brackets, the sample at 10 minutes gave undetectable conversion by 1H NMR. (B) THF SEC study of aliquots from reaction on sampling device. Inside the boxes, the reaction time is given to depict the normalised SEC traces. (C) Weight fraction% of peak 1 against peak 2 as a function of time. Two distinct Mn populations are observed, population 1 (conventional radical polymerisation) and population 2 (RAFT controlled), conversion at time points given in the boxes (molar ratio DDMAT/AIBN 2:1, 65 °C, 275 bar, 300 rpm stirring rate, 5 wt% of PDMS-MA as stabiliser (based on MMA)). | ||
In a perfectly controlled RAFT polymerisation, the slow growth of the polymer chains leads to Jcrit being achieved later than in conventional radical polymerisation, resulting in a delayed nucleation. In addition to the slower RAFT kinetics, inhibition and retardation are normally seen in RAFT polymerisation, in particular with dithiobenzoates. Inhibition is commonly attributed to the RAFT pre-equilibrium, but retardation is not well understood, although usually is associated with the intermediate radical.46,47 Three factors; kinetics; inhibition and retardation; have to be taken into account as possibly delaying nucleation in a RAFT controlled dispersed polymerisation.
It is important though, to reiterate that nucleation is also strongly related to the Mn of the polymer chains and not to monomer conversion. Once Jcrit is reached, the polymer will precipitate from solution and begin nucleation, regardless of the monomer conversion. However, Jcrit will be influenced by the solvency of the system with MMA working as a co-solvent.
We then repeated the polymerisation (Table 1 E1.2) with the sampling device in order to obtain aliquots on the time frame observed for the nucleation process. Interestingly, SEC analysis of aliquots taken immediately after turbidity onset (circa 10 minutes) show a dominant PMMA peak with high dispersity (Mn >400 kg mol−1; Ð = 1.5) (population 1), with a much smaller second peak (Mn ∼ 10 kg mol−1, Ð = 1.34 peak) (population 2) (Fig. 3B). As the reaction progresses, further kinetic sampling reveals that population 2 becomes the dominant species from 75 minutes into the reaction (3.8% monomer conversion), as clearly shown (Fig. 3C) by the SEC weight fraction (%) increase over time. Pop 2 was already >95% of the total weight fraction at the final sampling point (120 minutes) when monomer conversion was yet at 5.7% (Fig. 3C). In addition, population 2 was also found to present a UV signal at 300 nm, characteristic of CS bond on the trithiocarbonate chain end (Fig. S4†), indicating strongly that population 2 might correspond to living chains. Indeed, as the reaction progresses population 2 grows steadily to higher molecular weight (Mn = 62.2 kg mol−1; Ð = 1.22, 98.9% conversion after 24 hours). Additionally, we found that population 1 does not show a UV signal, which likely indicates that the corresponding chains are not carrying a trithiocarbonate chain end (Fig. S4†).
In a chemical sense, DDMAT is not supposed to be a good control agent for MMA polymerisation, as demonstrated in toluene (E1.1, Table 1). Thus, it seems reasonable to assume that RAFT control of the polymerisation of MMA in scCO2 will not be very efficient in the early stages of the process. At this point the medium is still homogeneous and the continuous phase is the reaction locus.44,45 As a result, some chains will certainly escape the expected RAFT equilibrium, and grow by conventional radical polymerisation to yield a high molecular weight population, which will nucleate very efficiently into PMMA particles and form a polymer rich phase. What then happens is that DDMAT, AIBN and MMA could begin to diffuse into those seed particles. These particles then become the main locus of the reaction where controlled polymerisation begins to take place. This also would explain the poor control observed at the beginning of the reaction in Fig. 2C.
It remains difficult to assess the concentration of each species in the different phases and the exact locus of the polymerisation in scCO2. The initiator (AIBN) is known to have high solubility in scCO2; DeSimone et al. studied the decomposition rate and efficiency of AIBN.38 Other studies also indicate that AIBN can equipartition between both phases,48,49 or yet have a partition coefficient (Kj) = 2,50 or Kj = 0.5,51 where Kj is the ratio of the concentration in the dispersed phase by the concentration in the continuous phase.
The solvent (CO2) and low molecular weight monomers such as MMA are known to diffuse into the PMMA particles very effectively under scCO2 conditions. This is evidenced by the observation of efficient chain extension and formation of block copolymers.13,19,36,52 Additionally, the swelling and sorption of PMMA films with MMA has been studied extensively in scCO2.53–55 As PMMA particles have a higher surface area than films, they are even more likely to swell, with a Kj estimated at 0.4 for MMA and 0.25 for CO2.56 No information on the partition of CTAs in scCO2 systems is currently available in the literature. However, the results here do show that controlled polymerisation starts when nucleation starts. In addition, the good agreement between expected and theoretical Mn values tell us that all the DDMAT is involved in the control, so its diffusion into the particles (either as CTA molecule or as PMMA-DDMAT oligomers) must be near quantitative.
Good polymerisation control with a poorly selected CTA has been very recently demonstrated in RAFT emulsion polymerisation.57 Such result, is only possible if a low MMA/CTA molar ratio is effectively established inside the particles throughout the reaction. In our system, as MMA is mainly consumed inside the particles, the monomer is continuously supplied to maintain an overall constant concentration of MMA within the particles (with respect to PMMA and CO2), establishing a limiting feed, until there is no more MMA in the continuous phase. This results in a low MMA/DDMAT molar ratio in the dispersed phase, which can overcome the low chain transfer constant of DDMAT, in a similar fashion as described by Perrier and co-workers.57 In this way we can rationalise the surprisingly good control observed with DDMAT.
As mentioned in the introduction, Winnik and Song in 200627 demonstrated that RAFT control in dispersion polymerisation in conventional solvent (ethanol and water/ethanol mixtures) could only be achieved by a delayed addition of the CTA. This creates a two-stage dispersion polymerisation, in which a high molecular weight polymer population was formed in the first stage via a conventional radical process. This yielded enough high molecular weight chains to induce nucleation. Then, after injection of the CTA, a lower molecular weight peak began to appear corresponding to living chains that are formed in the particles, and as the reaction progresses this second population becomes the dominant species.27 The final Mn obtained matches the theoretical value, with narrow dispersity, and the weight fraction of the original very high molecular weight chain remains low and are no longer detectable by SEC-THF.
Our results show that an a priori poor CTA in solution polymerisation can be used to achieve very good control in a single stage dispersion in scCO2 by effectively creating an in situ two-stage process. The poor efficiency of the CTA is counter balanced once the particles are formed as a result of the local modification of the monomer/CTA molar ratio.
RAFT polymerisation of MMA in scCO2 with a well-suited CTA
To broaden our understanding and corroborate our data we next looked at a range of other molecules that were thought to be good CTAs for MMA. In particular, 4-cyano-4-(phenylcarbonothioylthio) pentanoic acid (CPAB) (structure 2, Fig. 1) is known to be very well suited to control the polymerisation of MMA58 and in our hands performed well in conventional solution polymerisation in toluene (E2.1, Table 1).By contrast, in scCO2 control was poor with the Mn obtained almost 50% higher than the target (Mn = 57.9 kg mol−1vs. Mn,th = 84.9 kg mol−1) and a high dispersity (Ð = 1.49) (E2.2, Table 1). The reaction itself did appear to perform well, with high conversion (>90%) after 24 hours and formed a free-flowing powder (particle size ∼ 2.5 μm, Fig. S1 and Table S1†). The sampling of the reaction showed consistent growth of Mn with time (Fig. 4A). Looking in detail into the kinetics, after 30% conversion the plot of molecular weight against conversion (Fig. 4B) shows a deviation (*Mn) from the theoretical trend (black line) towards higher molecular weights, clearly showing sub-optimal control.
 | ||
| Fig. 4 Dispersion polymerisation of MMA in scCO2 using CPAB as CTA. (A) Evolution of MMA conversion versus time. (B) Evolution of Mn (blue) and Ð (red) versus conversion; solid trend line is the theoretical Mn and dashed trend line is the linear fitting of experimental data. (Molar ratio DDMAT/AIBN 2:1, 65 °C, 275 bar, 300 rpm stirring rate, 5 wt% of PDMS-MA as stabiliser (based on MMA)). A deviation from expected RDRP behaviour is observed after approximately 30% conversion (*Mn and Ð). | ||
What is also very interesting is that the reaction with CPAB shows initially very good dispersity (Fig. 4B), especially when compared with the reaction controlled with DDMAT (Fig. 2C). However, after 30% conversion (*Ð), the dispersity drifts upwards. Since CPAB has a very high chain transfer constant towards MMA we should expect better control in the early stages of the process, in the same way as is observed in toluene solution.
To investigate this unexpected result, the early stage of the scCO2 dispersion polymerisation with CPAB was analysed in the view cell (Fig. 5A). This time the onset of turbidity was observed after a much longer period of 43 minutes (conversion at 50 minutes = 2.9%), which can indicate that CPAB, a better CTA for methacrylates, is exerting control of the polymerisation. One might speculate that this is suppressing conventional radical polymerisation at the early stage and effectively preventing nucleation. The consequence of this might be that the in situ two-stage mechanism that we saw with DDMAT is not happening effectively.
 | ||
| Fig. 5 THF SEC study at reaction early stage (nucleation) of MMA dispersion polymerisation in scCO2 with CPAB. (A) Photographs of view cell study at different reaction times showing turbidity increase, with the monomer conversion presented in brackets. Aliquots from (B) 30 minutes to 2 hours and (C) 2 to 4 hours and final product at 24 hours. Two distinct populations are observed, population 1 (conventional radical polymerisation) and population 2 (RAFT controlled). Inside the boxes, the reaction time is given to depict the SEC traces. | ||
However, the THF-SEC analyses of the early stage of the CPAB controlled reaction (Fig. 5B and C) show a similar behaviour to that of DDMAT. A bimodal molecular weight distribution is observed, with two populations where the high molecular weight population (population 1) bears no UV signal (Fig. S6†) and this population becomes less dominant over time. In addition, the molecular weights at the early stage of the polymerisation with CPAB, ∼70 kg mol−1 (population 1), are much lower than those observed with DDMAT and closer to the expected final value for the polymerisation (Mn,th = 60 kg mol−1).
As mentioned earlier, it is important to remember that inhibition and retardation are other factors that might be at play in RAFT controlled polymerisation and can delay nucleation. In particular, inhibition and retardation are known to be frequently observed with dithiobenzoates such as CPAB.46,47 This might explain the delayed nucleation, but does not explain the kinetic behaviour observed, which better aligns to the theoretical Mn prior to 30% monomer conversion (Fig. 3B).
Another control agent that we have previously looked at in detail is 2-cyano-2-propyl(dodecyltrithiocarbonate) (CPDT) (structure 3, Fig. 1).42 This trithiocarbonate CTA is well-suited for MMA polymerisation in homogeneous medium, e.g. toluene (E3.1, Table 1). As an example of successful polymerisation in toluene, the kinetic plot of MMA polymerisation with CPDT is presented in the ESI (Fig. S12†). In contrast to CPAB we found that in scCO2 good control of MMA dispersion polymerisation is obtained with low dispersity (Ð = 1.20) and molecular weight on target (Mn = 60 kg mol−1vs. Mn,th = 58.6 kg mol−1) (E3.2, Table 1). CPDT is amongst the most active CTAs and is particularly good for controlling polymerisation of more activated monomers (MAMs) such as MMA;20 the cyanoalkyl –R group is an effective re-initiation group for MMA.34,59
The linear evolution of molecular weight with conversion has been also previously demonstrated using CPDT in scCO2 (Fig. S8†).42 In addition, investigation on the early stage of MMA polymerisation with CPDT, shows that turbidity and hence nucleation begins just 19 minutes into the reaction (Fig. S9†), and we also see a bimodal molecular weight distribution (Fig. S10†). One may keep in mind that, as a trithiocarbonate, CPDT would likely suffer lower impact from inhibition and retardation than CPAB.
So to summarise, we have confirmed that DDMAT, a poor choice of CTA, controls well MMA polymerisation in scCO2. However, CPAB, which should be a good choice of CTA, gives poor results; while CPDT, another good choice of CTA, gives good control. In addition to this, the Howdle group has previously reported good control of scCO2 dispersion of MMA with other well suited CTAs.28,29
Clearly, there must be other factors at play that are important in determining the final control we observe in these RAFT dispersion polymerisations. Independent of the RAFT agent, the mechanism of reaction appears to follow a two-stage mechanism. This highlights the importance of generating a high molecular weight species capable of nucleating particles at the start of the reaction. Furthermore, we identified that the limiting feed of MMA into the particles in the scCO2 dispersion polymerisation is responsible for establishing an MMA/CTA ratio that is low enough to overcome the low chain transfer constant of DDMAT. However, such a mechanism would not be limited to just this CTA. If this is a general trend, the partitioning of the CTA once the particles are formed must be crucial to control the polymerisation that is taking place now essentially inside the particles. Therefore, the solubility of the CTA in scCO2 and the subsequent effects on partitioning into the particles may be the cause of the unexpected behaviour we observed with this CTAs. Even though, previous computational simulations assumed that mobility of the CTA was effectively the same as the monomer or that the CTA was completely and instantaneously transported into the dispersed phase.51,60–62
In the following section we try to identify the solvent factors that might tune the behaviour of the CTAs. Computational solvation models are used to probe the affinities of the different CTAs towards toluene and scCO2 and we use the data to explain the disparity in control of our polymerisations.
Computational solvation model for CTAs in scCO2
Even though there are several models that attempt to explain the solvation mechanism of scCO2 (acid–base Lewis, π–π interactions),63,64 it is generally accepted that quadrupole–polar interactions play a significant role. To probe how the solvent influences the control of the polymerisation, pair distribution or radial distribution functions (RDF) were computed from molecular dynamics simulations. An RDF represents the relative probability of an interacting atom being found at a distance r from another reference atom. The RDFs of pairs of atoms that are strongly interacting through space will display two key features: (i) peaks shifted to shorter distances, since the atoms are closer, and (ii) a maximum greater than unity.The area under the first peak corresponds to the number of atoms directly interacting with the reference atom. For our purposes, the reference atoms were chosen to be C, O, N and S and the interacting atoms are the carbon atoms for both scCO2 and toluene. In our scCO2 polymerisation experiments with DDMAT, CPAB and CPDT we see positive, negative and no impact over the control of MMA polymerisation. Can the calculations pick out features that show such behaviour?
A recurring feature for all interactions involving scCO2 is its affinity for unsaturated polar bonds, as is the case for the CO and N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bonds that are present in the CTAs (Fig. 1). CO2 interacts more effectively with those groups than toluene (Fig. 6). For example, the CO interactions with scCO2 are more intense (Fig. 6A) compared to the interactions with toluene (Fig. 6B), as indicated by the peak shifted to shorter distances and the greater maximum at 1.2.
C bonds that are present in the CTAs (Fig. 1). CO2 interacts more effectively with those groups than toluene (Fig. 6). For example, the CO interactions with scCO2 are more intense (Fig. 6A) compared to the interactions with toluene (Fig. 6B), as indicated by the peak shifted to shorter distances and the greater maximum at 1.2.
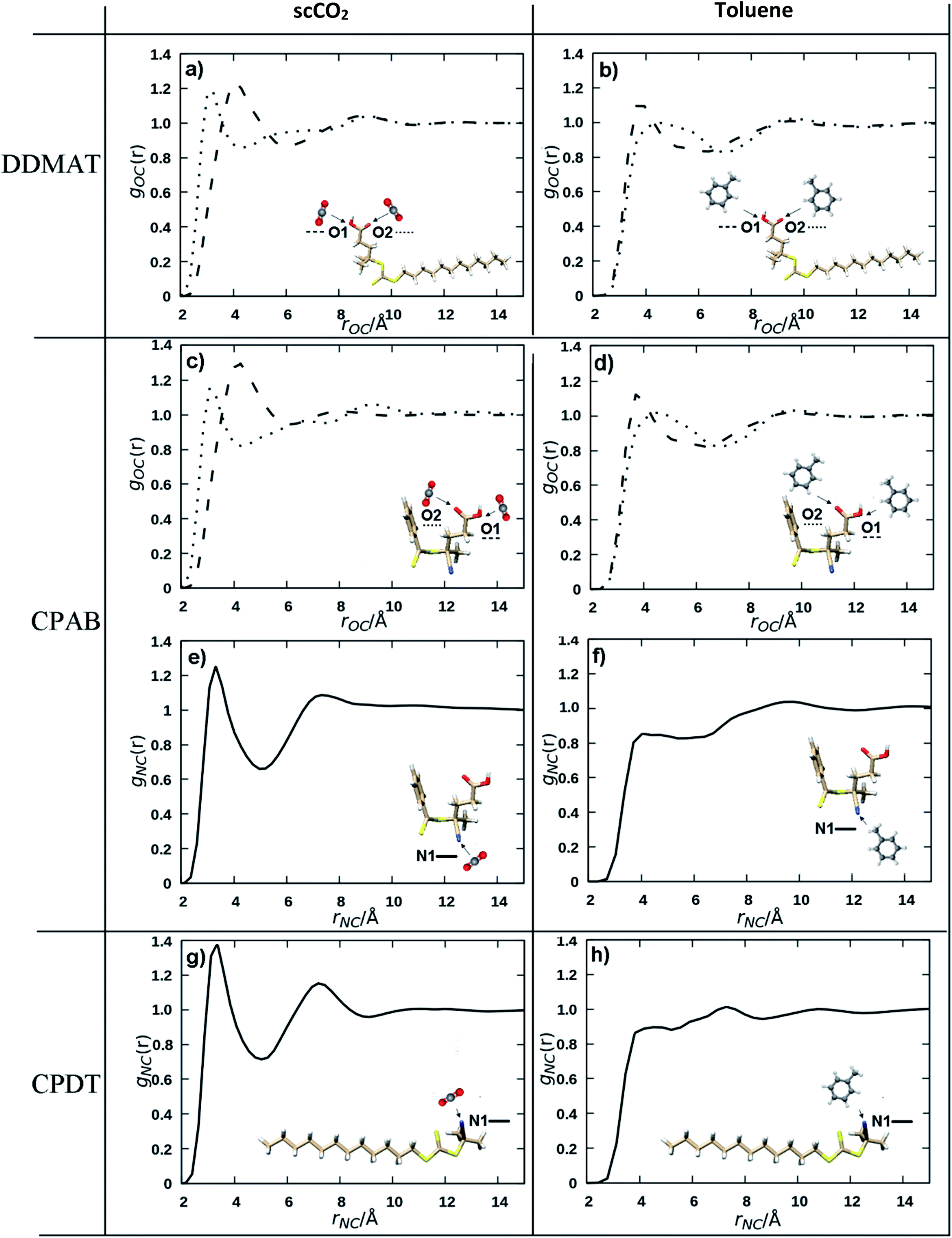 | ||
| Fig. 6 Radial distribution functions showing the functional groups interactions with the solvent, gij(r) (i = O1 - - -, O2 … or N1 — and j = carbon atoms of either CO2 or toluene) vs. interatomic distances for (a) oxygen atoms of DDMAT in scCO2, (b) oxygen atoms of DDMAT in toluene, (c) oxygen atoms of CPAB in scCO2, (d) oxygen atoms of CPAB in toluene, (e) nitrogen atom of CPAB in scCO2, (f) nitrogen atom of CPAB in toluene, (g) nitrogen atom of CPDT in scCO2, (h) nitrogen atom of CPDT in toluene. | ||
In particular, the NC group has a very strong affinity with an intense peak only seen for the scCO2 distributions (Fig. 6E and G) at around 2.5 Å, a value that is consistent with strong non-covalent interactions.65 scCO2 is well known to interact strongly with small apolar molecules and with polar unsaturated bonds.63,64 NC has much weaker interaction with toluene (Fig. 6F and H) as is evident from the less intense first maximum in the RDF at a longer distance. On the other hand, it is known that scCO2 solvation capacity diminishes rapidly for high molecular weight apolar groups and we would expect that the dodecyl Z group in DDMAT and CPDT would lower the solubility in scCO2 (Fig. S15 and S16†). By contrast, toluene has a high affinity for heavy hydrocarbons and does not form polar interactions.
The nature of the solvation mechanism of scCO2 is elusive and still a topic of debate. In order to propose a model based on the interactions we have seen from the molecular dynamics simulations we will consider the NC moiety to have a strong interaction as indicated by the intense peak in the RDFs (Fig. 6E and G) and the CO moiety to have a moderate interaction (Fig. 6A and C). Our aim is to provide a quick predictive tool to look at the behaviour of the RAFT agents in the scCO2 controlled dispersion polymerisations.
In light of this, we can rationalise the observation that CPAB controls the reaction poorly in scCO2 but performs well in toluene. CPAB possesses two unsaturated groups (CO and NC). This gives a high affinity for scCO2 that will move the partitioning of the CTA towards the continuous phase. This would move the radical fragment away from the primary reaction locus, the growing PMMA particles. In toluene, since these two unsaturated groups are not as sought by the toluene molecules, the radical fragment is allowed to fully act on the reaction locus, promoting polymer growth effectively. In fact, the only groups toluene displays affinity for is the phenyl ring.
By contrast, DDMAT possesses only one unsaturated group (CO) for which scCO2 has only a moderate affinity. DDMAT has the dodecyl –Z group, which is not favoured by scCO2 but will provide good affinity to the environment in the polymeric particles. In combination these factors will tend to push DDMAT to partition towards the dispersed phase. Therefore, the sole carbonyl group is not sufficient to cause the reaction to be delayed in scCO2. In toluene DDMAT interacts heavily with the solvent through its long dodecyl chain, so in addition to the low chain transfer constant towards MMA, the DDMAT molecule becomes shielded.
For CPDT, there is one NC moiety with a strong affinity for scCO2 and would tend to have good solubility in the scCO2 phase, but this is counterbalanced by the dodecyl –Z group which would again tend to present better affinity within the polymer particles. In toluene the dodecyl –Z group balances out the presence of the NC group and thus solvation does not disrupt the good reaction control expected for its reactive radical group, in addition to its high chain transfer constant towards MMA.
We emphasise that our solvation model focusses upon understanding the solubility of the CTAs at the beginning of the reaction. The RAFT reaction mechanism will unavoidably lead to the formation of an insoluble macro-CTA because of the addition of the monomer units as the reaction progresses.5 As a result, at a certain critical molar mass, the macro-CTA will become insoluble in the CO2-rich phase, and will then be restricted to the dispersed phase. Nevertheless, the solubility model of the low molecular weight CTAs provides a powerful comparison between the solubility of the CTAs and the macro-CTAs that they then become, whilst maintaining the simplicity of the model. In addition, the kinetic study shows that the early stage of the reaction could well be of great importance in defining the overall RAFT control, as both DDMAT and CPAB have very different reaction profiles below 30% conversion.
Furthermore, we earlier demonstrated that an in situ two-stage polymerisation mechanism is in place in the RAFT dispersion in scCO2, which provides a heterogeneous system to which the unreacted CTA and CTA-oligomers can partition whilst still below the critical molar mass. If the CTA and CTA-oligomers have a lower solubility in scCO2, they will enter the particles earlier, increasing the CTA concentration at the reaction locus. Hence, the study of the low molecular weight CTA solubility and behaviour is still very relevant to our system.
CTAs comparison: control and phase behaviour
In light of this we can then better understand the distinct results observed for dispersion polymerisation in scCO2 for each of the CTAs. The polymeric microparticles have previously been defined as the main locus for conventional radical polymerisation of MMA in scCO2.37,66 It is known that at the very beginning of the reaction, the locus is in the continuous phase; this was demonstrated by turbidimetry analysis.44,45 Mueller et al. modelled in detail MMA conventional radical polymerisation in scCO2 and evaluated the influence of the rates of interphase radical transport (diffusion of growing chains) and termination rates to define the reaction locus.48,67 They identified that after the initial period the particles are the main locus of the reaction, with any new radicals that are generated in the continuous phase rapidly migrating irreversibly into the dispersed phase prior to termination. However, their simulations also showed that if termination in the continuous phase was found to occur at a similar rate, or faster than the diffusion of these growing chains into particles, then a bimodal molecular weight distribution would be obtained. In this case, low molecular weight chains would be formed in the continuous phase and higher molecular weight chains would be formed in the particles of the dispersed phase.67Very few studies have tried to model RAFT dispersion polymerisation in scCO2.51,60,62,68 In that study, the particles were assumed to be the main locus of the reaction as for conventional radical polymerisation. Predici® software was used to study the RAFT polymerisation of styrene with AIBN initiator and S-thiobenzoyl thioglycolic acid (TBTGA) as the CTA in scCO2.51 Both the continuous and the dispersed phases were considered in the simulations. When the CTA was present, the simulations reported large dormant polymer chains to be produced in the continuous phase at a significant level, while lower molecular weight chains were formed in the dispersed phase, where they assumed the CTA would mainly partition.51 In these simulations, all chains were considered to be solely initiated via RAFT. However, our data show that this is not the case in reality. We do observe bimodal molecular weight distributions, but SEC-UV data show us that the higher molecular weight population does not carry a UV signal and indicates that this has grown in an uncontrolled manner via conventional radical polymerisation and not RAFT.
As previously mentioned, DDMAT shows surprising control. A high concentration of CTA in the particles and the slow feed of MMA into the particles as the reaction progresses would provide conditions for the unexpected good control. For DDMAT, the presence of only one CO (moderate interaction with CO2) and the presence of the dodecyl –Z group means that its solubility in scCO2 is considerably lower than CPAB and CPDT. Therefore, it is not unreasonable that it would diffuse more promptly into the particles and that the concentration of DDMAT with respect to MMA would be increased. This scenario would favour transfer reactions over propagation and this would counterbalance the known low chain transfer constant of DDMAT. Thus, the performance of DDMAT as a control agent will strongly be improved, and the MMA polymerisation will resume in a controlled way, generating living growing chains.
Reversibly, if the CTA is too soluble in CO2, as CPAB for example, it will be less partitioned into the particles and the higher concentration of MMA/CTA in the particles will result in loss of control. Even if the CTA has a high chain transfer constant, the high MMA/CTA ratio inside the particles will result in an artificially high theoretical Mn. In this way, it is the solubility of CPAB that will influence the level of control obtained.
The monomer conversion versus time plot provides important mechanistic insight. More specifically, the semilogarithmic plot clearly evidences the presence of different regimes, and has been previously used to show the onset of nucleation.69,70 As the polymer nuclei will swell with monomer, the high local concentration present in the particles can cause a significant increase of the reaction rate.
Two regimes can be observed when using CPAB (Fig. 7A). The nucleation onset is at 48 minutes, determined by the X axis intercept of the first regime (red), and this is in good agreement with the time at which turbidity was observed by naked eye in the view cell (i.e. 43 minutes).
 | ||
| Fig. 7 Pseudo-first-order kinetic plot of monomer conversion as a function of reaction time for the dispersion polymerisation of MMA in scCO2 with (A) CPAB, highlighting two distinct regimes (red and black) and (B) DDMAT, highlighting three distinct regimes (red, blue, black). | ||
Interestingly, the timing of the inflexion between the two regimes (i.e. 10 h, Fig. 7A) coincides with the deviation of Mn from Mn,th trend line (Fig. 4B), observed after 30% conversion. At 10 h the average conversion was 44% and Mn = 30.10 kg mol−1. Before this point, the molecular weights were all closer to the expected values. In addition, the final polymer obtained with CPAB was somewhat higher than the targeted Mn; this usually indicates incomplete usage of the CTA.21 Therefore, we hypothesise that the observed poor control of MMA polymerisation in scCO2 with CPAB results from poor partitioning of the newly initiated and PMMA oligomers into the polymer particles at the early stage of reaction. This would result in a smaller number of active dithiobenzoate moieties present inside each particle after nucleation. The mechanism for DDMAT appears to be more complex. There are three distinct regimes (Fig. 7B). The nucleation onset (first regime – red) appears at 27 minutes, while in the view cell turbidity was observed as early as 15 minutes. The second (blue) regime is from 4 to 8 hours, where the rate of polymerisation increases by 2.3-fold compared to first regime. Then there is a third (black) regime which shows a further 1.65-fold rate increase (Fig. 7B). The presence of three regimes might not be so surprising, since the dispersion polymerisation of MMA in scCO2 can be considered as a three stage polymerisation in terms of monomer conversion.62 At the first stage, polymerisation can only occur in the continuous phase, then in two phases (continuous and dispersed) and, finally, polymerisation occurs only in the dispersed phase.
For CPDT, we previously saw two regimes, similar to what we observed for CPAB, with nucleation onset calculated at 18 minutes (Fig. S11†), while turbidity was observed at 19 minutes in the view cell (Fig. S9†). With this CTA, the second regime starts at 6 hours and presents a 7.25-fold increase in the rate of polymerisation.42 However, different from the reaction in presence of CPAB, the final molecular weight was in agreement with the theoretical value. Furthermore, different from CPAB, the kinetics study did not show a deviation of Mn from Mn,th trend line (Fig. S8†).42 Therefore, the increase in the polymerisation rate is not likely caused by a small amount of CTA present in the dispersed phase. The solubility of CPDT is in between DDMAT and CPAB. The lower solubility compared to CPAB appears to be enough to allow a good partitioning of the CTA into the particles and therefore good control is obtained. While, the absence of a third inflexion as seen for DDMAT might indicate that CPDT solubility is high enough to prevent a step where polymerisation only can take place inside the particles, and therefore some polymerisation might still occur in the continuous phase.
Solubility effect over CTAs control and selection guideline
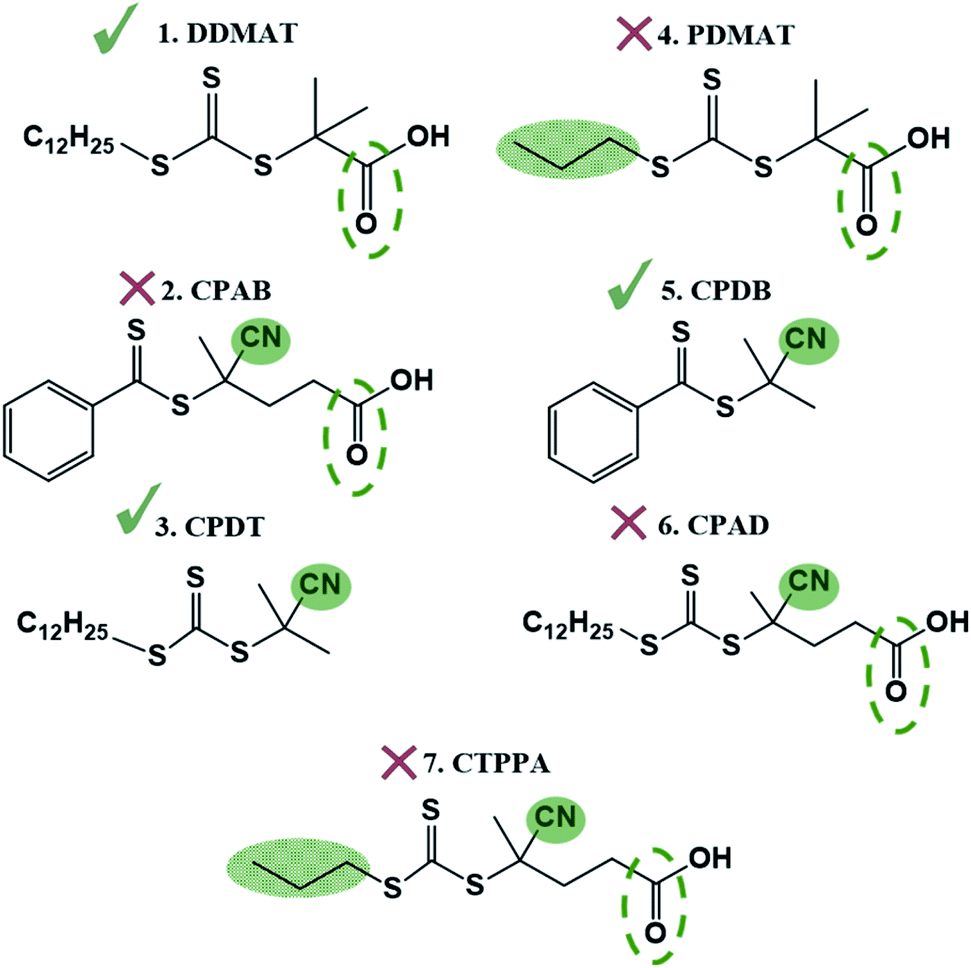
To test the ability of our model, we selected a range of further CTAs to probe the importance of partitioning and CTA solubility on RAFT control in scCO2. PDMAT is a trithiocarbonate that is almost identical to DDMAT but has a much shorter propyl stabilising group (structure 4, Fig. 8). | ||
| Fig. 8 Chain transfer agent (CTA) library with the R group in red and the Z group in blue: (4) PDMAT (2-(propylthiocarbonothioylthio)-2-methylpropionic acid); (5) CPDB (2-cyano-2-propyl benzodithioate); (6) CPAD (4-cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl]pentanoic acid); (7) CTPPA (4-cyano-4-[(propyllsulfanylthiocarbonyl)sulfanyl]pentanoic acid). | ||
n-Alkanes solubility in scCO2 has been well studied and its cloud-point pressures increases with chain length.71 In the same way, the chain length of the alkyl tail in acrylate monomers was shown to impact solubility in scCO2.72 In both cases, this arises from the reduction of polarity of the molecule with increasing alkyl chain length leading to a mismatch in the energy of solvation. As a result, PDMAT is very likely to be more soluble in scCO2 than DDMAT (Fig. S13†).
PDMAT was utilised to control polymerisation of MMA both in dispersion in scCO2 and in toluene (E4.1 and E4.2, Table 2). The results demonstrate poor control in toluene, just like the very similar DDMAT. However in scCO2, PDMAT showed much less control than DDMAT with Mn more than twice the predicted one and a broader dispersity (Ð = 1.50). Therefore, the increase in solubility of the CTA conferred by the shorter alkyl group does indeed negatively impact reaction control in accordance with our hypothesis.
| Expt. | CTA | Solvent | Conv.a (%) | M n,th | M n | Ð |
|---|---|---|---|---|---|---|
| a Conversion calculated from 1H NMR. b Theoretical Mn calculated relative to CTA and monomer concentration and given in kg mol−1. c Ð and Mn (in kg mol−1) obtained by THF-SEC with RI detector against PMMA standards. See Experimental section for reaction conditions used for scCO2 dispersion polymerisation. | ||||||
| E4.1 | 4-PDMAT | Toluene | 89 | 21.1 | 57.4 | 1.63 |
| E4.2 | 4-PDMAT | scCO2 | 98 | 60.6 | 101.2 | 1.50 |
| E5.1 | 5-CPDB | Toluene | 92 | 50.5 | 55.1 | 1.17 |
| E5.2 | 5-CPDB | scCO2 | 96 | 54.4 | 58.4 | 1.28 |
| E6.1 | 6-CPAD | Toluene | 50 | 30.0 | 24.9 | 1.30 |
| E6.2 | 6-CPAD | scCO2 | 99 | 59.4 | 84.1 | 1.59 |
| E7.1 | 7-CTPPA | Toluene | 65 | 39.5 | 53.1 | 1.29 |
| E7.2 | 7-CTPPA | scCO2 | 97 | 58.0 | 114.1 | 1.42 |
A further set of CTAs (structures 5–7, Fig. 8) was then tested for RAFT polymerisation in scCO2 (Table 2). The solvation analysis of these CTAs and their respective RDFs can be found in the ESI (Fig. S14–S24†).
In all cases a loss of control in scCO2 is systematically observed when a carboxylic acid is present in the CTA. For example, the pair CPAB/CPDB give similar good control in toluene but the CTA with a carboxylic acid (CPAB) shows significantly poorer control of the scCO2 dispersion (E2.2, Table 1 and E5.2, Table 2). One can rationalise this because the addition of the acid group makes the CTA more soluble in the continuous scCO2 medium and hence less likely to partition into the growing polymer particles, negatively impacting control. The same trend in behaviour can be observed for the CTA pair CPAD/CPDT (E6.2, Table 2 and E3.2, Table 1).
Again, the alkyl moiety in the –Z group has shown effect over control, e.g. the pair CTPPA/CPAD, with demonstrably poorer control in scCO2 when a shorter alkyl chain is present, CTPPA (E6.2 and E7.2, Table 2). This reinforces our earlier findings with DDMAT and PDMAT, where control was impaired due to the higher solubility in scCO2.
All of these data show us that for dispersion polymerisation in scCO2 the solubility of the CTA must not be so high that the ability to diffuse into the dispersed phase and control the polymerisation is impaired. But on the other hand, there must be an initial homogeneous system at reaction onset, so the CTA must have some solubility in scCO2. In fact, our data suggest a Goldilocks' principle; not too little, not too much, but just right.
To corroborate this, the selection of CTAs must be based on the balance of CO2-philic features to control phase behaviour (Table 3 and Fig. 9). The best CTAs for the dispersion polymerisation had one CO2-philic group, either NC or CO, and one polymer-philic group (dodecyl –Z group). When two or more CO2-philic groups were present (e.g. CPAB and CPAD with both CO and NC), then control was compromised. CPDT and DDMAT both have optimal solubility for controlling the reaction and we consider these two as the best choice of CTA for this reaction in scCO2, despite the DDMAT poor performance in homogeneous conditions in toluene.
| CTA | CO2-philic groups | Polymer-philic groups | Solubility in scCO2 | Control |
|---|---|---|---|---|
| DDMAT | 1 | 1 | Average | Good |
| CPAB | 2 | 0 | High | Poor |
| CPDT | 1 | 1 | Average | Good |
| PDMAT | 1 | 0 | High | Poor |
| CPDB | 1 | 0 | Average | Good |
| CPAD | 2 | 1 | High | Poor |
| CTPPA | 2 | 0 | High | Poor |
 | ||
| Fig. 9 Representation of CTAs groups that enhance solubility in scCO2. Block green fill: NC, dashed circle: CO and alternating fill: C3H5. This last does not provide specific interactions with CO2 molecules, however the low molecular weight makes it more soluble compared to the other Z groups here presented. CTAs that have more than one solubility enhancing group provided poor control in scCO2 (cross mark), while CTAs with only one group provided good control (tick mark). | ||
Conclusions
We confirm here the surprising control of DDMAT over the polymerisation of MMA in scCO2, despite the known poor control of this trithiocarbonate CTA over methacrylate polymerisation in conventional solvents.34 With the use of a recently developed sampling instrument, we have been able to accurately follow the kinetic evolution and uncover new insights into the early stage of RAFT dispersion polymerisation in scCO2. The good control of DDMAT was then attributed to the local modification of the monomer/CTA molar ratio inside the particles that are created under an in situ two-stage process. In order to broaden the palette of experimental observations and to corroborate our understanding, six more CTAs were studied for their ability to control MMA dispersion polymerisation in scCO2. We also present a novel solvation model based on molecular simulations to understand the effect of various moieties upon the solubility of the CTAs in scCO2. We have utilised this to identify correlations between polymerisation control and solubility of the CTA in scCO2. Our data also align with previous observations of two-stage dispersion polymerisation in conventional solvents.27We thus present a simple approach to identify the best CTA for dispersion polymerisation based upon solubility in scCO2 and the likely partitioning between the scCO2 continuous phase and the growing PMMA microparticle environment. We hope that these principles might be extended more broadly to controlled dispersion polymerisations in other solvents.
Experimental
Materials
MMA was purchased from ProSciTech (99%) and was filtrated through aluminium oxide to remove the stabiliser prior to polymerisation. 2,2′-Azobis(isobutyronitrile) (AIBN) was purchased from Sigma Aldrich (98%) and purified by recrystallisation in methanol prior to use. All other chemicals were used as received. All CTAs were purchased from Sigma-Aldrich, with exception of PDMAT and CTPPA. The synthesis of PDMAT is given here and CTPPA was synthesised according to previous work.73 Methacrylate terminated polydimethylsiloxane (PDMS-MA) 10 kg mol−1 was purchased from ABCR GmbH & Co. Heptane, toluene, tetrahydrofuran (HPLC grade), deuterated chloroform (CDCl3) and methanol were all purchased from Fischer Scientific.Standard RAFT solution polymerisation in toluene
A typical procedure in which PMMA with a molecular weight of 60 kg mol−1 was targeted, used AIBN (0.017 mmol), CTA (0.083 mmol), MMA (49.9 mmol) and 5 mL of toluene. All reactants were transferred to a 25 mL Schlenk tube with a magnetic stirrer, which was then sealed and degassed by at least three freeze–pump–thaw cycles. The tube was then heated to 65 °C in an oil bath and agitated by magnetic stirring. Samples were taken periodically with a syringe for analysis. After 24 hours, the vessel was cooled and the polymer was precipitated from solution in a ∼10-fold volume of methanol, filtered and dried in vacuum.Standard RAFT dispersion polymerisation in scCO2
A typical procedure used an in-house built high pressure MKIII autoclave (20 mL),74 which was degassed by purging with CO2 at 2 bar for 30 minutes. MMA (33 mmol), AIBN (0.028 mmol), PDMS-MA (5 wt% with respect to MMA) and the CTA (0.055 mmol) were degassed by bubbling with argon for 30 minutes. The reactants were then added to the autoclave through the keyhole against positive pressure of CO2. The vessel was then sealed and pressurised to 50 bar, heated to 65 °C, and the pressure topped up to 275 bar (4000 psi). The reaction mixture was stirred at 300 rpm with overhead magnet coupled stirrer. After 24 hours, heating was turned off and the reactor was cooled below supercritical conditions before being vented. All products were collected as dry free-flowing powders, unless stated differently, and taken for analysis.Standard polymerisation in sampling autoclave
A typical procedure used an in-house built high pressure sampling device consisting of an 60 mL MKIII clamp sealed autoclave74 with a cylinder sampling unit as described elsewhere,42 which was degassed by purging with CO2 at 2 bar for 30 minutes. MMA (0.1 mol), AIBN (0.08 mmol), PDMS-MA (5 wt% with respect to MMA) and the CTA (0.17 mmol) were degassed by bubbling with argon for 30 minutes. The reactants were then added to the autoclave through the keyhole against positive pressure of CO2. The vessel was then sealed and pressurised to 50 bar, heated to 65 °C, and the pressure topped up to 275 bar. The reaction mixture was stirred at 300 rpm with overhead magnet coupled stirrer. At sampling times, the cylinder was loaded with ∼5 mL deuterated chloroform and attached to the autoclave. A fraction of the reaction mixture was sampled into the small pipe space before the cylinder. The sample caused a small pressure drop, therefore pressure was topped up to an extra 13.5 bar to avoid fluctuations below reaction conditions. The content of the pipe was then sprayed into the cylinder and collected into chloroform. The samples were analysed via THF SEC and proton NMR.Standard solubility test in scCO2 in view cell
Solubility test of CTAs was carried out in a stainless steel view cell with two windows, which permitted visual observation of the phase behaviour. An accurately weighed amount of CTA and monomer (simulating reaction conditions) were added into a small glass vial and placed inside of the autoclave. The system was purged with CO2 and gradually heated to 65 °C and pressurised to 275 bar. Solubility was visually evaluated after allowing the system to stabilise. The process was repeated three times.Synthesis of 2-propylsulfanylthiocarbonylsulfanyl-2-methyl propionic acid (PDMAT)
The shorter Z-group CTA was synthesised according to Lai et al.35 The purification was done by acidifying the medium until pH < 2 with HCl, and then extracted with ethyl ether. The ether solution was dried over magnesium sulphate before removal of solvent. The yellow oily medium obtained was purified by chromatographic column eluting with 10% (v:v) ethyl acetate/hexane. The final product was a yellow oil. 1H NMR (400 MHz, CDCl3), δ (ppm): 3.27 (t, J = 7.4 Hz, 2H), 1.72 (m, 8H), 0.99 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, CDCl3), δ (ppm): 221.2, 178.3, 56.4, 39.2, 25.3, 25.0, 13.9.
Polymerisation in scCO2 in double-ended fast release static view cell
A typical procedure used an in-house built high pressure static view cell, which was degassed by purging with CO2 at 2 bar for 30 minutes. MMA (0.1 mol), AIBN (0.08 mmol), PDMS-MA (5 wt% with respect to MMA) and the CTA (0.2 mmol), if used, were degassed by bubbling with argon for 30 minutes. The reactants were then added to the autoclave through the keyhole against positive pressure of CO2. The vessel was then sealed and pressurised to 50 bar, heated to 65 °C, and the pressure topped up to 275 bar. The reaction mixture was stirred at 300 rpm with overhead magnet coupled stirrer. The reaction was monitored and recorded throughout the nucleation phase until complete blockage of back light.Polymer characterisation
The Mn and Ð of polymers were obtained by size exclusion chromatography (SEC) (PL-120, Polymer Labs) using a refractive index (RI) detector. The columns (30 cm PLgel Mixed-C, two in series) were eluted by THF and calibrated with PMMA standards. Calibration and analysis were performed at 40 °C with a flow rate of 1 mL min−1. In the case of the early stage aliquots, bimodal peaks molecular weights were determined by the integration of each peak, separately, present in the SEC chromatogram. Monomer conversion was determined by proton nuclear magnetic resonance (1H NMR). The spectra were recorded in CDCl3 using a Bruker DPX 400 MHz spectrometer, and referenced to CDCl3 at 7.26 ppm. Scanning electron microscopy (SEM) images were obtained using a JEOL 6060V SEM machine at various magnifications and an accelerating voltage of 10 kV. Samples were mounted on aluminium stubs using adhesive carbon tabs and sputter-coated with platinum before analysis. Mean particle diameter (Dn) was determined by measuring the diameter of 100 particles and taking a mean of these data. The coefficient of variance (Cv) was calculated by the ratio of the standard deviation (σ) by the mean particle diameter as by the equation: Cv = σ/Dn × 100.Computational methodology
To emulate the important quadrupolar moment of CO2 in the supercritical state, the EPM2 model was used,75 which is purely based on point charges. Hence, in our solvation model it is specifically fitted to the potential of supercritical CO2. The CTA molecules were described by the CHARMM general force field (CGenFF). The size of the cubic box was set as 10 nm initially. MD simulations were performed using the GROMACS package (v2019)76 with NPT ensemble at 338.15 K and 275 bar coupled by the Berendsen model. This resulted in a supercritical fluid with a density of 0.657 g cm−3. The time step was 1 fs. The cut-off length for intermolecular potential calculations was 1.2 nm. Ewald summation was adopted to compute the long-range electrostatic interactions. The system was simulated for 100 ns for the production dynamics.For toluene, the potential was obtained from http://virtualchemistry.org/. MD simulations were performed with NPT ensemble at 338.15 K and 14.5 psi (room pressure) coupled by the Berendsen model. The density of the solvent was thus 0.845 g cm−3. The other details of the simulation protocol were as for the CTA simulation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was co-funded through SINCHEM a Joint Doctorate program selected under the Erasmus Mundus Action 1 Program (FPA 2013-0037). We thank EPSRC for funding via prosperity partnership EP/S035990/1. We thank the University of Nottingham Green Chemicals Beacon for funding toward this research. We are grateful for access to the University of Nottingham High Performance Computer. JDH is supported by the Royal Academy of Engineering under the Chairs in Emerging Technologies scheme.Notes and references
- H. M. Woods, C. Nouvel, P. Licence, D. J. Irvine and S. M. Howdle, Macromolecules, 2005, 38, 3271–3282 CrossRef CAS.
- E. J. Beckman, J. Supercrit. Fluids, 2004, 28, 121–191 CrossRef CAS.
- P. B. Zetterlund, F. Aldabbagh and M. Okubo, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3711–3728 CrossRef CAS.
- P. B. Zetterlund, Y. Kagawa and M. Okubo, Chem. Rev., 2008, 108, 3747–3794 CrossRef CAS.
- L. H. Sperling, Introduction to Physical Polymer Science, Wiley Interscience, Bethlehem, Pennsylvania, 2001 Search PubMed.
- S. Shen, E. D. Sudol and M. S. El-Aasser, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 1087–1100 CrossRef CAS.
- J. L. Kendall, D. A. Canelas, J. L. Young and J. M. DeSimone, Chem. Rev., 1999, 99, 543–563 CrossRef CAS.
- J. M. DeSimone, E. E. Maury, Y. Z. Menceloglu, J. B. McClain, T. J. Romack and J. R. Combes, Science, 1994, 265, 356–359 CrossRef CAS.
- P. Christian, M. R. Giles, R. M. T. Griffiths, D. J. Irvine, R. C. Major and S. M. Howdle, Macromolecules, 2000, 33, 9222–9227 CrossRef CAS.
- M. R. Giles, J. N. Hay and S. M. Howdle, Macromol. Rapid Commun., 2000, 21, 1019–1023 CrossRef CAS.
- H. Shiho and J. M. DeSimone, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 2429–2437 CrossRef CAS.
- H. Shiho and J. M. DeSimone, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3783–3790 CrossRef CAS.
- J. Jennings, M. Beija, A. P. Richez, S. D. Cooper, P. E. Mignot, K. J. Thurecht, K. S. Jack and S. M. Howdle, J. Am. Chem. Soc., 2012, 134, 4772–4781 CrossRef CAS.
- T. D. McAllister, L. D. Farrand and S. M. Howdle, Macromol. Chem. Phys., 2016, 217, 2294–2301 CrossRef CAS.
- Z. Ma and P. Lacroix-Desmazes, Polymer, 2004, 45, 6789–6797 CrossRef CAS.
- R. McHale, F. Aldabbagh, P. B. Zetterlund, H. Minami and M. Okubo, Macromolecules, 2006, 39, 6853–6860 CrossRef CAS.
- B. Grignard, T. Phan, D. Bertin, D. Gigmes, C. Jerome and C. Detrembleur, Polym. Chem., 2010, 1, 837–840 RSC.
- J. Ryan, F. Aldabbagh, P. B. Zetterlund and M. Okubo, Polymer, 2005, 46, 9769–9777 CrossRef CAS.
- J. Jennings, G. He, S. M. Howdle and P. B. Zetterlund, Chem. Soc. Rev., 2016, 45, 5055–5084 RSC.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Strem Chem., 2011, 25, 1–10 Search PubMed.
- D. J. Keddie, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2012, 45, 5321–5342 CrossRef CAS.
- M. Hölderle, M. Baumert and R. Mülhaupt, Macromolecules, 1997, 30, 3420–3422 CrossRef.
- L. I. Gabaston, R. A. Jackson and S. P. Armes, Macromolecules, 1998, 31, 2883–2888 CrossRef CAS.
- P. J. Saikia, J. M. Lee, B. H. Lee and S. Choe, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 348–360 CrossRef CAS.
- P. B. Zetterlund, S. C. Thickett, S. Perrier, E. Bourgeat-Lami and M. Lansalot, Chem. Rev., 2015, 115, 9745–9800 CrossRef CAS.
- J.-S. Song and M. A. Winnik, Macromolecules, 2006, 39, 8318–8325 CrossRef CAS.
- A. M. Gregory, K. J. Thurecht and S. M. Howdle, Macromolecules, 2008, 41, 1215–1222 CrossRef CAS.
- K. J. Thurecht, A. M. Gregory, W. Wang and S. M. Howdle, Macromolecules, 2007, 40, 2965–2967 CrossRef CAS.
- Y. K. Chong, J. Kristina, T. P. T. Le, G. Moad, A. Postma, E. Rizzardo and S. H. Thang, Macromolecules, 2003, 36, 2256–2272 CrossRef CAS.
- J. Chiefari, R. T. A. Mayadunne, C. L. Moad, G. Moad, E. Rizzardo, A. Postma and S. H. Thang, Macromolecules, 2003, 36, 2273–2283 CrossRef CAS.
- C. Li and B. C. Benicewicz, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 1535–1543 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Acc. Chem. Res., 2008, 41, 1133–1142 CrossRef CAS.
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754–6756 CrossRef CAS.
- M. Alauhdin, T. M. Bennett, G. He, S. P. Bassett, G. Portale, W. Bras, D. Hermida-Merino and S. M. Howdle, Polym. Chem., 2019, 10, 860–871 RSC.
- M. L. O’Neill, M. Z. Yates, P. J. Johnston, C. D. Smith and S. P. Wilkinson, Macromolecules, 1998, 31, 2838–2847 CrossRef.
- Z. Guan, J. R. Combs, Y. Z. Menceloglu and J. M. DeSimone, Macromolecules, 1993, 26, 2663–2669 CrossRef CAS.
- M. R. Giles, J. N. Hay, S. M. Howdle and R. J. Winder, Polymer, 2000, 41, 6715–6721 CrossRef CAS.
- K. A. Shaffer, T. A. Jones, D. A. Canelas and J. M. DeSimone, Macromolecules, 1996, 29, 2704–2706 CrossRef CAS.
- J. Jennings, M. Beija, J. T. Kennon, H. Willcock, R. K. O'Reilly, S. Rimmer and S. M. Howdle, Macromolecules, 2013, 46, 6843–6851 CrossRef CAS.
- K. Kortsen, A. A. C. Pacheco, J. C. Lentz, V. Taresco and S. M. Howdle, J. Supercrit. Fluids, 2021, 167, 105047 CrossRef CAS.
- K. J. Thurecht, S. Villarroya, J. Zhou, S. M. Howdle, A. Heise, M. Degeus and M. F. Wyatt, Macromolecules, 2006, 39, 7967–7972 CrossRef CAS.
- U. Fehrenbacher, O. Muth, T. Hirth and M. Ballauff, Macromol. Chem. Phys., 2000, 201, 1532–1539 CrossRef CAS.
- U. Fehrenbacher and M. Matthias Ballauff, Macromolecules, 2002, 35, 3653–3661 CrossRef CAS.
- C. Barner-Kowollik, M. Buback, B. Charleux, M. L. Coote, M. Drache, T. Fukuda, A. Goto, B. Klumperman, A. B. Lowe, J. B. McLeary, G. Moad, M. J. Monteiro, R. D. Sanderson, M. P. Tonge and P. J. Vana, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5809 CrossRef CAS.
- G. Moad, Macromol. Rapid Commun., 2014, 215, 9–26 CrossRef CAS.
- P. A. Mueller, G. Storti and M. Morbidelli, Chem. Eng. Sci., 2005, 60, 377–397 CrossRef CAS.
- P. D. Condo, S. R. Sumpter, M. L. Lee and K. P. Johnston, Ind. Eng. Chem. Res., 1996, 35, 1115–1123 CrossRef CAS.
- J. J. Shim and K. P. Johnston, AIChE J., 1989, 3, 1097–1106 CrossRef.
- P. López-Domínguez, G. Jaramillo-Soto and E. Vivaldo-Lima, Macromol. React. Eng., 2018, 12, 1800011 CrossRef.
- T. M. Bennett, G. He, R. R. Larder, M. G. Fischer, G. A. Rance, M. W. Fay, A. K. Pearce, C. D. J. Parmenter, U. Steiner and S. M. Howdle, Nano Lett., 2018, 18, 7560–7569 CrossRef CAS.
- A. Rajendran, B. Bonavoglia, N. Forrer, G. Storti, M. Mazzotti and M. Morbidelli, Ind. Eng. Chem. Res., 2005, 44, 2549–2560 CrossRef CAS.
- B. Bonavoglia, G. Storti, M. Morbidelli, A. Rajendran and M. Mazzotti, J. Polym. Sci., Part B: Polym. Phys., 2006, 44, 1531–1546 CrossRef CAS.
- U. Fehrenbacher, T. Jakob, T. Berger, W. Knoll and M. Ballauff, Fluid Phase Equilib., 2002, 200, 147–160 CrossRef CAS.
- A. Quintero-Ortega, G. Jaramillo-Soto, P. R. García-Morán, M. L. Castellanos-Cárdenas, G. Luna-Bárcenas and E. M. R. E. Vivaldo-Lima, Macromol. React. Eng., 2008, 2, 304–320 CrossRef.
- R. A. E. Richardson, T. R. Guimarães, M. Khan, G. Moad, P. B. Zetterlund and S. Perrier, Macromolecules, 2020, 53, 7672–7683 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2012, 65, 985–1076 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- G. Jaramillo-Soto, M. L. Castellanos-Cárdenas, P. R. García-Morán, E. Vivaldo-Lima, G. Luna-Bárcenas and A. Penlidis, Macromol. Theory Simul., 2008, 17, 280–289 CrossRef CAS.
- G. Jaramillo-Soto, P. R. Garcia-Moran, F. J. Enriquez-Medrano, H. Maldonado-Textle, M. E. Albores-Velasco, R. Guerrero-Santos and E. Vivaldo-Lima, Polymer, 2009, 50, 5024–5030 CrossRef CAS.
- P. López-Domínguez, J. E. Rivera-Peláez, G. Jaramillo-Soto, J. F. Barragán-Aroche and E. Vivaldo-Lima, React. Chem. Eng., 2020, 5, 547–560 RSC.
- F. J. Gutiérrez Ortiz and A. Kruse, React. Chem. Eng., 2020, 5, 424–451 RSC.
- F. Ingrosso and M. F. Ruiz-López, ChemPhysChem, 2017, 18, 2560–2572 CrossRef CAS.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS.
- M. L. O'Neill, M. Z. Yates, K. P. Johnston, C. D. Smith and S. P. Wilkinson, Macromolecules, 1998, 31, 2848–2856 CrossRef.
- P. A. Mueller, G. Storti and M. Morbidelli, Chem. Eng. Sci., 2005, 60, 1911–1925 CrossRef CAS.
- G. Jaramillo-Soto, P. R. Garcia-Moran and E. Vivaldo-Lima, Macromol. Symp., 2010, 289, 149–154 CrossRef CAS.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS.
- L. A. Fielding, M. J. Derry, V. Ladmiral, J. Rosselgong, A. M. Rodrigues, L. P. D. Ratcliffe, S. Sugihara and S. P. Armes, Chem. Sci., 2013, 4, 2081 RSC.
- Q. Shi, L. Jing and W. Qiao, J. CO2 Util., 2015, 9, 29–38 CrossRef CAS.
- C. F. Kirby and M. A. McHugh, Chem. Rev., 1998, 99, 565–602 CrossRef.
- T. Boursier, I. Chaduc, J. Rieger, F. D'Agosto, M. Lansalot and B. Charleux, Polym. Chem., 2011, 2, 355–362 RSC.
- F. Furno, P. Licence, S. M. Howdle and M. Poliakoff, Actual. Chim., 2003, 62–66 CAS.
- J. G. Harris and K. H. Yung, J. Phys. Chem., 1995, 99, 12021–12024 CrossRef CAS.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc05281g |
| This journal is © The Royal Society of Chemistry 2021 |