
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMangana(III/IV)electro-catalyzed C(sp3)–H azidation†
Tjark H.
Meyer‡
 ab,
Ramesh C.
Samanta‡
ab,
Antonio
Del Vecchio
a and
Lutz
Ackermann
*ab
ab,
Ramesh C.
Samanta‡
ab,
Antonio
Del Vecchio
a and
Lutz
Ackermann
*ab
aInstitut für Organische und Biomolekulare Chemie, Georg-August-Universität Göttingen, Tammannstraße 2, 37077 Göttingen, Germany
bWoehler Research Institute for Sustainable Chemistry (WISCh), Georg-August-Universität Göttingen, Tammannstraße 2, 37077 Göttingen, Germany
First published on 28th December 2020
Abstract
Manganaelectro-catalyzed azidation of otherwise inert C(sp3)–H bonds was accomplished using most user-friendly sodium azide as the nitrogen-source. The operationally simple, resource-economic C–H azidation strategy was characterized by mild reaction conditions, no directing group, traceless electrons as the sole redox-reagent, Earth-abundant manganese as the catalyst, high functional-group compatibility and high chemoselectivity, setting the stage for late-stage azidation of bioactive compounds. Detailed mechanistic studies by experiment, spectrophotometry and cyclic voltammetry provided strong support for metal-catalyzed aliphatic radical formation, along with subsequent azidyl radical transfer within a manganese(III/IV) manifold.
Introduction
The development of selective, resource-economic methods for the direct conversion of otherwise unreactive C(sp3)–H bonds is one of the most demanding challenges in molecular sciences.1 Advances in this field will not only be of direct importance to petrochemical and polymer technologies,2 but will also enable step-economical syntheses of pharmaceutical relevant compounds3 by ideally late-stage drug diversification.4 Within this topical research arena, concepts for direct C(sp3)–H aminations are particularly prominent.5 Organic azides6 are key structural motifs, that are often key-intermediates in numerous molecules of interest to medicinal chemistry,7 materials sciences,8 peptide chemistry or molecular biology.9 Due to this key importance,10 a plethora of functional group interconversion strategies have been developed, exploiting inter alia organic halides, alcohols, epoxides, and aldehydes. However, more step-economical methods that directly install the azido-group into otherwise inert C(sp3)–H bonds continue to be scarce11 and generally require stoichiometric amounts of strong indiscriminate chemical oxidants, such as persulfates,12N-fluorobenzenesulfonimide (NFSI),13 hypervalent iodine reagents14 or photochemical15 irradiation.16 In previous reports on selective electrochemical vicinal diazidation of olefins, a metal-free approach was introduced by Schäfer in 1970 (Scheme 1a).17 Subsequently, Lin significantly improved the efficacy and selectivities upon the addition of a metal catalyst, that is able to mediate the transfer of an azidyl radical to the double bond.18 Here, particularly a manganese(II) pre-catalyst served as an efficient azide transfer reagent via a manganese(II/III) manifold.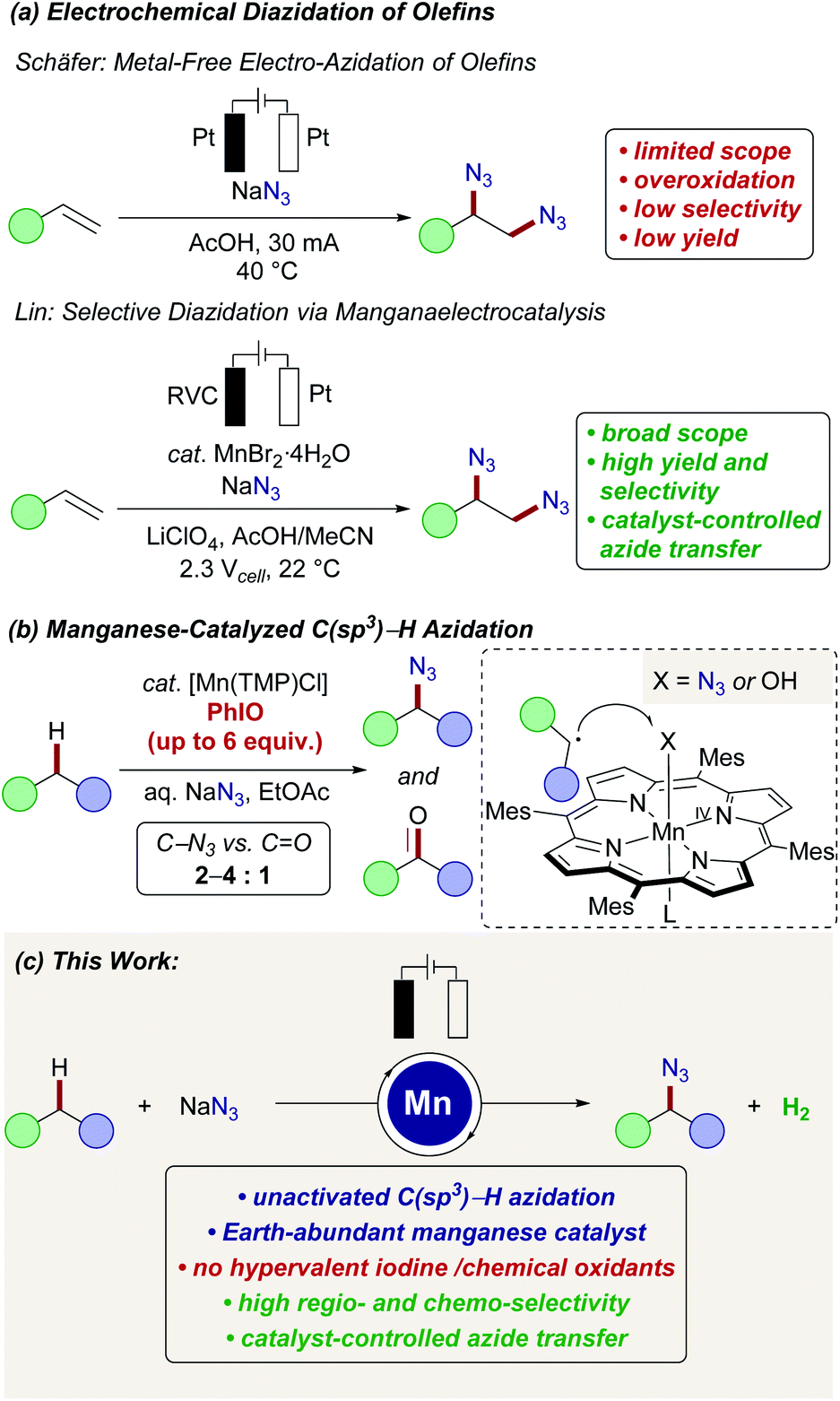 | ||
| Scheme 1 Manganaelectro-catalyzed azidation. | ||
Despite numerous reports on C(sp2)–H transformations catalyzed by manganese,19 reports on manganese-catalyzed C(sp3)–H functionalization remain underdeveloped.20 For the direct azidation of unactivated C(sp3)–H bonds, early reports by Hill and Groves indeed employed manganese complexes derived from porphyrin or a Schiff-base (Scheme 1b).21 Here, polymeric and potentially explosive iodosylbenzene was however used as the chemical oxidant to generate high-valent manganese(V)-oxo intermediates, which have been reported to undergo hydrogen-abstraction of aliphatic C(sp3)–H bonds to form the desired alkyl radical.22 Thereafter, the thus formed aliphatic radical is proposed to be captured by the Mn(IV)–X intermediate to form either azidated or undesired oxygenated products. Unfortunately, depending on the substrate, the reported azide to oxygen incorporation ratio was relatively low (2–4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).21a
:1).21a
Results and discussion
Within our program on sustainable organic electrosynthesis23 for strong bond activation,24 we envisioned the direct azidation of activated C(sp3)–H bonds via anodically formed azidyl-radicals in aqueous acidic media (Scheme 2).23g,25 The generation of transient azidyl-radicals by anodic oxidation was reported earlier,26 yet, direct electrochemical C(sp3)–H azidations are thus far unprecedented. The azide anion is thus proposed to be oxidized at the graphite-felt (GF) anode and hence results in nitrogen gas formation, while hydrogen gas is formed on the platinum cathode (Scheme 2a). Unfortunately, the direct oxidation of activated benzylic C(sp3)–H (BDE C–H = 82.9 ± 1.2 kcal mol−1,27Ep,a = 2.1 V vs. SCE28) occurred with only poor chemoselectivity, yet with high conversion of 1a (Scheme 2b). This unsatisfactory outcome, was however very promising since the desired organo-azide 2a was formed in 15% yield, motivated us to study envisioned electrochemical C(sp3)–H azidation in further detail.29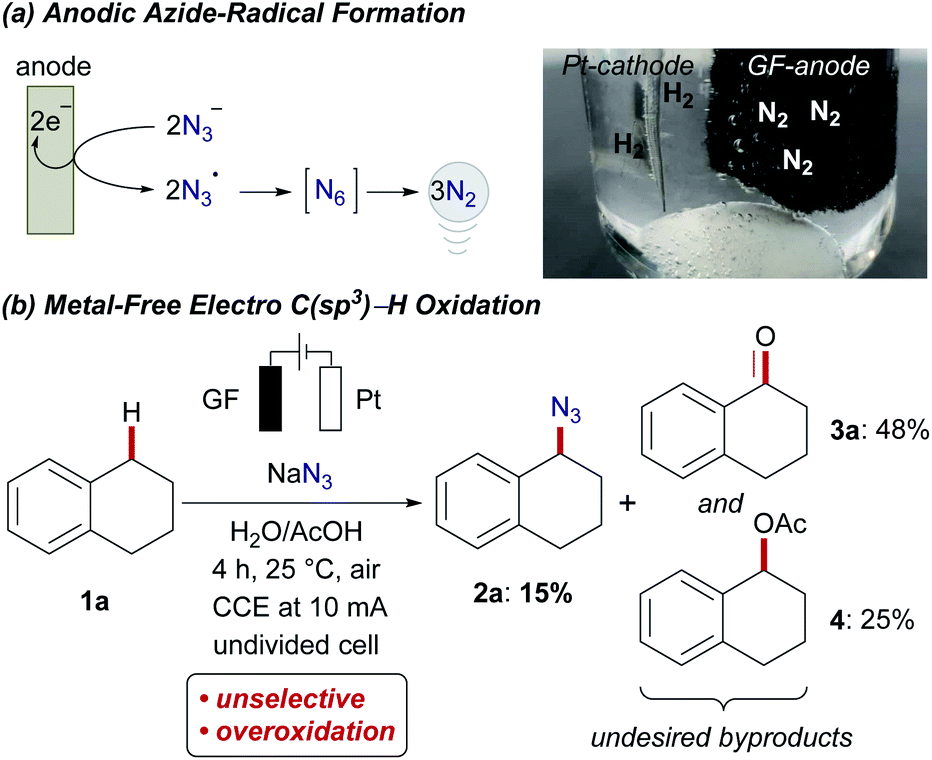 | ||
| Scheme 2 Proof of concept electro C(sp3)–H azidation. | ||
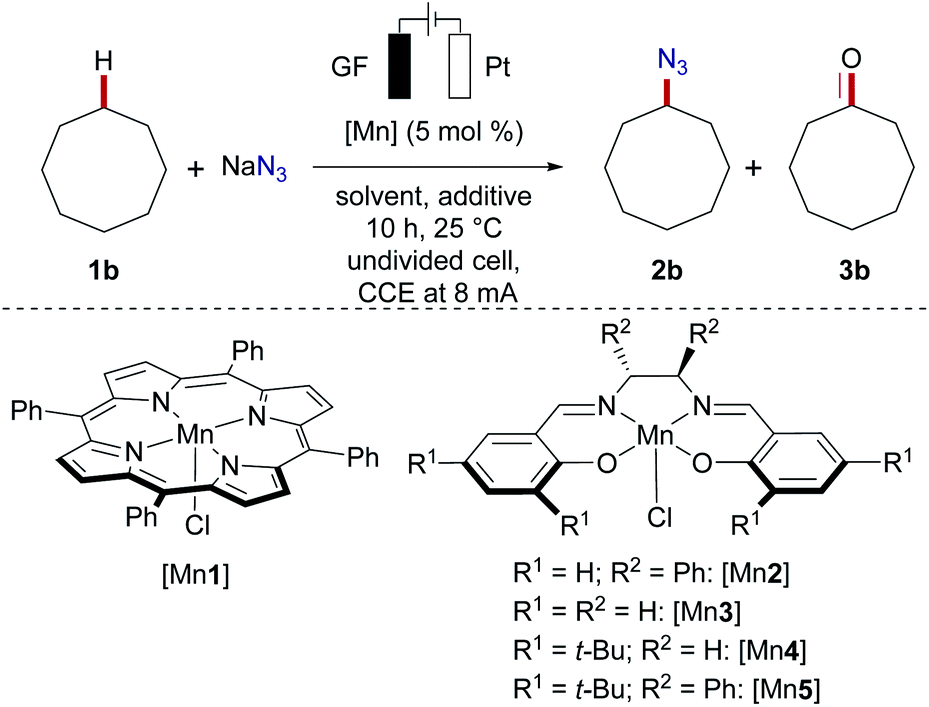
Herein, we report the manganaelectro-catalyzed C(sp3)–H azidation of unactivated secondary, tertiary and benzylic C–H bonds. Key features of our findings include (a) C(sp3)–H azidation via synergistic electrosynthesis and an Earth-abundant manganese catalyst, (b) mild and chemical oxidant-free reaction conditions, (c) high regio- and chemoselectivity, (d) and late-stage azidation of bioactive compounds. It is noteworthy that efficient manganese-catalyzed electro-azidation occurred in the absence of photochemical irradiation,15 with detailed mechanistic studies being supportive of a manganaelectro(III/IV) regime. We initiated our studies by probing various reaction parameters for the envisioned manganaelectro-catalyzed C(sp3)–H azidation of unactivated alkane 1b (BDE C–H: 95.7 kcal mol−1,30Eox ≥ 2.5 V31) (Tables 1 and S-2–S-5 in the ESI†).32
|
|
||||
|---|---|---|---|---|
| Entry | [Mn] | Solvent | Additive (equiv.) | Yield [%] (ratio 2b/3b) |
|
a Reaction conditions: undivided cell, 1b (0.5 mmol), [Mn] (5.0 mol%), NaN3 (8.0 equiv.), an additive (xx equiv.), solvent (1:1, 5 mL), 25 °C, constant current electrolysis (CCE) at 8 mA, a graphite felt (GF) anode, and a Pt-plate cathode. Yields are determined by GC-FID with n-dodecane as the internal standard.
b 5 h, 6 mA, 2.24 F.
c 10 h, 6 mA, 4.48 F.
d Under nitrogen atmosphere.
|
||||
| 1b | [Mn1] | AcOH/H2O | PhI (0.2) | 25 (2.6:1.0) |
| 2b | [Mn2] | AcOH/H2O | PhI (0.2) | 33 (2.0:1.0) |
| 3b | [Mn2] | AcOH/H2O | — | 30 (2.8:1.0) |
| 4b | [Mn2] | AcOH/MeCN | LiClO4 (1.0) | 31 (5.2:1.0) |
| 5c,d | [Mn2] | AcOH/MeCN | LiClO4 (1.0) | 40 (12.3:1.0) |
| 6d | [Mn2] | AcOH/MeCN | LiClO4 (1.0) | 45 (10.0:1.0) |
| Deviation from entry 6 | ||
|---|---|---|
| 7 | [Mn1] instead of [Mn2] | 44 (1.6:1.0) |
| 8 | [Mn3] instead of [Mn2] | 10 (4.0:1.0) |
| 9 | [Mn4] instead of [Mn2] | 39 (12.0:1.0) |
| 10 | [Mn5] instead of [Mn2] | 47 (8.4:1.0) |
| 11 | KN3 instead of NaN3 | 44 (5.3:1.0) |
| 12 | TMSN3 (4.0 equiv.) instead of NaN3 | 30 (8.0:1.0) |
| 13 | TsN3 (4.0 equiv.) instead of NaN3 | 0 |
| 14 | No current: under a N2 or air atmosphere | 0 |
| 15 | No [Mn] | 10 (2.0:1.0) |
| 16 | Light irradiation with blue LEDs | 30 (1.0:1.0) |
| 17 | In the dark | 42 (10.0:1.0) |
Preliminary optimization demonstrated that C–H azidation to afford 2b could indeed be achieved with a manganese(III) porphyrin33 or salen complex at 25 °C in acetic acid/H2O mixtures, under air, albeit with significant amounts of overoxidized ketone 3b as the side product (entries 1–3). After considerable experimentation, we found that the desired electrochemical C–H azidation was accomplished with high chemoselectivity and efficacy (5.97 F per mole 1b) when a mixture of MeCN/AcOH was used as the reaction medium under a N2 atmosphere (entries 4–6). Variation of the manganese catalyst did not improve the catalytic performance (entries 7–10). Nucleophilic azide sources proved to be beneficial (entries 11–13). Likewise, control experiments revealed the key role of electricity, as well as the importance of the high surface area of a carbon-based anode material (entry 14).32 The absence of the manganese catalyst resulted in a major nitrogen gas formation at the anode, translating into significantly reduced yield and chemoselectivity (entry 15). Light irradiation resulted in rather unselective product mixtures and a diminished yield (entry 16). However, the reaction efficacy remained unaffected under dark conditions (17).32
With the optimized manganese catalyst in hand, we evaluated manganaelectro-catalyzed C(sp3)–H azidation for benzylic, secondary and tertiary C–H bonds (Scheme 3). Azidation occurred in moderate to excellent yields with high selectivity for tertiary over secondary or primary C–H bonds. We also observed an unprecedented chemoselectivity for manganese-catalyzed C(sp3)–H azidation in terms of the azidation to oxygenation ratio. Functional groups including silyloxy, amides, ethers, esters, enolizable ketones and nitriles, were well tolerated, while the mass balance accounted for unreacted substrates 1. Cyclic, secondary hydrocarbons provided the organic azide in moderate yields 2b and 2c (32–40%). When multiple reactive sites were present, C–H azidation inherently took place at the tertiary C–H bond over secondary or primary C–H scission. Amide 1d and dihydrocitronellol derivatives 1e and 1f afforded the desired azidated products 2d–2f likewise, with good selectivity for the tertiary C–H bond. Leucine derivative 1g reacted efficiently under exceedingly mild reaction conditions. Similar to aliphatic substrates, the manganaelectro-catalyzed C(sp3)–H azidation of benzylic substrates preferentially selected tertiary over primary or secondary C–H bonds. Generally, electron-donating substituents facilitated C–H azidation; however, longer reaction times proved viable to convert electron-deficient substrates. Isobutylbenzene 1x and isopentylbenzene 1y containing both secondary benzylic and tertiary C–H bonds were next selected to examine site selectivity. Here, a preference for the benzylic C–H bond was detected, with a benzylic to tertiary ratio of about 3:1. The reaction of p-cymene 1z almost exclusively delivered azidation of tertiary C–H, highlighting the strong influence of C–H bond dissociation energies (BDE) over steric effects (2x–2z). Subsequently, we probed the manganaelectro-catalyzed C–H azidation of complex molecules and for late-stage functionalization of pharmaceutically active molecules 5.14a Biaryl 5a, an analogue of a retinoic acid receptor agonist34 was effectively converted into the corresponding azide 6a. Ibuprofen methyl ester 5b preferentially reacted at the secondary benzylic position (2.3:1, 42% yield), surprisingly without azidation of the tertiary C–H bond adjacent to the ester group. Azidation of celestolide 5c proceeded efficiently and afforded 6c in high yield (65%). Functionalization of acetyl(−)-menthol 5d, containing four tertiary C(sp3)–H bonds, afforded azide 6d, favoring the sterically more accessible isopropyl sidechain. Estrone acetate 5e was transformed into the desired azide 6e in 75% yield as a diastereomeric mixture. This result is supportive of a radical pathway, compared to previous reports where the proposed formation of benzylic carbocation resulted in different diastereomeric ratios.14a The user-friendly nature of our manganaelectro-catalyzed C(sp3)–H azidation was further illustrated by a gram-scale preparation of azidated product 2n with similar levels of efficiency (Scheme 4).
 | ||
| Scheme 3 Reaction scope of manganaelectro-catalyzed C(sp3)–H azidation. [a] Standard conditions: substrate (0.5 mmol), NaN3 (4.0 mmol), [Mn2] (2.5 or 5.0 mol%), LiClO4 (0.5 mmol), MeCN/AcOH (5 mL, 1:1), a nitrogen atmosphere, 10 h, 25 °C, and constant current electrolysis at 8.0 mA in an undivided cell. All yields are isolated products; ratios for site-selectivity are determined by 1H-NMR of the crude mixture. [b] At 50 °C. [c] 3,4-Dihydronaphthalen-1(2H)-one 3 was detected in 10% from crude 1H-NMR with 1,3,5-trimethoxy benzene as the internal standard. [d] Standard conditions for 20 h.32 | ||
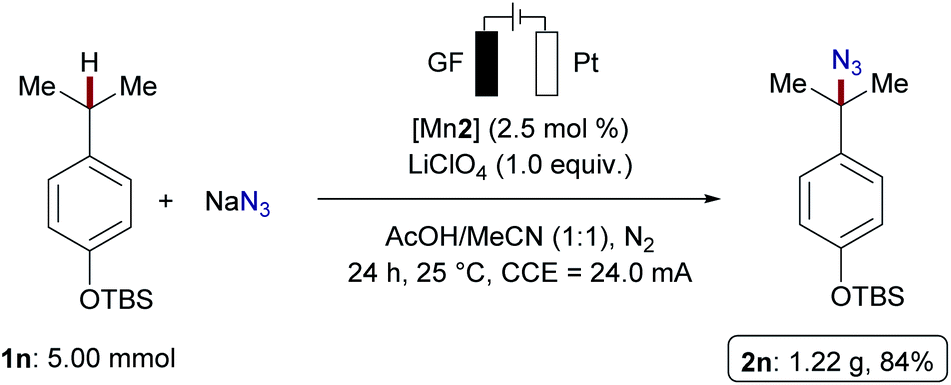 | ||
| Scheme 4 Gram-scale manganaelectro-catalyzed C–H azidation (4.30 F). | ||
Given the versatility of manganaelectro-catalyzed C–H azidation, we became attracted to probe its mode of action (Scheme 5). Upon the addition of persistent radical sources, such as TEMPO ((2,2,6,6-tetramethylpiperidin-1-yl)oxyl) or BHT (2,6-di-tert-butyl-4-methylphenol) the reaction was completely inhibited and no desired product 2r could be detected (Scheme 5a). Intermolecular competition reactions of 1o and 1q confirmed the general trend that electron-rich substrates reacted preferentially (Scheme 5b). Independent rate measurements revealed a significant kinetic isotope effect (KIE) of kH/kD = 3.0 (Scheme 5c). To unambiguously identify the involvement of a trans-diazidomanganese(IV) complex as the active catalyst for manganaelectro-catalyzed C–H azidation, the novel complexes Mn5(III)–N3 and Mn5(IV)–(N3)2 were prepared and fully characterized.32 Initially, Mn5(III)–N3 was synthesized by treating the corresponding chloride complex [Mn5] with sodium azide. Subsequently, the thus derived manganese(III) complex was oxidized to manganese(IV) in the presence of an excess of sodium azide to afford Mn5(IV)–(N3)2 (Scheme 5e). With the well-defined complexes in hand, spectrophotometric and electroanalytic studies were performed (Schemes 5f, 6 and Fig. S-2–S-17†).32 UV-vis spectroscopy of [Mn5], a mixture of [Mn5] and sodium azide, and the well-defined Mn5(III)–N3 complex resulted in similar spectra with characteristic absorption maxima at 244, 328, and 440 nm (Scheme 5f(i)). In comparison, Mn5(IV)–(N3)2 revealed significantly different absorption properties with maxima at 277, 342, 442, and 642 nm. The strong increased absorption at 442 nm can be assigned to the charge transfer bands from coordinating azide to the manganese(IV) metal center.35
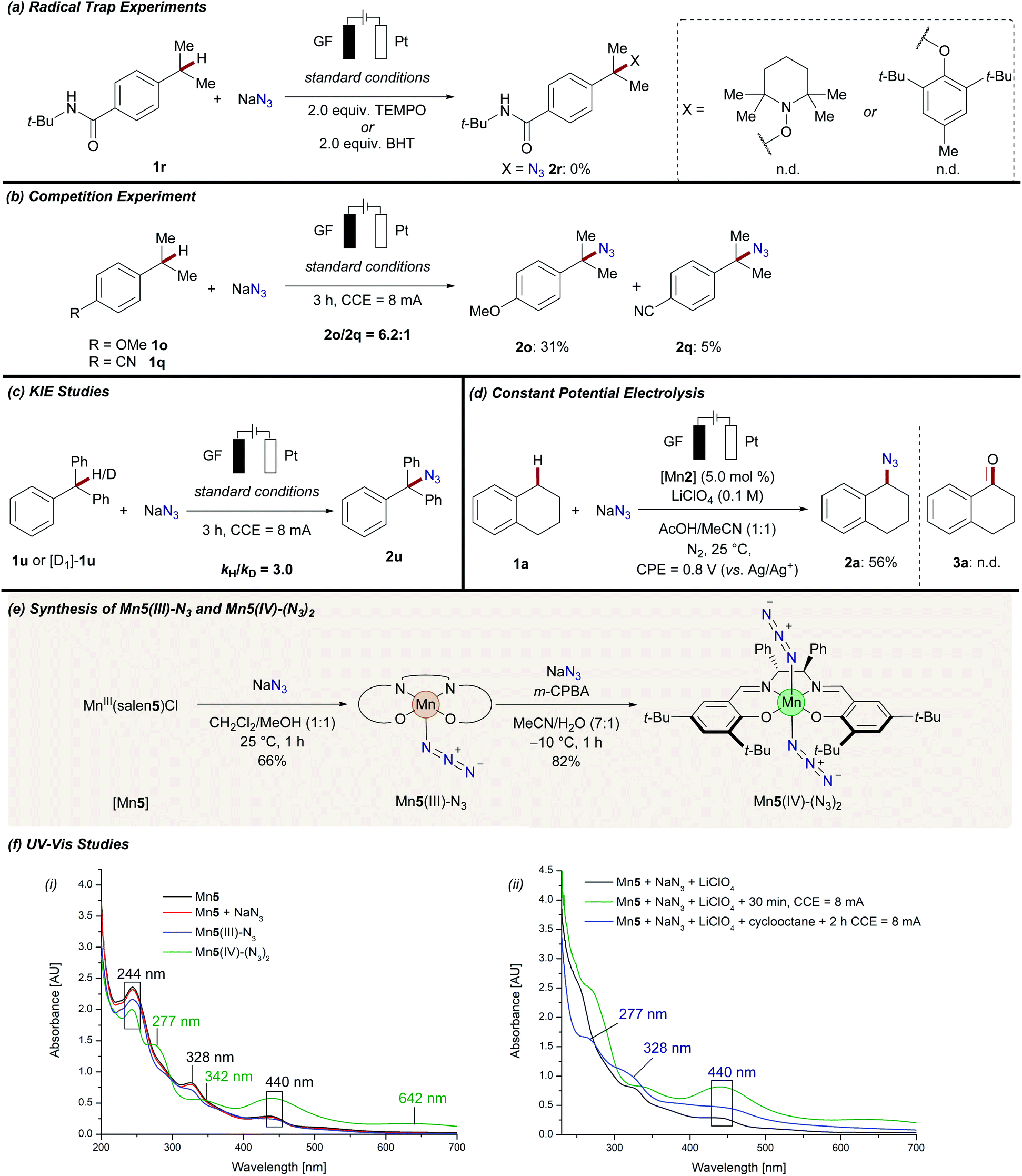 | ||
| Scheme 5 Mechanistic investigation of manganaelectro-catalyzed azidation of unactivated C(sp3)–H bonds. (a) Radical trap experiments. (b) Competition experiment. (c) Kinetic experiments. (d) Chronoamperometric studies at 0.8 V vs. Ag/Ag+. (e) Stoichiometric synthesis of well-defined manganese(III) azide and manganese(IV) diazide complexes. (f) UV-vis spectroscopic studies: (i) MeCN (0.04 mM) was used as the solvent at 25 °C. (ii) MeCN/AcOH (1:1, 0.05 mM) was used as the solvent at 25 °C. | ||
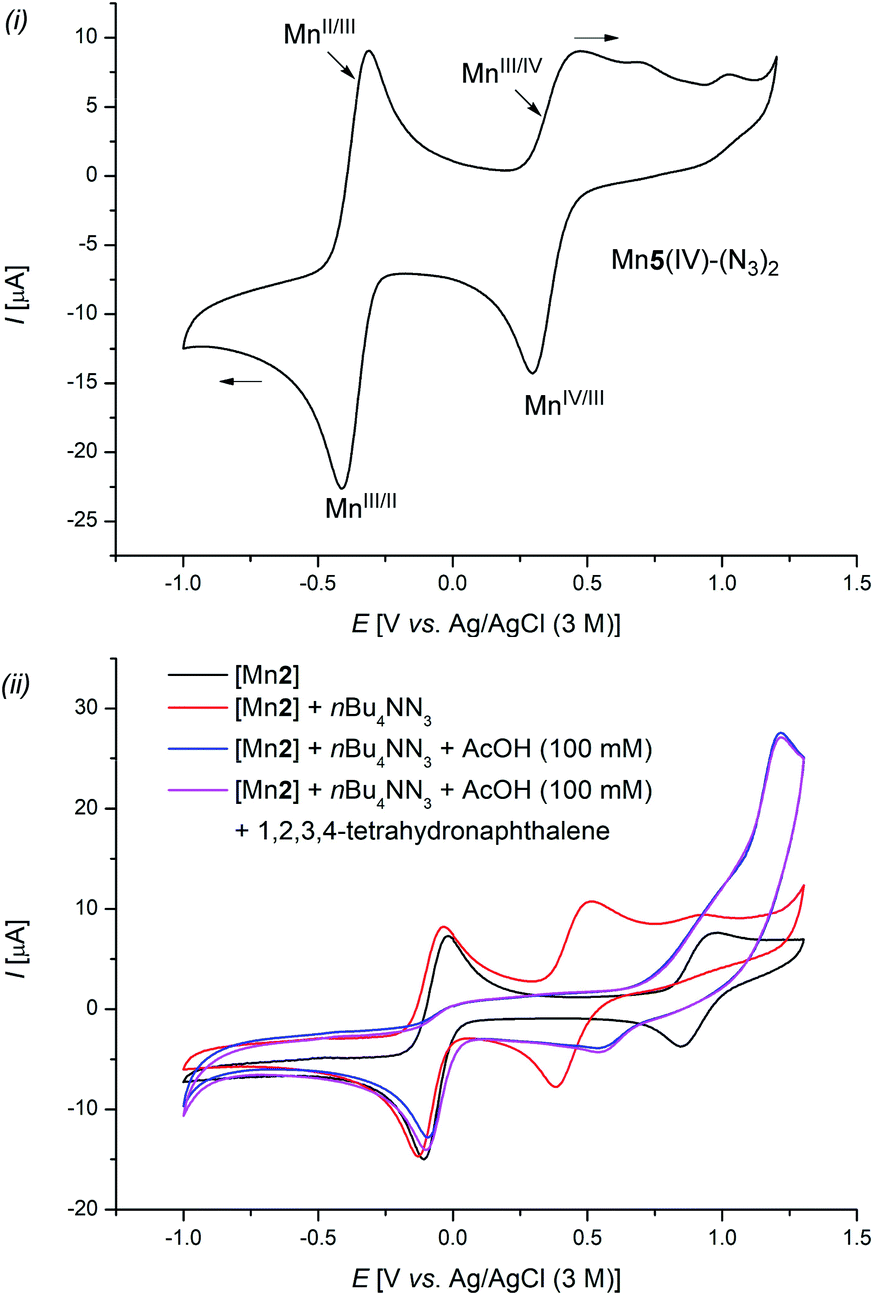 | ||
| Scheme 6 Cyclic voltammetry: MeCN (0.3 mm) and LiClO4 (0.1 m) was used as the electrolyte and Ag/AgCl (3 M) as the reference electrode, at 25 °C and a scanning rate of 100 mV s−1. | ||
Similar absorption maxima were detected when a mixture of [Mn5], NaN3, and LiClO4 in a MeCN/AcOH mixture was anodically oxidized for 30 min at 8 mA (Scheme 5f(ii), green line). However, when the crude reaction mixture was analyzed, mixed absorption maxima of both species, manganese(III) (328 nm) and manganese(IV) (277, and 440 nm), were detected, giving strong support for the catalytic formation and consumption of Mn5(IV)–(N3)2 (Scheme 5f(ii), blue line). These results were further supported by detailed cyclic voltammetric (CV) studies (Scheme 6). The well-defined complex Mn5(IV)–(N3)2 showed two characteristic redox-events at E1/2 = −0.37 V (vs. Ag/AgCl), which can be assigned to the Mn(II/III) redox couple and E1/2 = +0.37 V (vs. Ag/AgCl) for the formal Mn(III/IV) oxidation (Scheme 6(i)).36 Complex Mn5(III)–N3 showed similar values (Fig. S-5–S-7†).32 In stark contrast, a solution of the azide anion in MeCN showed irreversible oxidation at Ep,a = +0.89 V (vs. Ag/AgCl), which shifted to Ep,a = +1.45 V (vs. Ag/AgCl) when AcOH was added (Fig. S-3†). This was likely due to a protonation, which was substantially higher in potential, compared to the manganese-complexes. Mixtures of the manganese chloride complex with tetra-n-butylammonium azide showed comparable redox properties with the well-defined manganese azide complexes (Scheme 6(ii)).32 Finally, chronoamperometric experiments revealed selective product formations of tetralin 1a to the corresponding azide product 2a, at potentials as low as 0.8 V vs. Ag/Ag+, without the formation of oxygenated side-products 3a (Scheme 5d). This gives strong support that the manganese catalyst is involved in both steps of the reaction.
Based on our mechanistic studies, we propose the catalytic cycle to be initiated by facile ligand-exchange to form the active Mn(III)–N3 complex, followed by anodic oxidation to generate the manganese(IV) diazide complex (Fig. 1). The high-valent d3 manganese(IV) complex is prone35b to undergo hydrogen-atom-transfer (HAT)37 with the substrate, thus generating the aliphatic radical. Subsequently, the key C–N3 bond is formed via manganese-catalyzed azide radical transfer, yet alternative azide transfer scenarios, such as radical-polar crossover, can at this point not be fully ruled out.
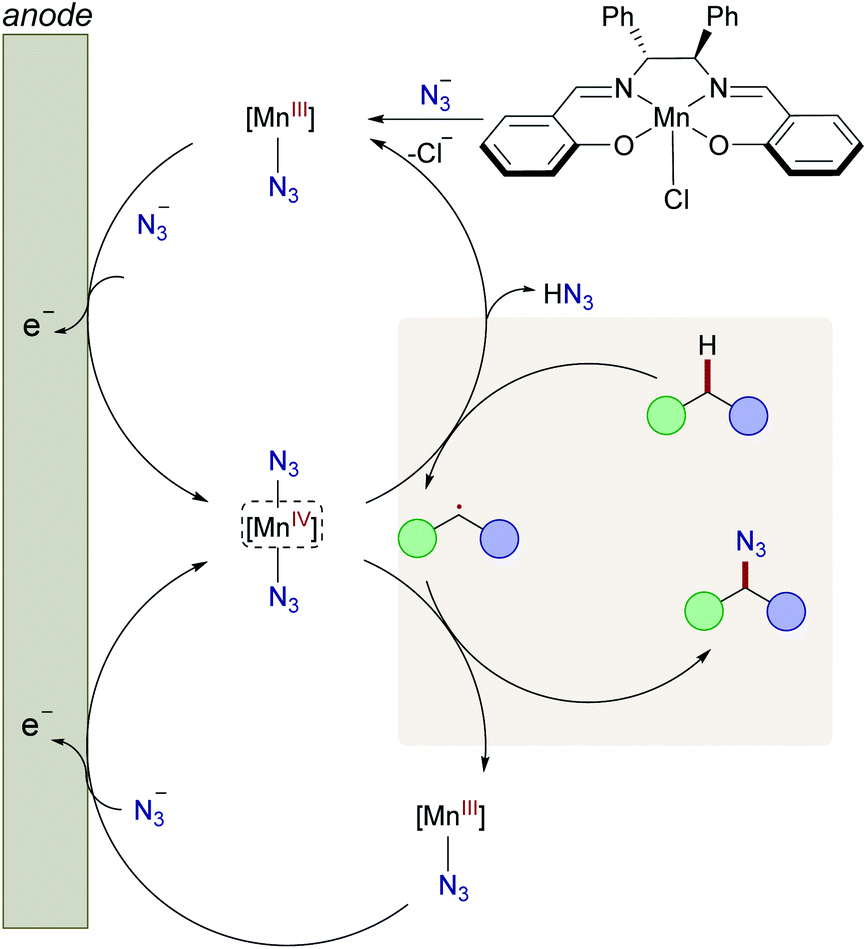 | ||
| Fig. 1 Proposed mechanism for manganaelectro-catalyzed C(sp3)–H azidation. | ||
Conclusions
In conclusion, we have reported the unprecedented manganese-electrocatalyzed C–H azidation of otherwise inert C(sp3)–H bonds devoid of chemical oxidants, hypervalent iodine reagents or photochemical irradiation. Detailed mechanistic studies by experiment, spectrophotometry and cyclic voltammetry are supportive of an unique manganese(III/IV) electrocatalysis, thus avoiding overoxidation to the carbocation, while minimizing undesired side-reactions to oxygenated products. The robustness and utility of the most user-friendly method was highlighted by azidation of bioactive and pharmaceutically-relevant compounds by late-stage diversification.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Generous support by the DFG (Gottfried-Wilhelm-Leibniz award to L. A.) and the Alexander von Humboldt Foundation (fellowship to R. C. S.) is gratefully acknowledged.Notes and references
- (a) S. M. Khake and N. Chatani, Trends Chem., 2019, 1, 524 CrossRef CAS; (b) P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192 CrossRef CAS; (c) J. C. K. Chu and T. Rovis, Angew. Chem., Int. Ed., 2018, 57, 62 CrossRef CAS; (d) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754 CrossRef CAS; (e) J. F. Hartwig and M. A. Larsen, ACS Cent. Sci., 2016, 2, 281 CrossRef CAS; (f) M. C. White, Science, 2012, 335, 807 CrossRef CAS; (g) T. Brückl, R. D. Baxter, Y. Ishihara and P. S. Baran, Acc. Chem. Res., 2012, 45, 826 CrossRef; (h) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS; (i) T. Satoh and M. Miura, Chem.–Eur. J., 2010, 16, 11212 CrossRef CAS; (j) R. G. Bergman, Nature, 2007, 446, 391 CrossRef CAS; (k) J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507 CrossRef CAS.
- (a) G. Laudadio, Y. Deng, K. van der Wal, D. Ravelli, M. Nuño, M. Fagnoni, D. Guthrie, Y. Sun and T. Noël, Science, 2020, 369, 92 CrossRef CAS; (b) A. Bunescu, S. Lee, Q. Li and J. F. Hartwig, ACS Cent. Sci., 2017, 3, 895 CrossRef CAS; (c) J.-R. Pouliot, F. Grenier, J. T. Blaskovits, S. Beaupré and M. Leclerc, Chem. Rev., 2016, 116, 14225 CrossRef CAS; (d) D. J. Schipper and K. Fagnou, Chem. Mater., 2011, 23, 1594 CrossRef CAS.
- (a) O. Baudoin, Angew. Chem., Int. Ed., 2020, 59, 17798 CrossRef CAS; (b) R. Hili and A. K. Yudin, Nat. Chem. Biol., 2006, 2, 284 CrossRef CAS.
- (a) J. Börgel and T. Ritter, Chem, 2020, 6, 1877 CrossRef; (b) F. Wu, J. Zhang, F. Song, S. Wang, H. Guo, Q. Wei, H. Dai, X. Chen, X. Xia, X. Liu, L. Zhang, J.-Q. Yu and X. Lei, ACS Cent. Sci., 2020, 6, 928 CrossRef CAS; (c) W. Wang, M. M. Lorion, J. Shah, A. R. Kapdi and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 14700 CrossRef CAS; (d) T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546 RSC; (e) A. F. M. Noisier and M. A. Brimble, Chem. Rev., 2014, 114, 8775 CrossRef CAS.
- For selected examples, see: (a) R. Singh and A. Mukherjee, ACS Catal., 2019, 9, 3604 CrossRef CAS; (b) J. R. Clark, K. Feng, A. Sookezian and M. C. White, Nat. Chem., 2018, 10, 583 CrossRef CAS; (c) S. M. Paradine, J. R. Griffin, J. Zhao, A. L. Petronico, S. M. Miller and M. Christina White, Nat. Chem., 2015, 7, 987 CrossRef CAS; (d) A. McNally, B. Haffemayer, B. S. L. Collins and M. J. Gaunt, Nature, 2014, 510, 129 CrossRef CAS; (e) J. L. Jeffrey and R. Sarpong, Chem. Sci., 2013, 4, 4092 RSC; (f) J. L. Roizen, M. E. Harvey and J. Du Bois, Acc. Chem. Res., 2012, 45, 911 CrossRef CAS; (g) F. Collet, R. H. Dodd and P. Dauban, Chem. Commun., 2009, 5061 RSC; (h) H. M. L. Davies and M. S. Long, Angew. Chem., Int. Ed., 2005, 44, 3518 CrossRef CAS; (i) X.-Q. Yu, J.-S. Huang, X.-G. Zhou and C.-M. Che, Org. Lett., 2000, 2, 2233 CrossRef CAS.
- For reviews, see: (a) M. Goswami and B. de Bruin, Eur. J. Org. Chem., 2017, 1152 CrossRef CAS; (b) W. Song, S. I. Kozhushkov and L. Ackermann, Angew. Chem., Int. Ed., 2013, 52, 6576 CrossRef CAS; (c) S. Chiba, Synlett, 2012, 2012, 21 CrossRef; (d) N. Jung and S. Bräse, Angew. Chem., Int. Ed., 2012, 51, 12169 CrossRef CAS; (e) S. Bräse, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188 CrossRef; (f) M. Köhn and R. Breinbauer, Angew. Chem., Int. Ed., 2004, 43, 3106 CrossRef.
- (a) P. P. Geurink, W. A. van der Linden, A. C. Mirabella, N. Gallastegui, G. de Bruin, A. E. M. Blom, M. J. Voges, E. D. Mock, B. I. Florea, G. A. van der Marel, C. Driessen, M. van der Stelt, M. Groll, H. S. Overkleeft and A. F. Kisselev, J. Med. Chem., 2013, 56, 1262 CrossRef CAS; (b) D. B. Smith, G. Kalayanov, C. Sund, A. Winqvist, T. Maltseva, V. J. P. Leveque, S. Rajyaguru, S. L. Pogam, I. Najera, K. Benkestock, X.-X. Zhou, A. C. Kaiser, H. Maag, N. Cammack, J. A. Martin, S. Swallow, N. G. Johansson, K. Klumpp and M. Smith, J. Med. Chem., 2009, 52, 2971 CrossRef CAS; (c) G. C. Tron, T. Pirali, R. A. Billington, P. L. Canonico, G. Sorba and A. A. Genazzani, Med. Res. Rev., 2008, 28, 278 CrossRef CAS; (d) A. G. Habeeb, P. N. Praveen Rao and E. E. Knaus, J. Med. Chem., 2001, 44, 3039 CrossRef CAS.
- (a) W. Xi, T. F. Scott, C. J. Kloxin and C. N. Bowman, Adv. Funct. Mater., 2014, 24, 2572 CrossRef CAS; (b) R. Kluger, J. Am. Chem. Soc., 2010, 132, 6611 CrossRef CAS.
- (a) P. Thirumurugan, D. Matosiuk and K. Jozwiak, Chem. Rev., 2013, 113, 4905 CrossRef CAS; (b) L. A. Canalle, D. W. P. M. Löwik and J. C. M. van Hest, Chem. Soc. Rev., 2010, 39, 329 RSC; (c) M. D. Best, Biochemistry, 2009, 48, 6571 CrossRef CAS; (d) D. M. Huryn and M. Okabe, Chem. Rev., 1992, 92, 1745 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- For reviews, see: (a) L. Ge, M.-F. Chiou, Y. Li and H. Bao, Green Synthesis and Catalysis, 2020, 1, 86 CrossRef; (b) P.-G. Ding, X.-S. Hu, F. Zhou and J. Zhou, Org. Chem. Front., 2018, 5, 1542 RSC; (c) X. Huang and J. T. Groves, ACS Catal., 2016, 6, 751 CrossRef CAS.
- X. Zhang, H. Yang and P. Tang, Org. Lett., 2015, 17, 5828 CrossRef CAS.
- S.-E. Suh, S.-J. Chen, M. Mandal, I. A. Guzei, C. J. Cramer and S. S. Stahl, J. Am. Chem. Soc., 2020, 142, 11388 CrossRef CAS.
- (a) R. R. Karimov, A. Sharma and J. F. Hartwig, ACS Cent. Sci., 2016, 2, 715 CrossRef CAS; (b) P. T. G. Rabet, G. Fumagalli, S. Boyd and M. F. Greaney, Org. Lett., 2016, 18, 1646 CrossRef CAS; (c) A. Sharma and J. F. Hartwig, Nature, 2015, 517, 600 CrossRef CAS; (d) C. Viuf and M. Bols, Angew. Chem., Int. Ed., 2001, 40, 623 CrossRef CAS; (e) V. V. Zhdankin, A. P. Krasutsky, C. J. Kuehl, A. J. Simonsen, J. K. Woodward, B. Mismash and J. T. Bolz, J. Am. Chem. Soc., 1996, 118, 5192 CrossRef CAS; (f) P. Magnus, J. Lacour, P. A. Evans, M. B. Roe and C. Hulme, J. Am. Chem. Soc., 1996, 118, 3406 CrossRef CAS; (g) Y. Kita, H. Tohma, T. Takada, S. Mitoh, S. Fujita and M. Gyoten, Synlett, 1994, 1994, 427 CrossRef; (h) P. Magnus, J. Lacour and W. Weber, J. Am. Chem. Soc., 1993, 115, 9347 CrossRef CAS.
- L. Niu, C. Jiang, Y. Liang, D. Liu, F. Bu, R. Shi, H. Chen, A. D. Chowdhury and A. Lei, J. Am. Chem. Soc., 2020, 142, 17693 CrossRef CAS.
- (a) K. A. Margrey, W. L. Czaplyski, D. A. Nicewicz and E. J. Alexanian, J. Am. Chem. Soc., 2018, 140, 4213 CrossRef CAS; (b) S. Kamijo, M. Watanabe, K. Kamijo, K. Tao and T. Murafuji, Synthesis, 2016, 48, 115 CrossRef; (c) Y. Wang, G.-X. Li, G. Yang, G. He and G. Chen, Chem. Sci., 2016, 7, 2679 RSC.
- H. Schäfer, Angew. Chem., Int. Ed. Engl., 1970, 9, 158 CrossRef.
- (a) J. C. Siu, J. B. Parry and S. Lin, J. Am. Chem. Soc., 2019, 141, 2825 CrossRef CAS; (b) N. Fu, G. S. Sauer and S. Lin, Nat. Protoc., 2018, 13, 1725 CrossRef CAS; (c) J. C. Siu, G. S. Sauer, A. Saha, R. L. Macey, N. Fu, T. Chauviré, K. M. Lancaster and S. Lin, J. Am. Chem. Soc., 2018, 140, 12511 CrossRef CAS; (d) N. Fu, G. S. Sauer, A. Saha, A. Loo and S. Lin, Science, 2017, 357, 575 CrossRef CAS.
- For reviews, see: (a) Y. Hu, B. Zhou and C. Wang, Acc. Chem. Res., 2018, 51, 816 CrossRef CAS; (b) W. Liu and L. Ackermann, ACS Catal., 2016, 6, 3743 CrossRef CAS; for selected examples, see: (c) N. Kaplaneris, T. Rogge, R. Yin, H. Wang, G. Sirvinskaite and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 3476 CrossRef CAS; (d) C. Wang, A. Wang and M. Rueping, Angew. Chem., Int. Ed., 2017, 56, 9935 CrossRef CAS; (e) B. Zhou, Y. Hu and C. Wang, Angew. Chem., Int. Ed., 2015, 54, 13659 CrossRef CAS; (f) W. Liu, D. Zell, M. John and L. Ackermann, Angew. Chem., Int. Ed., 2015, 54, 4092 CrossRef CAS; (g) Y. Kuninobu, Y. Nishina, T. Takeuchi and K. Takai, Angew. Chem., Int. Ed., 2007, 46, 6518 CrossRef CAS.
- For selected examples, see: (a) K. Feng, R. E. Quevedo, J. T. Kohrt, M. S. Oderinde, U. Reilly and M. C. White, Nature, 2020, 580, 621 CrossRef CAS; (b) J. Zhao, T. Nanjo, E. C. de Lucca and M. C. White, Nat. Chem., 2019, 11, 213 CrossRef CAS; (c) M. Milan, M. Salamone, M. Costas and M. Bietti, Acc. Chem. Res., 2018, 51, 1984 CrossRef CAS.
- (a) X. Huang, T. M. Bergsten and J. T. Groves, J. Am. Chem. Soc., 2015, 137, 5300 CrossRef CAS; (b) C. L. Hill, J. A. Smegal and T. J. Henly, J. Org. Chem., 1983, 48, 3277 CrossRef CAS; (c) C. L. Hill and B. C. Schardt, J. Am. Chem. Soc., 1980, 102, 6374 CrossRef CAS.
- (a) M. Guo, T. Corona, K. Ray and W. Nam, ACS Cent. Sci., 2019, 5, 13 CrossRef CAS; (b) M. Guo, M. S. Seo, Y.-M. Lee, S. Fukuzumi and W. Nam, J. Am. Chem. Soc., 2019, 141, 12187 CrossRef CAS; (c) X. Huang and J. T. Groves, Chem. Rev., 2018, 118, 2491 CrossRef CAS; (d) W. Liu and J. T. Groves, Acc. Chem. Res., 2015, 48, 1727 CrossRef CAS; (e) W. Liu, X. Huang, M.-J. Cheng, R. J. Nielsen, W. A. Goddard and J. T. Groves, Science, 2012, 337, 1322 CrossRef CAS; (f) V. Lyaskovskyy, A. I. O. Suarez, H. Lu, H. Jiang, X. P. Zhang and B. de Bruin, J. Am. Chem. Soc., 2011, 133, 12264 CrossRef CAS; (g) M. Costas, Coord. Chem. Rev., 2011, 255, 2912 CrossRef CAS; (h) J. E. Lyons, P. E. Ellis and S. N. Shaikh, Inorg. Chim. Acta, 1998, 270, 162 CrossRef CAS; (i) P. Battioni, J. P. Renaud, J. F. Bartoli, M. Reina-Artiles, M. Fort and D. Mansuy, J. Am. Chem. Soc., 1988, 110, 8462 CrossRef CAS; (j) J. T. Groves, W. J. Kruper and R. C. Haushalter, J. Am. Chem. Soc., 1980, 102, 6375 CrossRef CAS.
- (a) R. D. Little, J. Org. Chem., 2020, 85, 13375 CrossRef CAS; (b) K.-J. Jiao, Y.-K. Xing, Q.-L. Yang, H. Qiu and T.-S. Mei, Acc. Chem. Res., 2020, 53, 300 CrossRef CAS; (c) F. Wang and S. S. Stahl, Acc. Chem. Res., 2020, 53, 561 CrossRef CAS; (d) J. C. Siu, N. Fu and S. Lin, Acc. Chem. Res., 2020, 53, 547 CrossRef CAS; (e) J. Liu, L. Lu, D. Wood and S. Lin, ACS Cent. Sci., 2020, 6, 1317 CrossRef CAS; (f) P. Xiong and H.-C. Xu, Acc. Chem. Res., 2019, 52, 3339 CrossRef CAS; (g) Y. Yuan and A. Lei, Acc. Chem. Res., 2019, 52, 3309 CrossRef CAS; (h) J.-i. Yoshida, A. Shimizu and R. Hayashi, Chem. Rev., 2018, 118, 4702 CrossRef CAS; (i) M. D. Kärkäs, Chem. Soc. Rev., 2018, 47, 5786 RSC; (j) A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594 CrossRef CAS; (k) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230 CrossRef CAS; (l) R. Feng, J. A. Smith and K. D. Moeller, Acc. Chem. Res., 2017, 50, 2346 CrossRef CAS; (m) R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492 RSC.
- (a) L. Ackermann, Acc. Chem. Res., 2020, 53, 84 CrossRef CAS; (b) T. H. Meyer, I. Choi, C. Tian and L. Ackermann, Chem, 2020, 6, 2484 CrossRef CAS; (c) T. H. Meyer, L. H. Finger, P. Gandeepan and L. Ackermann, Trends Chem., 2019, 1, 63 CrossRef CAS; (d) N. Sauermann, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Catal., 2018, 8, 7086 CrossRef CAS.
- V. Plzak and H. Wendt, Ber. Bunsen-Ges. Phys. Chem., 1979, 83, 481 CrossRef CAS.
- G. A. Ward and C. M. Wright, J. Electroanal. Chem., 1964, 8, 302 CAS.
- X.-S. Xue, P. Ji, B. Zhou and J.-P. Cheng, Chem. Rev., 2017, 117, 8622 CrossRef CAS.
- M. Zhang, Z.-F. Lu, Y. Liu, G. Grampp, H.-W. Hu and J.-H. Xu, Tetrahedron, 2006, 62, 5663 CrossRef CAS.
- (a) S. Herold, D. Bafaluy and K. Muñiz, Green Chem., 2018, 20, 3191 RSC; (b) Y. Kawamata, M. Yan, Z. Liu, D.-H. Bao, J. Chen, J. T. Starr and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 7448 CrossRef CAS; (c) G. Balavoine, D. H. R. Barton, J. Boivin, A. Gref, N. Ozbalik and H. Rivière, Tetrahedron Lett., 1986, 27, 2849 CrossRef CAS.
- A. Mukherjee, M. Martinho, E. L. Bominaar, E. Münck and L. Que Jr, Angew. Chem., Int. Ed., 2009, 48, 1780 CrossRef CAS.
- H. J. Schäfer, Electrochemical Conversion of Alkanes, in Alkanes and Cycloalkanes, ed. S. Patai and Z. Rappoport, Wiley & Sons, New York, 1992, pp. 781–808 Search PubMed.
- For detailed information see the ESI.†.
- (a) L. Gaillon, F. Bedioui, P. Battioni and J. Devynck, J. Mol. Catal., 1993, 78, L23 CrossRef CAS; (b) P. Leduc, P. Battioni, J. F. Bartoli and D. Mansuy, Tetrahedron Lett., 1988, 29, 205 CrossRef CAS; (c) J. C. Lennox and R. W. Murray, J. Am. Chem. Soc., 1978, 100, 3710 CrossRef CAS.
- B. W. Lund, F. Piu, N. K. Gauthier, A. Eeg, E. Currier, V. Sherbukhin, M. R. Brann, U. Hacksell and R. Olsson, J. Med. Chem., 2005, 48, 7517 CrossRef CAS.
- (a) T. Kurahashi, M. Hada and H. Fujii, J. Am. Chem. Soc., 2009, 131, 12394 CrossRef CAS; (b) T. Kurahashi and H. Fujii, Inorg. Chem., 2008, 47, 7556 CrossRef CAS.
- T. Kurahashi and H. Fujii, J. Am. Chem. Soc., 2011, 133, 8307 CrossRef CAS.
- For reviews, see: (a) L. Capaldo and D. Ravelli, Eur. J. Org. Chem., 2017, 2017, 2056 CrossRef CAS; (b) J. M. Mayer, Acc. Chem. Res., 2011, 44, 36 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc05924b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |