
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydration breaking and chemical ordering in a levitated NaCl solution droplet beyond the metastable zone width limit: evidence for the early stage of two-step nucleation†
Hyerim
Hwang
 a,
Yong Chan
Cho
a,
Sooheyong
Lee
ab,
Yun-Hee
Lee
ab,
Seongheun
Kim
c,
Yongjae
Kim
a,
Wonhyuk
Jo
a,
Patrick
Duchstein
d,
Dirk
Zahn
*d and
Geun Woo
Lee
*ab
a,
Yong Chan
Cho
a,
Sooheyong
Lee
ab,
Yun-Hee
Lee
ab,
Seongheun
Kim
c,
Yongjae
Kim
a,
Wonhyuk
Jo
a,
Patrick
Duchstein
d,
Dirk
Zahn
*d and
Geun Woo
Lee
*ab
aDivision of Industrial Metrology, Korea Research Institute of Standards and Science, Daejeon 34113, Republic of Korea. E-mail: gwlee@kriss.re.kr
bDepartment of Nano Science, University of Science and Technology, Daejeon 34113, Republic of Korea
cPohang Accelerator Laboratory, POSTECH, Pohang 37673, Republic of Korea
dComputer Chemistry Center, Friedrich-Alexander University of Erlangen-Nuremberg, 91052 Erlangen, Germany. E-mail: dirk.zahn@fau.de
First published on 9th December 2020
Abstract
For over two decades, NaCl nucleation from a supersaturated aqueous solution has been predicted to occur via a two-step nucleation (TSN) mechanism, i.e., two sequential events, the formation of locally dense liquid regions followed by structural ordering. However, the formation of dense liquid regions in the very early stage of TSN has never been experimentally observed. By using a state-of-the-art technique, a combination of electrostatic levitation (ESL) and in situ synchrotron X-ray and Raman scatterings, we find experimental evidence that indicates the formation of dense liquid regions in NaCl bulk solution at an unprecedentedly high level of supersaturation (S = 2.31). As supersaturation increases, evolution of ion clusters leads to chemical ordering, but no topological ordering, which is a precursor for forming the dense disordered regions of ion clusters at the early stage of TSN. Moreover, as the ion clusters proceed to evolve under highly supersaturated conditions, we observe the breakage of the water hydration structure indicating the stability limit of the dense liquid regions, and thus leading to nucleation. The evolution of solute clusters and breakage of hydration in highly supersaturated NaCl bulk solution will provide new insights into the detailed mechanism of TSN for many other aqueous solutions.
Introduction
Nucleation plays a fundamental role in phase formation, and thus its characteristics are utilized in eminent applications such as engineering materials in a desired form, pharmaceuticals, cosmetics, living cells, and so on.1,2 According to classical nucleation theory (CNT), crystal nuclei form through spontaneous density fluctuation (i.e., simultaneous densification and structural ordering) in metastable liquids.3 Although CNT has successfully described the essential feature of nucleation, such single-step pathways cannot cover the whole complexity of possible phase formation processes. Recent studies have revealed that nucleation can occur via multi-step stages, of which the phenomenon is addressed as two-step nucleation (TSN),4–6 multiple pathways of nucleation7,8 or, in more general terms, Ostwald's step rule.9–11 In the case of TSN, single density fluctuation in CNT is divided into two processes, i.e., density fluctuation forming locally concentrated regions or pre-nucleation clusters as an intermediate phase, and sequential structural fluctuation triggering the crystal nuclei within the intermediate phase. Although such clusters preceeding the nucleation event have often been detected in liquids through a cryogenic transmission electron microscope (c-TEM),12–14 the formation processes of these clusters are still obscure due to the effect of the electron beam and limited spatial dimension. Moreover, scarce information on the specific structure and stability of dense liquids (or clusters) in highly supersaturated bulk solution prevents from unveiling the detailed mechanism of TSN.Revealing the TSN mechanism in sodium chloride (NaCl) solution has been an ongoing challenge for decades. Simulation studies have shown that solute-cluster aggregation as a precursor of dense liquid regions forms in undersaturated NaCl solution through the evolution of ion clustering with more than 10 ions;15,16 the ion clusters evolve from solvent-separated ion pairs (SSIP) to contact ion pairs (CIP) in solutions. They manifest themselves as molten salt-like aggregates at higher concentrations.17,18 Moreover, other simulation studies have predicted the structural evolution of ion clusters in supersaturated NaCl solution, which strongly suggests the existence of dense liquid regions in the early stage of TSN.17–21 Although the imprint of ion clustering has been experimentally captured in structural studies in undersaturation,22,23 no clear experimental evidence has been provided for either the existence and stability of the ion clusters or their structural evolution in highly supersaturated NaCl solution. The experimental difficulties resulted from the narrow metastable zone width of NaCl solution (i.e., a narrow supersaturation zone, typically less than S = 1.1) which leads to easy nucleation occurring just above saturation. This makes one hardly explore and prove the structural evolution of NaCl solution with supersaturation. Furthermore, this prevents from verifying recent theoretical suggestions (i.e., one- or two-step nucleation and its transition) of nucleation mechanisms in extremely supersaturated NaCl solution.24,25 Therefore, the direct detection of ion clustering and investigation of cluster stability in extremely supersaturated NaCl solution are highly required to propose the TSN mechanism.
We here measure the structural evolution of the highly supersaturated NaCl bulk solution by using a newly developed apparatus, combined with a containerless levitation technique and X-ray and micro-Raman scattering devices. Electrostatic levitation (ESL) enables levitation of a bulk solution droplet with a millimeter size in a containerless environment, minimizing heterogeneous nucleation sites and thus facilitating high supersaturation by gradual water evaporation. Such extremely water-depleted circumstances provide great advantages to unveil the atomic and molecular structures of solutes in the solution by in situ synchrotron X-ray and micro-Raman scatterings.26,27 In the present study, we find evidence of dense liquid regions with nm-sized solute-rich regions (i.e., dense liquid regions), showing an extended chemical ordering of Na+ and Cl− ions, which is also confirmed by molecular dynamics (MD) simulation studies. As the clustering further evolves, we find that the hydration structure is broken due to significant water depletion under extremely high supersaturation. This indicates the stability limit of the dense liquid regions. The formation of a locally-concentrated region and its stability at a highly supersaturated state are the crucial features of the first step in TSN, which will provide a key insight into understanding TSN in many other solution systems.
Results and discussion
Extremely supersaturated NaCl solution
A NaCl solution droplet (∼2.5–3 mm diameter) is levitated between a pair of electrodes in an ESL device under ambient conditions26,27 (Fig. 1(a), see the detailed experimental procedure in the Experimental section and the ESI†). The levitated droplet gradually shrinks with time, due to water evaporation, resulting in increasing concentration, as shown in Fig. 1(b). In the containerless environment provided by ESL, the solute concentration can easily exceed the solubility limit (i.e., saturation concentration, S = 1.0) without crystallization and approaches high supersaturation (Movie S1†). The highest supersaturation that we achieve in this study is S = 2.31 (Fig. 1(c), blue-solid line) which has never been obtained with NaCl bulk solution. If we use a container-based conventional method to achieve such highly supersaturated solution, undissolved NaCl (85 g) in water (100 g) should be heated up to about 600 °C to completely dissolve the solute (Fig. 1(c), red-dotted line) and slowly cooled down about ΔT = 575 °C without crystallization (Fig. 1(c), green-dotted line). This is hardly expected, since the container wall acts as heterogeneous nucleation sites on supercooling. At the highest supersaturation S = 2.31, the solution contains a significantly lower number of water molecules, 1.95 per ion on average, as compared to the solution at S = 0.1 with 44.43 water molecules and at S = 1.0 with 4.51 water molecules for each ion (Fig. 1(c) and S2 of the ESI†). The reduced water environment offers a great advantage to clearly detect the distinct solute structures and further look into micro-hydration in extremely supersaturated solutions.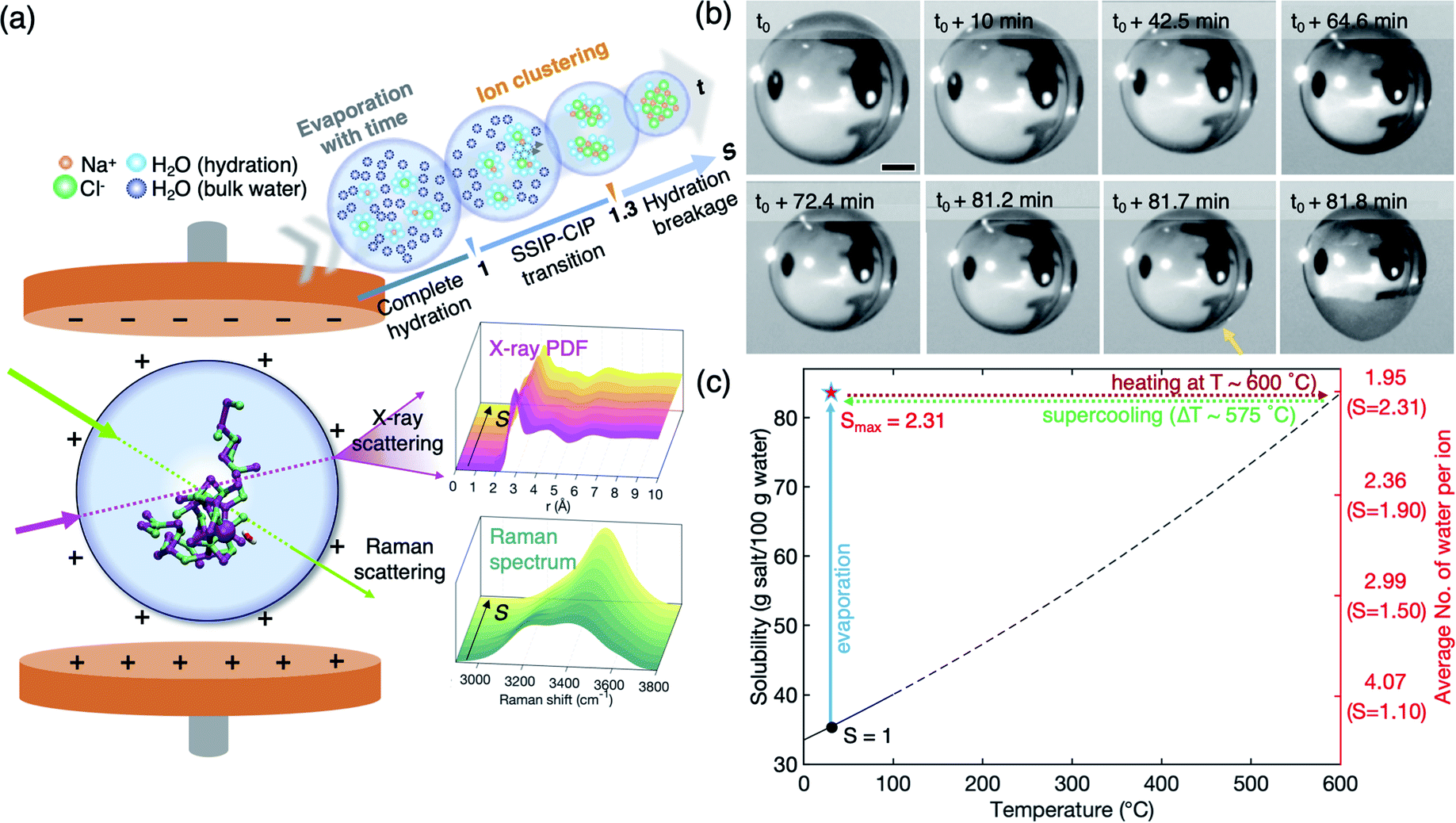 | ||
| Fig. 1 (a) Schematic diagram of electrostatic levitation (ESL) and the levitated solution droplet (side view). Evolution of the atomic and molecular structures of the solution as a function of supersaturation is probed by in situ X-ray and micro-Raman scatterings, respectively. (b) A time series of images of a levitated droplet during the evaporative process. The saturations at t0 (1st image) and t0 + 81.7 min (7th image) are S = 1.16 and S = 2.31, respectively, where the degree of supersaturation is determined by S = C/Ce with Ce = 360 g L−1. The last two images show the crystallization of the solution (marked by an arrow). Scale bar, 500 μm. (c) Solubility curve of NaCl. Dashed-line portions are extrapolations beyond the range of the known data (0–100 °C). The highest supersaturation (Smax = 2.31) corresponds to supercooling the droplet by ΔT = 575 °C. The average number of water molecules per ion and corresponding supersaturation under experimental conditions (24 °C) are noted on the right axis. | ||
Structural evolution of the supersaturated NaCl solution: an in situ synchrotron X-ray scattering study
We successfully measure the structural evolution of NaCl bulk solution under high supersaturation conditions using synchrotron X-ray scattering, combined with ESL (Fig. S1†). Fig. 2 shows the structure factors S(q) and pair distribution functions g(r) of the solution with concentration, which has been for the first time obtained over S = 1.0 for aqueous NaCl solution. The structure factors of water and undersaturated NaCl solutions are consistent with previous X-ray scattering results that were obtained at a low concentration around S = 0.65.22,28–30 In Fig. 2(a), the peak at 2.06 Å−1 in S(q) of pure water slightly shifts to higher q as salinity increases, while the peak at 2.89 Å−1 does not show a q-shift, but significantly reduced intensity. The third and fourth peaks clearly move towards shorter wavenumbers until S = 1.02 with concentration. The changes in the peaks indicate that different types of short-range- and medium-range-orderings appear and become dominant as water evaporates.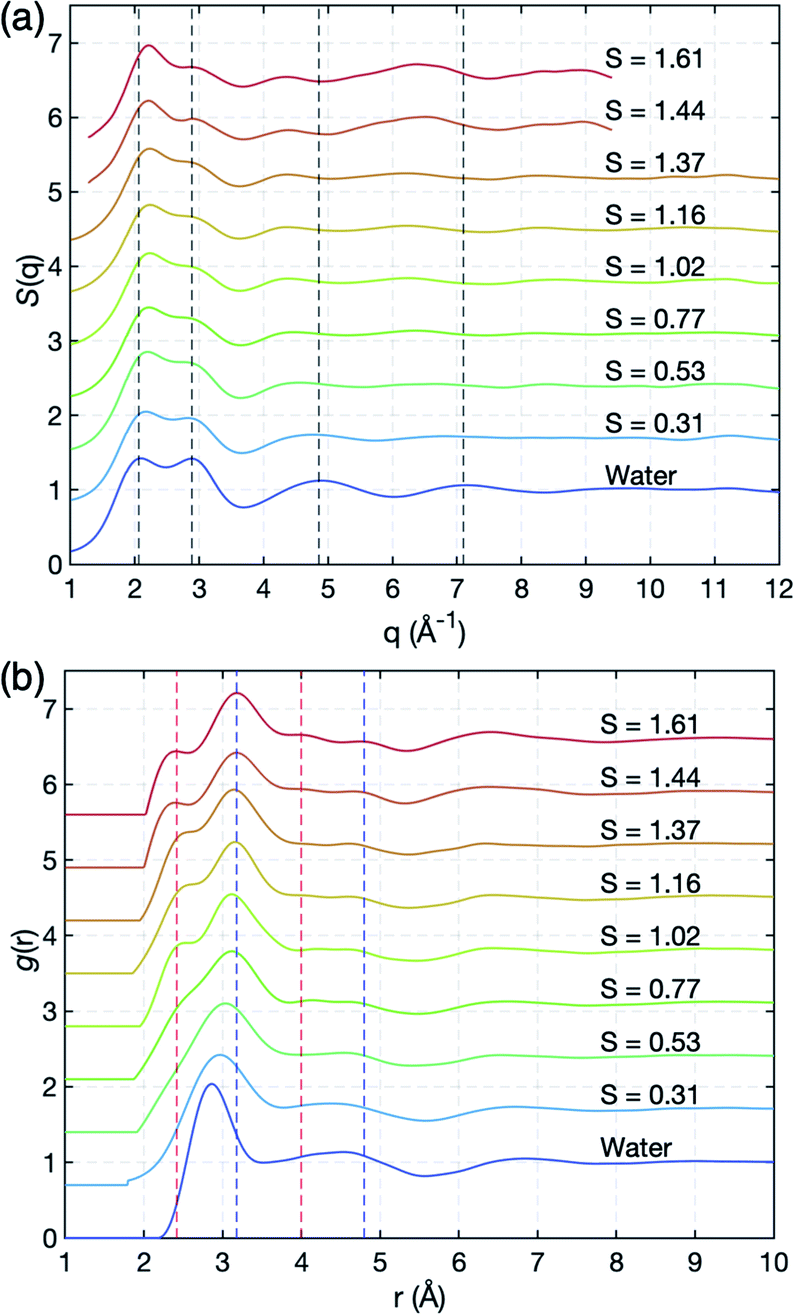 | ||
| Fig. 2 Experimental X-ray scattering measurements. (a) Structure factors, S(q), and (b) pair correlation functions, g(r), of pure water and aqueous NaCl solutions at various salt concentrations. Red and blue dashed lines around 2.4 Å and 3.2 Å at the first nearest distance correspond to O–Na+ and O–Cl−, respectively, and those (around 4 Å and 4.8 Å) at the second nearest distances indicate the extended chemical ordering of –Na+–Cl−–Na+- and –Cl−–Na+–Cl−-alignments (see Fig. 3(c) and (d)). | ||
More detailed spatial information is provided by the pair distribution functions (PDF), g(r), in Fig. 2(b), via Fourier transformations of S(q). As the ion concentration increases, g(r) of the NaCl solution clearly deviates from that of pure water. The first peak displays an asymmetric feature even at a low concentration S = 0.31, and is resolved into two peaks near 2.42 Å and 3.18 Å at S = 1.02 which are identified as sodium (O–Na+) and chloride (O–Cl−) ions, respectively, in a previous study;22,31,32 see also the calculated partial g(r) for O–Na+ and O–Cl− obtained from MD simulation in Fig. S3 of the ESI.† As water further evaporates, the positions of the two peaks gradually move to 2.36 Å and 3.17 Å at the highest supersaturation S = 1.61.
Interestingly, additional two peaks appear at 4.0 Å and 4.8 Å at S = 1.61 in g(r) due to the significant reduction in water scattering (Fig. 2(b)). These peaks are the result of extended chemical ordering –Na+–Cl−–Na+– and –Cl−–Na+–Cl−– alignments, since ion–ion interactions should be more dominant than water–ion interactions in such water-depleted environments. This is confirmed by the MD simulation study which we will discuss later in Fig. 3(c) and (d). Although simulation studies have implicitly shown the new peaks by Na+–Cl− ion pairs in undersaturation,21,28,33–37 and the existence of extended chemical ordering by the formation of more complex and dynamic ion networks with increasing S,17,38,39 such a clear appearance has never been experimentally observed to date. The average size of this ordering extends up to more than 1 nm, based on the location of the minimum valley around 5.3 Å in g(r) in highly supersaturated solution (Fig. 2(b)). Since it takes longer than 30 minutes to reach S = 1.61 from saturation, this extended chemical ordering is quite stable under supersaturation. Accordingly, the extended chemical ordering, but no topological ordering, strongly supports the existence of dense liquid regions at the supersaturated state prior to NaCl crystal nucleation.40–42
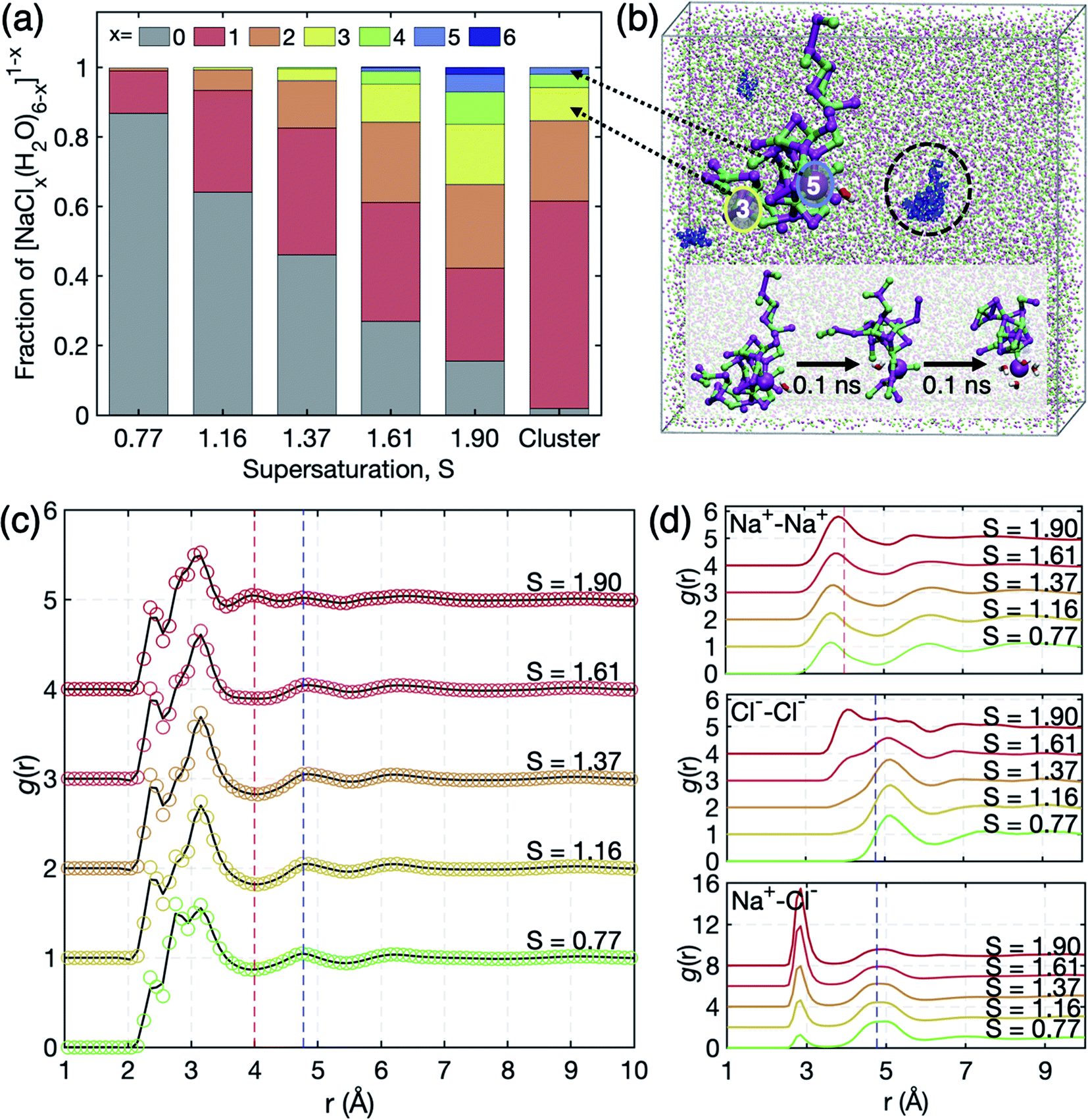 | ||
| Fig. 3 (a) Occurrence profile of Na+ coordination by Cl− ions. [NaClx(H2O)6−x]1−x complexes persist for x = 0–6. (b) Illustration of the overall simulation cell of S = 1.61 (Na+: magenta, Cl−: green). The snapshot highlights the networks of the nearest neighbor ion–ion contacts embedding a [NaCl5(H2O)]4− motif. The cluster in a black circle is magnified on its left, where red and blue circles indicate Na+ coordination by 3 and 5 Cl− ions, respectively. Such highly charged clusters do not persist as stand-alone, but are connected to further ions in a 1–2 nm sized network which experiences shape fluctuation and connectivity reorganization on the 0.1 ns scale. The inset shows the 0.1 ns scale dynamics of network reorganization involving water association and dissociation around [NaCl5(H2O)]4−. (c) Total pair correlation functions g(r) of the solution. The peaks around 2.4 Å, 2.8 Å, and 3.2 Å correspond to O–Na+, O–O, and O–Cl− distances. The O–O peak at 2.8 Å is smeared out due to the short q range in the experiment in Fig. 2(b). (d) Partial pair correlation functions g(r) of Na+–Na+, Cl−–Cl−, and Na+–Cl−. Extended chemical ordering is marked by red and blue dashed vertical lines. | ||
Evolution of ion clusters in high supersaturation: a molecular dynamics simulation study
To further scrutinize the structural evolution in microscopic viewpoint under such extreme supersaturation, we carried out MD simulations on NaCl solutions at S = 0.77–1.90 (see S4 of the ESI† for more details on the MD simulations). As the concentration increases, ions aggregate and form more SSIPs (solvent-separated ion pairs) and CIPs (contact ion pairs). Therefore, we examine the coordination statistics of ion clusters (i.e., [NaClx(H2O)6−x]1−x, x ≥ 1) as a function of S in the solution (Fig. 3(a)). The fractions of the ion clusters increase with supersaturation and isolated ions by water molecules (x = 0) remain less than 50% at S = 1.37. At the highest supersaturation S = 1.91, the fraction of ion clusters is more than 80%. This reflects that the supersaturated NaCl bulk solution retains lots of the dense liquid regions acting as intermediates to crystal nucleation. The intermediates are observed to be not completely dehydrated even at high supersaturation and the outmost level of spontaneous ion dehydration was found as [NaCl5(H2O)]4− motifs which reflect deformed octahedra with Na–Na and Cl–Cl distances of 3.9 Å that are close to those of the NaCl crystal (3.98 Å). Moreover, the dehydrated cluster composed of mostly Na+ and Cl− ions exhibits long chemical ordering which can yield the dense liquid regions in Fig. 2(b). This feature is clearly displayed in the total g(r) of S = 1.90 in Fig. 3(c); the peaks at 2.4 Å, 2.8 Å, and 3.2 Å are of O–Na+, O–O, and O–Cl−, respectively. Here, the O–O peak in experimental data is somewhat smeared out during Fourier transformation of S(q) due to the short q range.Interestingly, the peaks at 4 Å and 4.8 Å in Fig. 2(b) are reproduced in simulated g(r) at S = 1.90, which are contributed by the Na+–Na+, Na+–Cl−, and Cl−–Cl− distances with 3.9 Å, 4.8 Å and 5.1 Å, respectively, from partial g(r) in Fig. 3(d). The appearance of the peaks at different concentrations in the MD simulation study may reflect that the formation of dense liquid regions is slow as compared to the 10 ns scale of the MD runs. This may require the more drastic supersaturation to speed up the ordering in the 10 ns scale of the MD simulations, causing different degrees of supersaturation between simulations and experiments. Consequently, both scattering and MD studies state that the extended chemical ordering of ions underlies the formation of dense liquid regions in the supersaturated NaCl solution.
Evolution and breakage of the hydration structure: an in situ micro-Raman scattering study
As supersaturation increases, the extended chemical ordering of the ions should be accompanied by the rearrangement of water molecules in the solution which is important for understanding the role of the hydration structure (i.e., a structure of water molecules surrounding the ion clusters) on ion clustering and crystal nucleation. We investigate the change of the hydration structure using Raman scattering (Fig. 4). Since H2O molecules make bonds with their neighbour by a proton donor (D), proton acceptor (A), or their combinations, the Raman spectra of water are composed of mainly five sub-bands – DDAA, DDA, DAA, DA, and free-OH – that are engaged in different hydrogen bondings.43 A good agreement is observed between our data and previous studies in undersaturation;43–46 the Raman spectrum of water varies with weakened intensity in a low wavenumber region (<3300 cm−1) and elevated intensity in a higher wavenumber region (>3300 cm−1) in Fig. 4(a).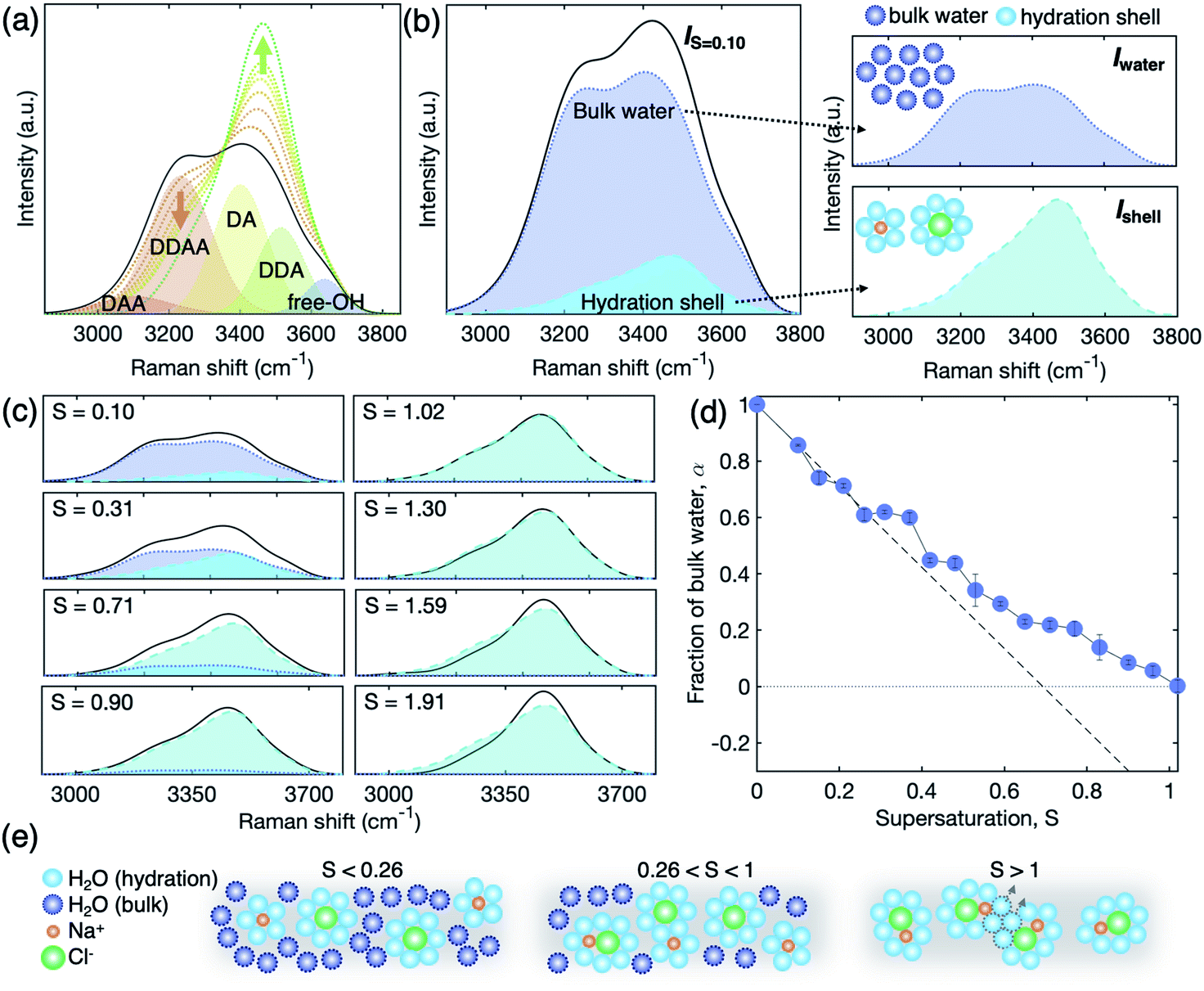 | ||
| Fig. 4 Spectral changes in the Raman OH-stretching vibration. (a) Raman spectra of pure water with 5-Gaussian fitted curves for hydrogen-bonded water clusters (DAA, DDAA, DA, DDA, and free-OH) and NaCl solutions at different salinity (black solid line: water, dotted lines with a color gradient: S = 0.21 (orange), 0.42, 0.65, 0.90, 1.16, 1.30, and 1.91 (green)). (b and c) Relative contribution of bulk water (Iwater, blue dotted line) and the hydration shell (Ishell, light blue dashed line) to the total Raman signal of a dilute solution (IS=0.1, black solid line). (c) Raman spectra (black solid line) of solutions at S = 0.10, 0.31, 0.71, 0.90, 1.02, 1.30, 1.59, and 1.91 with the signal of bulk water (blue) and the hydration shell (light blue). (d) Fraction of bulk water α in the solution: estimated values (blue circles) and predicted values for full hydration (black dashed line). (e) Schematic diagram of representative solution structures at different regimes of undersaturation. S < 0.26: complete hydration. 0.26 < S < 1: coexistence of SSIPs and CIPs. S > 1: transition from SSIPs to CIPs. Here, orange, green, light blue, and dark blue circles represent Na+ ion, Cl− ions, hydrated water molecules, and bulk water molecules, respectively. Over S > 1, only hydrated water molecules remained and can further be removed by continuous evaporation resulting in growing CIPs. | ||
To further scrutinize the evolution of the hydration structure, the signal of the hydration structure should be extracted from the total Raman signal that includes both contributions of bulk water and hydrated water as schematically described in Fig. 4(b). Based on previous studies,22,44,47 the hydration shell is composed of 5.5 and 7.5 water molecules in average, surrounding Na+ and Cl− ions, respectively. Therefore, we consider that the rest of the water molecules are assigned to the bulk water; the total Raman spectra are given by Itotal = α × Iwater + (1 − α) × Ishell, where Iwater and Ishell are the Raman scattering intensity of bulk water and the hydration shell in Fig. 4(b), respectively, and α is the fraction of bulk water (see S8 of the ESI† for the details). Since we can measure Iwater from pure bulk water, Ishell can be extracted from a dilute solution by considering the fraction of Iwater, with an assumption that the ions are completely hydrated and homogeneously suspended in the dilute solution (S = 0.1).
I water changes inversely with Ishell as a function of salinity and finally disappears at S = 1.02 (Fig. 4(c)). This signifies no bulk water in the solution at S = 1.02 and the Raman signal comes solely from the hydration shell. The resulting α, however, differs from calculations based on complete hydration. That is, if all the solute ions are fully hydrated with water molecules of 5.5 for Na+ and 7.5 for Cl− in average at undersaturation, it is expected that the bulk water is fully consumed at S = 0.7 (see a broken line for the fraction of bulk water, α, in Fig. 4(d)). Moreover, the experimental α starts deviating from the calculated line at S = 0.25, and becomes zero at S = 1.02. This illustrates that there is unexpected excess bulk water in the NaCl solution from S = 0.25 to S = 1.02. The excess bulk water reflects that the individual solute ions are not completely separated by hydration, but rather likely form partially hydrated ion clusters with SSIPs or CIPs even in undersaturation. We schematically describe the evolution of the hydration structure in Fig. 4(e). We assume that Ishell of the SSIPs does not differ from that of complete hydrated ions. This observation supports further the existence of solute-rich regions or spatial heterogeneity in the NaCl solution even at undersaturation, which has been suspected with the coexistence of CIPs and SSIPs in experiments23 and simulation studies.38,40–42
At S = 1.02–1.30, we observe no significant change in the Raman spectra Ishell (Fig. 4(c)). This might be caused by progressive aggregation among the ion pairs, which is called SSIP–CIP transition; the ion clusters at above S = 1.02 are possibly united with neighbor clusters by excluding a few water molecules from the nearest-neighbour ion coordination shells. This would compensate for the lower amount of water molecules in the system, whilst maintaining the nature of the hydration structure of the solutes.
Above S = 1.30, the Raman spectra continuously deviate from Ishell at S = 1.02 (Fig. 4(c)), which reflects the change of the hydration structure with supersaturation. Since the water molecules are extremely depleted in this supersaturation regime, the change of Raman spectra signifies the breakage of the hydration shell. In this stage, the ion–ion interaction is stronger than the ion–water interaction,33 and thus, the ion cluster may not sustain its stability and possibly leads to NaCl crystal nucleation.
From an analysis of the OH-stretching vibration spectra of liquid water and solution, we can further deduce a detailed picture of hydration evolution in solution as a function of supersaturation (see S7 and Fig. S4 of the ESI† for the detailed analysis). Fig. 5 depicts the sub-band changes of the Raman spectra with concentration, of which the fraction is calculated from the integrated area of the sub-bands. As the concentration increases up to S = 0.9, the tetrahedral hydrogen-bonded structure (DDAA) between H2O molecules is broken, resulting in decreasing population of DDAA and an increasing number of less hydrogen-bonded water structures, such as DA and DDA. We note the pair changes of the fraction (f) (i.e., DDAA–DDA and DAA–DA) with increasing salinity, implying that O–Na+ and H–Cl− interactions replace the hydrogen bonds of water molecules. However, this behavior does not persist above S = 1.0 where the ion–ion interaction is dominant rather than the ion–water interaction with supersaturation. Above S = 1.0, almost no change of the pairs is observed up to S = 1.3, and then a noticeable change of the pairs occurs in DDAA–DDA above S = 1.3 (Fig. 5(a) and (b)). This illustrates that the evolution of the hydration structure at high supersaturation differs from that at undersaturation and low supersaturation.
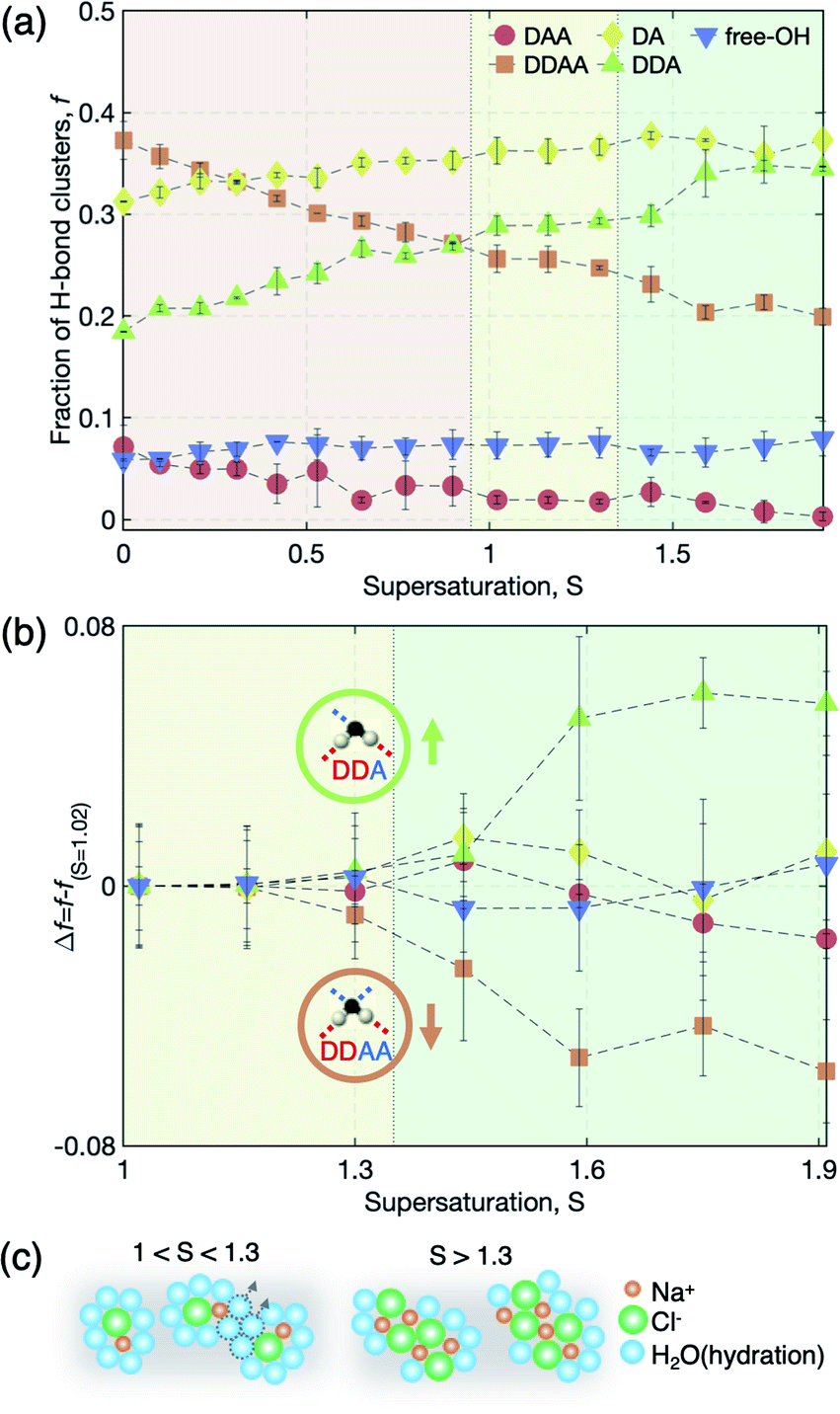 | ||
| Fig. 5 Population changes in hydrogen-bonded water clusters (DAA, DDAA, DA, DDA, and free-OH). (a) Fraction f of five water clusters in water and solution at varying saturations. (b) Changes in fraction Δf = f − fS=1.02 at S > 1.02. (c) Schematic diagram of the representative solution structures at supersaturation: 1 < S < 1.3 and S > 1.3. Here, orange, green, and light blue spheres represent the Na+ ion, Cl− ion, and water molecule, respectively. | ||
The pair change of DDAA–DDA suggests that the structure of the hydration shells may be altered by further water depletion from the hydration shells, which yields larger ion clusters by aggregating among CIPs. This is also reflected by the molecular dynamics simulations showing the increased occurrence of [NaClx(H2O)6−x]1−x motifs with x ≥ 2 for S > 1.3 (Fig. 3(a)). In general, Na+ and Cl− ions and their ion clusters are stable with hydration as charge balance is maintained. Therefore, the breakage of the hydration structure increases the instability of the ion clusters and reinforces the ion–ion interaction, and thus can result in structural ordering for nucleation. In other words, the stability limit of dense liquid regions is presumably determined by the breakage of the hydration structure in the solution.
It is worth noting that the three interest regions based on Raman spectra show quite similar aspects to the results of ion pairing in the vapor state of salt water;18 for NaCl(H2O)n, CIPs and SSIPs coexist at n = 9–12, and CIPs are dominant at n < 9. Since the average number of water molecules per ion is about 1.95 at supersaturation S = 2.31, this result is relatively comparable to that of the vapor state and thus the change of the hydration structure in this study will be useful to understand micro-hydration in the vapor state.
Nucleation of NaCl crystals from the solution
We now discuss crystal nucleation from the supersaturated NaCl solution, which still opens challenges in the study of aqueous NaCl solution. Since there is no clear theoretical framework for TSN, the nucleation process is still ambiguous in NaCl solution. For example, interfacial free energy as the nucleation barrier has been reported by only a few studies which show large different values.48,49 In the present study, the evolution and stability of ion clusters in highly supersaturated solution imply that nucleation possibly occurs in the dense liquid domains with chemical ordering of rich solutes. Therefore, if at least one nucleus with a critical size is needed to initiate the nucleation and a quasi-static equilibrium is maintained during supersaturation, CNT gives a following relation at a given droplet volume V, nucleation rate Is, and time t at a certain supersaturation S on continuous water evaporation,where time t = 0 refers to the onset of the saturated state (S = 1). This method has been successfully applied to estimate crystal-liquid interfacial free energy (IFE) for supersaturated potassium dihydrogen phosphate (KDP) solution recently,26 metallic liquids in supercooling experiments,50–53 and high density water in supercompression experiments54 (the details for the calculations are described in S9 of the ESI†). The estimated IFE, σ on NaCl solution is 46.17 mJ m−2 at S = 2.31 in the present study, which is in good agreement with a reported value σ = 47 mJ m−2 at 1.29 < S < 1.62 for NaCl nucleus-brine solution from computer simulations,44 but much lower than 87.31 mJ m−2 at S = 2.31 obtained from a micro-droplet solution49 and 63 mJ m−2 for NaCl-solution in a simulation study.55 From the IFE, we deduce the critical radius of the nucleus r* = 1.20 nm at S = 2.31 from r* = 2σvm/kBT
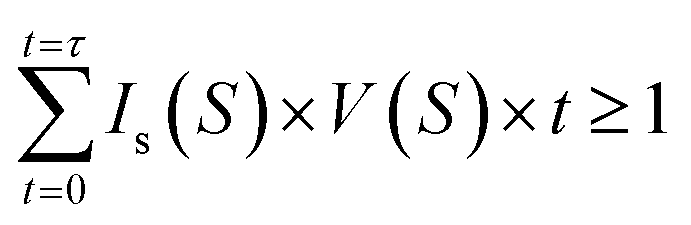
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnS, where vm and kB are the molar volume and the Boltzmann constant, respectively. Recall that the average size of the dense solute regions is 1.06 nm at S = 1.61 with chemical ordering by the sequential Na+ and Cl− ions in Fig. 2(b), which is comparable to, but smaller than that of the critical nucleus. Therefore, the aggregation between CIPs should continue to occur until the critical size nuclei is formed via breakage of the hydration structure, indicating the stability limit of the dense solute regions under high supersaturation conditions.
lnS, where vm and kB are the molar volume and the Boltzmann constant, respectively. Recall that the average size of the dense solute regions is 1.06 nm at S = 1.61 with chemical ordering by the sequential Na+ and Cl− ions in Fig. 2(b), which is comparable to, but smaller than that of the critical nucleus. Therefore, the aggregation between CIPs should continue to occur until the critical size nuclei is formed via breakage of the hydration structure, indicating the stability limit of the dense solute regions under high supersaturation conditions.
A recent simulation study predicted a spinodal point where transition from a one-step to two-step nucleation process of NaCl crystals occurs in highly supersaturated solution;24 above the transition concentration, ∼15.0 mol kg−1 (S = 2.44), at which the stability limit of the aqueous NaCl solution for TSN is expected. In the present study, we approached such extremely high supersaturation and our observations of hydration breaking and chemical ordering strongly support TSN in NaCl solution. All the concentrations are lower than those predicted by the simulation due to the system size.
Conclusion
In summary, we report the structural evolution of NaCl solution and ion (de)hydration characteristics spanning from undersaturated to highly supersaturated regions using in situ synchrotron X-ray and micro-Raman scatterings combined with ESL as well as MD simulations. We find that 1–2 nm scale solute-rich liquid regions with chemically-ordered ions are evidently formed in supersaturated solution prior to crystal nucleation. Moreover, Raman studies unveil the detailed structural evolution of hydration as a function of supersaturation, indicating complete ion hydration below S < 0.26, coexistence of SSIPs and CIPs in 0.26 < S < 1, transition from SSIPs to CIPs in 1 < S < 1.3, and increasing water depletion of hydration shells (or aggregation of CIPs) above S > 1.3, causing the breakage of the hydration structure. These findings strongly support the proposed mechanism of TSN, i.e., the formation of dense liquid regions at the early stage of nucleation in NaCl solutions at high supersaturation which has been suggested by simulation studies only.18,19,28 Moreover, the stability limit of the dense liquid regions is governed by the evolution of the hydration structure, and the breakage of the hydration structure can trigger nucleation in the dense liquid regions. These results provide a new perspective for detailed understanding of the early stage of two-step nucleation in aqueous solutions with monatomic ions, and will impact many research fields such as solution chemistry, electrochemistry, mineralogy, geochemistry, and molecular biology.Experimental section
Sample preparation
We prepared aqueous brine solution by dissolving NaCl (Aldrich, 99%) in deionized water in concentrations ranging from 252 to 288 g L−1 (NaCl solute in grams per water solvent in liters). Under the ambient conditions (i.e., 24 ± 0.5 °C and relative humidity of 46 ± 1%), the solution was permeated through a syringe filter with a pore size of 220 nm (JEL BIOFIL) to eliminate impurities which can act as potential nucleation sites. We inject the filtered solution through a needle (Hamilton 8646-01) which is present in the bottom electrode.Electrostatic levitation (ESL)
The ESL apparatus is composed of two copper electrodes that are parallel-aligned and separated by ∼2 cm (Fig. 1(a)). A solution droplet on a hydrophobically coated metal tip (Hamilton, 8646-01) by injection is levitated when applied electric voltage is larger than the wetting force on the tip. The electric voltage is precisely adjusted so as to maintain the droplet at a desired position using a position-voltage feedback system. During levitation, the applied voltage is typically 800 to 1700 V, depending on the droplet size. The details of the ESL apparatus and experimental process are given in the ESI† and ref. 26.In situ synchrotron X-ray and micro-Raman scattering
The ESL is installed at the sector 1C and 5A beamlines at the Pohang Light Source II so as to study the structural change of the NaCl solution with supersaturation. An X-ray detector (Pilatus 300K-W) is placed behind the ESL chamber to collect scattered signals from the levitated solution droplet. Once the levitated droplet approaches the desired supersaturation, the X-ray shutter opens and the scattering data are obtained within 1–3 seconds exposure time. For the study of the water vibration mode in the levitated NaCl solution, the ESL is also combined with a micro-Raman spectroscope (DONGWOO OPTRON; DM500i, a 2D detector (ANDOR DV401A-Bv)). In this experiment, a laser power of 1.5 mW was used to minimize the heating of the solution. The typical exposure time to gather the scattered light is about 40 s.Molecular dynamics (MD) simulations
While the transition path sampling study in ref. 38 was designed to trigger density fluctuations, unbiased MD simulations were performed for much larger systems and longer relaxation time scales without artificially driving solute dehydration in this study. The MD simulations were carried out with the LAMPPS package using a time step of 1 fs and the Langevin thermostat–barostat combination.56 Periodic boundary conditions are applied to mimic bulk solutions. Accordingly, cut-off (1.2 nm) potentials are used in combination with Ewald summation (with a damping constant of 0.5 nm−1). Before sampling, each simulation system was relaxed for 15 ns to ensure convergence, monitored by average system energy, the volume, and the solute distribution. The assessment of pair correlation functions (g(r)) was performed from 10 ns MD runs after relaxation, which ensures well-converged g(r) profiles sampled with a resolution of 0.01 nm bins. g(r) compiled from MD trajectories is intrinsically deconvoluted and even further scrutinized by discriminating Na+ in SSIPs in selective statistics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Research Foundation of Korea (NRF-2014M3C1A8048818, NRF-2014M1A01030128 and NRF-2019K1A3A7A09033397) and funded by the Ministry of Science and ICT (MSIT) and Korea Research Institute of Standards and Science (KRISS-2017-GP2017-0029).References
- J. Anwar and D. Zahn, Angew. Chem., Int. Ed., 2011, 50, 1996 CrossRef CAS PubMed.
- D. Erdemir, A. Y. Lee and A. S. Myerson, Acc. Chem. Res., 2009, 42, 621 CrossRef CAS PubMed.
- P. G. Debenedetti, Metastable Liquids: Concepts and Principles, Princeton Univ. Press, USA 1997 Search PubMed.
- P. G. Vekilov, Nanoscale, 2010, 2, 2346 RSC.
- P. G. Vekilov, Cryst. Growth Des., 2004, 4, 671 CrossRef CAS.
- P. R. ten Wolde and D. Frenkel, Science, 1997, 277, 1975 CrossRef CAS PubMed.
- S.-Y. Chung, Y.-M. Kim, J.-G. Kim and Y.-J. Kim, Nat. Phys., 2008, 5, 68 Search PubMed.
- R. Demichelis, P. Raiteri, J. D. Gale, D. Quigely and D. Gebauer, Nat. Commun., 2011, 590, 2 Search PubMed.
- W. Ostwald, Z. Physiol. Chem., 1897, 22, 289 Search PubMed.
- N. Nývlt, Cryst. Res. Technol., 1995, 30, 443 CrossRef.
- T. Threlfall, Org. Process Res. Dev., 2003, 7, 1017 CrossRef CAS.
- M. H. Nielsen, S. Alonni and J. J. De Yoreo, Science, 2014, 345, 1158 CrossRef CAS PubMed.
- X. Wang, I.-M. Chou, W. Hu and R. C. Burruss, Geochim. Cosmochim. Acta, 2013, 103, 1 CrossRef CAS.
- N. D. Loh, S. Sen, M. Bosman, M. S. F. Tan, J. Zhong, C. A. Nijhuis, P. Král, P. Matsudaira and U. Mirsaidov, Nat. Chem., 2017, 9, 77 CrossRef CAS PubMed.
- S. A. Hassan, J. Chem. Phys., 2011, 134, 114508 CrossRef PubMed.
- S. A. Hassan, J. Phys. Chem. B, 2008, 112, 10573 CrossRef CAS.
- S. Kim, H. Kim, J.-H. Choi and M. Cho, J. Chem. Phys., 2014, 141, 124510 CrossRef.
- G.-L. Hou, C.-W. Liu, R.-Z. Li, H.-G. Xu, Y. Q. Gao and W.-J. Zheng, J. Phys. Chem. Lett., 2016, 8, 13 CrossRef.
- L. Degrève and F. L. B. da Silva, J. Chem. Phys., 1999, 111, 5150 CrossRef.
- F. Giberti, G. A. Tribello and M. Parrinello, J. Chem. Theory Comput., 2013, 9, 2526 CrossRef CAS.
- L. Degrève and F. L. B. da Silva, J. Chem. Phys., 1999, 110, 3070 CrossRef.
- S. Bouazizi, S. Nasr, N. Jaidane and M.-C. Bellissent-Funel, J. Phys. Chem. B, 2006, 110, 23515 CrossRef CAS.
- Y. Georgalis, A. M. Kierzek and W. Saenger, J. Phys. Chem. B, 2000, 104, 3405 CrossRef CAS.
- H. Jiang, P. G. Debenedetti and A. Z. Panagiotopoulos, J. Chem. Phys., 2019, 150, 124502 CrossRef PubMed.
- D. James, S. Beairsto, C. Hartt, O. Zavalov, I. Saika-Voivod, R. K. Bowles and P. H. Poole, J. Chem. Phys., 2019, 150, 074501 CrossRef PubMed.
- S. Lee, H. S. Wi, W. Jo, Y. C. Cho, H. H. Lee, S.-Y. Jeong, Y.-I. Kim and G. W. Lee, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 13618 CrossRef CAS PubMed.
- H. Hwang, Y. C. Cho, S. Lee, T. M. Choi, S.-H. Kim and G. W. Lee, Small, 2020, 88, 055101 Search PubMed.
- R. Mancinelli, A. Botti, F. Bruni and M. A. Ricci, J. Phys. Chem. B, 2007, 111, 13570 CrossRef CAS PubMed.
- R. Mancinelli, A. Botti, F. Bruni, M. A. Ricci and A. K. Soper, Phys. Chem. Chem. Phys., 2007, 9, 2959 RSC.
- G. Hura, D. Russo, R. M. Glaeser, T. Head-Gordon, M. Krack and M. Parrinello, Phys. Chem. Chem. Phys., 2003, 5, 1981 RSC.
- A. H. Narten and H. A. Levy, J. Chem. Phys., 1971, 55, 2263 CrossRef CAS.
- A. K. Soper, J. Phys.: Condens. Matter, 2007, 19, 335206 CrossRef CAS PubMed.
- J. M. Khalack and A. P. Lyubartsev, Condens. Matter Phys., 2004, 7, 683 CrossRef.
- A. Mizoguchi, Y. Ohshima and Y. Endo, J. Am. Chem. Soc., 2003, 125, 1716 CrossRef CAS PubMed.
- D. E. Smith and L. X. Dang, J. Chem. Phys., 1994, 100, 3757 CrossRef CAS.
- S. Chowdhuri and A. Chandra, J. Chem. Phys., 2001, 115, 3732 CrossRef CAS.
- A. C. Belch, M. Berkowitz and J. A. McCammon, J. Am. Chem. Soc., 1986, 108, 1755 CrossRef CAS.
- D. Zahn, Phys. Rev. Lett., 2004, 92, 040801 CrossRef.
- Q. Zhang, T. Wu, C. Chen, S. Mukamel and W. Zhuang, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 10023 CrossRef CAS PubMed.
- D. Chakraborty and G. N. Patey, Chem. Phys. Lett., 2013, 587, 25 CrossRef CAS.
- D. Chakraborty and G. N. Patey, J. Phys. Chem. Lett., 2013, 4, 573 CrossRef CAS.
- G. Lanaro and G. N. Patey, J. Phys. Chem. B, 2016, 120, 9076 CrossRef CAS PubMed.
- Q. Sun, Vib. Spectrosc., 2012, 62, 110 CrossRef CAS.
- R. Li, Z. Jiang, F. Chen, H. Yang and Y. Guan, J. Mol. Struct., 2004, 707, 83 CrossRef CAS.
- Q. Hu, H. Guo, W. Lu, X. Lu, Y. Chen and L. Lim, J. Mol. Liq., 2014, 199, 83 CrossRef CAS.
- Q. Sun, L. Zhao, N. Li and J. Liu, Chem. Geol., 2010, 272, 55 CrossRef CAS.
- M. Llano-Restrepo and W. G. Chapman, J. Chem. Phys., 1994, 100, 8321 CrossRef CAS.
- N. E. Zimmermann, B. Vorselaars, D. Quigley and B. Peters, J. Am. Chem. Soc., 2015, 137, 13352 CrossRef CAS PubMed.
- H.-S. Na, S. Arnold and A. S. Myerson, J. Cryst. Growth, 1997, 139, 104 CrossRef.
- K. F. Kelton, Solid State Physics, Academic, Boston, MA, USA, 1991 Search PubMed.
- K. F. Kelton, G. W. Lee, A. K. Gangopadhyay, R. W. Hyers, T. J. Rathz, J. R. Rogers, M. B. Robinson and D. S. Robinson, Phys. Rev. Lett., 2003, 90, 195504 CrossRef CAS PubMed.
- G. W. Lee, A. K. Gangopadhyay, T. K. Croat, T. J. Rathz, R. W. Hyers, J. R. Rogers and K. F. Kelton, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 174107 CrossRef.
- D. H. Kang, S. Jeon, H. Yoo, T. Ishikawa, J. T. Okada, P. F. Paradis and G. W. Lee, Cryst. Growth Des., 2014, 14, 1103 CrossRef CAS.
- G. W. Lee, W. J. Evans and C. S. Yoo, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 134112 CrossRef.
- R. Bahadur, L. M. Russel and S. Alavi, J. Phys. Chem. B, 2007, 111, 11989 CrossRef CAS PubMed.
- S. Plimpton, J. Comput. Phys., 1995, 117, 1 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc04817h |
| This journal is © The Royal Society of Chemistry 2021 |