
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Transition-metal-mediated reduction and reversible double-cyclization of cyanuric triazide to an asymmetric bitetrazolate involving cleavage of the six-membered aromatic ring†
Shivaiah
Vaddypally
 a,
Vitaly G.
Kiselev
bcde,
Alex N.
Byrne
a,
C. Franklin
Goldsmith
*b and
Michael J.
Zdilla
*a
a,
Vitaly G.
Kiselev
bcde,
Alex N.
Byrne
a,
C. Franklin
Goldsmith
*b and
Michael J.
Zdilla
*a
aTemple University, 1901 N. 13th St., Philadelphia, PA 19122, USA. E-mail: mzdilla@temple.edu
bSchool of Engineering, Brown University, 184 Hope St., Providence, RI 02912, USA. E-mail: franklin_goldsmith@brown.edu
cNovosibirsk State University, 1 Pirogova Str., 630090 Novosibirsk, Russia
dInstitute of Chemical Kinetics and Combustion SB RAS, 3 Institutskaya Str., 630090 Novosibirsk, Russia
eSemenov Federal Research Center for Chemical Physics RAS, 4 Kosygina Str., 119991 Moscow, Russia
First published on 8th December 2020
Abstract
Cyanuric triazide reacts with several transition metal precursors, extruding one equivalent of N2 and reducing the putative diazidotriazeneylnitrene species by two electrons, which rearranges to N-(1′H-[1,5′-bitetrazol]-5-yl)methanediiminate (biTzI2−) dianionic ligand, which ligates the metal and dimerizes, and is isolated from pyridine as [M(biTzI)]2Py6 (M = Mn, Fe, Zn, Cu, Ni). Reagent scope, product analysis, and quantum chemical calculations were combined to elucidate the mechanism of formation as a two-electron reduction preceding ligand rearrangement.
Introduction
Novel cycloaddition reactions are of fundamental interest to synthetic chemistry, as (hetero)cyclic compounds play important roles in tuning stability,1–12 electronic properties,13–15 and biological activity of molecules.16–20 Access to specific or novel approaches for cyclization are of fundamental interest for enhancing the synthetic chemist's toolbox. Nitrogen-rich heterocycles are of fundamental interest in synthetic chemistry as biological mimics and pharmaceuticals,13,16–20 and as energetic materials such as explosives and propellants.21–26 In this latter category, heterocyclization reactions have been crucial to the realization of structures with improved kinetic stability and physical properties.1–12,21–26Among the most important cycloadditions is the so-called Huisgen [2 + 3] cycloaddition, which results in the joining of reactive and (frequently) impact sensitive azides with reactive alkynes into kinetically stable five-membered 1,2,3-triazole rings.27–29 Azole based systems are of interest as novel high explosives due to the remarkable stability of the five-membered heteroaromatic ring, which makes this class of molecules generally more insensitive than energetic materials with comparable nitrogen content.21,30,31 The original Huisgen cycloaddition is generally agreed to be a concerted [3 + 2] cycloaddition that gives two possible stereochemical outcomes. The copper-catalyzed variant, discovered approximately at the same time by Meldal32 and Sharpless,33 operates by a different mechanism (nicknamed the “click” reaction due to its success in selective biorthogonal chemistry), yielding exclusively the 1,4-substituted product. Azides may also be cyclized with nitriles to give the four-nitrogen tetrazole aromatic ring.23
Other cycloadditions of azide reactions are of interest as well. Our group34,35 and others36–41 have explored the cycloaddition of two azide units, coupled to metal-mediated extrusion of N2, to give five-membered tetrazene metallacycles. In these complexes, a redox-non-innocent four-nitrogen chain of catenated nitrogen ligands acts as a bidentate ligand.34,35,39–41 A highly relevant cycloaddition reaction of azide occurs on ortho-azidoazines, viz., nitrogen-substituted benzene rings with an azide group ortho to a ring nitrogen. This species can undergo a reversible isomerization to a indole-like fused bicycle containing a tetrazole ring fused to the six-membered azine.3,7,42–46 Oligomerized small molecules with high-nitrogen–carbon ratio are of interest in astrochemistry, where small molecules such as nitrogen and HCN in deep space may be activated by high energy radiation and aggregate into complex organic molecules.47 Here we report a related double cycloaddition rearrangement reaction of cyanuric triazide (2,4,6-triazido-1,3,5-triazine, 1) following reduction of the substrate to release N2, concomitant with the cleavage of the 6-membered ring to give N-([1,5′-bitetrazol]-5-yl)methanediiminate ligand in a dimeric metal complex. This asymmetrically linked bitetrazole nucleus is uncommon, limited to a few reports with structurally characterized examples: one with amino and nitramido side chains,48 and one on metal complex chemistry of the parent bitetrazole.49 The variant reported in this paper contains a pendant carbodiimide group bridging two metal centers. Upon demetallation, this ligand undergoes a similar reverse reaction to regenerate the azidotriazine ring.
Results and discussion
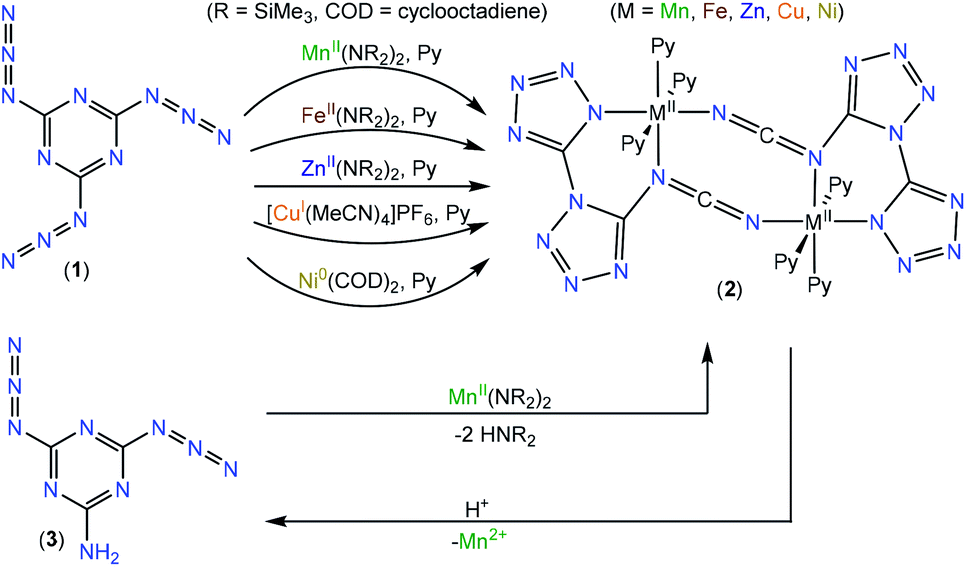
We intended to extrapolate our work on the formation of metallotetrazene species34–41 from addition of azides at manganese centers to cyanuric triazide50 by reaction with Mn(NR2)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 51) (R = SiMe3). However, this experiment resulted in an entirely different type of chemical reaction. Single-crystal X-ray diffraction analysis of the crystalized product of this reaction revealed the structure to be that of a dimeric metal complex consisting of two dibasic N-([1,5′-bitetrazol]-5-yl)methanediiminate ligands (biTzI), two MnII ions, and capping pyridine ligands. The resulting product, [Mn(biTzI)Py3]2 (2a) is shown in Scheme 1 and Fig. 1. The drastic ligand rearrangement resulting in the loss of the six-membered aromatic ring and the appearance of two new five-membered aromatic heterocycles was unexpected, but may be rationalized by a ligand reduction to extrude N2, accompanied by a rearrangement of the resulting imido species involving N–C cleavage, and closure of the azides with ortho azine nitrogen atoms (Scheme 2). This isomerization of a sensitive cyanuric polyazide into more stable tetrazole ring systems represents a novel transformation into a new type of energetic moiety: one involving the cleavage of a six member aromatic ring, which is an exceedingly rare phenomenon usually discussed in the context of the 2-aminophenol dioxygenase enzyme52 and model chemistry thereof.53,54
51) (R = SiMe3). However, this experiment resulted in an entirely different type of chemical reaction. Single-crystal X-ray diffraction analysis of the crystalized product of this reaction revealed the structure to be that of a dimeric metal complex consisting of two dibasic N-([1,5′-bitetrazol]-5-yl)methanediiminate ligands (biTzI), two MnII ions, and capping pyridine ligands. The resulting product, [Mn(biTzI)Py3]2 (2a) is shown in Scheme 1 and Fig. 1. The drastic ligand rearrangement resulting in the loss of the six-membered aromatic ring and the appearance of two new five-membered aromatic heterocycles was unexpected, but may be rationalized by a ligand reduction to extrude N2, accompanied by a rearrangement of the resulting imido species involving N–C cleavage, and closure of the azides with ortho azine nitrogen atoms (Scheme 2). This isomerization of a sensitive cyanuric polyazide into more stable tetrazole ring systems represents a novel transformation into a new type of energetic moiety: one involving the cleavage of a six member aromatic ring, which is an exceedingly rare phenomenon usually discussed in the context of the 2-aminophenol dioxygenase enzyme52 and model chemistry thereof.53,54
 | ||
| Scheme 1 Metal mediated conversions between azidotriazenes and metal–biTzI complexes. | ||
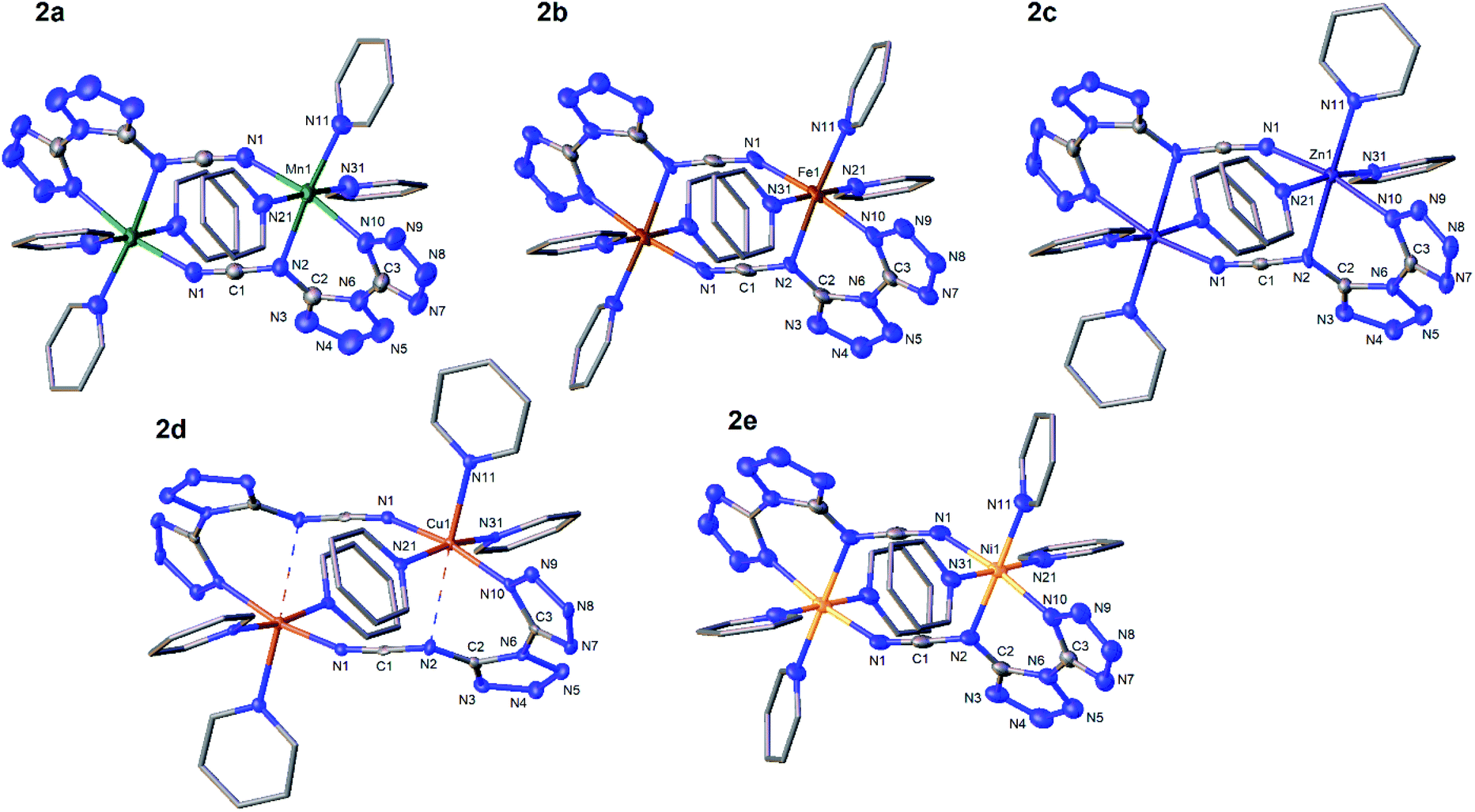 | ||
| Fig. 1 Thermal ellipsoid plots of [MII(biTzI)Py3]2 (2). M = Mn (2a), M = Fe (2b), M = Zn (2c), M = Cu (2d), M = Ni (2e). Ellipsoids set at 50% probability. Pyridine carbon atoms are shown in stick mode, and hydrogen atoms and lattice solvents omitted for clarity. | ||
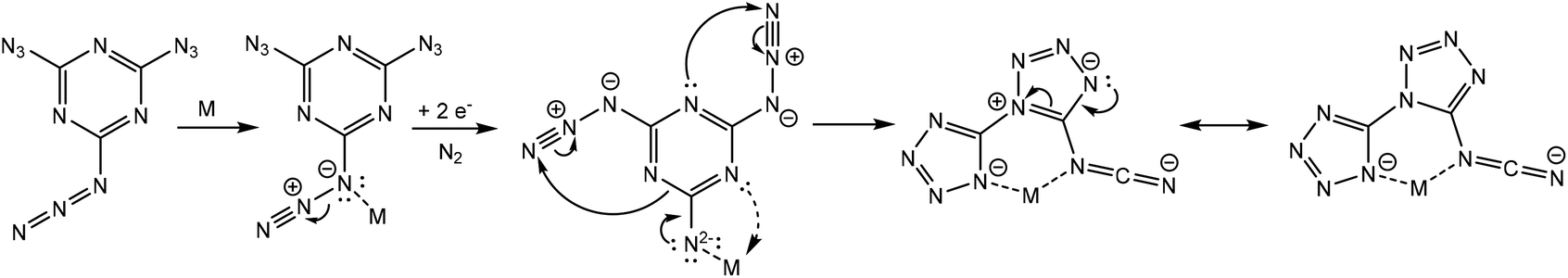 | ||
| Scheme 2 Proposed mechanism of metal mediated reaction/rearrangement of 1 into biTzI ligand. | ||
However, charge counting considerations lead to a formal oxidation state of 2+ on the manganese ions, suggesting that the metal ion has not been oxidized. We initially hypothesized that additional equivalents of MnII act as a sacrificial reductant to form MnIII or MnIV, and that the resulting reduced ligand framework then assembles with additional equivalents of MnII to generate 2. However, we were unable to identify any other byproducts of the reaction containing oxidized manganese. As a test of the hypothesis that the metal serves two roles (reductant and divalent metal source) we performed the reaction with a more easily oxidized metal ion, FeII, using the analogous Fe(NR2)251 reagent, which is highly reducing and favors formation of FeIII. However, the isolated product of this reaction was the analogous ferrous complex [Fe(biTzI)Py3]2 (2b) also containing formal FeII ions (Scheme 1, Fig. 1). Again, we were unable to isolate any oxidized iron species from this reaction.
Challenged by the question of the origin of the reducing equivalents, we considered next the synthesis of an analog of 2 using a strictly redox innocent metal, Zn2+, an approach that should entirely preclude metal-based redox reactions. To our surprise, the reaction of Zn(NR2)2 with cyanuric triazide resulted in the isolation of the analogous zinc complex, [Zn(biTzI)Py3]2 (2c) in good yield, and which comprised the same two-electron reduced biTzI ligand complexed to ZnII (Scheme 2). In addition to crystallographic characterization, the diamagnetic 2c could be characterized by 13C NMR (other compounds experience paramagnetic broadening of resonances and are thus invisible). The NMR spectrum shows three large signals corresponding to pyridine ligands, and three small signals corresponding to quaterinary carbons, with the carbodiimide signal shifted significantly upfield to 119 ppm due to its location in two shielding cones of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bonds (Fig. S4†).
N double bonds (Fig. S4†).
Inspired by the successful synthesis of an analog of 2 with a redox-innocent metal, we considered the possibility that the ligand reduction is not a prerequisite for ligand rearrangement, and whether instead a pathway involving a rearrangement of the ligand prior to reduction (for example, by adventitious oxidation of solvent) could result in product formation. To address this question, we performed quantum chemical calculations using modern, reliable DLPNO-CCSD(T)55,56 and CCSD(T)-F12 methodologies57,58 along with the polarized continuum model (PCM)59 to account for the solvent. We considered several possibilities for the rearrangement chemistry: (1) the extrusion of N2 to generate either singlet or triplet neutral nitrene, followed by a ligand rearrangement. (2) one-electron reduction with the extrusion of N2 to generate an anionic nitrenyl radical, or (3) a two-electron reduction to extrude N2 and generate a metal–ligated imido species.
The mechanism of (1) is disfavored based on theoretical considerations, as the activation barrier for two types of cyclization rearrangements is high: viz., the formation of the double-cyclized product (without a cleavage of the six-membered ring) has an effective activation barrier around 31 kcal mol−1 (this corresponds to a reaction time trxn ∼30 years). Moreover, the activation barrier of the thermal extrusion of N2 yielding a nitrene intermediate, is predicted to be 38 kcal mol−1, and the subsequent reaction cascade did not favor the formation of the biTzI ligand system or a similar species (see Fig. S1†).
The one-electron reduction of 1 renders the N2 elimination yielding a nitrenyl radical anion to be an extremely fast process (in agreement with the literature data for similar species,60 the activation barrier is close to zero, Fig. S2†). The latter compound has a lower-barrier entrance channel to the rearrangement chemistry of only 16 kcal mol−1, which could cascade in several steps to the biTzI˙− anion radical, whose formation is exothermic by 9 kcal mol−1 (Fig. S2†). However, this is not the most stable rearrangement product in the cascade; a bis(tetrazolo)[1,5-a:5′,1′-d][1,3,5]triazinyliminyl radical anion is ca. 5 kcal mol−1 more favorable thermodynamically than the latter species (Fig. S2†).
The most favorable pathway of 1 transformation involves a two-electron reduction preceding a molecular nitrogen elimination followed by a rearrangement of the nitrene intermediate. The examination of an N2-eliminated dianionic imide coordinated to a model Zn2+ center predicted a favorable rearrangement to the expected [biTzI]2− ligand product in a sequence of three irreversible reactions (Fig. 2). The last rate-limiting step occurring via TS3 has an activation barrier ∼18 kcal mol−1, which is affordable at room temperature (the reaction time of a few seconds). The lowest-energy product predicted in the cascade, is exothermic by 29.6 kcal mol−1 (Fig. 2), and represents an essentially identical mechanism to that proposed in Scheme 2, proceeding in three low-barrier steps.
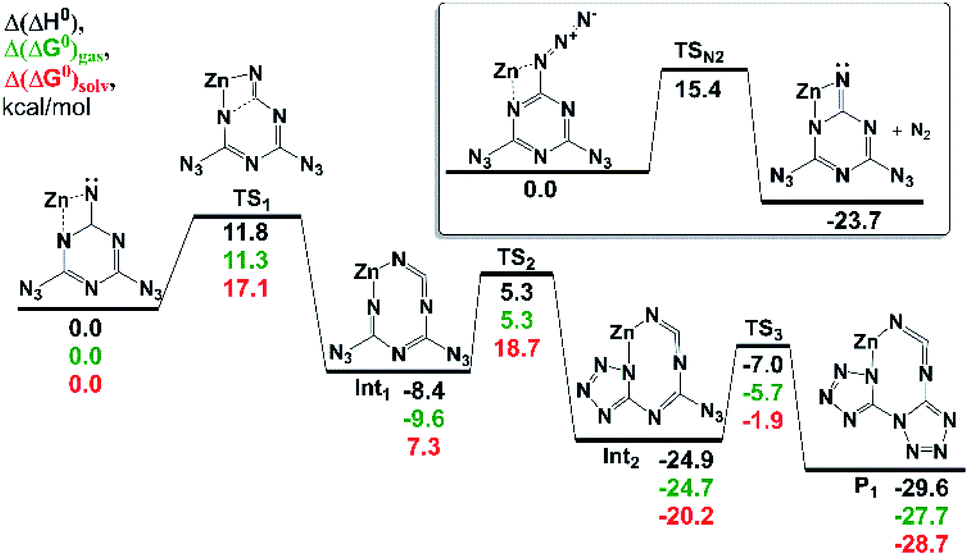 | ||
| Fig. 2 The stationary points on the PES corresponding to the rearrangements of Zn2+ complex with a dianion intermediate. Inset: the N2 elimination from the initial complex of Zn and 1. All values are calculated at the DLPNO-CCSD(T)/aVQZ//M06-2X/6-311++G(2df,p) level of theory and are given in kcal mol−1. The PCM free energies of solvation are calculated at the same DFT level of theory using tetrahydrofuran as a solvent. | ||
As a further test of the proposed mechanism where reduction precedes a ligand rearrangement, we synthesized 2a using the already-reduced variant of 1: viz., 2-amino-4,6-diazido-1,3,5-triazine (3).61 This precursor was combined with Mn(NR2)2 in pyridine to determine whether simple deprotonation of already-reduced ligand 3 by protolysis with 1 would result in the proposed ligand arrangement. Indeed, this reaction resulted in the isolation of 2a, which was confirmed by single-crystal X-ray crystallographic analysis. To our surprise, the attempt to demetallate 1a by acid hydrolysis resulted not in the isolation of (biTzI)H2, but in a reverse rearrangement of the hydrolyzed ligand back to 3 (Scheme 1) which was isolated and identified by single-crystal XRD (Fig. S16†). This result suggests that the six-membered-ring aryl bond cleavage and rearrangement to the bitetrazole-based [biTzI] ligand is favored in basic media, but is reversible and may be converted back by cleavage of the two tetrazole rings and reformation of the six-membered azine ring in acidic media (Scheme 1).
Given the predicted favorableness of the ligand reduction preceding rearrangement, we considered the hypothesis that trimethylsilylamido (NR2−) ligands of the precursor M(NR2)2 could act as the reductant, undergoing a reductive elimination and forming tetrakis(trimethylsilylamido)hydrazine (R2N–NR2) as a byproduct. The latter compound is isolable, stable, and crystalline,62–64 and its formation is plausible due to the known redox relationships between amines/amides and hydrazine.65 R2N–NR2 has a reported 1H NMR upfield chemical shift of 0.2 ppm in benzene-d6 solvent, distinct from the chemical shifts of hexamethyldisiloxane (0.12)66 and hexamethyldisilazane (0.10).67 The analysis of the crude reaction mixture of the synthesis of 2c reveals the formation of R2N–NR2, (Fig. S3†) and provides an explanation for the observed redox processes, and implies the following reaction stoichiometry:
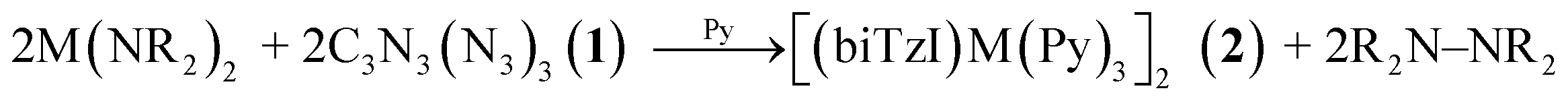 | (1) |
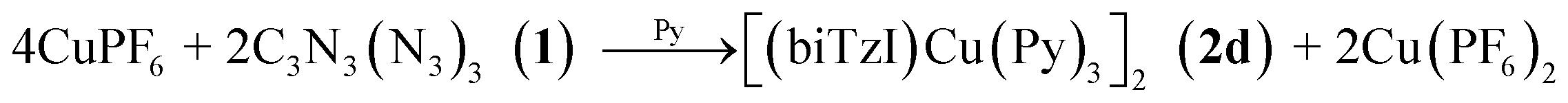 | (2) |
Finally, in order to test for the generation of a variant of 2 with a metal-based two-electron reductant, we tested the reaction of the strong reducing agent bis(cyclooctadiene)nickel(0) (COD2Ni0) with 1. The expected product [Ni(bTzNCN)Py3]2 (2e) is isolated in good yield according to eqn (3):
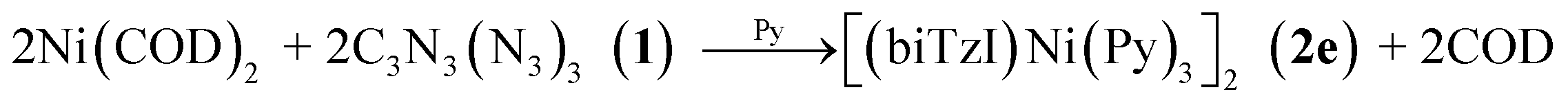 | (3) |
The scope of synthetic chemistry including the results from quantum chemical calculations indicate that the reduction of 1 is favorable if coupled to the described cycloaddition rearrangement, and can be mediated by metal based reduction (both one- and two-electron reductants) as well as by ligand-based reduction mediated at redox innocent transition metals.
Experimental and computational details
Precautions
The low-carbon azide and tetrazole-based molecules presented in this report are explosive materials. They should be stored in solution or isolated only in quantities of a few milligrams. Shock, spark, heat from exposure to air can result in explosion. All syntheses were performed in microscale for maximum safety. Proper precautions should be taken, including use of standard laboratory PPE plus explosion proof mask, shield, Kevlar gloves, use of plastic spatulae, and electrical grounding while handling these materials. While we never experienced any accidental initiation of compounds 2a–2e resulting from physical stimuli (e.g., spatula scraping), appropriate caution is recommended. Detonation was, however, observed in these materials when irradiated by a pulsed laser.Synthesis
Mn(NR2)2(THF)2,51 Fe(NR2)2(THF)2,51 (R = SiMe3), cyanuric triazide (1),50 were prepared according to literature protocol.Method A. 0.10 g (0.15 mmol) of Mn(NR)2(THF)2 is dissolved in tetrahydrofuran followed by addition of 0.065 g (0.32 mmol) of cyanuric triazide (1). After dissolving these two reactants 200 μL of tBuNH2 was added, which results in a color change to red. This reaction mixture is stirred for one hour and the solvent is removed in vacuo. The dried product is washed with pentane and to remove HNR2. The residue is redissolved by addition of ∼ 2 mL of equal amounts of anhydrous pyridine and acetonitrile. This is filtered to remove undissolved residue, and the filtrate is crystallized by storing the filtrate at −35 °C yielding colorless crystals of 2a, which were isolated by decanting the supernatant. Yield 0.050 g (68%).
Method B. 21 mg (0.032 mmol) of Mn(NR2)2(THF)2 and 10 mg (0.056 mmol) of 3,5-diazido-1-amino-1,3,4-triazine were dissolved in THF. The reaction mixture stirred overnight and dried in vacuo. The residue was dissolved in ∼2 mL of equal amounts of anhydrous pyridine and acetonitrile and crystalized by slow evaporation under inert atmosphere. A single crystal was removed and identified by single-crystal X-ray diffraction. Unit cell: triclinic, a = 11.495(3) Å b = 11.697(3) Å c = 11.753(3)Å, α = 72.463(4)°, β = 82.359(4)°, γ = 71.422(3)° V = 1427.1(6) Å3. FTIR (cm−1): ν 2206, 2140 (N
CN asymmetric stretch) 1545, 1351 (C–N, N–N stretches), 1245, 1211, 1071, 1038 (C–H bends), 841 (NCN bend), 753, 702 (Ph out-of-plane bend).
:1 mixture of anhydrous pyridine and acetonitrile and kept at −35 °C from which colorless crystals of 2b were obtained in trace yield after two weeks at low temperature. Unit cell: triclinic, a = 11.034(4) Å b = 13.414(6) Å c = 18.758(8) Å, α = 91.221(6)°, β = 106.188(6)°, γ = 99.178(7)° V = 2625.8(19)Å3.
:1 anhydrous pyridine and acetonitrile. Crystals of 2c are obtained after two days of evaporation at room temperature under anerobic atmosphere. Yield: 0.02 g, (40%). Unit cell: monoclinic P, a = 9.580(5) Å b = 13.076(8) Å c = 15.904(9) Å, β = 97.636(13)°, V = 1974.6(19) Å3. 13C NMR [ppm] (500 MHz, DMSO-d6): δ(ppm): 118.86 (NCN), 124.917, 137, 150 (Py), 154, 159 (Tz). FTIR (cm−1): ν 2198, 2064 (NCN asymmetric stretch), 1597, 1545 (C–N, N–N stretches), 1242, 1211 (C–H bends), 756, 696 (Ph out-of-plane bend).
:1 pyridine and acetonitrile, and filtered to remove undissolved residue. The filtrate is recrystallized by vapor diffusion with pentane over several days to give green crystals. Yield: 0.02 g (100% based on eqn (2)). Unit cell: monoclinic P, a = 9.332(3) Å b = 13.337(5) Å c = 15.845(6) Å, β = 97.844(7)°, V = 1953.6(12) Å3. FTIR (cm−1): ν 2204 (NCN asymmetric stretch), 1600, 1575, 1558 1542, 1351 (C–N, N–N stretch), 1257, 1214, 1211, 1090, 1071, 1042 (C–H bend), 838 (NCN bend), 762, 699, (Ph out-of-plane bend). UV-vis: nm (ε, M−1 cm−1), 700 (27000).
:1 anhydrous pyridine and acetonitrile and filtered to remove undissolved residue. This solution is evaporated open to anaerobic atmosphere. Colorless crystals are obtained within 3–5 days. Yield: 0.007 g (18%). Unit cell: monoclinic P, a = 11.927(2) Å b = 20.144(4) Å c = 10.560(2) Å, β = 98.269(4)°, V = 2510.7(8) Å3.
Quantum chemical calculations
Electronic structure calculations were performed using the Gaussian 09,68 Molpro 2010,69 and ORCA 4.070 program packages. The geometries of all structures corresponding to the stationary points on the potential energy surface (PES) of the species studied were fully optimized using density functional theory at the M06-2X/6-311++G(2df,p).71 All the equilibrium and transition state structures were ascertained to be minima and saddle points, respectively, on the potential energy surfaces. The nature of all localized transition states was verified using the intrinsic reaction coordinate (IRC) procedure. Zero-point energies and thermal corrections to enthalpy and Gibbs free energy were computed at the same DFT level of theory. Single-point electronic energies were afterward refined using the DLPNO-CCSD(T) methodology (the “NormalPNO” truncation thresholds were set)55 along with the aug-cc-pVQZ (aVQZ) basis set.72 The RIJK density fitting (DF) approximation73 was used to accelerate the convergence of the SCF components of DLPNO-CCSD(T) energy. The corresponding auxiliary basis sets (aug-cc-pVQZ/JK and aug-cc-pVQZ/C in the ORCA nomenclature)71 were used in the DF calculations of the SCF and correlation energies. In several cases, an explicitly correlated coupled-cluster formalism CCSD(T)-F12b was employed.57 Both DLPNO-CCSD(T) and CCSD(T)-F12 techniques have recently been shown to perform quite well for thermochemistry and kinetics of nitrogen-rich heterocycles.74–77 To account for the solvent effects, the free energies of solvation were calculated at the M06-2X/6-311++G(2df,p) level of theory using the polarized continuum model (PCM)59 for isotropic media with ε = 7.4 corresponding to tetrahydrofuran solution used in all syntheses. We used the gas-phase vibrational partition functions and single-point solvation energies for gas-phase optimized geometries, as was earlier benchmarked.78 The infrared spectrum of 2d (Fig. S9†) was calculated using the B3LYP/6-31G(d) level of theory.79–81
Conclusions
In summary, a unique rearrangement of a six-membered aromatic polyazide 1 is reported that results from initial ligand reduction and elimination of N2 followed by cleavage of the six-membered triazine ring concomitant with formation of two new five-membered aromatic tetrazole rings. The initiating ligand reduction may be achieved via a ligand-based oxidation of trimethylsilylamido ligands to hydrazine, or by using reducing metal (CuI or Ni0) precursors. This product type appears to be stable and amenable at a number of transition metals, being isolable with at least five mid-late 3d transition metals and stabilizing them in their +2 oxidation states. Upon exposure to acid, the ligand product of the rearrangement undergoes a reverse reaction with the cleavage of the two five-membered azole rings regenerating the initial six-membered triazine ring. Future work will focus on the energetics of the acid/base dependent rearrangement, and the energetic properties of biTzI-type compounds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. J. Z. and C. F. G. gratefully acknowledge financial support from ONR (N00014-19-1-2087 and N00014-1-61-2054). V. G. K. acknowledges the Supercomputer Center of Novosibirsk State University and the Russian Science Foundation for a financial support of the computational part of this work (project 19-73-20217). S. V. and M. J. Z. are supported by the National Science Foundation under award 1800105. This work was also supported by Brown University through the use of the facilities of its Center for Computation and Visualization. The authors thank Prof. Steven A. Fleming for helpful conversations surrounding cycloaddition mechanisms, and Charles Debrosse for helpful conversations concerning NMR assignments. Finally, the authors thank Prof. Edward G. Gillon of the University of Iowa for helpful conversations on the preparation and handling of cyanuric triazide.Notes and references
- D. E. Chavez, J. C. Bottaro, M. Petrie and D. A. Parrish, Angew. Chem., Int. Ed., 2015, 54, 12973–12975 CrossRef CAS.
- Y. Tang, D. Kumar and J. n. M. Shreeve, J. Am. Chem. Soc., 2017, 139, 13684–13687 CrossRef CAS.
- D. E. Chavez, D. A. Parrish, L. Mitchell and G. H. Imler, Angew. Chem., Int. Ed., 2017, 56, 3575–3578 CrossRef CAS.
- A. M. Churakov, S. L. Ioffe, Y. A. Strelenko and V. A. Tartakovskii, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1990, 39, 639–640 CrossRef.
- A. M. Churakov, S. L. Ioffe and V. A. Tartakovsky, Mendeleev Commun., 1995, 5, 227–228 CrossRef.
- T. Kaihoh, T. Itoh, K. Yamaguchi and A. Ohsawa, J. Chem. Soc., Chem. Commun., 1988, 1608–1609 RSC.
- H. Wei, J. Zhang and J. n. M. Shreeve, Chem.–Asian J., 2015, 10, 1130–1132 CrossRef CAS.
- E. Alcalde, J. De Mendoza and J. Elguero, J. Heterocycl. Chem., 1974, 11, 921–928 CrossRef CAS.
- E. Alcalde, R. M. Claramunt, J. Elguero and C. P. S. Huber, J. Heterocycl. Chem., 1978, 15, 395–399 CrossRef CAS.
- G. Ege, R. Heck, K. Gilbert, H. Irngartinger, U. Huber-Patz and H. Rodewald, J. Heterocycl. Chem., 1983, 20, 1629–1639 CrossRef CAS.
- S. Chen, Y. Liu, Y. Feng, X. Yang and Q. Zhang, Chem. Commun., 2020, 56, 1493–1496 RSC.
- T. M. Klapötke, D. G. Piercey and J. Stierstorfer, Chem.–Eur. J., 2011, 17, 13068–13077 CrossRef.
- L. D. Lavis and R. T. Raines, ACS Chem. Biol., 2008, 3, 142–155 CrossRef CAS.
- W. R. Dawson and M. W. Windsor, J. Phys. Chem., 1968, 72, 3251–3260 CrossRef CAS.
- N. N. Makhova, L. I. Belen'kii, G. A. Gazieva, I. L. Dalinger, L. S. Konstantinova, V. V. Kuznetsov, A. N. Kravchenko, M. M. Krayushkin, O. A. Rakitin, A. M. Starosotnikov, L. L. Fershtat, S. A. Shevelev, V. Z. Shirinian and V. N. Yarovenko, Russ. Chem. Rev., 2020, 89, 55–124 CrossRef CAS.
- D. W. Woolley and E. Shaw, J. Biol. Chem., 1951, 189, 401–410 CrossRef CAS.
- J. J. Biesele, Cancer, 1952, 5, 787–791 CrossRef CAS.
- E. Shaw and D. W. Woolley, J. Biol. Chem., 1952, 194, 641–654 CrossRef CAS.
- C. Grundmann and H. Ulrich, J. Org. Chem., 1959, 24, 272–274 CrossRef CAS.
- V. Papesch and R. M. Dodson, J. Org. Chem., 1963, 28, 1329–1331 CrossRef CAS.
- G. I. Koldobskii and V. A. Ostrovskii, Russ. Chem. Rev., 1994, 63, 797–814 CrossRef.
- C. G. Neochoritis, T. Zhao and A. Dömling, Chem. Rev., 2019, 119, 1970–2042 CrossRef CAS.
- F. R. Benson, Chem. Rev., 1947, 41, 1–61 CrossRef CAS.
- V. Ji Ram, A. Sethi, M. Nath and R. Pratap, in The Chemistry of Heterocycles, ed. V. Ji Ram, A. Sethi, M. Nath and R. Pratap, Elsevier, 2019, pp. 149–478, DOI:10.1016/B978-0-08-101033-4.00005-X.
- G. Aromí, L. A. Barrios, O. Roubeau and P. Gamez, Coord. Chem. Rev., 2011, 255, 485–546 CrossRef.
- Q. Zhang and J. M. Shreeve, Angew. Chem., Int. Ed., 2013, 52, 8792–8794 CrossRef CAS.
- R. Huisgen and I. Ugi, Angew. Chem., 1956, 68, 705–706 CrossRef CAS.
- R. Huisgen and I. Ugi, Chem. Ber., 1957, 90, 2914–2927 CrossRef CAS.
- I. Ugi and R. Huisgen, Chem. Ber., 1958, 91, 531–537 CrossRef CAS.
- J. P. Agrawal, High Energy Materials, WILEY-VCH Verlag GmbH & Co., Weinheim, Germany, 2010 Search PubMed.
- T. Klapötke, Energetic Materials Encyclopedia, de Gruyter, Boston, 2001 Search PubMed.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- S. Vaddypally, W. Tomlinson, O. T. O'Sullivan, R. Ding, M. M. Van Vliet, B. B. Wayland, J. P. Hooper and M. J. Zdilla, J. Am. Chem. Soc., 2019, 141, 5699–5709 CrossRef CAS.
- S. Vaddypally, I. G. McKendry, W. Tomlinson, J. P. Hooper and M. J. Zdilla, Chem.–Eur. J., 2016, 22, 10548–10557 CrossRef CAS.
- R. I. Michelman, R. G. Bergman and R. A. Andersen, Organometallics, 1993, 12, 2741–2751 CAS.
- A. H. Obenhuber, T. L. Gianetti, X. Berrebi, R. G. Bergman and J. Arnold, J. Am. Chem. Soc., 2014, 136, 2994–2997 CAS.
- K. E. Meyer, P. J. Walsh and R. G. Bergman, J. Am. Chem. Soc., 1995, 117, 974–985 CrossRef CAS.
- M. T. Mock, C. V. Popescu, G. P. A. Yap, W. G. Dougherty and C. G. Riordan, Inorg. Chem., 2008, 47, 1889–1891 CrossRef CAS.
- R. E. Cowley, E. Bill, F. Neese, W. W. Brennessel and P. L. Holland, Inorg. Chem., 2009, 48, 4828–4836 CrossRef CAS.
- A. C. Bowman, A. M. Tondreau, E. Lobkovsky, G. W. Margulieux and P. J. Chirik, Inorg. Chem., 2018, 57, 9634–9643 CrossRef CAS.
- D. Zhao, M. Lv and C. Cai, Propellants, Explos., Pyrotech., 2015, 40, 627–631 CrossRef CAS.
- Q. Wang, Y. Shao and M. Lu, Chem. Commun., 2019, 55, 6062–6065 RSC.
- A. Hammerl, T. M. Klapötke and R. Rocha, Eur. J. Inorg. Chem., 2006, 2006, 2210–2228 CrossRef.
- J. Yang, X. Gong and G. Wang, RSC Adv., 2015, 5, 9503–9509 RSC.
- E. Cubero, M. Orozco and F. J. Luque, J. Am. Chem. Soc., 1998, 120, 4723–4731 CrossRef CAS.
- K. I. Öberg, Chem. Rev., 2016, 116, 9631–9663 CrossRef.
- B. Wang, X. Qi, W. Zhang, K. Wang, W. Li and Q. Zhang, J. Mater. Chem. A, 2017, 5, 20867–20873 RSC.
- N. Fischer, D. Izsák, T. M. Klapötke and J. Stierstorfer, Chem.–Eur. J., 2013, 19, 8948–8957 CrossRef CAS.
- E. Keßenich, T. M. Klapötke, J. Knizek, H. Nöth and A. Schulz, Eur. J. Inorg. Chem., 1998, 1998, 2013–2016 CrossRef.
- R. A. Andersen, K. Faegri, J. C. Green, A. Haaland, M. F. Lappert, W. P. Leung and K. Rypdal, Inorg. Chem., 1988, 27, 1782–1786 CrossRef CAS.
- G. Dong, J. Lu and W. Lai, ACS Catal., 2016, 6, 3796–3803 CrossRef CAS.
- T. Funabiki, H. Sakamoto, S. Yoshida and K. Tarma, J. Chem. Soc., Chem. Commun., 1979, 754–755 RSC.
- B. Chakraborty and T. K. Paine, Angew. Chem., Int. Ed., 2013, 52, 920–924 CrossRef CAS.
- D. G. Liakos, M. Sparta, M. K. Kesharwani, J. M. L. Martin and F. Neese, J. Chem. Theory Comput., 2015, 11, 1525–1539 CrossRef CAS.
- C. Riplinger, P. Pinski, U. Becker, E. F. Valeev and F. Neese, J. Chem. Phys., 2016, 144, 024109 CrossRef.
- G. Knizia, T. B. Adler and H.-J. Werner, J. Chem. Phys., 2009, 130, 054104 CrossRef.
- L. Kong, F. A. Bischoff and E. F. Valeev, Chem. Rev., 2012, 112, 75–107 CrossRef CAS.
- B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 106, 5151–5158 CrossRef CAS.
- N. P. Gritsan, E. A. Pritchina, I. I. Barabanov, G. T. Burdzinski and M. S. Platz, J. Phys. Chem. C, 2009, 113, 11579–11589 CrossRef CAS.
- C. V. Hart, J. Am. Chem. Soc., 1928, 50, 1922–1930 CrossRef CAS.
- N. Wiberg and W. Uhlenbrock, Angew. Chem., Int. Ed. Engl., 1970, 9, 70–71 CrossRef CAS.
- N. Wiberg and W. Uhlenbrock, J. Organomet. Chem., 1974, 70, 239–248 CrossRef CAS.
- S. K. Vasisht, M. Sood, P. K. Verma, T. Kaur and K. Usha, Phosphorus, Sulfur Silicon Relat. Elem., 1990, 47, 349–355 CrossRef CAS.
- R. A. Cox and E. Buncel, in The Chemistry of the Hydrazo, Azo, and Azoxy Groups, ed. S. Patai, John Wiley & Sons, London, 1975, pp. 775–805 Search PubMed.
- Z. Zheng, O. S. Trofymchuk, T. Kurogi, E. Varela, D. J. Mindiola and P. J. Walsh, Adv. Synth. Catal., 2020, 362, 659–666 CrossRef CAS.
- G. E. Martinez, C. Ocampo, Y. J. Park and A. R. Fout, J. Am. Chem. Soc., 2016, 138, 4290–4293 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision D.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz, P. Celani, W. Györffy, D. Kats, T. Korona, R. Lindh, A. Mitrushenkov, G. Rauhut, K. R. Shamasundar, T. B. Adler, R. D. Amos, S. J. Bennie, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, E. Goll, C. Hampel, A. Hesselmann, G. Hetzer, T. Hrenar, G. Jansen, C. Köppl, S. J. R. Lee, Y. Liu, A. W. Lloyd, Q. Ma, R. A. Mata, A. J. May, S. J. McNicholas, W. Meyer, T. F. Miller III, M. E. Mura, A. Nicklass, D. P. O'Neill, P. Palmieri, D. Peng, K. Pflüger, R. Pitzer, M. Reiher, T. Shiozaki, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson, M. Wang and M. Welborn, MOLPRO, version 2010.1, 2010 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- R. A. Kendall, T. H. Dunning Jr and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- S. Kossmann and F. Neese, Chem. Phys. Lett., 2009, 481, 240–243 CrossRef CAS.
- M. V. Shakhova, N. V. Muravyev, N. P. Gritsan and V. G. Kiselev, J. Phys. Chem. A, 2018, 122, 3939–3949 CrossRef CAS.
- V. G. Kiselev and C. F. Goldsmith, J. Phys. Chem. A, 2019, 123, 9818–9827 CrossRef CAS.
- M. V. Gorn, N. P. Gritsan, C. F. Goldsmith and V. G. Kiselev, J. Phys. Chem. A, 2020, 124, 7665–7677 CrossRef CAS.
- V. G. Kiselev and C. F. Goldsmith, J. Phys. Chem. A, 2019, 123, 4883–4890 CrossRef CAS.
- R. F. Ribeiro, A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2011, 115, 14556–14562 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- V. A. Rassolov, J. A. Pople, M. A. Ratner and T. L. Windus, J. Chem. Phys., 1998, 109, 1223–1229 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of quantum chemical calculations, NMR, UV-vis and IR spectra, X-ray crystallographic figures and tables. CCDC under deposition numbers 2024699–2024704. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc04949b |
| This journal is © The Royal Society of Chemistry 2021 |