
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence2-Mercaptomethyl-thiazolidines use conserved aromatic–S interactions to achieve broad-range inhibition of metallo-β-lactamases†
Maria-Agustina
Rossi‡
 a,
Veronica
Martinez‡
b,
Philip
Hinchliffe‡
c,
Maria F.
Mojica
def,
Valerie
Castillo
b,
Diego M.
Moreno
gj,
Ryan
Smith
c,
Brad
Spellberg
h,
George L.
Drusano
i,
Claudia
Banchio
aj,
Robert A.
Bonomo
eklm,
James
Spencer
c,
Alejandro J.
Vila
*aj and
Graciela
Mahler
*b
a,
Veronica
Martinez‡
b,
Philip
Hinchliffe‡
c,
Maria F.
Mojica
def,
Valerie
Castillo
b,
Diego M.
Moreno
gj,
Ryan
Smith
c,
Brad
Spellberg
h,
George L.
Drusano
i,
Claudia
Banchio
aj,
Robert A.
Bonomo
eklm,
James
Spencer
c,
Alejandro J.
Vila
*aj and
Graciela
Mahler
*b
aInstituto de Biología Molecular y Celular de Rosario (IBR, CONICET-UNR), Ocampo and Esmeralda, S2002LRK Rosario, Argentina. E-mail: vila@ibr-conicet.gov.ar
bLaboratorio de Química Farmacéutica, Departamento de Química Orgánica, Facultad de Química, Universidad de la República (UdelaR), Avda. General Flores 2124, CC1157, Montevideo, Uruguay. E-mail: gmahler@fq.edu.uy
cSchool of Cellular and Molecular Medicine, University of Bristol, Biomedical Sciences Building, University Walk, Bristol BS8 1TD, UK
dInfectious Diseases Department, School of Medicine, Case Western Reserve University, Cleveland, OH, USA
eResearch Service, Louis Stokes Cleveland Department of Veterans Affairs Medical Center, Cleveland, OH, USA
fGrupo de Resistencia Antimicrobiana y Epidemiología Hospitalaria, Universidad El Bosque, Bogotá, DC, Colombia
gInstituto de Química de Rosario (IQUIR, CONICET-UNR), Suipacha 570, S2002LRK Rosario, Argentina
hLos Angeles County and University of Southern California (LAC + USC) Medical Center, Los Angeles, CA, USA
iCenter for Pharmacometrics and Systems Pharmacology, Department of Pharmaceutics, College of Pharmacy, University of Florida, Orlando, FL, USA
jFacultad de Ciencias Bioquímicas y Farmacéuticas, Universidad Nacional de Rosario, S2002LRK Rosario, Argentina
kDepartments of Medicine, Pharmacology, Molecular Biology and Microbiology, Biochemistry, and Proteomics and Bioinformatics, Case Western Reserve University School of Medicine, Cleveland, OH, USA
lMedical Service, GRECC, Louis Stokes Cleveland Department of Veterans Affairs Medical Center, Cleveland, OH, USA
mCWRU-Cleveland VAMC Center for Antimicrobial Resistance and Epidemiology (Case VA CARES), Cleveland, OH, USA
First published on 5th January 2021
Abstract
Infections caused by multidrug resistant (MDR) bacteria are a major public health threat. Carbapenems are among the most potent antimicrobial agents that are commercially available to treat MDR bacteria. Bacterial production of carbapenem-hydrolysing metallo-β-lactamases (MBLs) challenges their safety and efficacy, with subclass B1 MBLs hydrolysing almost all β-lactam antibiotics. MBL inhibitors would fulfil an urgent clinical need by prolonging the lifetime of these life-saving drugs. Here we report the synthesis and activity of a series of 2-mercaptomethyl-thiazolidines (MMTZs), designed to replicate MBL interactions with reaction intermediates or hydrolysis products. MMTZs are potent competitive inhibitors of B1 MBLs in vitro (e.g., Ki = 0.44 μM vs. NDM-1). Crystal structures of MMTZ complexes reveal similar binding patterns to the most clinically important B1 MBLs (NDM-1, VIM-2 and IMP-1), contrasting with previously studied thiol-based MBL inhibitors, such as bisthiazolidines (BTZs) or captopril stereoisomers, which exhibit lower, more variable potencies and multiple binding modes. MMTZ binding involves thiol coordination to the Zn(II) site and extensive hydrophobic interactions, burying the inhibitor more deeply within the active site than D/L-captopril. Unexpectedly, MMTZ binding features a thioether–π interaction with a conserved active-site aromatic residue, consistent with their equipotent inhibition and similar binding to multiple MBLs. MMTZs penetrate multiple Enterobacterales, inhibit NDM-1 in situ, and restore carbapenem potency against clinical isolates expressing B1 MBLs. Based on their inhibitory profile and lack of eukaryotic cell toxicity, MMTZs represent a promising scaffold for MBL inhibitor development. These results also suggest sulphur–π interactions can be exploited for general ligand design in medicinal chemistry.
Introduction
β-Lactams are the most frequently prescribed antibiotics as they are well tolerated, bioavailable and broadly active against multiple Gram-positive and -negative bacteria.1,2 Despite their unrivalled therapeutic efficacy, the increasing occurrence of multidrug-resistant (MDR) bacteria in clinical environments has become a major public health problem.3 This situation is aggravated by growing numbers of susceptible, compromised patients, including patients with COVID-19 for whom coinfections with antibiotic-resistant bacteria are frequently reported.4,5Carbapenems are highly potent anti-infective agents that are often used to treat MDR Gram-negative bacteria.6 Among Gram-negative bacteria, the main mechanism of carbapenem resistance is the expression of β-lactamases, enzymes that hydrolyse the β-lactam ring, rendering these compounds inactive against their targets.1,7,8 Presently, the most worrisome β-lactamases are the metallo-β-lactamases (MBLs), Zn(II)-dependent enzymes that display a broad substrate profile, being capable of inactivating penicillins, cephalosporins and carbapenems.9–12 Furthermore, these enzymes are refractory to the action of all currently available β-lactamase inhibitors, including mechanism-based compounds such as clavulanic acid, tazobactam and sulbactam, the diazabicyclooctane avibactam, and the boronate vaborbactam.1,7 MBLs are becoming increasingly widespread in clinically relevant Gram-negative pathogens including diverse Enterobacterales species, Pseudomonas aeruginosa and Acinetobacter spp., on which they confer carbapenem resistance.13 Since carbapenem resistant Gram-negative bacteria are usually also extensively drug resistant (XDR), the World Health Organization (WHO) has assigned them the “highest priority for antibacterial drug research and development”.14 The worldwide dissemination of genes encoding for MBLs, and the continuing emergence of new enzymes and variants, is rapidly making them leading contributors to the global antibiotic resistance crisis.
MBLs are classified into three subclasses, with the most clinically relevant enzymes of subclass B1 active with two Zn(II) ions bound to their active site.10,15 Although the metal coordination sphere is conserved in all B1 enzymes, little homology is present between the active sites of different enzymes even within this subclass. For example, the most important B1 enzymes (those of the IMP, VIM and NDM families) share approximately 30% sequence identity.9,10,16 This structural diversity makes the development of a universal MBL inhibitor an exceptionally challenging task.17–19 As a result, the use of chelating compounds as MBL inhibitors has been pursued,20,21 despite the risk of off-target activity arising from indiscriminate chelation of metal ions.
One strategy to address the challenge of MBL inhibition is to exploit our current knowledge regarding common features of β-lactam antibiotic binding and hydrolysis that are shared by different MBLs despite their structural differences.19,22 Based on this approach, we recently reported a series of bisthiazolidines (BTZs), bicyclic substrate mimics decorated with a zinc-binding thiol moiety (Fig. 1), that are cross-class MBL inhibitors active in vitro and against bacterial pathogens expressing MBLs.23–25 X-ray crystallographic analyses of MBL:BTZ complexes revealed that this scaffold mimics the structure of bicyclic substrates, and that different stereochemistries can elicit different coordination modes within the active site, that nevertheless result in equally effective inhibition.25
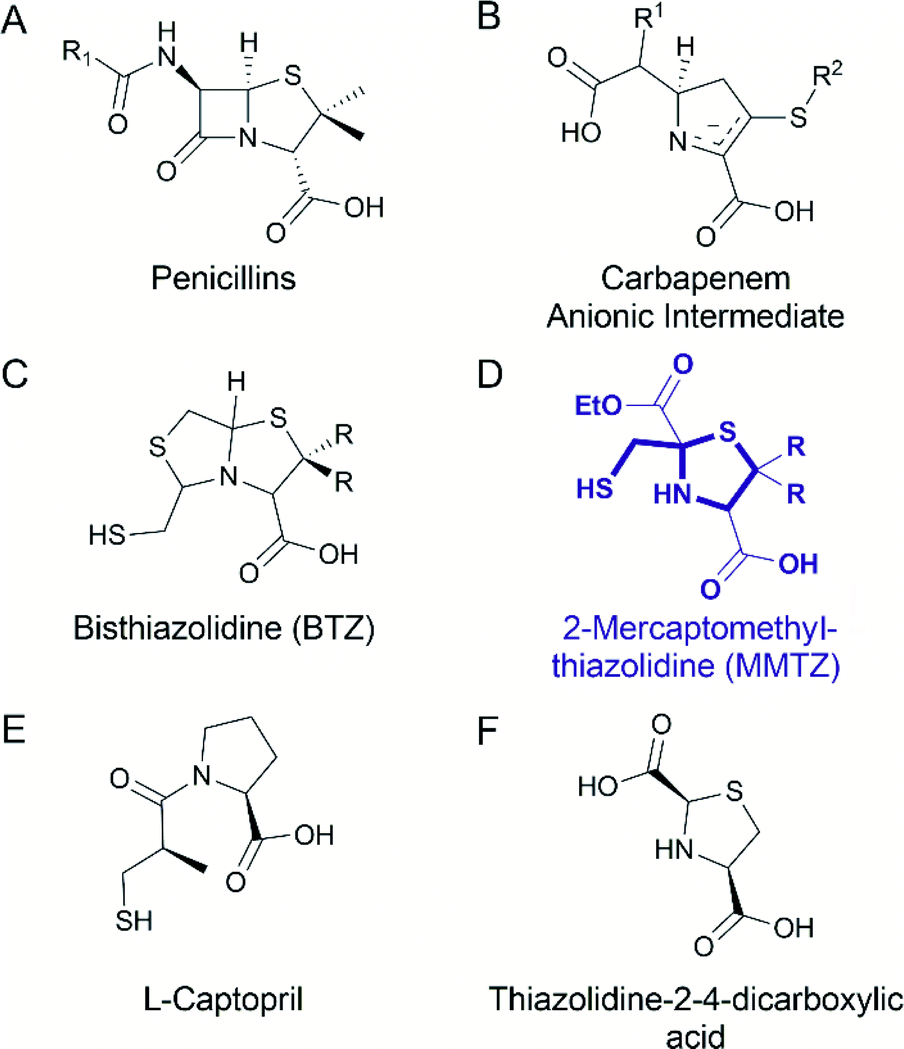 | ||
| Fig. 1 Chemical structure of the (A) penicillins, (B) carbapenem anionic intermediate, (C) bisthiazolidine (BTZ), (D) MMTZ scaffold herein designed, (E) L-captopril reported in ref. 30 and 31 as an MBL inhibitor, (F) thiazolidine-2-4-dicarboxylic acid reported in ref. 32 as an MBL inhibitor. | ||
Common mechanistic features of MBLs are identified by kinetic, spectroscopic and structural studies. MBLs of multiple subtypes have been shown to hydrolyse carbapenems via a common catalytic intermediate, in which the β-lactam ring has already been cleaved, that interacts with the metal site in a manner that is consistent across different enzyme targets.19,22 The binding mode of these intermediates resembles that reported for several enzyme-product adducts described by X-ray crystallography.7,26 Capturing features of the interactions of hydrolytic intermediates, and related hydrolysed products, with MBLs, can therefore guide the design of compounds aimed at inhibiting multiple MBL targets.
Based on this knowledge, we here report a novel 2-mercaptomethyl-thiazolidine (MMTZ) scaffold, a prototype that partially resembles MBL-bound intermediates and product species, whilst retaining the metal-binding thiol group present in the BTZs. This “second advanced” series of inhibitors displays more potent inhibitory activity than the earlier, “first generation” BTZs and can restore the activity of imipenem against MBL-producing carbapenem resistant Enterobacterales (MBL-CREs). Importantly, 2-mercaptomethyl-thiazolidines (MMTZs) represent a synthetically accessible series of pharmacophores that can be easily prepared by a condensation reaction of aminothiols with mercapto-carbonyl compounds masked under a dithiane form. All synthesised and tested MMTZs show similar inhibitory potency against different B1 MBLs, consistent with a conserved mode of binding. MMTZ stereoisomers display a less variable range of potencies than stereoisomers of a chemically related compound, D/L-captopril, a clinically approved anti-hypertensive agent previously investigated27 as an MBL inhibitor.28,29 Crystal structures of NDM-1, VIM-2 and IMP-1 complexed with MMTZs reveal that this is likely due to the reduced conformational flexibility of MMTZs, compared to the captopril series, and to the presence of a conserved interaction between the sulphur atom of the thiazolidine (TZ) ring and an aromatic residue present at the base of a mobile active site loop present in the different B1 enzymes. Thus, MMTZs represent a novel scaffold able to inhibit the clinically relevant B1 MBLs in vitro and in situ through a conserved binding mode and constitute a promising new avenue in the continuing search for MBL inhibitors.
Results
Design and synthesis of MMTZs
BTZs are bicyclic MBL inhibitors, designed as substrate mimics, decorated with two zinc binding groups: a thiol and a carboxylate (Fig. 1).23 Based on these features we envisioned that a simpler, monocyclic compound, mimicking the reaction intermediate or hydrolysis product, could allow us to better explore stereochemical preferences in the in the MBL active site, retaining the main binding features of the BTZs, but being more versatile with respect to stereochemistry. In this regard, L-captopril captures these features (Fig. 1), partially mimicking the five-membered ring present in hydrolysed penicillins and conserving the carboxylate moiety, with the thiol group providing an anchoring point to the metal binding site.29 However, different captopril stereoisomers demonstrate a range of inhibitory potencies towards B1 MBLs.30,31 We reasoned that a TZ scaffold bearing a thiol group and carboxylate moiety (Fig. 1) could outperform or improve upon captopril due to the shorter thiol-bearing side chain, that would restrict conformational mobility within the active site and provide a more rigid metal-binding scaffold. Since a TZ containing two carboxylate groups was shown to be a potent inhibitor of the B1 enzyme CcrA (Fig. 1),32 we hypothesised that a MMTZ molecule would elicit even more potent inhibition due to the presence of the zinc-binding thiol moiety.Thiazolidines are versatile motifs broadly used in medicinal chemistry33 that can be easily obtained by the condensation between carbonyls and 1,2-aminothiols. Based on the required structural features, two series of 2-ethoxycarbonyl-2-mercaptomethyl thiazolidine-4-carboxylic acids were designed (Fig. 2), 1a with R = H, and 1b with R = Me, thus including the gem dimethyl group present in penicillins. For each series, we prepared the L and D enantiomers starting from the corresponding L and D amino acids 2. The mercapto-ketone needed for its preparation was used under the dithiane form 3, and synthesised as previously reported.34 MMTZs were next prepared by cyclocondensation of 1,2-aminothiols 2 and dithiane 3 (Fig. 2), affording the syn/anti diastereomeric mixtures, that were easily purified and evaluated as pure compounds. In the case of MMTZ 1a, only the anti diastereomer could be isolated, although both isomers were observed in NMR spectra of the crude reaction. The two diastereomers of thiazolidine 1b were isolated and characterised independently. MMTZs (L-anti-1a, D-anti-1a, L-anti-1b, D-anti-1b, L-syn-1b, D-syn-1b) were prepared and isolated (Fig. 2). The stability of the compound D-anti-1b was assessed in PBS (pH 7.2, 27 °C). We observed that, under these conditions, 88% of intact MMTZ remains after 6 hours (Fig. S1†). Analysis by LCMS indicates that the MMTZ did not hydrolyse to penicillamine and the main degradation product was the disulphide derivative (Fig. S2†).
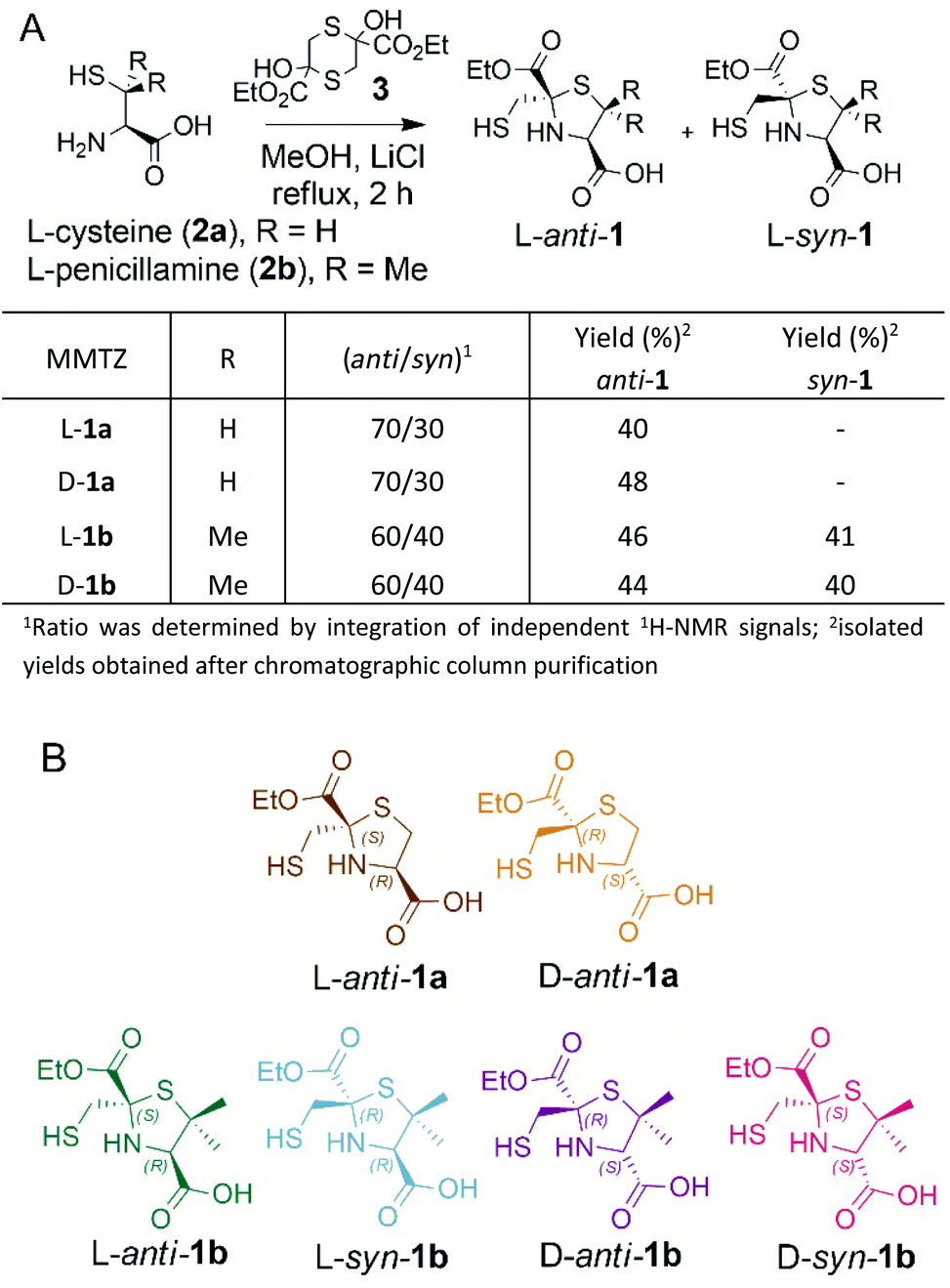 | ||
| Fig. 2 (A) Synthetic scheme of 2-mercaptomethyl-thiazolidines (MMTZs) and reaction yields. (B) Chemical structure of the MMTZ inhibitors: L-anti-1a (brown), D-anti-1a (orange), L-anti-1b (green), L-syn-1b (light blue), D-anti-1b (purple) and D-syn-1b (pink). The stereocentres of the thiazolidine ring are labelled according to their absolute configuration as S or R. | ||
MMTZs are potent MBL inhibitors
We assessed the inhibitory potency of the synthesised MMTZs against three representatives, clinically important, subclass B1 MBLs: NDM-1, IMP-1, and VIM-2.17 Hydrolysis rates for the carbapenem imipenem were determined under steady-state conditions in the presence of MMTZs, with progress curves able to be fitted to a competitive inhibition model (Fig. S3 and S4†) yielding inhibition constants (Ki) in the low-to sub-μM range (Table 1).| Inhibition potency (μM) | |||||
|---|---|---|---|---|---|
| Inhibitor | Chemical structure | NDM-1 | IMP-1 | VIM-2 | Ref. |
| L-anti-1a |
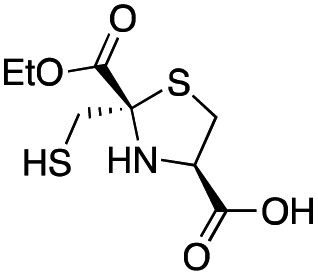
|
5.2 ± 0.7 | 1.0 ± 0.2 | 0.38 ± 0.05 | This work |
| D-anti-1a |
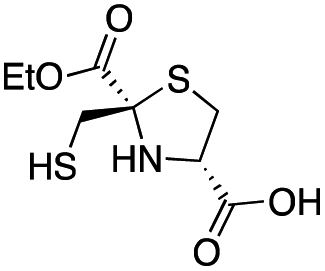
|
2.5 ± 0.5 | 1.3 ± 0.1 | 0.39 ± 0.04 | This work |
| L-anti-1b |
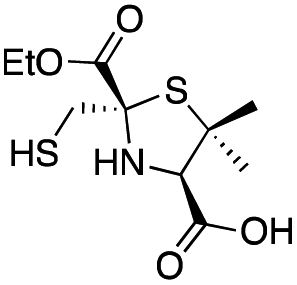
|
0.44 ± 0.06 | 0.46 ± 0.05 | 0.75 ± 0.09 | This work |
| L-syn-1b |
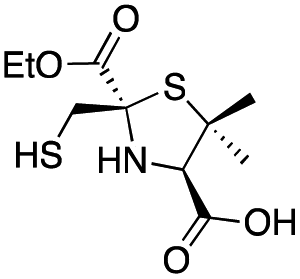
|
8 ± 1 | 6.0 ± 0.6 | 3.6 ± 0.4 | This work |
| D-anti-1b |
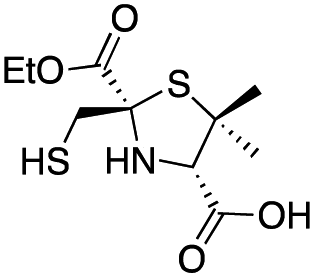
|
3.1 ± 0.3 | 0.93 ± 0.08 | 0.9 ± 0.1 | This work |
| D-syn-1b |
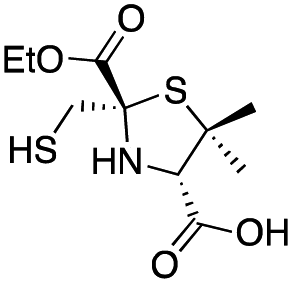
|
0.60 ± 0.05 | 2.0 ± 0.2 | 1.9 ± 0.1 | This work |
| L-BTZ-1 |
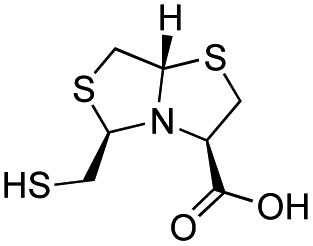
|
7 ± 1 | 8 ± 2 | 2.9 ± 0.4 | 23 and 25 |
| D-BTZ-1 |
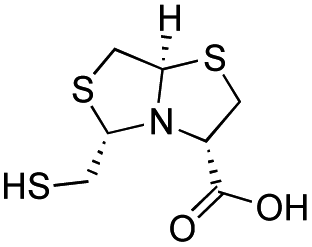
|
19 ± 3 | 6 ± 1 | 3.2 ± 0.4 | 23 and 25 |
| L-BTZ-2 |
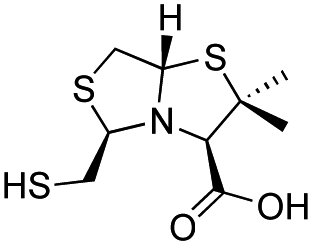
|
18 ± 3 | 15 ± 3 | 6 ± 1 | 23 and 25 |
| D-BTZ-2 |
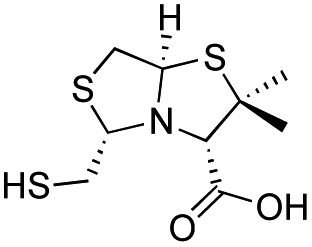
|
12 ± 1 | 14 ± 3 | 10 ± 2 | 23 and 25 |
| L-Captopril |
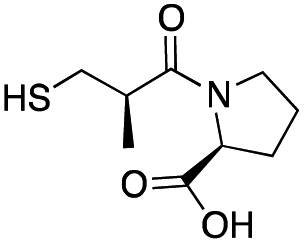
|
157 ± 1 | 23 ± 1 | 4.4 ± 0.8 | 31 |
| D-Captopril |
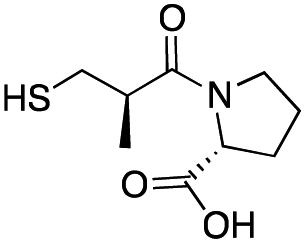
|
20 ± 2 | 7 ± 1 | 0.07 ± 0.01 | 31 |
| L-epi-Captopril |
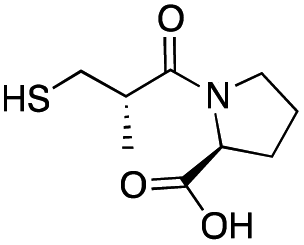
|
>500 | 436 ± 1 | 6 ± 2 | 31 |
| D-epi-Captopril |
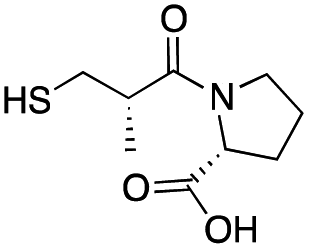
|
65 ± 1 | 173 ± 1 | 5.5 ± 0.7 | 31 |
All MMTZs were potent inhibitors of all three assayed MBLs, with Ki values ranging between 0.38 and 8 μM. The two anti-enantiomers of MMTZ 1a showed similar potencies against each enzyme (e.g. 5.2 and 2.5 μM against NDM-1 for L-anti-1a and D-anti-1a, respectively) despite their differing enantiomeric configurations. In contrast, for the MMTZs 1b, bearing a gem-dimethyl group, the stereochemistry has an impact (though modest) upon inhibition. In the case of NDM-1, Ki values for these four compounds varied between 0.44 and 8 μM. Of the six compounds tested, the two most potent NDM-1 inhibitors are L-anti-1b (0.44 μM) and D-syn-1b (0.60 μM), both with the gem-dimethyl group. Within the series of compounds 1b, the heterocycles with the mercaptomethyl group in the 2S configuration (L-anti-1b and D-syn-1b) were better inhibitors than those in the 2R configuration: MMTZ L-anti-1b was 7-fold more active against NDM-1 than its enantiomer D-anti-1b; and D-syn-1b was 13 times more active than the enantiomer L-syn-1b. The potencies displayed by MMTZs with an R stereocentre were comparable to the Ki values of compounds from the 1a series.
The MMTZ inhibitors performed best against VIM-2 (Ki values between 0.38 and 3.6 μM), with the two 1a compounds showing the highest potency. For IMP-1, Ki values ranged between 0.46 and 6 μM. Among compounds containing the gem-dimethyl group (series 1b), MMTZs with an anti configuration showed generally lower Ki values than their syn counterparts for both VIM-2 and IMP-1. MMTZ L-anti-1b showed the best inhibition profile against all three MBLs. Overall, the inhibitory activity against MBLs is clearly better than that of BTZs against the B1 enzymes (Table 1). In the case of NDM-1, L-anti-1b and D-syn-1b are 12-fold more potent inhibitors than L-BTZ-1 (the best compound within the BTZ series), with all other MMTZs displaying equal lower or lower Ki values to those measured for L-BTZ-1 (Table 1).
We then assessed the ability of these compounds to penetrate the bacterial outer membrane and inhibit NDM-1 within the bacterial periplasm.35 To this end, we used 1H NMR to follow imipenem hydrolysis by E. coli cells expressing NDM-1 in the presence of the different inhibitors (Fig. S5†). NDM-1 in the E. coli periplasm hydrolyses a solution of 500 μM imipenem completely in 12–15 min (Fig. S5†). All MMTZs were able to protect imipenem from the hydrolytic activity of NDM-1. Table 2 summarises the in-cell IC50 values for E. coli cells expressing NDM-1. In E. coli, D-syn-1b was the most potent inhibitor (in-cell IC50 = 10 μM, Fig. S5 and S6†). MMTZs lacking the gem dimethyl group, L-anti-1a and D-anti-1a, were 5 and 6 times less potent than D-syn-1b. However, MMTZs bearing the gem dimethyl group and different stereochemistry than D-syn-1b displayed lower potencies. These relatively elevated IC50 values likely arise from differences in cell penetration and in the stability of the inhibitor thiol in the cellular environment. In comparison, L-BTZ-1, displayed an in-cell IC50 for NDM-1-catalysed imipenem hydrolysis of 23 μM,23 in the equivalent low micromolar range to the value for D-syn-1b.
| Inhibitor | In-cell IC50 (μM) |
|---|---|
| L-anti-1a | 54 ± 4 |
| D-anti-1a | 64 ± 5 |
| L-anti-1b | 160 ± 10 |
| L-syn-1b | 230 ± 30 |
| D-anti-1b | 210 ± 10 |
| D-syn-1b | 10 ± 1 |
MMTZs adopt a uniform binding mode to class B1 MBLs
To understand the interactions of MMTZs with the different B1 MBLs, we obtained the crystal structures of NDM-1, VIM-2 and IMP-1 in complex with both L-anti-1b and D-syn-1b (resolutions 1.4–1.87 Å, Table S1†), the most potent compounds in the inhibition studies. In all cases clear positive Fo − Fc difference density was observed in electron density maps derived from inhibitor-exposed crystals. Compounds could be modelled into these features (Fig. S7†) with real space correlation coefficients (RSCC values, calculated during PDB submission) ≥0.91 (Table S2†) giving high confidence in the presence and correct fitting of bound inhibitor.36,37 For all enzymes inhibitors were modelled into all molecules of the asymmetric unit (ASU), with the exception of NDM-1:L-anti-1b, in which the inhibitor was modelled in only one of the two chains in the ASU. Evidence of inhibitor dimerization in the active sites was not present.In all cases, the thiol moiety of the inhibitors binds equidistant between the two zinc ions, displacing the hydrolytic water/hydroxide (Fig. 3 and Table S3†), and maintaining a Zn–Zn distance of 3.57–3.79 Å (Table S3†). This distance is consistent with that reported for the uncomplexed forms of MBLs and for some complexes with products and inhibitors.25,30,38–40 Additionally, several hydrophobic residues lining the active site significantly stabilise inhibitor binding. These include residues on the mobile L3 loop, with NDM-1 Leu59, Met61 and Val67 contributing to binding in both inhibitor complexes (Fig. 3A and B), while in VIM-2 Phe61 and Tyr67 at the base of the L3 loop are in close proximity to both inhibitors (Fig. 3C and D). In IMP-1, due to a small conformational change in the flexible L3 loop,25 either two (Val61, Phe87, L-anti-1b, Fig. 3E) or three (Val61, Trp64, Phe87, D-syn-1b, Fig. 3F) hydrophobic residues stabilise binding. Furthermore, the residue Trp87 (in NDM-1 and VIM-2) or Phe87 (IMP-1) is positioned close (approximately 4 Å) from the ethoxy sidechain in all the MBL:MMTZ complexes, providing a strong hydrophobic interaction. In NDM-1 only, an additional hydrophobic residue, Ile29 situated close to the N-terminus, also contributes to binding.
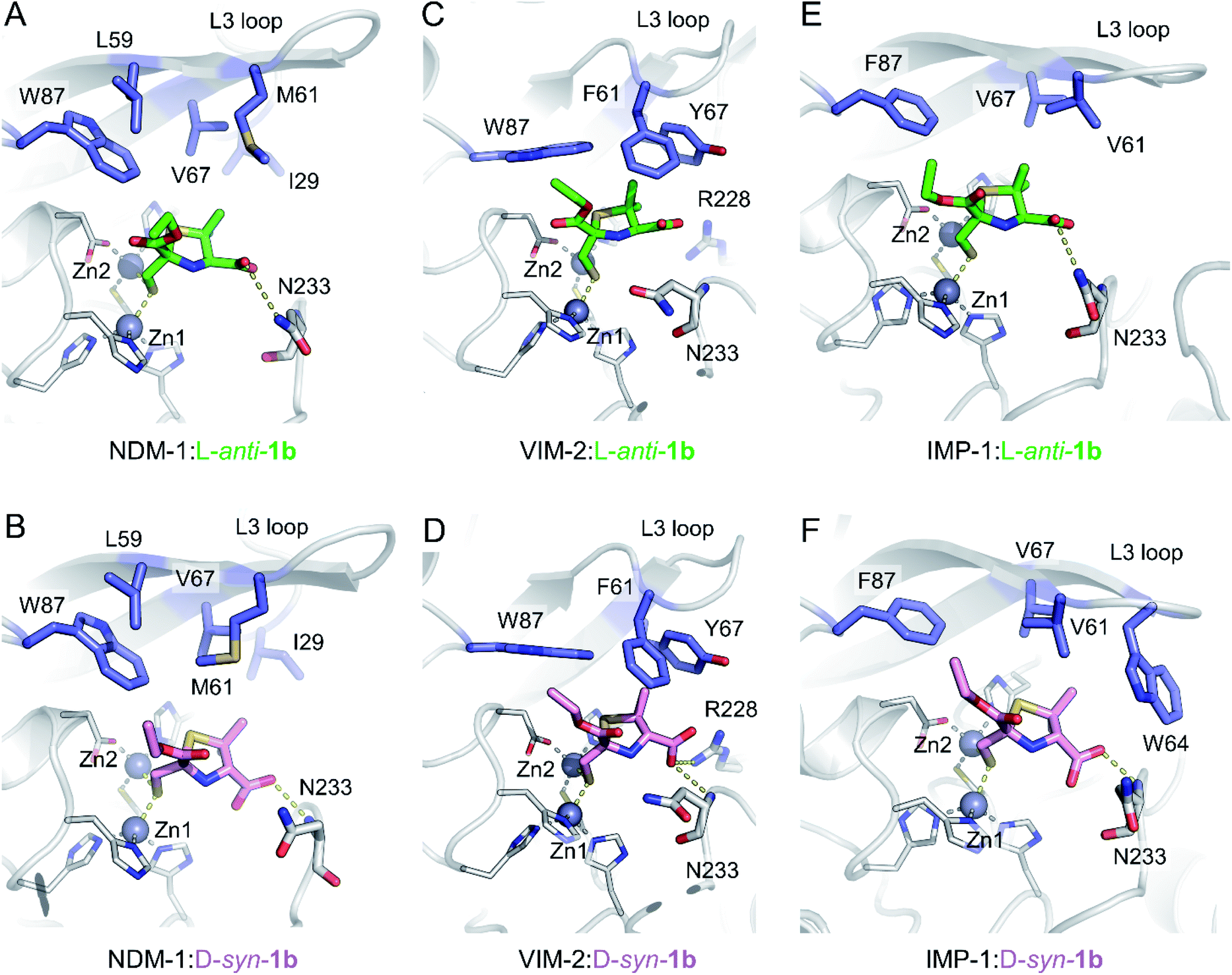 | ||
| Fig. 3 Interactions of MMTZs in the active sites of B1 MBLs. Crystal structures for complexes of L-anti-1b (green) with NDM-1 (A), VIM-2 (B) and IMP-1 (C); and for complexes of D-syn-1b (pink) with NDM-1 (D), VIM-2 (E) and IMP-1 (F). Zinc ions are shown as grey spheres, blue side chains denote hydrophobic residues involved in inhibitor interactions, thick sticks denote residues involved in hydrogen bonding interactions with inhibitor, hydrogen bonding and metal-coordinating interactions are shown as yellow dashed lines. Zinc-coordinating residues are shown as thin sticks. | ||
The inhibitors form few hydrogen bonds within the active site (Table S3†). For L-anti-1b, the carboxylate interacts weakly with Asn233 on the L10 loop in NDM-1 and IMP-1 (3.46 and 3.28 Å, respectively), but this is not observed in the VIM-2 complex. In complexes with D-syn-1b the inhibitor interacts more closely with the backbone amide of Asn233 (2.56–3.07 Å, Table S3†), while in VIM-2 there is an additional weak interaction with Arg228 (3.39 Å) that is not present in NDM-1 and IMP-1.
Overlays of complexes of L-anti-1b with NDM-1, VIM-2 and IMP-1 highlight the similarities in binding (Fig. S8A†), particularly between NDM-1 and IMP-1, with differences in the positioning of the L10 loop in VIM-2 resulting in a slightly different orientation for the bound inhibitor. The structures also highlight some flexibility of binding for the ethoxy carbonyl side chain, which is rotated 180° about the carbonyl carbon in the IMP-1:L-anti-1b structure compared to NDM-1 or VIM-2, largely due to the lack of hydrogen bonds between the inhibitor side chain and the protein backbone. This flexibility is also apparent in the electron density, which is more poorly defined for the ethoxy –CH3 atoms in the NDM-1 structures (Fig. S7†).
The inhibitor D-syn-1b binds very similarly to NDM-1 and IMP-1 (Fig. S8B†) but has a slightly different orientation in VIM-2 due to interactions of the carboxylate with Arg228. Despite these small differences in binding, however, both L-anti-1b and D-syn-1b display broadly consistent binding modes, making few (and weak) interactions with the protein backbone and with the thiol:zinc interaction dominating binding.
Role of a common S–π interaction on MMTZ binding
Notably, the sulphur atom of the thiazolidine ring of MMTZs is located at 3.8 Å from the edge of the indole group of NDM-1 Trp87, at the base of loop L3 (Fig. 4). A similar sulphur–π interaction with an aromatic group at this position (Trp in NDM-1 and VIM-2, Phe in IMP-1) is observed in all 6 complex structures with the two MMTZs (Fig. 4 and Table S3†). In NDM-1 and IMP-1, the sulphur atom is located in a coplanar manner to the edge of the aromatic ring, while in VIM-2, the sulphur forms a stacking interaction with Trp87. The conservation of this interaction does not seem to be incidental. Unfortunately, attempts to test its role by preparing oxazolidine analogs, using the same synthetic methodology, were unsuccessful (data not shown). Therefore, we decided to explore its role in silico. Based on the crystal structures of NDM-1 bound to L-anti-1b and D-syn-1b, we constructed structural models of the analogous 2-mercaptomethyl oxazolidines (MMOZs) bound to NDM-1 by replacing the sulphur atom of the thiazolidine ring with an oxygen atom. The experimental geometries of the bound MMTZs and the simulated geometries of the bound MMOZs were minimised by hybrid quantum mechanics molecular calculations (QM/MM). The adducts of MMTZs with NDM-1 yielded final geometries similar to those in the crystal structure, i.e., preserving the S–π distances (Fig. S9A and B†). Instead, in the theoretical complexes with MMOZs, the interaction distance between the O atom and Trp87 was increased upon geometry optimization (Fig. S9C and D†). The difference is most prominent in the NDM-1:L-anti-1b complex which showed an increase of 1.10 Å when the O is present and significant movement of the ligand. In the NDM-1:D-syn-1b adduct, the O–π distance is 0.34 Å longer than the S–π distance. We conclude that the presence of the thioether S favours the interaction with the aromatic groups in all studied MBLs, contributing to binding.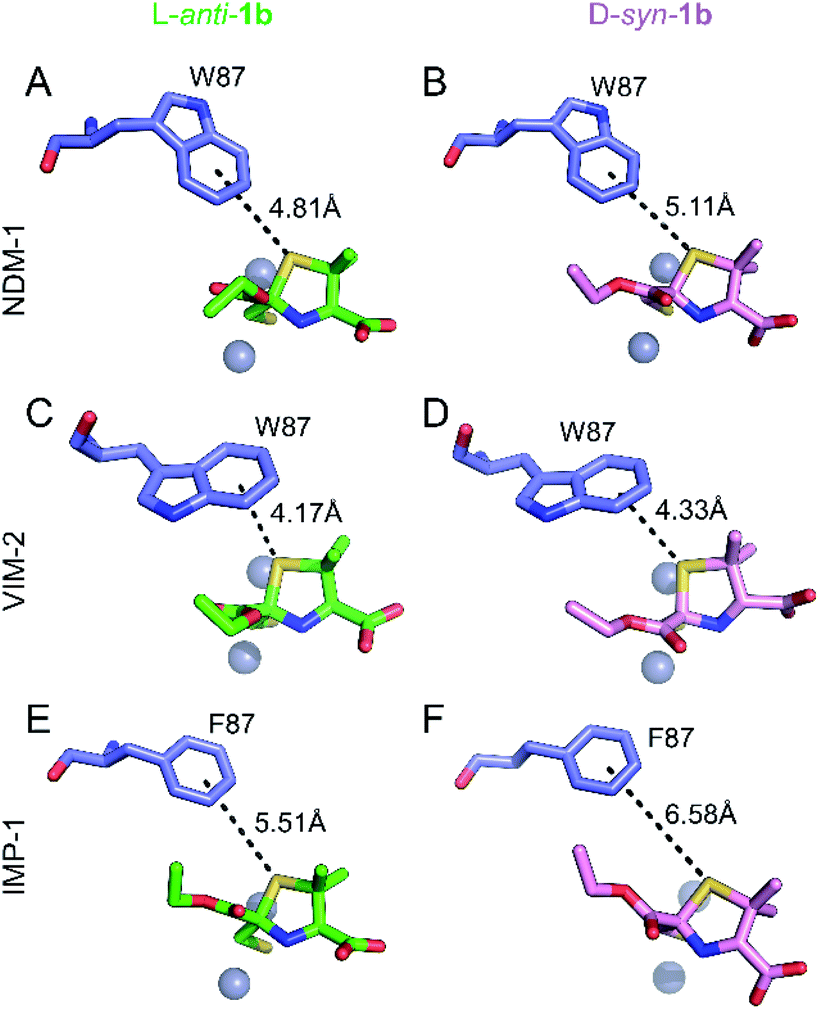 | ||
| Fig. 4 Interactions of the thiazolidine ring sulphur atom with the aromatic group of the residue at position 87. Interactions (black dashes) between the benzene ring of Trp87 or Phe87 (blue sticks) and the thiazolidine sulphur of L-anti-1b (top, green sticks) or D-syn-1b (bottom, pink sticks) in the active sites of NDM-1 (A and B), VIM-2 (C and D) and IMP-1 (E and F). Distances shown in Å are measured from the centre of the benzene ring to the thiazolidine sulphur atom. | ||
MMTZs restore the action of imipenem against Enterobacterales and are not toxic against eukaryotic cells
We analysed the activity of MMTZs against clinical isolates producing MBLs by measuring their impact on the minimal inhibitory concentration (MICs) of imipenem (Table 3). MMTZs, particularly compound D-anti-1b, were able to successfully inhibit the MBLs present in Enterobacterales, as evident by a reduction of up to 16-fold in MIC values. In the absence of the carbapenem antibiotic, none of the MMTZs inhibitors has a detrimental effect on the bacterial growth, indicating that reduction of the MIC values is due to the restoration of susceptibility to imipenem through inhibition of the MBLs by the MMTZ inhibitors.| MIC (mg L−1) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Isolate | MBL | IMI | IMI + L-anti-1a | IMI + D-anti-1a | IMI + L-anti-1b | IMI + L-syn-1b | IMI + D-anti-1b | IMI + D-syn-1b |
| a Due to intrinsic reduced susceptibility to imipenem, strains from the Morganellaceae family were tested using meropenem. | ||||||||
| K. pneumoniae (1.37) | NDM | 32 | 8 | 8 | 8 | 16 | 8 | 8 |
| E. coli (8.68) | NDM-1 | 64 | 16 | 16 | 16 | 32 | 16 | 32 |
| K. pneumoniae (LC 82) | IMP-13 | 1 | 0.5 | 0.25 | 0.25 | 1 | 0.25 | 0.5 |
| Enterobacter spp. (42713) | IMP | 16 | 16 | 8 | 8 | 16 | 8 | 8 |
| P. mirabilis (UNC KPC 170)a | IMP | 16 | 8 | 8 | 4 | 16 | 1 | 8 |
| K. pneumoniae (6907) | VIM-2 | 4 | 2 | 2 | 1 | 4 | 1 | 4 |
| K. aerogenes (6915) | VIM-2 | 8 | 4 | 2 | 2 | 4 | 2 | 4 |
| K. pneumoniae (5639) | VIM-24 | 8 | 8 | 4 | 4 | 8 | 1 | 8 |
Finally, we evaluated the cytotoxicity of the six MMTZs against cultured HEK293, L929 and VERO cell lines at 260, 355 and 530 μM (Fig. 5). Encouragingly, compounds bearing the gem dimethyl group did not show any toxicity under the assayed conditions. However, MMTZ D-anti-1a induced a decline in cell viability percentages in almost all conditions, while L-anti-1a was more selectively toxic towards the HEK293 cell line. Overall, however, the combined impact of MMTZs in lowering the MICs of MBL-CRE, and their lack of or low toxicity, make them promising compounds for development as MBL inhibitors.
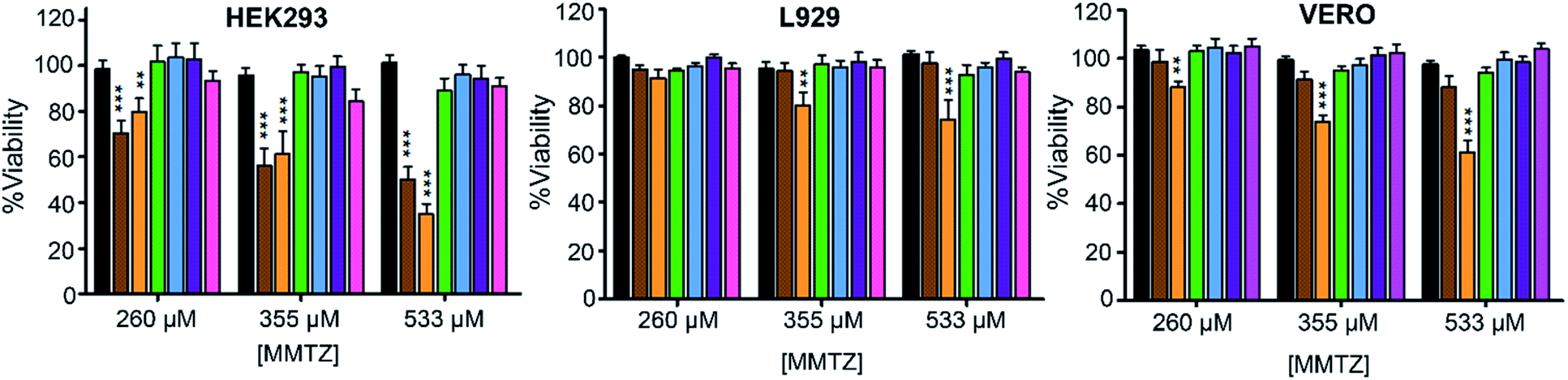 | ||
| Fig. 5 Cytotoxicity for the MMTZs in HEK293, L929 and VERO cell lines. Control (black), L-anti-1a (brown), D-anti-1a (orange), L-anti-1b (green), L-syn-1b (light blue), D-anti-1b (purple), and D-syn-1b (pink). | ||
Discussion
Here we report a series of MMTZs as potent inhibitors of B1 MBLs. These compounds showed comparable potencies against the three representative MBLs enzymes (NDM-1, VIM-2 and IMP-1, the most clinically important resistance determinants). MMTZs were able to enter the periplasm of E. coli, as evidenced by inhibition of NDM-1 catalysed imipenem hydrolysis in situ, and restore the antimicrobial effect of imipenem against clinical Enterobacterales. The discrepancies between the Ki and in-cell IC50 values can be attributed either to the stability of the thiol groups in the different strains or to differences in permeability between the clinical strains. Despite differences in the stereochemistry of individual inhibitors, the MMTZ scaffold utilises a similar binding mode to bind to the active site of the different MBLs tested.As previously reported for most thiolate MBL inhibitors,25,30 the driving force for MMTZ binding to the active site is the interaction with the metal centre of the thiolate, that bridges the two Zn(II) ions and replaces the hydroxide/water nucleophile. MMTZs share this binding mode with the related BTZs and with the captopril stereoisomers. Notably, however, MMTZs differ from these compound classes through the absence of electrostatic interactions, such as those of the carboxylate groups of BTZs or captopril stereoisomers with Asn233 (conserved in most B1 MBLs) and Lys224 (NDM-1 and IMP-1) or Arg228 (VIM-2) (Fig. S10 and S11†). These interactions have been also identified as critical for substrate recognition by MBLs. Notwithstanding, MMTZs show better inhibition potencies against the B1 MBLs tested than BTZs or captopril stereoisomers. Most importantly, our data reveal that the different MMTZs adopt a similar, common binding mode to their different B1 MBL targets, contrasting with other thiol-based inhibitors where binding is both enzyme and stereospecific.25,30
MMTZs were designed to mimic product-like reaction intermediates (Fig. 1) or replicate enzyme:product interactions, with the addition of a thiol to strengthen binding to the bi-Zn(II) site. Therefore, we compared the binding modes of MMTZs with some reported enzyme:product complexes, such as those of NDM-1 with hydrolysed meropenem (PDB 5ypm41), cefuroxime (PDB 5o2e42) and ampicillin (PDB 5zgr39). The five-membered rings of the MMTZs lies perpendicular in the active site with respect to the rings of the hydrolysed antibiotics (Fig. S12†) as a result of the electrostatic interactions of the β-lactam carboxylate group with Lys224 that are not replicated by MMTZs.
The conserved sulphur–π interaction (Fig. 4) plays a significant role in enhancing MMTZ binding, as confirmed by QM/MM calculations. This interaction is not present in the enzyme–product complexes of penicillins with MBLs,43 since the thiazolidine ring of ampicillin is perpendicular to the MMTZ thiazolidine ring (Fig. S12†). Therefore, MMTZs provide stronger sulphur–aromatic interactions than penicillins hydrolysis products.
In the case of the complex with the cephalosporin cefuroxime, the six-membered dihydrothiazine ring lies perpendicular to the five-membered ring of MMTZs. However, the geometry of the cefuroxime dihydrothiazine ring enables the interaction of the sulphur with Trp87 to be maintained (Fig. S12†). It is also notable that, in MMTZ complexes with NDM-1, the position of the L3 loop more closely resembles that observed in the complex with hydrolysed meropenem than in those with either ampicillin or cefuroxime. This may reflect the absence in MMTZs of a group equivalent to the relatively bulky C6/C7 substituents of these substrates (as opposed to the much smaller C6 hydroxyethyl group of meropenem), and the consequent avoidance of possible steric clashes.
MMTZs also resemble the captopril stereoisomers (Table 1), since both families of compounds have a carboxylate group bound to a five-membered ring (mimicking the intermediate/product of penicillin hydrolysis) and an additional thiol moiety. However, the different captopril stereoisomers exhibit a wide range of inhibitory potencies towards B1 MBLs (Table 1). For example, epi-D- and epi-L-captopril are only efficient inhibitors of VIM-2, while showing greatly reduced potency against NDM-1 and IMP-1.30 The adducts of D- and L-captopril with B1 MBLs show a similar binding mode in the active site, but the D isomer is much more potent and targets a wider range of MBLs. This is not the case for the different stereoisomers of the MMTZs reported here, whose potencies across the range of targets fall consistently within the low- to sub-μM range. Selectivity of captopril D isomers has been explained by appreciating the more extensive electrostatic interactions of the carboxylate moiety with Lys224 (NDM-1, IMP-1) or Arg228 (VIM-2) (Fig. S10†).30 A comparison of the binding modes of the MMTZs with captopril reveals that the five-membered ring of MMTZs is positioned more deeply within the active site (i.e. closer to the metal centre), despite the smaller number of electrostatic interactions and hydrogen bonds formed by MMTZs (Fig. S10†). This finding confirms our prediction that a shorter side chain connecting the thiol group with the five-membered ring could enhance binding with respect to the captopril variants.
We conclude that the consistent involvement in binding of sulphur–π interactions (not possible for the pyrrolidine ring of captopril) explains the better and more uniform inhibitory potency of MMTZs against the range of B1 MBLs, compared to the more variable potency of the different captopril isomers.
Similarly, although the related, but less potent, BTZs show more consistent inhibitory potency across different MBL targets, they do so by means of different binding modes that, while overall conserving the electrostatic interactions of the carboxylate group with residues at positions 224, 228 and 233, vary markedly between MBL target and BTZ stereoisomer. Due to their rigid bicyclic scaffold, none of the BTZs tested can adopt a mode of binding that shows significant sulphur–π interactions, supporting our hypothesis that such an interaction both enhances inhibitory potency and controls the conformation in the MBL active site of bound MMTZ.
Thioether–aromatic interactions can stabilise ligand binding by up to ca. 3.2 kcal mol−1, an energy comparable to a salt bridge, with the further advantage of being insensitive to pH changes.44–46 The distances and orientations observed in these MBL:MMTZ complexes (Fig. 4) are, based on previous experimental and theoretical studies, expected to maximise these interactions.46 This interaction orients the five-membered ring of the MMTZ within the active site, overriding the steering effect of the electrostatic interactions that may be formed by the carboxylate group. We conclude that the MMTZ scaffold improves the affinity of the thiol binding group by favouring hydrophobic contacts with the active site of B1 MBLs, specifically by means of a sulphur–π interaction with the conserved aromatic residue at position 87.
Conclusions
MMTZs were iteratively designed as a novel chemical scaffold to inhibit B1 MBLs. The six purified inhibitors displayed inhibition constants in the low- to sub-μM range for the B1 enzymes, with D-syn-1b as overall the most consistently potent inhibitor of both the different purified enzymes in vitro and, in combination with carbapenem antibiotics, in antibacterial assays against MBL-expressing strains of clinical microorganisms such as E. coli. These compounds are versatile and can be easily prepared with high yields. Since we report that the carboxylate group of MMTZs is not involved in significant electrostatic interactions, the possibility of introducing other substituents at the 4-position could result in even more potent inhibitors to improve the development of this scaffold. Furthermore, our discovery that the sulphur–π interaction apparently plays a key role in inhibitor binding strongly supports the suggestion that this feature can be exploited as a general concept both in MBL inhibition, taking advantage of the availability of conserved hydrophobic residues in accessible positions across multiple target enzymes, and more widely in ligand design in medicinal chemistry.Experimental methods
General methods
Reactions were monitored by analytical thin layer chromatography (TLC) on 0.25 mm silica gel coated plastic sheets (SIL G/UV 254). Flash chromatography on silica gel 60 (40 μm average particle diameter) was used to purify the crude reaction mixtures. All NMR spectra were recorded in CDCl3, at 300 K on a Bruker Avance 400 MHz spectrometer equipped with a 5 mm QXI probe and on an Avance III 400 MHz spectrometer equipped with a 5 mm BBO probe. Chemical shifts (δ) are expressed in ppm using tetramethylsilane (TMS) as internal reference, and multiplicities are indicated as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), br (broad). All reactions were carried out in dry, freshly distilled solvents under anhydrous conditions unless otherwise stated. HRMS-ESI spectra were recorded on MicroTOF-QTM (Bruker Daltonics) mass spectrometers. Optical rotation was measured using a Kruss Optronic GmbH P8000 polarimeter with a 0.5 mL cell (concentration c given as g/100 mL). The purity of final tested compounds was ≥95% as determined by 1H-NMR, and HPLC.Synthesis of diethyl 2,5-dihydroxy-1,4-dithiane-2,5-dicarboxylate 3
To a stirred solution of ethyl 3-bromopyruvate (14 g, 0.071 mol) in MeOH (125 mL) cooled at −10 °C, was added a cooled (0 °C) suspension of NaHS (11 g, 0.20 mol) in MeOH (125 mL) at 2 mL per minute. A white solid is formed during the addition, and cooling with stirring was continued for 1 h. The solid was collected, washed with Et2O (50 mL) and dried to give 3 as white solid (7.8 g, yield 73%). 1H-NMR (400 MHz, DMSO-d6) δ 6.98 (s, 2H), 4.18 (q, J = 7.1 Hz, 4H), 3.65 (d, J = 14.1 Hz, 2H), 2.87 (d, J = 14.1 Hz, 2H), 1.23 (t, J = 7.1 Hz, 6H) according to reported values in literature.34Synthesis of (2S,4R)-2-(ethoxycarbonyl)-2-(mercaptomethyl)thiazolidine-4-carboxylic acid (L-anti-1a)
To a stirred suspension of L-cysteine (L-2a) (1 g, 8.3 mmol) in MeOH (20 mL) was added under N2, dithiane 3 (1.4 g, 2.76 mmol), LiCl (35 mg, 0.82 mmol) and heated at reflux for 2 h. The reaction mixture was concentrated under reduced pressure. The residue was poured into H2O (60 mL), the pH was adjusted to 3 with HCl 1 M, and extracted with AcOEt (3 × 60 mL). The combined organic layers were dried (Na2SO4), filtered and concentrated. A crude reaction was obtained as a diastereomeric mixture of L-anti/syn-1a 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30. The crude was purified by SiO2 using nHex:AcOEt:AcOH (70:29:1) to afford L-anti-1a as white solid (0.32 g, yield 40%), mp 99.1–101 °C. 1H-NMR (400 MHz, CDCl3) δ 4.36–4.29 (m, 2H), 4.11 (dd, J = 9.0, 6.2 Hz, 1H), 3.43 (dd, J = 10.7, 6.2 Hz, 1H), 3.06–3.01 (m, 1H), 3.03 (d, J = 8.4 Hz, 2H), 1.97 (t, J = 8.4 Hz, 1H), 1.34 (t, J = 7.1 Hz, 3H), 0.86 (br, NH). 13C-NMR (100 MHz, CDCl3) δ 174.1, 171.0, 79.3, 65.0, 62.9, 39.2, 33.6, 14.0. [α]21D = −94.12 (c 2.6, AcOEt).
:30. The crude was purified by SiO2 using nHex:AcOEt:AcOH (70:29:1) to afford L-anti-1a as white solid (0.32 g, yield 40%), mp 99.1–101 °C. 1H-NMR (400 MHz, CDCl3) δ 4.36–4.29 (m, 2H), 4.11 (dd, J = 9.0, 6.2 Hz, 1H), 3.43 (dd, J = 10.7, 6.2 Hz, 1H), 3.06–3.01 (m, 1H), 3.03 (d, J = 8.4 Hz, 2H), 1.97 (t, J = 8.4 Hz, 1H), 1.34 (t, J = 7.1 Hz, 3H), 0.86 (br, NH). 13C-NMR (100 MHz, CDCl3) δ 174.1, 171.0, 79.3, 65.0, 62.9, 39.2, 33.6, 14.0. [α]21D = −94.12 (c 2.6, AcOEt).
Synthesis of (2R,4S)-2-(ethoxycarbonyl)-2-(mercaptomethyl)thiazolidine-4-carboxylic acid (D-anti-1a)
Prepared in analogous route as described for L-anti-1a, starting from D-cysteine D-2a and dithiane 3. The crude was purified by SiO2 using nHex:AcOEt:AcOH (70:29:1) to afford D-anti-1a as a white solid, yield 48%. The spectroscopic properties were identical to those obtained for L-anti-1a. HRMS calculated for C8H13NO4S2 [MNa]+ 274.0184, found 274.0178.
Synthesis of (2S/R,4R)-2-(ethoxycarbonyl)-2-(mercaptomethyl)-5,5-dimethylthiazolidine-4-carboxylic acid (L-anti/syn-1b)
Prepared in analogous route as described for L-anti-1a, starting from L-penicillamine L-2b and dithiane 3. A crude reaction was obtained as a diastereomeric mixture (L-anti/syn-1b 60:40). The crude was purified by SiO2 using nHex:AcOEt:AcOH (70:29:1) to afford L-anti-1b 46% yield, and L-syn-1b 41% yield.
Synthesis of (2S/R,4S)-2-(ethoxycarbonyl)-2-(mercaptomethyl)-5,5-dimethylthiazolidine-4-carboxylic acid (D-anti/syn-1b)
Prepared in analogous route as described for L-anti-1a, starting from D-penicillamine D-2b, and dithiane 3. The residue was purified by SiO2 using nHex:AcOEt:AcOH (60:40:0.5) to afford D-anti-1b 44% yield, and D-syn-1b 40% yield.
Author contributions
MAR purified the proteins and performed the Ki determination and in-cell IC50 measurements. VM and VC synthesised, purified and characterised the MMTZ compounds. MAR and CB performed the eukaryotic cell cultures and cytotoxicity assays. PH and RS purified the proteins and performed the crystallisation and structure determination. DMM performed the QM/MM calculations. MFM performed the antimicrobial susceptibility test. GM and AJV designed the compounds. MAR, PH, MFM, BS, GLD, RAB, JS, AJV and GM analysed and discussed the data. MAR, PH, MFM, RAB, JS, AJV and GM wrote the paper, and all authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (NIH) to R. A. B. under Award Numbers R01AI063517 and R01AI072219, to R. A. B., G. M., J. S. and A. J. V. under Award Number R01AI100560 and to B. S. under Award Numbers R01 AI130060 and AI117211. This study was also supported in part by funds and/or facilities provided by the Cleveland Department of Veterans Affairs, Award Number 1I01BX001974 to R. A. B. from the Biomedical Laboratory Research & Development Service of the VA Office of Research and Development, and the Geriatric Research Education and Clinical Center VISN 10. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or the Department of Veterans Affairs. GM thanks M. Macías and L. Suescun from Cryssmat-Lab/DETEMA for the single crystal X-ray diffraction structure determination and data deposition. VM is recipient of a fellowship from Comisión Académica de Posgrado (CAP-Udelar). We thank Diamond Light Source for beamtime (proposal MX17212), and the staff of beamlines I03 and I04 for assistance with crystal testing and data collection. Funding for R. S. was provided by the University of Bristol. This work was supported by grant PICT-2016-1657 from ANPCyT to A. J. V. C. B., D. M. M. and A. J. V. are staff members from CONICET. M. A. R. is recipient of a fellowship from CONICET.Notes and references
- K. Bush and P. A. Bradford, Nat. Rev. Microbiol., 2019, 17, 295–306 CrossRef CAS.
- E. Y. Klein, T. P. Van Boeckel, E. M. Martinez, S. Pant, S. Gandra, S. A. Levin, H. Goossens and R. Laxminarayan, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E3463–E3470 CrossRef CAS.
- P. Nordmann, L. Dortet and L. Poirel, Trends Mol. Med., 2012, 18, 263–272 CrossRef CAS.
- S. Reardon, Science, 2020 DOI:10.1126/science.abc2995.
- Antimicrobial resistance in the age of COVID-19, Nat. Microbiol., 2020, 5, 779 Search PubMed.
- K. M. Papp-Wallace, A. Endimiani, M. A. Taracila and R. A. Bonomo, Antimicrob. Agents Chemother., 2011, 55, 4943–4960 CrossRef CAS.
- C. L. Tooke, P. Hinchliffe, E. C. Bragginton, C. K. Colenso, V. H. A. Hirvonen, Y. Takebayashi and J. Spencer, J. Mol. Biol., 2019, 431, 3472–3500 CrossRef CAS.
- M. S. Wilke, A. L. Lovering and N. C. J. Strynadka, Curr. Opin. Microbiol., 2005, 8, 525–533 CrossRef CAS.
- T. Palzkill, Ann. N. Y. Acad. Sci., 2013, 1277, 91–104 CrossRef CAS.
- M. W. Crowder, J. Spencer and A. J. Vila, Acc. Chem. Res., 2006, 39, 721–728 CrossRef CAS.
- C. Bebrone, Biochem. Pharmacol., 2007, 74, 1686–1701 CrossRef CAS.
- M.-R. R. Meini, L. I. Llarrull and A. J. Vila, FEBS Lett., 2015, 589, 3419–3432 CrossRef CAS.
- CDC, Antibiotic Resistance Threats in the United States, U.S. Department of Health and Human Services, CDC, Atlanta, GA, 2019 Search PubMed.
- E. Tacconelli, E. Carrara, A. Savoldi, S. Harbarth, M. Mendelson, D. L. Monnet, C. Pulcini, G. Kahlmeter, J. Kluytmans, Y. Carmeli, M. Ouellette, K. Outterson, J. Patel, M. Cavaleri, E. M. Cox, C. R. Houchens, M. L. Grayson, P. Hansen, N. Singh, U. Theuretzbacher, N. Magrini, A. O. Aboderin, S. S. Al-Abri, N. Awang Jalil, N. Benzonana, S. Bhattacharya, A. J. Brink, F. R. Burkert, O. Cars, G. Cornaglia, O. J. Dyar, A. W. Friedrich, A. C. Gales, S. Gandra, C. G. Giske, D. A. Goff, H. Goossens, T. Gottlieb, M. Guzman Blanco, W. Hryniewicz, D. Kattula, T. Jinks, S. S. Kanj, L. Kerr, M. P. Kieny, Y. S. Kim, R. S. Kozlov, J. Labarca, R. Laxminarayan, K. Leder, L. Leibovici, G. Levy-Hara, J. Littman, S. Malhotra-Kumar, V. Manchanda, L. Moja, B. Ndoye, A. Pan, D. L. Paterson, M. Paul, H. Qiu, P. Ramon-Pardo, J. Rodríguez-Baño, M. Sanguinetti, S. Sengupta, M. Sharland, M. Si-Mehand, L. L. Silver, W. Song, M. Steinbakk, J. Thomsen, G. E. Thwaites, J. W. van der Meer, N. Van Kinh, S. Vega, M. V. Villegas, A. Wechsler-Fördös, H. F. L. Wertheim, E. Wesangula, N. Woodford, F. O. Yilmaz and A. Zorzet, Lancet Infect. Dis., 2018, 18, 318–327 CrossRef.
- K. Bush, J. Infect. Chemother., 2013, 19, 549–559 CrossRef CAS.
- M. R. Meini, L. I. Llarrull and A. J. Vila, Antibiotics, 2014, 3, 285–316 CrossRef.
- M. F. Mojica, R. A. Bonomo and W. Fast, Curr. Drug Targets, 2016, 17, 1029–1050 CrossRef CAS.
- C. M. Rotondo and G. D. Wright, Curr. Opin. Microbiol., 2017, 39, 96–105 CrossRef CAS.
- A. R. Palacios, M.-A. Rossi, G. S. Mahler and A. J. Vila, Biomolecules, 2020, 10, 854 CrossRef CAS.
- R. Wang, T. P. Lai, P. Gao, H. Zhang, P. L. Ho, P. C. Y. Woo, G. Ma, R. Y. T. Kao, H. Li and H. Sun, Nat. Commun., 2018, 9, 439 CrossRef.
- A. M. King, S. A. Reid-yu, W. Wang, D. T. King, G. De Pascale, N. C. Strynadka, T. R. Walsh, B. K. Coombes and G. D. Wright, Nature, 2014, 510, 503–506 CrossRef CAS.
- M.-N. Lisa, A. R. Palacios, M. Aitha, M. M. González, D. M. Moreno, M. W. Crowder, R. A. Bonomo, J. Spencer, D. L. Tierney, L. I. Llarrull and A. J. Vila, Nat. Commun., 2017, 8, 538 CrossRef.
- M. M. González, M. Kosmopoulou, M. F. Mojica, V. Castillo, P. Hinchliffe, I. Pettinati, J. Brem, C. J. Schofield, G. Mahler, R. A. Bonomo, L. I. Llarrull, J. Spencer and A. J. Vila, ACS Infect. Dis., 2016, 1, 544–554 CrossRef.
- M. F. Mojica, S. G. Mahler, C. R. Bethel, M. A. Taracila, M. Kosmopoulou, K. M. Papp-Wallace, L. I. Llarrull, B. M. Wilson, S. H. Marshall, C. J. Wallace, M. V. Villegas, M. E. Harris, A. J. Vila, J. Spencer and R. A. Bonomo, Biochemistry, 2015, 54, 3183–3196 CrossRef CAS.
- P. Hinchliffe, M. M. González, M. F. Mojica, J. M. González, V. Castillo, C. Saiz, M. Kosmopoulou, C. L. Tooke, L. I. Llarrull, G. Mahler, R. A. Bonomo, A. J. Vila and J. Spencer, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E3745–E3754 CrossRef CAS.
- D. T. King, L. J. Worrall, R. J. Gruninger and N. C. J. J. Strynadka, J. Am. Chem. Soc., 2012, 134, 11362–11365 CrossRef CAS.
- D. W. Cushman and M. A. Ondetti, Nat. Med., 1999, 5, 1110–1112 CrossRef CAS.
- I. García-Sáez, P. S. Mercuri, C. Papamicael, R. Kahn, J. M. Frère, M. Galleni, G. M. Rossolini and O. Dideberg, J. Mol. Biol., 2003, 325, 651–660 CrossRef.
- U. Heinz, R. Bauer, S. Wommer, W. Meyer-Klaucke, C. Papamichaels, J. Bateson and H.-W. Adolph, J. Biol. Chem., 2003, 278, 20659–20666 CrossRef CAS.
- J. Brem, S. S. Van Berkel, D. Zollman, S. Y. Lee, O. Gileadi, P. J. McHugh, T. R. Walsh, M. A. McDonough and C. J. Schofield, Antimicrob. Agents Chemother., 2016, 60, 142–150 CrossRef CAS.
- Y. Yusof, D. T. C. Tan, O. K. Arjomandi, G. Schenk and R. P. McGeary, Bioorg. Med. Chem. Lett., 2016, 26, 1589–1593 CrossRef CAS.
- L. Feng, K.-W. Yang, L.-S. Zhou, J.-M. Xiao, X. Yang, L. Zhai, Y.-L. Zhang and M. W. Crowder, Bioorg. Med. Chem. Lett., 2012, 22, 5185–5189 CrossRef CAS.
- N. Sahiba, A. Sethiya, J. Soni, D. K. Agarwal and S. Agarwal, Top. Curr. Chem., 2020, 378, 34 CrossRef CAS.
- H. T. Nagasawa, D. J. W. W. Goon, D. L. Crankshaw, R. Vince and S. E. Patterson, J. Med. Chem., 2007, 50, 6462–6464 CrossRef CAS.
- J. Ma, S. McLeod, K. MacCormack, S. Sriram, N. Gao, A. L. Breeze and J. Hu, Angew. Chem., Int. Ed. Engl., 2014, 53, 2130–2133 CrossRef CAS.
- O. S. Smart, V. Horský, S. Gore, R. S. Vařeková, V. Bendová, G. J. Kleywegt and S. Velankar, Acta Crystallogr., Sect. D: Struct. Biol., 2018, 74, 228–236 CrossRef CAS.
- M. C. Deller and B. Rupp, J. Comput.-Aided Mol. Des., 2015, 29, 817–836 CrossRef CAS.
- J. Spencer, J. Read, R. B. Sessions, S. Howell, A. G. Michael Blackburn and S. J. Gamblin, J. Am. Chem. Soc., 2005, 127, 14439–14444 CrossRef CAS.
- H. Zhang, G. Ma, Y. Zhu, L. Zeng, A. Ahmad, C. Wang, B. Pang, H. Fang, L. Zhao and Q. Hao, Antimicrob. Agents Chemother., 2018, 62, e01579 Search PubMed.
- M. Aitha, A. R. Marts, A. Bergstrom, A. J. Møller, L. Moritz, L. Turner, J. C. Nix, R. A. Bonomo, R. C. Page, D. L. Tierney and M. W. Crowder, Biochemistry, 2014, 53, 7321–7331 CrossRef CAS.
- H. Feng, X. Liu, S. Wang, J. Fleming, D. C. Wang and W. Liu, Nat. Commun., 2017, 8, 2242 CrossRef.
- J. E. Raczynska, I. G. Shabalin, W. Minor, A. Wlodawer and M. Jaskolski, Drug Resist. Updates, 2018, 40, 1–12 CrossRef.
- H. Zhang and Q. Hao, FASEB J., 2011, 25, 2574–2582 CrossRef CAS.
- E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210–1250 CrossRef CAS.
- B. R. Beno, K. S. Yeung, M. D. Bartberger, L. D. Pennington and N. A. Meanwell, J. Med. Chem., 2015, 58, 4383–4438 CrossRef CAS.
- A. L. Ringer, A. Senenko and C. D. Sherrill, Protein Sci., 2007, 16, 2216–2223 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials and methods for protein preparation, inhibitory measurements in vitro and in-cell, antimicrobial susceptibility testing, cell toxicity and protein crystallization and structure determination. X-ray data collection and structure refinement data, NMR spectra of the synthesized inhibitors, and additional figures. CCDC 2017238. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc05172a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |