
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermo- and light-triggered reversible interconversion of dysprosium–anthracene complexes and their responsive optical, magnetic and dielectric properties†
Xin-Da
Huang
,
Ge-Hua
Wen
,
Song-Song
Bao
,
Jia-Ge
Jia
and
Li-Min
Zheng
 *
*
State Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Collaborative Innovation Centre of Advanced Microstructures, Nanjing University, Nanjing 210023, P. R. China. E-mail: lmzheng@nju.edu.cn
First published on 11th November 2020
Abstract
Artificial smart materials with switchable multifunctionality are of immense interest owing to their wide application in sensors, displays and memory devices. Lanthanide complexes are promising multifunctional materials integrating optical and magnetic characteristics. However, synergistic manipulation of different physical properties in lanthanide systems is still challenging. Herein we designed and synthesized a mononuclear complex [DyIII(SCN)3(depma)2(4-hpy)2] (1), which incorporates 9-diethylphosphonomethylanthracene (depma) as a photo-active component and 4-hydroxypyridine (4-hpy) as a polar component. This compound shows several unusual features: (a) reversible thermo-responsive phase transition associated with the order–disorder transition of 4-hpy and SCN−, which leads to thermochromic behavior and dielectric anomaly; (b) reversible photo-induced dimerization of anthracene groups, which leads to synergistic switching of luminescence, magnetic and dielectric properties. To our knowledge, compound 1 is the first example of lanthanide complexes that show stimuli-triggered synergistic and reversible switching of luminescence, magnetic and dielectric properties.
Introduction
Artificial smart materials that possess switchable and cooperative multifunctionality have garnered considerable attention in recent years, owing to their wide application in switches, sensors, displays and memory devices.1–3 The responsive dynamics of these materials often involve two distinct states, which can be optical, magnetic, conductive and dielectric, that switch upon exposure to an external stimulus.4–7 Among various types of multifunctional smart materials, metal–organic systems combining magnetism with optical8–12 and/or dielectric13–16 properties are of particular interest because of their importance in information storage and spintronic devices. However, despite many reports concerning the coexistence of two functions, simultaneous manipulation of different physical properties has been a challenging task. For example, luminescent lanthanide-based single molecule magnets (Ln-SMMs) are promising bifunctional materials.17–20 Although their SMM behavior can be sensitive to light,21 solvent,22–24 redox,25,26 and mechanical force,27,28 it is extremely difficult to modulate the luminescence and magnetism of Ln-SMMs synergistically. In particular, switchable Ln-SMMs with additional dielectric properties remain elusive.Chemists have succeeded in designing and synthesizing a variety of smart materials. Among them, photo-controllable materials are rather attractive not only because light can provide convenient and precise operation but also due to the change of their optical properties.29–32 Apart from the well-known photoswitches such as diarylethenes, azobenzene and olefins, anthracene and its derivatives have potential to exhibit a reversible [4 + 4] photocycloaddition reaction in the solid state when the anthracene groups are face-to-face π⋯π interacted.33 The reversible photocycloaddition reaction causes remarkable structural changes leading to drastic alteration of the physical properties of the corresponding materials.34–36 Noting that both the luminescence and SMM behavior of Ln-SMMs can be significantly affected by the structural changes arising from photochemical reactions37–41 and dielectric properties are related to the charge distribution of the material, we speculate that designing lanthanide complexes containing both photo-responsive and polar components can be a good solution to achieve optical, magnetic and dielectric triple switches using light as the trigger.
In this paper, we report a mononuclear complex [DyIII(SCN)3(depma)2(4-hpy)2] (1) containing both 9-diethylphosphonomethylanthracene (depma) as a photo-active component and 4-hydroxypyridine (4-hpy) as a polar component. In our design, the neutral ligand 4-hydroxypyridine was chosen due to not only its polar nature but also its capability to provide a short axial Dy–O bond as it possesses a resonant structure with the proton bound to the pyridine nitrogen atom. Three iso-thiocyanate anions and two photoactive depma ligands can afford N or O donors to occupy the five equatorial positions to complete a coordination sphere with D5h symmetry for the dysprosium ion which favors SMM behavior.42 Furthermore, the anthracene groups of depma from the neighboring molecules are face-to-face π⋯π interacted, favoring photocycloaddition reactions.33 Consequently, compound 1 is unique in the following aspects: (a) it shows reversible thermo-responsive phase transition associated with the order–disorder transition of 4-hpy and SCN−, which leads to thermochromic behavior and dielectric anomaly; (b) it shows reversible photo-induced dimerization of anthracene groups, which leads to synergistic switching of luminescence, magnetic and dielectric properties; (c) it shows SMM behavior under a zero dc field with an energy barrier of 141 K, surpassing the other known dysprosium–anthracene systems which show slow magnetic relaxation only under an external dc field.40,41,43 As far as we are aware, compound 1 is the first example of lanthanide complexes that display thermo- and/or light-triggered synergistic and reversible switching of luminescence, magnetic and dielectric properties.
Results and discussion
Crystal structures
Complex 1 was obtained as block yellow crystals from an acetonitrile solution of as-prepared Dy(SCN)3, depma and 4-hpy at room temperature. Single crystal structural analyses of 1 were performed at 300 K (1RT) and 193 K (1LT), respectively. The room-temperature phase 1RT crystalizes in a monoclinic space group P21/m (Table S1†). The asymmetric unit is composed of 0.5 DyIII, 1.5 SCN−, one depma and two half 4-hpy. Both 4-hpy are disordered over two sites (N3 and N3′ with occupancies of 0.55 and 0.46, N4 and N4A with half occupancy, and symmetry codes: A = x, 1/2 − y, z). The whole molecule is bisected by a mirror plane running through the DyIII ion, one disordered SCN− (S2, S2′), and one disordered 4-hpy (N3, N3′) (Fig. 1a). The DyIII ion is seven-coordinated with a pentagon-bipyramidal geometry slightly deviated from the ideal one [CShM = 0.96(4), Table S2†].44 The five equatorial positions are occupied by three nitrogen atoms (N1, N1A, and N2) from three SCN− anions and two oxygen atoms (O1 and O1A) from two depma [Dy–N(O): 2.312(4) to 2.433(5) Å, Table S3†]. The axial sites are filled with two oxygen atoms (O4/O4′ and O5/O5A) from two 4-hpy [Dy–O: 2.246(12)/2.188(15) and 2.207(6)]. The shorter axial Dy–O distances compared with the equatorial ones and the pseudo-linear arrangement of the axial atoms [O4/O4′–Dy1–O5 angles: 174.9(3) and 173.9(4)°] can enhance the magnetic anisotropy and favor the single ion magnet behavior (vide infra). | ||
| Fig. 1 (a and c) Molecular structures and (b and d) packing diagrams of compound 1 at 300 K (1RT) and 193 K (1LT). Weak intermolecular interactions such as π⋯π stacking, hydrogen bonding and S⋯S interactions are represented by dashes lines. | ||
Upon cooling down to 193 K, the crystals of 1 cracked. Fortunately, single crystallinity remained in a few crystals, one of which was subjected to the structural determination. Unlike 1RT, the low-temperature phase 1LT crystallizes in a triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The two 4-hpy ligands are completely ordered, and the molecular symmetry is lowered. As a result, the coordination geometry around the dysprosium ion in 1LT deviates significantly from that of an ideal pentagon-bipyramid (CShM = 2.019). Surprisingly, both the equatorial Dy–O(N) [2.347(12)–2.489(14) Å] and axial Dy–O [2.258(13), 2.251(12) Å] bond lengths in 1LT are slightly elongated compared with those in 1RT.
. The two 4-hpy ligands are completely ordered, and the molecular symmetry is lowered. As a result, the coordination geometry around the dysprosium ion in 1LT deviates significantly from that of an ideal pentagon-bipyramid (CShM = 2.019). Surprisingly, both the equatorial Dy–O(N) [2.347(12)–2.489(14) Å] and axial Dy–O [2.258(13), 2.251(12) Å] bond lengths in 1LT are slightly elongated compared with those in 1RT.
Extensive H-bonds are present in both phases between protonated pyridine nitrogen and SCN− anions forming supramolecular layers in the ac plane for 1RT or the ab plane for 1LT (Fig. 1 and Table S4†). The layers are stacked and further stabilized by face-to-face π–π interactions of the anthracene groups. In 1RT, there exists only one type of π–π interaction between the anthracene groups [plane-to-plane: 3.495 Å, center-to-center: 3.796 Å and nearest intermolecular C2–C9′: 3.786 Å]. But in 1LT, there are two types (plane-to-plane distance: 3.521 and 3.556 Å, center-to-center distance: 3.910 and 3.808 Å, C21–C28′: 3.877 Å, and C2–C9′: 3.775 Å). In both cases, the face-to-face π⋯π interacted anthracene groups meet the Schmidt principle for the [4 + 4] photocycloaddition reaction.33,45 The closest Dy⋯Dy distances within the H-bonded layer and between the layers are 10.081 and 13.039 Å in 1RT, and 9.592 and 13.152 Å in 1LT.
Thermo-induced phase transition
The fact that 1RT and 1LT crystallize in two different space groups demonstrates a thermo-triggered phase transition associated with the order-disorder transition of the 4-hpy and SCN− ligands. Differential scanning calorimetry (DSC) measurements on a polycrystalline sample of 1 revealed that it underwent a phase transition with an endothermic peak at ca. 297.5 K on heating, and an exothermic peak at ca. 240.2 K on cooling (Fig. 2). The thermal hysteresis (ΔT = 57.3 K) is extremely large compared with other metal–organic systems associated with order–disorder phase transition.7,46 To our knowledge, the only examples with wider hysteresis than 1 are perovskite compounds (Me3NCH2Cl)MnCl3 (ΔT = 72 K)47 and (CH3CH2NH3)[Mg(HCOO)3] (ΔT = 58 K for the first transition).48 According to the endothermal peak upon heating, the enthalpy change ΔH and entropy change ΔS are estimated to be 9.05 kJ mol−1 and 30.5 J mol−1 K−1, respectively. Based on the Boltzmann equation ΔS = R![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(N), where R is the gas constant and N represents the proportion of numbers of possible orientations for the disordered system, the value N is calculated to be 30.9. Such large values of ΔS and N indicate a severe order–disorder phase transition.
ln(N), where R is the gas constant and N represents the proportion of numbers of possible orientations for the disordered system, the value N is calculated to be 30.9. Such large values of ΔS and N indicate a severe order–disorder phase transition.
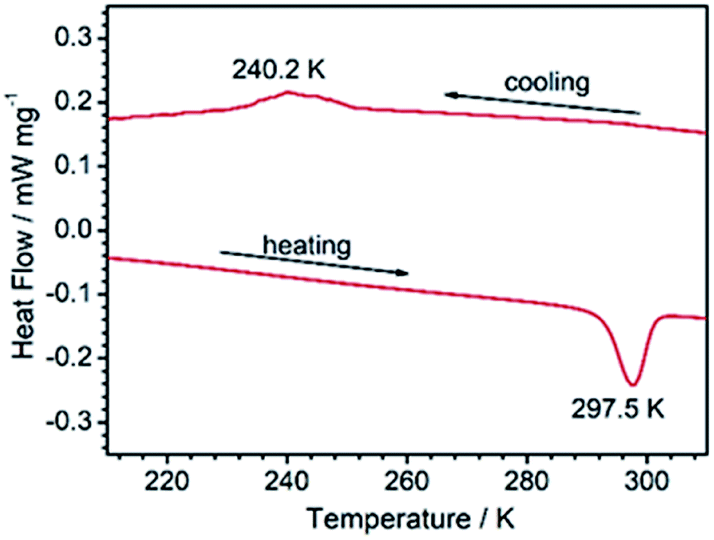 | ||
| Fig. 2 The DSC curves of 1 collected between 213 and 313 K at a sweeping rate of 5 K min−1 under a 150 mL min−1 N2 flow. | ||
A careful structural analysis reveals that the phase transition upon cooling is concomitant with not only the disorder-to-order transition of 4-hpy and SCN− ligands, but also the rearrangements of the SCN− and depma ligands. As shown in Fig. 3, there appears a flipping for the disordered SCN− accompanied by remarkable changes of Dy–N–S angles from 173.8, 155.7° (av. 164.8°) in 1RT to 144.1° in 1LT. The other two Dy–N–S angles are 159.4 and 159.4° for 1RT, and 151.8 and 161.9° for 1LT. The ethyl groups of depma also rotate somehow when the temperature is decreased, as evidenced by the torsion angles of P–O–C–C. For 1RT, there is only one crystallographically distinguished depma. The P–O–C–C torsion angles are 136.8 and 172.4°. For 1LT, two types of depma are found and their P–O–C–C torsion angles are 142.7 and 168.7° and 168.2 and 170.4°, respectively. The latter changes more significantly. All these structural rearrangements together with the order–disorder transition of the 4-hpy and SCN− ligands should contribute to the wide thermal hysteresis of the DSC curve and large ΔS and N values.
 | ||
| Fig. 3 Structural transformation from 1RT (a) into 1LT (b) upon cooling. The anthracene groups are omitted for clarity. | ||
Moreover, the DSC curve suggests that the isolated low temperature phase can be stable up to ca. 290 K, thus making the characterization of both 1RT and 1LT at near room temperature possible. Indeed, the powder X-ray diffraction pattern of the polycrystalline sample of 1LT obtained at 293 K is in line with the one simulated from the single crystal data obtained at 193 K except slight shifts of 2θ angles (ca. +0.15°) due to the difference of measuring temperature (Fig. S1†), thus confirming the purity of the low temperature phase.
Thermo- and light-triggered optical switch
Structural analyses revealed that the thermo-induced phase transition led to a slight change in the π⋯π stacking of the anthracene moieties, e.g. one type in 1RT and two types in 1LT. The phase transition is reflected by their optical properties. As shown in Fig. 4a and S2,†1RT emits a broad and strong band peaking at 535 nm with a lifetime of 37.3 ns (Table S5†), stemming from excimer emission through pairing of the neighbouring anthracene.49 An identical emission spectrum is observed for 1LT except a small hypsochromic shift of the peak (530 nm) by 5 nm owing to slightly different arrangements of the anthracene pairs. The lifetime is 44.7 ns. The spectrum can be completely reversed after warming up 1LT above 298 K, manifesting a reversible thermochromic behaviour.The presence of distinct π⋯π interactions between the anthracene groups favors photocycloaddition reactions. Time-dependent photoluminescence spectra were recorded by exposing a 2 mg powder sample of 1RT to 365 nm UV light at a power of 185 mW cm−2 for different periods of time. The peak intensity at 535 nm declines with increasing irradiation time, concomitant with the emergence of two peaks at 424 and 446 nm and two shoulder peaks at 485 and 575 nm (Fig. S3a†). The lifetimes for the peaks at 424 and 446 nm (1.2 and 1.3 ns, Table S5†) are on the nanosecond scale. They belong to the π*–π transition of dianthracene moieties after photodimerization.40,50 The emission peaks at 485 and 575 nm possess a microsecond lifetime (21.2 and 20.7 μs), and are ascribed to the characteristic transition of DyIII ions from 4F9/2 to 6H15/2 and 6H13/2 (Fig. 4a). Apparently the cycloaddition of face-to-face anthracene moieties raises the energy of the 3T state of the ligand center and sensitizes the f–f transition of the dysprosium ion. After 20 min of irradiation, the intensities of all peaks become constant, indicating that the photocycloaddition reaction is complete.
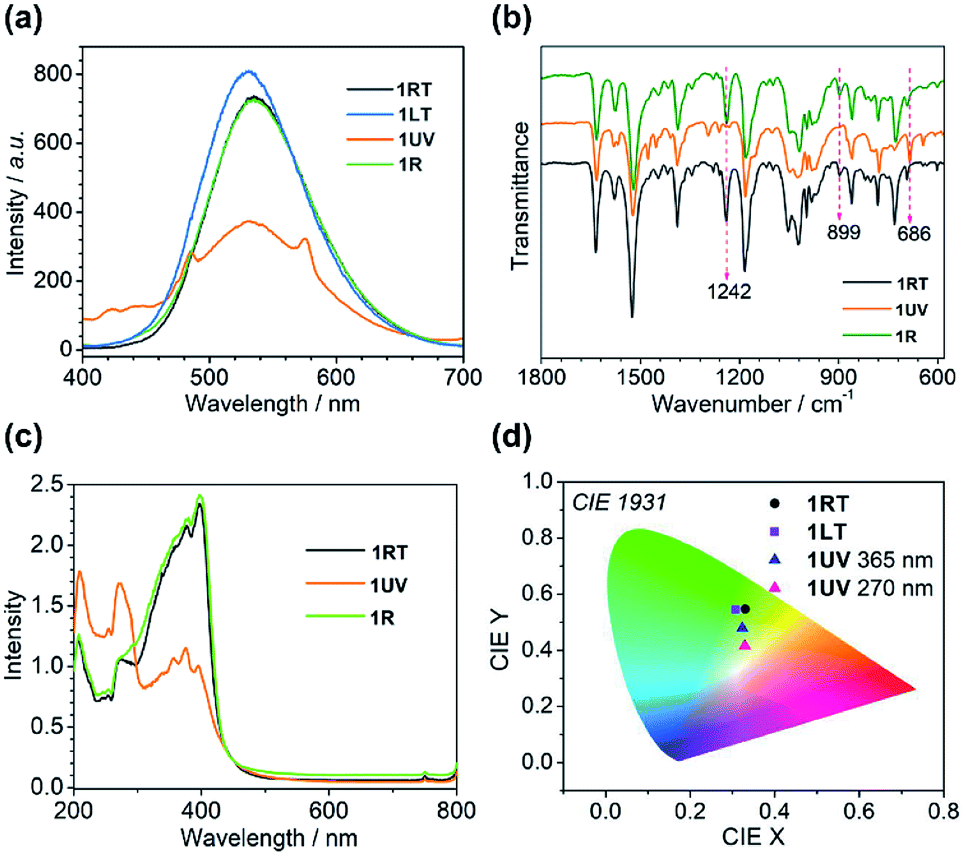 | ||
| Fig. 4 (a) The emission spectra excited at 365 nm for 1RT, 1LT, 1UV and 1R. (b) The IR spectra of 1RT, 1UV and 1R. (c) The absorption spectra of 1RT, 1LT, 1UV and 1R in the solid state. (d) The 1931 CIE graphs of 1RT, 1LT and 1UV. | ||
The time-dependent IR spectra can provide structural information of the photocycloaddition reaction. Upon 365 nm UV light irradiation, the intensities of the vibration bands peaking at 1242 and 899 cm−1, assigned to the bending vibration of C–H bonds of the anthracene moieties, decline and finally disappear after 40 min (Fig. S4†).51,52 In contrast, a new peak emerges at 686 cm−1, characteristic of dianthracene, the intensity of which reaches a constant after 40 min of irradiation. The result again supports the occurrence of a photocycloaddition reaction.
A complete photocycloaddition reaction of 1RT would result in a one-dimensional coordination polymer, named 1UV. To prepare 1UV for other physical measurements, a 20 mg polycrystalline sample of 1RT was irradiated by UV light for 20 hours to ensure a complete photocycloaddition reaction, confirmed by luminescence and IR measurements (Fig. S3† and 4b). Unfortunately, single crystals of 1RT were cracked after UV light irradiation with the loss of single crystallinity, making it impossible to determine the single crystal structure of 1UV (Fig. S5†). It is noted, however, that the PXRD pattern of 1UV is distinguished from that of 1RT but close to that of 1LT, suggesting that the 4-hpy ligands become ordered after the photocycloaddition reaction (Fig. S6†). This surmise is supported by DSC measurements which show no peak upon heating and cooling (Fig. S7†). The observed diffraction peaks of 1UV can be well fitted by the TOPAS program53 using the same space group P as that of 1LT (Fig. S8†). The resulted parameters are a = 8.55 Å, b = 11.96 Å, c = 25.84 Å, α = 89.85°, β = 92.26°, γ = 112.15°, and V = 2731.4 Å3.
The photocycloaddition reaction was also performed for 1LT at 273 or 253 K. The product shows an identical PXRD pattern and IR and luminescence spectra to 1UV (Fig. S6 and S9†). The results imply that UV light irradiation of both 1RT and 1LT results in the same photodimerized phase 1UV.
Obviously, the photocycloaddition reaction of 1 causes a significant structural change, accompanied by a switch of its luminescence properties. The 1931 CIE (Commission Internationale de l' Eclairage) chromaticity coordinates change from (0.33, 0.55) for 1RT to (0.32, 0.48) for 1UV as a consequence of multiple bands in the luminescence from both the ligand and DyIII. The colour is yellowish green. Interestingly, the excitation of 1UV at 270 nm leads to a nearly complete energy transfer from the ligand to DyIII with the observation of two predominant emission peaks at 485 and 575 nm (Fig. S3†). Accordingly, the color changes to yellow with CIE coordinates (0.33, 0.42) (Fig. 4d).
The UV-vis absorption spectra are also different for 1RT and 1UV (Fig. 4c). The former shows three peaks around 207, 271 and 398 nm giving a characteristic green-yellow color attributed to the π–π* transition of the anthracene group. Meanwhile, one weak peak at 756 nm is ascribed to the f–f transitions of the DyIII ion from 6H15/2 to 6F3/2. For 1UV, the absorption band between 300 and 440 nm displays several weak structural peaks while the band around 207 and 271 nm becomes very strong, in agreement with the formation of dianthracene after photodimerization.54
The photodimerization process can be reversed by annealing of 1UV at 120 °C for 10 min. The resultant product, 1R, shows identical luminescence and IR and UV-vis spectra as well as the PXRD pattern to 1RT (Fig. 4, S10 and S11†). The thermo-induced de-dimerization process can be monitored by thermogravimetric analysis (TGA) and DSC measurements. As shown in Fig. 5, there is no weight loss for 1UV below 160 °C. However, a sharp exothermic peak appears at 106 °C in the DSC curve (Fig. 5, inset). The enthalpy is estimated to be −47.8 kJ mol−1 per molecule, which is comparable to those for  (NO3)6(hmpa)4 (−46.8 kJ mol−1 for both de-dimerization and conformation change of depma2),40 and Dy2L2(depma2)Cl2 [H2L = N1,N3-bis(salicylideneimino)diethylenetriamine] (−42.4 kJ mol−1).55
(NO3)6(hmpa)4 (−46.8 kJ mol−1 for both de-dimerization and conformation change of depma2),40 and Dy2L2(depma2)Cl2 [H2L = N1,N3-bis(salicylideneimino)diethylenetriamine] (−42.4 kJ mol−1).55
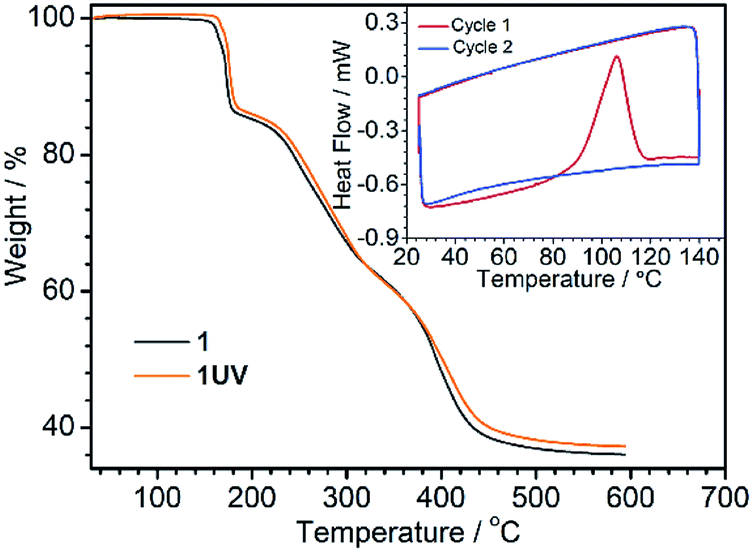 | ||
| Fig. 5 TGA curves of 1 (grey) and 1UV (yellow) measured under a nitrogen flow at a heating rate of 5 °C min−1. Inset: the DSC curves for 1UV between 25 and 140 °C at a scan speed of 5 °C min−1 for two cycles. | ||
Fig. 6 highlights the structural transformation of compound 1RT into 1LT by cooling and into 1UV by light irradiation, both of which are reversible by simply warming up to room temperature or annealing at 120 °C. Light illumination of 1LT results in the same product of 1UV, but this process is not reversible.
 | ||
| Fig. 6 Structural transformation among 1RT, 1LT and 1UV. A proposed structure of 1UV is presented for clarity. | ||
It is worth mentioning that only a handful metal–anthracene complexes (Ru,56 Re,57 Cu,36 Ag,58 Pt,35 and Ln40,41) are known to undergo a [4 + 4] photocycloaddition reaction upon UV irradiation either in solution or in the solid state. Metal–organic systems showing both reversible thermochromism and photodimerization of anthracene have never been documented thus far.
Thermo- and light-triggered dielectric switch
The thermo-induced order–disorder transition of 1 can be monitored by dielectric measurements. Variable temperature dielectric spectra were recorded on a pressed powder sample of 1 in the range 193–323 K and 1.1–1328 kHz. Fig. 7 shows clearly that the real part (ε′) of the relative permittivity of 1 remains almost constant above 260 K with a value of 26.8, corresponding to a high-dielectric state of 1RT. Then a rapid drop is followed to a value of 26.1 at 193 K, ascribed to a transition to the low-dielectric state of 1LT. This dielectric anomaly is reversible upon heating. The cooling and heating processes lead to an obvious hysteresis in the temperature range of 200–290 K, in agreement with the DSC result (Fig. 2 and S12†).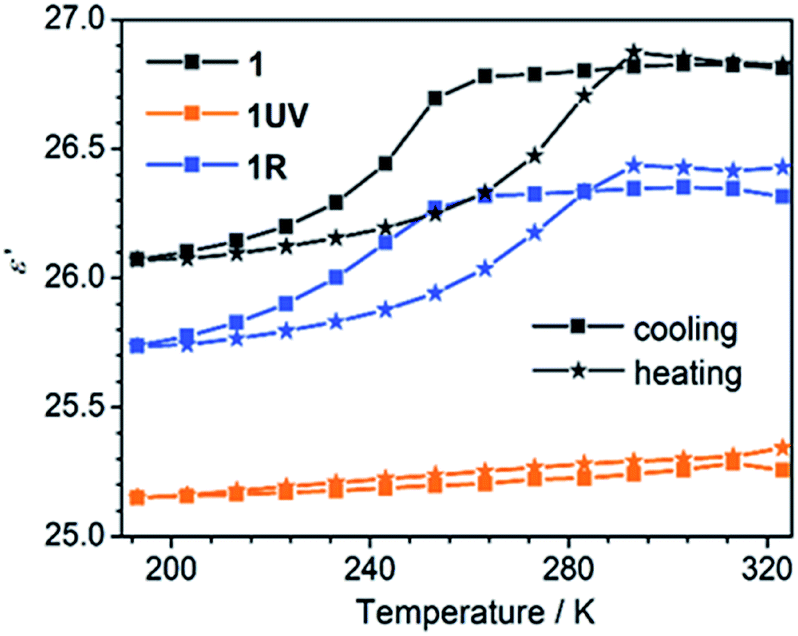 | ||
| Fig. 7 Temperature-dependent dielectric constants for 1, 1UV and 1R in 193–323 K at a frequency of 1328 kHz. | ||
The dielectric response of 1 is associated with the local dynamic changes of polar components due to the dipolar orientation transformation between ordered and disordered states, as found in many other molecular phase transition systems.46,59,60 For the room temperature phase 1RT, there are two kinds of 4-hpy ligands showing rotor-like and pendulum-like motions, respectively (Fig. 6). While for the low temperature phase 1LT, the 4-hpy ligands are in the static state without disorder. The polar motion of 4-hpy can be affected by the intermolecular hydrogen bonds involving 4-hpy as well as the temperature. As shown in Fig. 1, the 4-hpy forms N–H⋯S hydrogen bonds with the SCN− anions from neighboring molecules. Noting that these H-bond interactions in 1RT [N⋯S: 2.63–3.45 Å] are comparable to or slightly stronger than those in 1LT [N⋯S: 3.26–3.42 Å] (Table S4†), the polar motion of the disordered state (1RT) can be mainly a result of thermal agitation. No evident frequency dependence of the dielectric constant is observed, suggesting that the polar motion is much faster than 1.3 MHz.
After the photocycloaddition reaction, the ε′ value of 1UV drops to ca. 25.2 and keeps nearly constant in the whole measured temperature range. The result manifests that the relative permittivity ε′ of 1 is reduced and its thermo-induced dielectric transition behavior is switched off after the photochemical reaction. The DSC measurement also supports this conclusion, which shows no thermal anomaly in the range 193–323 K (Fig. S7†). Interestingly, annealing of 1UV leads to the recovery of the pristine phase (1R), accompanied by the switch-on of the thermo-induced dielectric anomaly (Fig. 7).
Notably, although thermo-induced dielectric switch was observed in a number of molecular systems,7,46 there are only two compounds reported exhibiting light-triggered dielectric switch. One is a diarylethene derivative having imidazoline rings which isomerizes between open- and closed-ring isomers upon light irradiation,61a and the other is a zinc–olefin coordination polymer showing a [2 + 2] photocycloaddition reaction.61b Thus compound 1 provides the first example of metal–organic complexes showing both thermo- and light-triggered dielectric switches.
Light-triggered magnetic switch
The dc susceptibilities were measured for 1 in the temperature range of 300–2 K. The χMT product is 14.14 cm3 K mol−1 at 300 K (Fig. 8), in accordance with the expected value of 14.17 cm3 K mol−1 for one isolated DyIII ion (6H15/2, S = 5/2, L = 5, and gJ = 4/3). Upon lowering the temperature, it slightly decreases to a minimum of 12.81 cm3 K mol−1 at 14 K, ascribed to the depopulation of Stark sublevels. The upturn of χMT below 14 K is indicative of dominant ferromagnetic interaction between DyIII ions.62 Notably, the change of χMT in the temperature range 240–300 K associated with the order–disorder phase transition is not distinct. Magnetization measurement at 2 K reveals a rapid increase below 10 kOe followed by a gradual increase up to 5.21 μB (Fig. S13 and S14†). The value is far from the saturated value of 10 μB, attributed to the presence of magnetic anisotropy and/or low lying excited states which is further ensured by the non-superposition of the M vs. H/T plots at different temperatures (Fig. S14†).63 Remarkably, a butterfly shaped hysteresis loop is observed at 2.5 K or lower (Fig. 9a), implying a SMM behavior.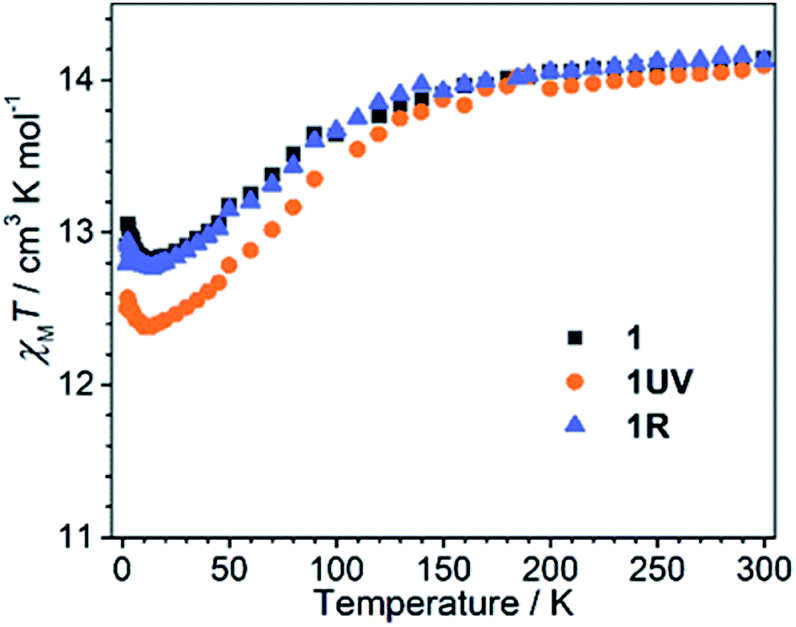 | ||
| Fig. 8 The χMT vs. T plots for 1RT, 1UV and 1R measured under a 1 kOe dc field. | ||
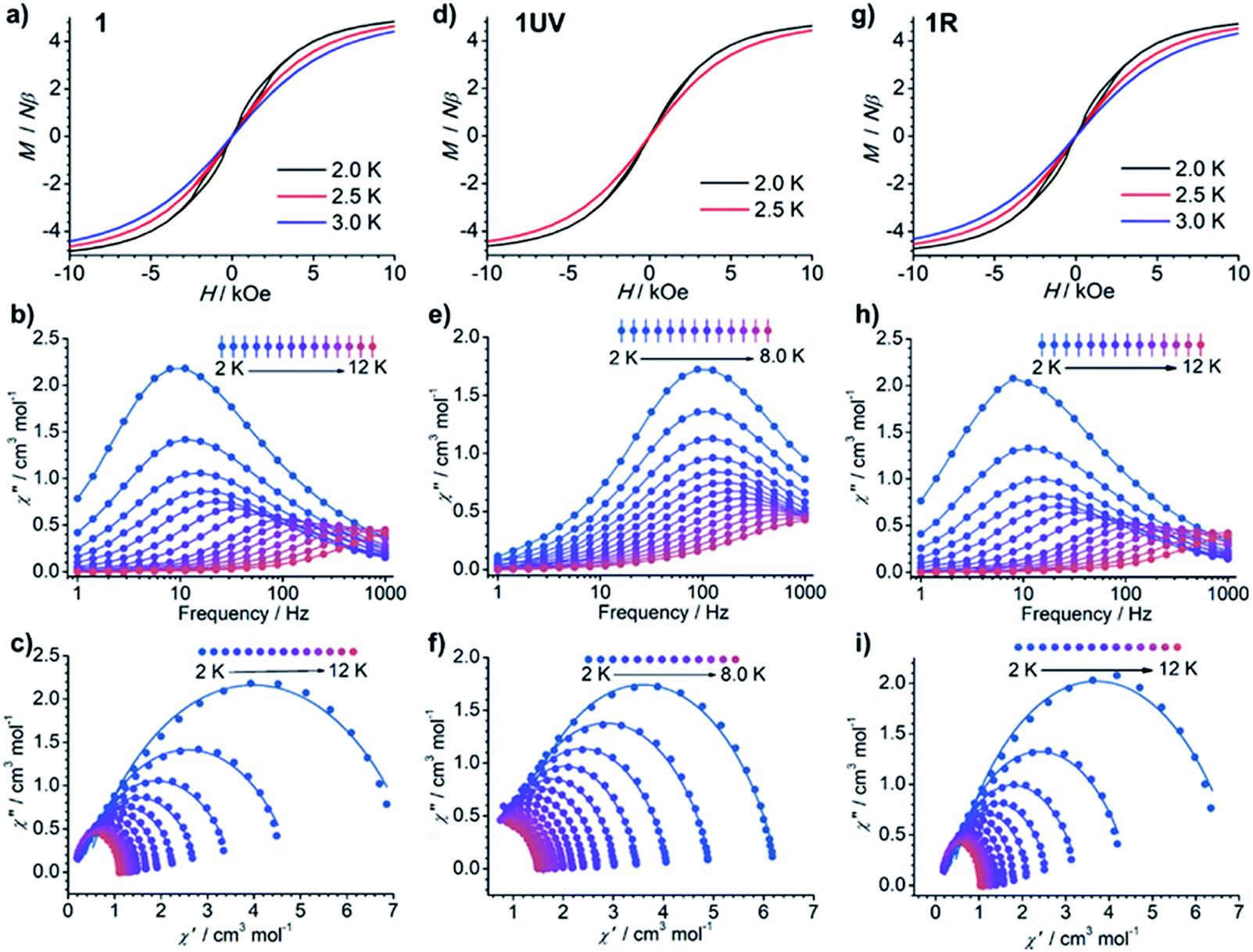 | ||
| Fig. 9 (a, d and g) The hysteresis curves, (b, e and h) out-of-phase ac susceptibilities (χ′′), and (c, f and i) corresponding Cole–Cole plots for 1 (a–c), 1UV (d–f) and 1R (g–i). | ||
The alternating current (ac) susceptibility measurements of 1 demonstrate evident frequency (ν) dependent in-phase (χ′) and out-of-phase (χ′′) ac susceptibilities under a zero dc field (Fig. 9b and S15†). The ac susceptibility curves and Cole–Cole plots (Fig. 9c) were fitted to a generalized Debye model to extract the relaxation time (τ) and distribution coefficient α (Table S6†).64,65 The lnτ vs. T−1 plot can be well fitted by eqn (1)66 which contains quantum tunneling of magnetization (QTM), and Raman-like and Orbach relaxation processes, and the resultant parameters are given in Table 1.
| τ−1 = τQTM−1 + CTn + τ−1exp(−Ueff/kBT) | (1) |
| Compound | χ M T/cm3 K mol−1 | M/μB | U eff/K | τ 0/10−9 s | C/K−n s−1 | n | τ QTM/s |
|---|---|---|---|---|---|---|---|
| 1 | 14.14 (300 K) | 5.21 (2 K, 7 T) | 141 (0 Oe) | 1.1 | 0.3 | 3.4 | 0.013 |
| 12.92 (2 K) | 181 (1 kOe) | 0.04 | 2.4 × 10−4 | 6.4 | — | ||
| 1UV | 14.09 (300 K) | 5.18 (2 K, 7 T) | 101 (0 Oe) | 1.0 | 1.5 | 3.9 | 0.0014 |
| 12.50 (2 K) | 123 (1 kOe) | 1.0 | 4.7 | 7.2 | — | ||
| 1R | 14.12 (300 K) | 5.18 (2 K, 7 T) | 153 (0 Oe) | 0.4 | 0.18 | 3.6 | 0.013 |
| 12.78 (2 K) | — | — | — | — | — |
The best fit leads to an energy barrier (Ueff) of 141 K (τ0 = 1.1 × 10−9 s). When an external field of 1 kOe is applied, the QTM process is suppressed (Fig. S16 and S17; Tables S7 and S8†). The lnτ vs. T−1 plot can be fitted by considering Raman-like and Orbach processes,67 leading to parameters Ueff = 181 K and τ0 = 4.0 × 10−11. The energy barrier is increased by ca. 40 K compared to that obtained under a zero dc field. It is about nine times higher than that of DyIII(NO3)3(depma) (hmpa)2 (Ueff = 20.4 K),40 demonstrating significantly improved SMM performance when the DyIII ion is placed in a D5h environment with a short Dy–O axial distance.
Dramatic magnetic changes are observed when 1 is subjected to UV light irradiation. As shown in Fig. 8, although the overall profiles of χMT vs. T are quite similar for 1UV and 1, the χMT value declines more quickly upon cooling for 1UV than that for 1. Furthermore, the hysteresis loop is not obvious for 1UV down to 2 K (Fig. 9d), unlike compound 1 which shows butterfly hysteresis below 2.5 K. The ac susceptibilities of 1UV also show frequency dependent signals under a zero dc field, but the peaks in the χ′′(ν) curves appear at much higher frequencies than those for 1 at the same temperature (Fig. 9e, S18–S20 and Tables S9–S11†). The energy barriers for 1UV are 101 K under a zero dc field and 123 K at 1 kOe, both are much lower than those for 1. Clearly, photodimerization of the anthracene groups in 1 results in a reduction of the magnetization reversal barrier and the acceleration of the magnetic relaxation, which can be related to the symmetry lowering around the DyIII ion after the photocycloaddition reaction. Interestingly, annealing of 1UV leads to 1R with the recovery of the butterfly-like hysteresis loop (Fig. 9g) and the ac profile (Fig. 9h) with an energy barrier of 153 K under a zero dc field (Fig. S21 and Table S12†), implying that the photo-induced magnetic switching is reversible.
Reversible light-responsive SMMs with a high energy barrier and open magnetic hysteresis loop are anxiously desired for their applications in molecular spintronics and devices. As shown in Table S13,† photo-induced reversible magnetic switches were achieved only in three Dy complexes so far,21,39,40 among which [Dy(Tppy)F(Lc)](PF6) is the only one showing a light-modulable butterfly-like hysteresis loop in response to the photoisomerization of the bridging dithienylethene ligand. But the energy barrier remains the same (225.9 K, 157 cm−1) before and after light irradiation (ΔUeff = 0).21 Compound 1, which also shows a butterfly-like hysteresis loop below 2.5 K, has an advantage over [Dy(Tppy)F(Lc)](PF6) in the big difference of the energy barrier before and after light irradiation (ΔUeff = 40 K). Moreover, its photoluminescence and dielectric properties can be modulated by thermal or light treatment. All these characteristics make compound 1 stand out in the responsive Ln-SMMs.
Conclusions
In summary, we successfully designed a new lanthanide-based single molecule magnet, [DyIII(SCN)3(depma)2(4-hpy)2] (1), which combines the SMM behavior with photo-reactivity and polar characteristics. The compound shows thermo-responsive phase transition associated with the order–disorder transition of 4-hpy and SCN− leading to reversible thermochromic behavior and dielectric anomaly and light-responsive photodimerization of anthracene groups leading to reversible and synergistic switching of luminescence, magnetic and dielectric properties. Simultaneous manipulation of tri-functions appears extremely rare in molecular systems, and the current work provides a feasible strategy for the design of multifunctional molecular switches, especially those with more than two functions. The work may also shed light on the development of smart molecular materials with reconfigurable properties for applications in molecular devices and sensors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the National Key R&D Program of China (2017YFA0303203 and 2018YFA0306004) and the National Natural Science Foundation of China (21731003). We thank Dr Diming Xu at Peking University for valuable discussions.Notes and references
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef.
- O. Sato, Nat. Chem., 2016, 8, 644–656 CrossRef CAS.
- F. Bigdeli, C. T. Lollar, A. Morsali and H.-C. Zhou, Angew. Chem., Int. Ed., 2020, 59, 4652–4669 CrossRef CAS.
- P. Chen, Q. Li, S. Grindy and N. Holten-Andersen, J. Am. Chem. Soc., 2015, 137, 11590–11593 CrossRef CAS.
- E. Coronado, Nat. Rev. Mater., 2020, 5, 87–104 CrossRef.
- S. Garg, H. Schwartz, M. Kozlowska, A. B. Kanj, K. Mgller, W. Wenzel, U. Ruschewitz and L. Heinke, Angew. Chem., Int. Ed., 2019, 58, 1193–1197 CrossRef CAS.
- W. Zhang and R.-G. Xiong, Chem. Rev., 2012, 112, 1163–1195 CrossRef CAS.
- M. Arczyński, J. Stanek, B. Sieklucka, K. R. Dunbar and D. Pinkowicz, J. Am. Chem. Soc., 2019, 141, 19067–19077 CrossRef.
- Y.-S. Meng, O. Sato and T. Liu, Angew. Chem., Int. Ed., 2018, 57, 12216–12226 CrossRef CAS.
- J. Yuan, S.-Q. Wu, M.-J. Liu, O. Sato and H.-Z. Kou, J. Am. Chem. Soc., 2018, 140, 9426–9433 CrossRef CAS.
- M. M. Paquette, D. Plaul, A. Kurimoto, B. O. Patrick and N. L. Frank, J. Am. Chem. Soc., 2018, 140, 14990–15000 CrossRef CAS.
- J.-Y. Ge, Z. Chen, L. Zhang, X. Liang, J. Su, M. Kurmoo and J.-L. Zuo, Angew. Chem., Int. Ed., 2019, 58, 8789–8793 CrossRef CAS.
- Y. Tian, S. Shen, J. Cong, L. Yan, S. Wang and Y. Sun, J. Am. Chem. Soc., 2016, 138, 782–785 CrossRef CAS.
- G.-C. Xu, W. Zhang, X.-M. Ma, Y.-H. Chen, L. Zhang, H.-L. Cai, Z.-M. Wang, R.-G. Xiong and S. Gao, J. Am. Chem. Soc., 2011, 133, 14948–14951 CrossRef CAS.
- (a) P.-P. Shi, Q. Ye, Q. Li, H.-T. Wang, D.-W. Fu, Y. Zhang and R.-G. Xiong, Chem. Mater., 2014, 26, 6042–6049 CrossRef CAS; (b) D. Li, X.-M. Zhao, H.-X. Zhao, L.-S. Long and L.-S. Zheng, Inorg. Chem., 2019, 58, 655–662 CrossRef CAS.
- (a) Y.-X. Wang, W. Shi, H. Li, Y. Song, L. Fang, Y. Lan, A. K. Powell, W. Wernsdorfer, L. Ungur, L. F. Chibotaru, M. Shen and P. Cheng, Chem. Sci., 2012, 3, 3366–3370 RSC; (b) B. Huang, J.-Y. Zhang, R.-K. Huang, M.-K. Chen, W. Xue, W.-X. Zhang, M.-H. Zeng and X.-M. Chen, Chem. Sci., 2018, 9, 7413–7418 RSC.
- D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110–5148 CrossRef CAS.
- F. Pointillart, B. Guennic, O. Cador, O. Maury and L. Ouahab, Acc. Chem. Res., 2015, 48, 2834–2842 CrossRef CAS.
- (a) J. Long, Y. Guari, R. A. S. Ferreira, L. D. Carlos and J. Larionova, Coord. Chem. Rev., 2018, 363, 57–70 CrossRef CAS; (b) J.-H. Jia, Q.-W. Li, Y.-C. Chen, J.-L. Liu and M.-L. Tong, Coord. Chem. Rev., 2019, 378, 365–381 CrossRef CAS.
- R. Marin, G. Brunet and M. Murugesu, Angew. Chem., Int. Ed., 2019 DOI:10.1002/anie.201910299.
- M. Hojorat, H. A. Sabea, L. Norel, K. Bernot, T. Roisnel, F. Gendron, B. L. Guennic, E. Trzop, E. Collet, J. R. Long and S. Rigaut, J. Am. Chem. Soc., 2020, 142, 931–936 CrossRef CAS.
- Y. Xin, J. Wang, M. Zychowicz, J. J. Zakrzewski, K. Nakabayashi, B. Sieklucka, S. Chorazy and S.-i. Ohkoshi, J. Am. Chem. Soc., 2019, 141, 18211–18220 CrossRef CAS.
- X. Zhang, V. Vieru, X. Feng, J.-L. Liu, Z. Zhang, B. Na, W. Shi, B.-W. Wang, A. K. Powell, L. F. Chibotaru, S. Gao, P. Cheng and J. R. Long, Angew. Chem., Int. Ed., 2015, 54, 9861–9865 CrossRef CAS.
- H. Tian, J.-B. Su, S.-S. Bao, M. Kurmoo, X.-D. Huang, Y.-Q. Zhang and L.-M. Zheng, Chem. Sci., 2018, 9, 6424–6433 RSC.
- J. D. Rinehart, M. Fang, W. J. Evans and J. R. Long, Nat. Chem., 2011, 3, 538–542 CrossRef CAS.
- C. M. Dickie, A. L. Laughlin, J. D. Wofford, N. S. Bhuvanesha and M. Nippe, Chem. Sci., 2017, 8, 8039–8049 RSC.
- W.-B. Chen, Y.-C. Chen, J.-L. Liu, J.-H. Jia, L.-F. Wang, Q.-W. Li and M.-L. Tong, Inorg. Chem., 2017, 56, 8730–8734 CrossRef CAS.
- X.-D. Huang, M. Kurmoo, S.-S. Bao, K. Fan, Y. Xu, Z.-B. Hu and L.-M. Zheng, Chem. Commun., 2018, 54, 3278–3281 RSC.
- D.-H. Qu, Q.-C. Wang, Q.-W. Zhang, X. Ma and H. Tian, Chem. Rev., 2015, 115, 7543–7588 CrossRef CAS.
- A. M. Rice, C. R. Martin, V. A. Galitskiy, A. A. Berseneva, G. A. Leith and N. B. Shustova, Chem. Rev., 2020, 120, 8790–8813 CrossRef CAS.
- J. J. Vittal and H. S. Quah, Dalton Trans., 2017, 46, 7120–7140 RSC.
- S.-L. Huang, T. S. A. Hor and G.-X. Jin, Coord. Chem. Rev., 2017, 346, 112–122 CrossRef CAS.
- G. M. J. Schmidt, Pure Appl. Chem., 1971, 27, 647–678 CAS.
- M. Tu, H. Reinsch, S. Rodríguez-Hermida, R. Verbeke, T. Stassin, W. Egger, M. Dickmann, B. Dieu, J. Hofkens, I. F. J. Vankelecom, N. Stock and R. Ameloot, Angew. Chem., Int. Ed., 2019, 58, 2423–2427 CrossRef CAS.
- Q. Yu, M. Li, J. Gao, P. Xu, Q. Chen, D. Xing, J. Yan, M. J. Zaworotko, J. Xu, Y. Chen, P. Cheng and Z. Zhang, Angew. Chem., Int. Ed., 2019, 58, 18634–18640 CrossRef CAS.
- M. Castellano, J. Ferrando-Soria, E. Pardo, M. Julve, F. Lloret, C. Mathonière, J. Pasán, C. Ruiz-Pérez, L. Cañadillas-Delgado, R. Ruiz-GarcÍa and J. Cano, Chem. Commun., 2011, 47, 11035–11037 RSC.
- L.-F. Wang, J.-Z. Qiu, Y.-C. Chen, J.-L. Liu, Q.-W. Li, J.-H. Jia and M.-L. Tong, Inorg. Chem. Front., 2017, 4, 1311–1318 RSC.
- H. Meng, C. Zhao, M. Nie, C. Wang and T. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 32607–32612 CrossRef CAS.
- Y.-J. Ma, J.-X. Hu, S.-D. Han, J. Pan, J.-H. Li and G.-M. Wang, J. Am. Chem. Soc., 2020, 142, 2682–2689 CrossRef CAS.
- X.-D. Huang, Y. Xu, K. Fan, S.-S. Bao, M. Kurmoo and L.-M. Zheng, Angew. Chem., Int. Ed., 2018, 57, 8577–8581 CrossRef CAS.
- X.-D. Huang, J.-G. Jia, M. Kurmoo, S.-S. Bao and L.-M. Zheng, Dalton Trans., 2019, 48, 13769–13779 RSC.
- J.-L. Liu, Y.-C. Chen and M.-L. Tong, Chem. Soc. Rev., 2018, 47, 2431–2453 RSC.
- Q. Zou, X.-D. Huang, J.-C. Liu, S.-S. Bao and L.-M. Zheng, Dalton Trans., 2019, 48, 2735–2740 RSC.
- (a) M. Pinsky and D. Avnir, Inorg. Chem., 1998, 37, 5575–5582 CrossRef CAS; (b) D. Casanova, P. Alemany, J. M. Bofill and S. Alvarez, Chem. – Eur. J., 2003, 9, 1281–1295 CrossRef CAS.
- I. Zouev, D.-K. Cao, T. V. Sreevidya, M. Telzhensky, M. Botoshanskya and M. Kaftory, CrystEngComm, 2011, 13, 4376–4381 RSC.
- C. Shi, X.-B. Ha and W. Zhang, Coord. Chem. Rev., 2019, 378, 561–576 CrossRef CAS.
- Q. Wang, Y.-W. Zhang, W.-Y. Zhang, P.-P. Shi, Q. Ye and D.-W. Fu, J. Mater. Chem. C, 2019, 7, 2994–3002 RSC.
- R. Shang, G.-C. Xu, Z.-M. Wang and S. Gao, Chem. – Eur. J., 2014, 20, 1146–1158 CrossRef CAS.
- H.-C. Liu, Y. Gao and B. Yang, Chin. Sci. Bull., 2017, 62, 4099–4112 CrossRef.
- P. Payamyar, K. Kaja, C. Ruiz-Vargas, A. Stemmer, D. J. Murray, C. J. Johnson, B. T. King, F. Schiffmann, J. VandeVondele, A. Renn, S. Götzinger, P. Ceroni, A. Schütz, L.-T. Lee, Z. Zheng, J. Sakamoto and A. D. Schlüter, Adv. Mater., 2014, 26, 2052–2058 CrossRef CAS.
- R. Bhola, P. Payamyar, D. J. Murray, B. Kumar, A. J. Teator, M. U. Schmidt, S. M. Hammer, A. Saha, J. Sakamoto, A. D. Schlüter and B. T. King, J. Am. Chem. Soc., 2013, 135, 14134–14141 CrossRef CAS.
- N. Huang, X. Ding, J. Kim, H. Ihee and D. Jiang, Angew. Chem., Int. Ed., 2015, 54, 8704–8707 CrossRef CAS.
- TOPAS, Version 5.0, Bruker AXS Inc., Madison, WI, 2014 Search PubMed.
- S. R. Jezowski, L. Zhu, Y. Wang, A. P. Rice, G. W. Scott, C. J. Bardeen and E. L. Chronister, J. Am. Chem. Soc., 2012, 134, 7459–7466 CrossRef CAS.
- J.-C. Liu, X.-D. Huang, Q. Zou, S.-S. Bao, X.-Z. Wang, J.-Y. Ma and L.-M. Zheng, J. Mater. Chem. C, 2020, 8, 7369–7377 RSC.
- (a) T. D. Trouts, D. S. Tyson, R. Pohl, D. V. Kozlov, A. G. Waldron and F. N. Castellano, Adv. Funct. Mater., 2003, 13, 398–402 CrossRef CAS; (b) V. Ferri, M. Scoponi, C. A. Bignozzi, D. S. Tyson, F. N. Castellano, H. Doyle and G. Redmond, Nano Lett., 2004, 4, 835–839 CrossRef CAS; (c) E. E. Karslyan, A. I. Konovalov, A. O. Borissova, P. V. Petrovskii and A. R. Kudinov, Mendeleev Commun., 2011, 21, 309–331 CrossRef CAS.
- A. Beyeler, P. Belser and L. De Cola, Angew. Chem., Int. Ed. Engl., 1997, 36, 2779–2781 CrossRef CAS.
- F. Spinelli, S. d'Agostino, P. Taddei, C. D. Jones, J. W. Steed and F. Grepioni, Dalton Trans., 2018, 47, 5725–5733 RSC.
- P.-P. Shi, Y.-Y. Tang, P.-F. Li, W.-Q. Liao, Z.-X. Wang, Q. Ye and R.-G. Xiong, Chem. Soc. Rev., 2016, 45, 3811–3827 RSC.
- H.-Y. Zhang, Y.-Y. Tang, P.-P. Shi and R.-G. Xiong, Acc. Chem. Res., 2019, 52, 1928–1938 CrossRef CAS.
- (a) M. Morimoto and M. Irie, Chem. Commun., 2011, 47, 4186–4188 RSC; (b) J.-Y. Sima, H.-X. Li, D. J. Young, P. Braunstein and J.-P. Lang, Chem. Commun., 2019, 55, 3532–3535 RSC.
- A. B. Canaj, M. K. Singh, E. R. Marti, M. Damjanović, C. Wilson, O. Céspedes, W. Wernsdorfer, G. Rajaraman and M. Murrie, Chem. Commun., 2019, 55, 5950–5953 RSC.
- K. L. M. Harriman, J. L. Brosmer, L. Ungur, P. L. Diaconescu and M. Murugesu, J. Am. Chem. Soc., 2017, 139, 1420–1423 CrossRef CAS.
- K. S. Cole and R. H. Cole, J. Chem. Phys., 1941, 9, 341–351 CrossRef CAS.
- Y.-N. Guo, G.-F. Xu, Y. Guo and J. Tang, Dalton Trans., 2011, 40, 9953–9963 RSC.
- G. Brunet, R. Marin, M.-J. Monk, U. Resch-Genger, D. A. Gálico, F. A. Sigoli, E. A. Suturina, E. Hemmer and M. Murugesu, Chem. Sci., 2019, 10, 6799–6808 RSC.
- K. R. McClain, C. A. Gould, K. Chakarawet, S. J. Teat, T. J. Groshens, J. R. Long and B. G. Harvey, Chem. Sci., 2018, 9, 8492–8503 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, crystallographic details, and full characterization (PXRD, photoluminescence, IR and UV-vis spectra, thermal analysis, and dielectric and magnetism measurements) of all the described compounds. CCDC 2014614 and 2014615. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc04851h |
| This journal is © The Royal Society of Chemistry 2021 |