
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNear-infrared fluorescent probes: a next-generation tool for protein-labeling applications
Shahi Imam
Reja
 a,
Masafumi
Minoshima
a,
Yuichiro
Hori
ab and
Kazuya
Kikuchi
*abc
a,
Masafumi
Minoshima
a,
Yuichiro
Hori
ab and
Kazuya
Kikuchi
*abc
aGraduate School of Engineering, Osaka University, Suita, Osaka 565-0871, Japan. E-mail: kkikuchi@mls.eng.osaka-u.ac.jp
bImmunology Frontier Research Center, Osaka University, Osaka 565-0871, Japan
cQuantum Information and Quantum Biology Division, Osaka University, Suita, Osaka 565-0871, Japan
First published on 23rd October 2020
Abstract
The development of near-infrared (NIR) fluorescent probes over the past few decades has changed the way that biomolecules are imaged, and thus represents one of the most rapidly progressing areas of research. Presently, NIR fluorescent probes are routinely used to visualize and understand intracellular activities. The ability to penetrate tissues deeply, reduced photodamage to living organisms, and a high signal-to-noise ratio characterize NIR fluorescent probes as efficient next-generation tools for elucidating various biological events. The coupling of self-labeling protein tags with synthetic fluorescent probes is one of the most promising research areas in chemical biology. Indeed, at present, protein-labeling techniques are not only used to monitor the dynamics and localization of proteins but also play a more diverse role in imaging applications. For instance, one of the dominant technologies employed in the visualization of protein activity and regulation is based on protein tags and their associated NIR fluorescent probes. In this mini-review, we will discuss the development of several NIR fluorescent probes used for various protein-tag systems.
 Shahi Imam Reja | Shahi Imam Reja is a postdoctoral fellow in the group of Prof. Kazuya Kikuchi at Osaka University, Japan. He received his PhD in 2017 from the Department of Chemistry, Guru Nanak Dev University, India, supervision of Prof. Manoj Kumar. He worked as a postdoctoral fellow at University of Guelph, Canada (2017–2018) under the guidance of Prof. Richard A. Manderville and then joined Prof. Kikuchi's group at Osaka University in 2018. His research interest is focused on the development of chemical probes for imaging protein functions. |
 Masafumi Minoshima | Masafumi Minoshima is an assistant professor at the Graduate School of Engineering, Osaka University. He received his PhD in 2010 from the Graduate School of Science, Kyoto University. After two-year work at the University of Tokyo as a specially appointed assistant professor under the direction of Prof. Kazuhito Hashimoto, he joined Prof. Kikuchi's group at Osaka University in 2012 as a specially appointed assistant professor and became his current position in 2016. His research interests are to develop chemical tools for imaging and manipulating biological function. |
 Yuichiro Hori | Yuichiro Hori is an associate professor at the Graduate School of Engineering, Osaka University. He received his PhD in 2004 from the Graduate School of Pharmaceutical Sciences, Kyoto University. After performing postdoctoral research at the Rockefeller University, New York, USA under the direction of Prof. Tom W. Muir, he became an assistant professor at Osaka University in 2006 and attained his current position in 2016. His research interests involve the development of chemical biology tools using organic chemistry techniques in combination with peptide/protein engineering. |
 Kazuya Kikuchi | Kazuya Kikuchi is a professor at the Graduate School of Engineering, Osaka University. He received his PhD in 1994 from the Graduate School of Pharmaceutical Sciences at the University of Tokyo. After conducting postdoctoral research at the University of California in San Diego with Prof. Roger Y. Tsien and at the Scripps Research Institute with Prof. Donald Hilvert, he became an assistant professor at the University of Tokyo in 1997 and was promoted to an associate professor in 2000. In 2005, he was appointed as a full professor at Osaka University. His research interests are in using chemistry-based techniques to perform biological research, especially in designing imaging probes with chemical switches. |
Introduction
Fluorescence imaging is an attractive method to investigate various biological phenomena in real-time.1 In this regard, fluorescence imaging is often preferred because it provides precise spatio-temporal information on biomolecules with high sensitivity.2 Although this technique is widely used, its use is restricted by autofluorescence, light absorption, and light scattering, which reduce the signal-to-background ratio and the contrast of fluorescence images, particularly when imaging deep tissues and animals. Visible light is strongly scattered in deep tissues, and thus its penetration is limited. Recently, to overcome the limitations of visible-range fluorophores, near-infrared (NIR) fluorophores were developed to minimize background fluorescence signals and to visualize deep tissues (Fig. 1).3 NIR fluorescence emission provides several advantages in the application of imaging to fundamental scientific research, owing to its reduced photon scattering, autofluorescence, and excitation energy.4 The autofluorescence of the cellular system depends on various endogenous molecules, such as reduced nicotinamide adenine dinucleotide (NADH), flavins, and reticulin, whose emission spectra fall in the visible region.5 NIR fluorophores minimize spectral overlap with the autofluorescence background signals, which is especially suited for single-molecule imaging in cells. In addition, endogenous chromophores, such as hemoglobin, strongly absorb light in the visible region; thus, visible light has limited capability to penetrate tissues.6 Moreover, the wavelength range of 650–900 nm enables high-resolution imaging through deep tissue penetration. Most biological components contain water and lipids, which are generally transparent to light from the visible to the NIR region but strongly absorb light in the infrared region.7 In addition to the benefits to tissue or in vivo imaging, an extension of the fluorophore wavelength to the NIR region is also advantageous in cell imaging. The addition of the NIR region enables more options for fluorophores with different fluorescent colors that are required for multicolor imaging. Thus, the development of NIR fluorescent dyes is one of the most promising areas of research is not only in vivo imaging, but also in cell imaging. | ||
| Fig. 1 Emission regions and chemical structures of commonly used fluorophores. | ||
To date, various NIR-based fluorophores have been developed based on small organic molecules, inorganic materials, or organic–inorganic hybrid materials, for various applications in material sciences, clinical research, or chemical biology.8,9 Among the above mentioned NIR fluorophores, small organic molecule-based NIR fluorophores offer attractive imaging tools owing to their small size, biocompatibility, and high design flexibility.10 Many small molecule-based NIR fluorophores have been developed, using various scaffolds such as cyanine, squaraine, bodipy, xanthene, and benzobisthiadiazole.11 The cyanine-based fluorophores are commonly used in imaging applications, and although their photostability is a constraint, modifications have been made to create a highly photostable cyanine platform for high-contrast imaging applications.12 Squaraine dyes are NIR fluorophores with a high molar extinction coefficient and are an alternative to cyanine dye.13 Bodipy core is a promising scaffold for creating NIR fluorophores. Aza-bodipy is one of the key derivatives designed for NIR probe development.14 The xanthene-based fluorophore, where the 10 position oxygen atom of the xanthene core is replaced by various heteroatoms such as Si (Si-rhodamine; SiR), Ge (GeR), Te (TeR), or P (PR), emits NIR fluorescence with photostability.15 Another unique cyanine-based dye was reported by Yuan et al., where they combined cyanine and rhodamine fluorophores to develop a NIR dye.16 This example reveals the possibility of creating various NIR fluorophores by modifying a parent fluorophore.
NIR fluorescent probes were applied to image metal ions, signaling molecules, reactive species, nucleic acids, and proteins.17,18 Moreover, there is growing interest in the detection of an enzyme using NIR probes, such as the detection of aminopeptidase N (APN), γ-glutamyltransferase, β-galactosidase, alkaline phosphatases, nitroreductase, and cysteine proteases.19–24 Furthermore, NIR probes are now used for clinical research, such as in fluorescence imaging-guided surgery and detection of cancer.25 These NIR fluorophores were also used to find new cellular events or explore the cellular structure with fluorescence nanoscopy techniques.26
Live cell imaging of the dynamics and functions of proteins is a critical area of research that discovers new roles of proteins in cellular activities. Particularly, fluorescent proteins (FPs) are extensively used to monitor the localization and dynamics of fused proteins using fluorescence microscopy.27 However, limitations to the use of FPs in fluorescence imaging are due to low photostability and brightness, which hinder the use of FPs in high-resolution imaging techniques.28 Thus, various improvements have been made to enhance the photostability and brightness of FPs. Alternatively, the use of self-labeling protein tags with synthetically developed fluorescent probes is an advantageous strategy because of the easy tuning of small molecule-based fluorescent probes with favorable photophysical properties (Fig. 2).29 In this technique, a protein tag is fused to a protein of interest and specifically labeled using synthetic fluorescent probes, providing a fluorescence image of protein localization. One of the significant advantages of using protein tags over FPs is their possible application in pulse-chase experiments, in which the fluorescent probe can label a protein at specific time points.30 Moreover, the high photostability and color variability of the synthetic probes make the protein-labeling technique a suitable alternative to FPs. To date, various NIR fluorescent probes with high photostability and brightness have been developed for labeling protein tags to provide chemical tools for pulse-chase imaging, multicolor imaging, single-molecule imaging, and super-resolution imaging. Thus, in the present review, we focus on the recent development of NIR fluorescent probes for various protein-labeling tags, describe their design principle and biological applications, and discuss the current challenges and future directions of this field.
 | ||
| Fig. 2 Schematic representation of self-labeling protein tag with chemical probe. | ||
Protein labeling using various NIR fluorescent probes
To date, a variety of protein tags have been employed to generate protein-labeling systems, such as SNAP-tag,31 CLIP-tag,32 Halo-tag,33 PYP-tag,34 BL-tag,35 eDHFR/TMP-tag,36 FAP-tag,37 and FAST tag.38 For bioorthogonal labeling in eukaryotic cells, these tags are commonly acquired by exploring bacterial sources, mutating eukaryotic proteins through directed evolution, or by combining these two approaches. NIR fluorescent probes that specifically bind to these tags have been designed using a variety of chemical strategies to enhance brightness, show fluorogenic response upon protein labeling, or contain unique fluorescence properties for super-resolution imaging. We summarize how these tags were obtained, what chemical principles were used for probe design, and how these protein-labeling systems were applied for biological studies.SNAP-tag and CLIP-tag based systems
SNAP-tag is a self-labeling protein tag (19.4 kDa), developed by Johnsson's group from the mutant version of the human O6-alkylguanine-DNA alkyltransferase (hAGT), which is a DNA repair protein.31 Cysteine 145 is an active amino acid that transfers the alkyl group from the O6 of the alkylguanine substrate via an SN2 reaction (Fig. 3A). Currently, SNAP-tag-based fluorescent probes are commercially available for monitoring protein localization and dynamics. The SNAP-tag-based strategy has the advantage of being a general method for protein labeling. Moreover, CLIP-tag is an AGT-based tag that reacts specifically with O2-benzylcytosine (BC) derivatives.32 The distinct specificity of these tags makes it possible to label two different proteins simultaneously.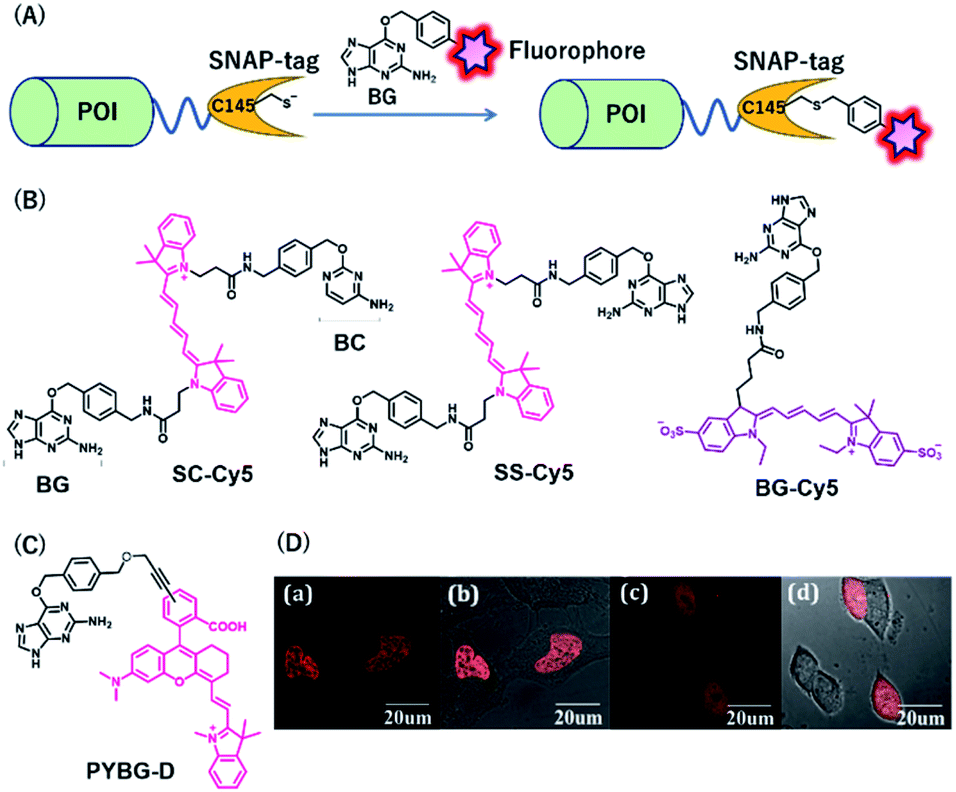 | ||
| Fig. 3 (A) Schematic representation of a SNAP-tag-based protein tag with chemical probe. (B) Structures of cyanine (Cy5)-based chemical probes connecting with BG/BC. (C) Structure of Changsha fluorophore-based chemical probe for SNAP-tag. (D) COS-7 cells stained with 5 μM PYBG-D for 1 hour; (a) fluorescence image of histone 2B proteins fused with SNAP-tag; (b) overlay of bright field and fluorescence image of (a); (c) fluorescence image of SNAP-tag proteins; (d) overlay of bright field and fluorescence image of (c). POI, protein of interest. BG, O6-benzylguanine; BC, O2-benzylcytosine. Reproduced from ref. 41 with permission from [The Royal Society of Chemistry], copyright [2017]. | ||
NIR fluorescent probes, SC-Cy5 and SS-Cy5, were developed using a Cy5 scaffold that is connected with BG, BC, or both to conjugate SNAP-tag, CLIP-tag, or both (Fig. 3B), allowing selective cross-linking of proteins in cell lysates for the investigation of protein–protein interactions.39 This approach is more convenient than that by using commercially available bismaleimide cross-linker. Indeed, this system can detect weak protein–protein interactions, even with a KD value of 30 μM. Campos et al. used a Cy5-conjugated NIR probe, BG-Cy5, for SNAP-tag labeling to verify cell lineages in zebrafish (Fig. 3B).40 Using this probe, the authors demonstrated that their system could be useful to image zebrafish subcellular structures, such as the nucleus, cell membranes, and endosomal membranes.
Considering the advantages of NIR fluorescent probes, Song et al. developed a SNAP-tag probe, PYBG-D, using a Changsha NIR fluorophore (Fig. 3C).41 The probe showed a large molar extinction coefficient and a high quantum yield value with maximum wavelength >700 nm. Interestingly, this probe was permeable to cellular membranes and was shown to specifically label histone 2B proteins fused to SNAP-tags in the nuclei (Fig. 3D). This probe was also used to image mitochondrial proteins, indicating that the probe could serve as a potential tool to reveal mitochondrial protein dysfunction or translocation.
Rhodamine-based fluorophores are widely used for the development of many fluorescent probes owing to their high photostability and brightness. The fluorophore can extend the emission wavelength by the incorporation of silicon heteroatoms into the xanthene core, generating Si-rhodamine (SiR) that emits NIR fluorescence at around 660–670 nm.42 SiR carboxy derivatives linked to protein tag ligands are preferentially present as a nonfluorescent closed spirolactone conformation because of reversible aggregation in aqueous solution (Fig. 4A).43 When bound to the polar surface of proteins, the fluorophore forms an open conformation that displays a high fluorescence intensity, resulting in the detection of proteins with high signal-to-background ratio. Owing to this fluorogenic property, SiR-based chemical probes, SiR-SNAP and SiR-CLIP, which label SNAP-tag and CLIP-tag, respectively (Fig. 4B), gave clear images of nuclear, mitochondrial, and actin proteins.43 Interestingly, this fluorescence response triggered by the spirocyclization was also shown when the fluorophore was linked to the other protein ligands for HaloTag, actin, and tubulin.43 Further, SiR-SNAP was applied for super-resolution microscopy using ground state depletion microscopy followed by individual molecular return/stochastic optical reconstruction microscopy (GSDIM/STORM)44,45 and stimulated emission depletion (STED)46 techniques that require stable blinking and high photostability of fluorophores, respectively. SiR-SNAP showed such photophysical properties necessary for these microscopic techniques and successfully imaged the precise localization of centrosomal proteins in living cells using STED microscopy. This study demonstrated the advantages of synthetic fluorescent probes with desirable photophysical properties in super-resolution imaging of proteins.
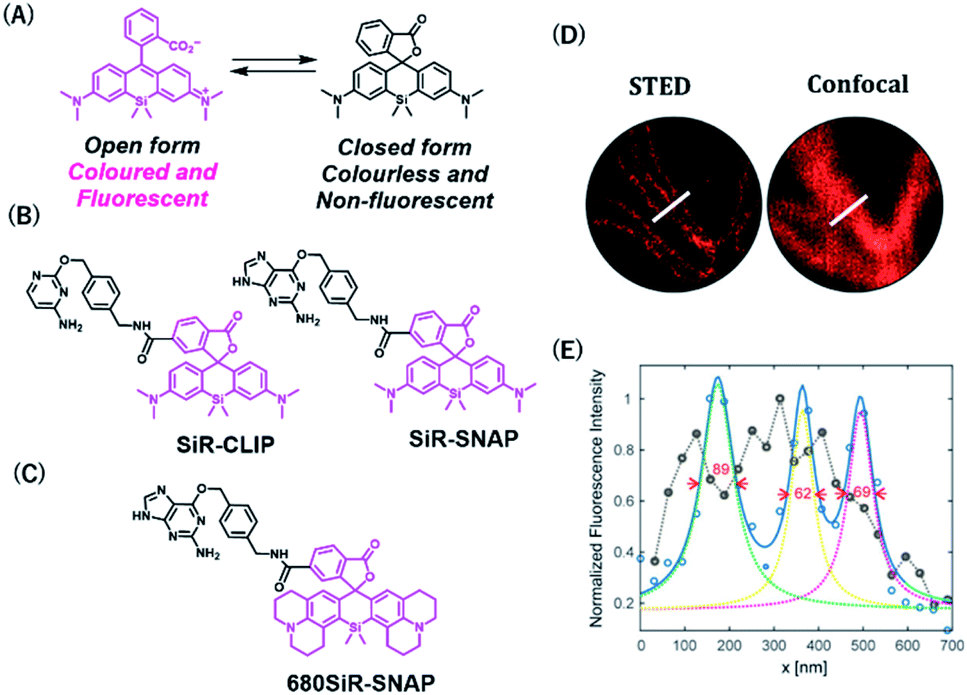 | ||
| Fig. 4 (A) Structures of SiR (Silicon-rhodamine) open and closed forms. (B) Structures of SiR based chemical probes for SNAP-tag-and CLIP-tag. (C) Structure of 680SiR-SNAP. (D) Live-cell STED and confocal images of vimentin-SNAP obtained using 680SiR-SNAP. (E) Line profile of the corresponding image (D). Reproduced from ref. 47 with permission from [The American Chemical Society], copyright [2018]. | ||
Butkevich et al. developed a SiR derivative, 680SiR, which showed improved fluorescence intensity.47 Xanthene fluorophores with N,N-dialkylamino groups in the excited state were found to induce TICT (twisted internal charge transfer), which reduces the fluorescence quantum yield.48 680SiR was designed by introducing a conformationally rigid julolidine into the amino groups of a SiR fluorophore, resulting in suppression of TICT and enhancement of fluorescence intensity. This dye was connected to a SNAP-tag ligand to create 680SiR-SNAP (Fig. 4C) and was used for STED imaging of vimentin. The NIR spectral property of this probe enabled two-color STED imaging, in which a carbopyronine-based probe emitting red fluorescence is used together with 680SiR-SNAP for imaging tubulin and vimentin (Fig. 4D and E).
Halo-tag-based systems
Halo-tag is a mutant of the bacterial enzyme haloalkane dehydrogenase, in which the histidine 272 is replaced by phenylalanine. Its aspartic acid 106 is an active residue that forms an ester bond upon SN2 reaction with haloalkane (Fig. 5A).33 Halo-tag is widely used as a protein tag for imaging applications, although its size (33 kDa) is larger than that of other protein tags. Kosaka et al. developed a NIR fluorescent probe, HaloTagL-IR800, consisting of a HaloTag ligand and an IR800 fluorophore for in vivo cancer imaging (Fig. 5B).49 To demonstrate the potential of this system, the authors performed in vivo fluorescence imaging of mice with implanted tumors stably expressing β1-integrin-HaloTag fusion proteins. Interestingly, the use of HaloTagL-IR800, together with HaloTag ligands containing the other fluorophores, resulted in the multi-colored depiction of the tumors. Pulse-chase in vivo spectral fluorescence imaging using HaloTagL-IR800 and tetramethylrhodamine (TMR)-linked HaloTag ligand showed that fluorescence from tumor nodules was observed for at least 96 h in anesthetized live mice after a single injection of either ligand. | ||
| Fig. 5 (A) Schematic representation of Halo-tag-based protein tag with chemical probe. (B) Structures of NIR Halo-tag chemical probes. (C) Reversible nucleophilic addition of glutathione to SiP650. (D) Live-cell SMLM image of β-tubulin-Halo obtained using SiP650-Halo. POI, protein of interest. Reproduced from ref. 56 with permission from [The American Chemical Society], copyright [2020]. | ||
Lesiak et al. developed NIR HaloTag probes containing a phosphinate-based fluorophore, Nebraska Red, in which the bridging oxygen of TMR is replaced by phosphinate (Fig. 5B).50,51 The fluorophore emits fluorescence at approximately 685 nm, which is slightly red-shifted compared to that in SiR. A phosphinate-based probe named HT-NR666 could selectively label membrane proteins because of its cell membrane-impermeable properties and was used for real-time monitoring of the internalization of G protein-coupled receptors (GPCRs) upon stimulation with its agonist.
Wang et al. recently developed a general strategy to produce cell membrane-permeable probes containing rhodamine fluorophores by attaching different electron-withdrawing groups at the spirolactam of the fluorophores.52 In the free state, the probes form spirolactam, which is nonfluorescent and cell membrane permeable, while protein labeling reactions induce the opening of the lactam ring, resulting in fluorescence enhancement. The authors demonstrated that the acylcyanamide derivative of SiR700 appended with a Halo-tag ligand (MaP700-Halo, Fig. 5B) exhibited a 650-fold increase in fluorescence intensity upon reaction with Halo-tag. MaP700-Halo enabled no-wash imaging of proteins and showed a higher signal-to-background ratio of the image compared to that in SiR700-Halo, which consists of a lactone derivative.
To improve the brightness of rhodamine fluorophores, Grimm et al. developed Janelia fluorophores using a four-membered azetidine ring.48 TICT formation was indicated to reduce the fluorescence quantum yield of rhodamine fluorophores with N,N-dialkylamino groups. Introduction of azetidine into the amino groups was thought to suppress TICT, resulting in an increase in fluorescence intensity of the fluorophores. Through this strategy, the authors developed a JF646 fluorophore from a SiR scaffold, showing that the fluorophore was brighter than that of the original SiR. JF646 was linked with a HaloTag ligand to produce JF646-Halo that showed larger fluorescence enhancement (21-fold) upon HaloTag labeling reaction than that with the SiR-linked HaloTag ligand (6.8-fold). Additionally, the same group also added a single fluorine atom on each azetidine ring to create a probe, JF635-Halo, in which the spectral and chemical properties were optimized (Fig. 5B).53 It was shown that JF635-Halo could visualize Halo-tag fusion proteins in living cells with a higher signal-to-background ratio (9.2-fold) than that did JF646-Halo (6.7-fold). Furthermore, JH635-Halo was used to label myristoylated Halo-tagged proteins expressed in the living brain tissue of Drosophila larvae.
Uno et al. reported spontaneously blinking fluorophores, which generally exist in an inactive nonfluorescent state and occasionally emit fluorescence for a short time.54 This switching property is modulated by the thermal equilibrium between spirocyclized and open forms of rhodamine fluorophores and was shown to be useful for single-molecule localization microscopy (SMLM).55 Among the rhodamine derivatives, a hydroxymethyl SiR (HMSiR) derivative showed photophysical properties suitable for SMLM. HMSiR appended with a HaloTag ligand, HMSiR-Halo (Fig. 5B), enabled super-resolution imaging of β-tubulin fused with HaloTag. Importantly, HMSiR-Halo allowed SMLM without the use of high-power laser irradiation or any additives, such as thiol that is potentially toxic.
To expand the color range of the spontaneously blinking fluorophores that work in live cells, the Morozumi et al. developed green and NIR fluorophores, CP550, and SiP650, consisting of carbopyronine and silicon pyronine, respectively.56 These fluorophores were designed to undergo reversible nucleophilic addition of intracellular glutathione, resulting in fluorescence blinking in living cells (Fig. 5C).56 HaloTag ligands with these fluorophores (CP550-Halo and SiP650-Halo) specifically labeled β-tubulin–HaloTag fusion proteins and allowed SMLM imaging (Fig. 5D). Moreover, CP550 linked with a SNAP-tag ligand was also developed and was used together with HMSiR-Halo for dual-color SMLM imaging of SNAP- and Halo-tagged proteins in live cells.
PYP-tag-based system
PYP-tag is a photoactive yellow protein derived from purple bacteria and is a small monomeric protein (14 kDa).57 It binds to the thioester derivatives of 4-hydroxycinnamic acid or coumarin via a trans-thioesterification reaction with cysteine 69, dissociating thiophenol as a leaving group (Fig. 6A).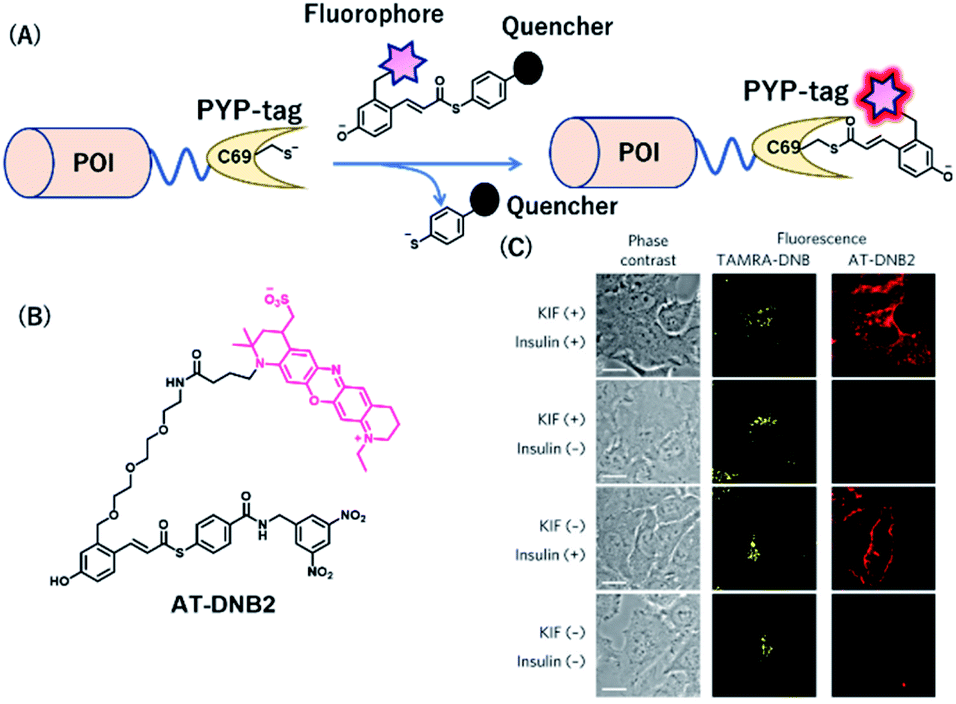 | ||
| Fig. 6 (A) Schematic representation of a PYP-tag-based protein tag with chemical probe. (B) Structure of AT-DNB2, a NIR PYP-tag chemical probe. (C) Dual-color imaging of GLUT4 expressed in HeLa cells treated with kifunensine (KIF), which inhibits glycan maturation. Orange and red fluorescence signals in the images are derived from GLUT4 labeled with the TAMRA probe and AT-DNB2, respectively. POI, protein of interest. Reproduced from ref. 58 with permission from [Springer Nature], copyright [2016]. | ||
A NIR fluorogenic probe, AT-DNB2, targeting the PYP-tag (Fig. 6B) was created by incorporating a dinitrobenzene (DNB) quencher into a 4-hydroxycinnamic acid ligand connected to a phenoxazine fluorophore, ATTO655.58 The fluorophore moiety intramolecularly interacts with the DNB quencher, resulting in fluorescence quenching of AT-DNB2 in the free state. AT-DNB2 was covalently bound to PYP-tag and simultaneously released the DNB quencher, thereby emitting a 16-fold brighter fluorescence. This probe design strategy was also applied to other xanthene fluorophores, fluorescein and TAMRA, to create fluorogenic probes emitting visible fluorescence. AT-DNB2 does not react with intracellular PYP-tagged proteins but selectively labels cell-surface PYP-tagged proteins because of the cell membrane impermeability of the probe; other fluorogenic probes allowed intracellular labeling. Using these fluorogenic probes, intracellular trafficking of glucose transporter 4 (GLUT4) fused to the PYP-tag was visualized. GLUT4 is a vital transmembrane glycoprotein that maintains healthy blood sugar levels by mediating insulin-regulated glucose import into cells.59 In the presence of insulin, GLUT4 is translocated from the intracellular storage vesicle to the plasma membrane (Fig. 6C) and is retained on the membrane for glucose uptake. Meanwhile, the removal of insulin promotes the internalization of GLUT4. We conducted multicolor imaging of intracellular GLUT4 containing N-glycan, and cell-surface GLUT4 lacking the native glycan, using the cell-permeable TAMRA probe and cell-impermeant AT-DNB2, respectively, and verified how N-glycan affected the trafficking of GLUT4. Taking advantage of the fluorogenic property of AT-DNB2, no-wash imaging of GLUT4 was conducted. As a result, transient exocytosis and subsequent internalization of GLUT4 without the native glycan was detected even in the presence of insulin, giving a new finding that the N-glycan plays an important role in the retention of GLUT4 on the cell membrane.
BL-tag-based system
BL-tag is a self-labeling protein tag that was developed by our group, by modifying β-lactamase. The BL-tag is a mutant version of TEM-1 in which glutamic acid at position 166 is replaced by glutamine, thus inhibiting the deacylation step, whereas serine 70 acts as an active residue (Fig. 7A) that forms ester bonds with the β-lactam ring.35 | ||
| Fig. 7 (A) Schematic representation of a BL-tag-based protein tag with chemical probe. (B) Structure of SiRcB and SiRcB(n) (n = 2, 4, 6) as NIR BL-tag chemical probes. (C) Emission spectra of the probes and fluorescence images of BL-NLS expressed in HEK293T cells. POI, protein of interest. Reproduced from ref. 60 with permission from [The American Chemical Society], copyright [2017]. | ||
Our group developed SiR-based NIR fluorescent probes for intracellular protein labeling using BL-tag as a protein tag and applied them for multicolor single-molecule imaging inside living cells.60 We developed a series of membrane-permeable probes SiRcB(n) (n = 2, 4, 6) (Fig. 7B), where SiR attached as a NIR fluorophore, is conjugated to bacampicillin, a prodrug of penicillin, as a BL-tag ligand. With these tools, NIR fluorescence signals from the nucleus were observed, indicating that the probes were permeable to the intracellular membrane (Fig. 7C). It is noteworthy that the fine-tuning of the hydrophilicity and the cell permeability of the probes by modifying the length of the oligoethylenoxy linker was critical for single-molecule imaging of labeled intracellular proteins. Moreover, using both the NIR BL-tag probe, SiRcB4, and a red Halo-tag probe, we revealed the dynamics of interaction between toll-like receptor 4 (TLR4) and TIR domain-containing adaptor protein (TIRAP) using multicolor single-molecule imaging upon stimulation with lipopolysaccharide (LPS).
eDHFR/TMP-tag-based systems
Together with the development of covalent protein-tag systems, non-covalent-based protein-labeling strategies have also emerged.61 Unlike covalent-based protein-labeling strategies, non-covalent labeling provides some advantages, such as extremely fast labeling owing to the lack of need for covalent bond formation. Miller et al. developed a small molecule-based inhibitor tag from an Escherichia coli enzyme, dihydrofolate reductase (eDHFR). A trimethoprim (TMP)-conjugated fluorophore was used as a probe for selective labeling of eDHFR-tagged fusion proteins (Fig. 8A). The small size (18 kDa) of this protein tag and its high affinity for the TMP ligand characterize eDHFR as a promising alternative non-covalent self-labeling protein tag.36 | ||
| Fig. 8 (A) Schematic representation of an eDHFR/TMP-tag-based protein tag with chemical probe. (B) Structure of NIR probe for eDHFR/TMP-tag. (C) Representation of covalent eDHFR/TMP-tag. (D) Structure of covalent NIR probe for eDHFR/TMP-tag. (E) Fluorescence images of H2B-eDHFR:L28C and PMLS-eDHFR:L28C in HEK293T cells using A-TMP-Atto655. POI, protein of interest. Reproduced from ref. 64 with permission from [The American Chemical Society], copyright [2012]. | ||
Furthermore, Wombacher et al. developed super-resolution imaging using an eDHFR/TMP tag combined with a synthetic NIR fluorophore.62 They exploited ATTO655 as an ideal fluorophore for super-resolution imaging because of its efficient photo-switching properties under the reducing environment of the cellular conditions. Using TMP-ATTO655 as a fluorescent TMP ligand (Fig. 8B), they labeled histone H2B proteins and visualized the protein in nano-dimensions by direct stochastic optical reconstruction microscopy (dSTORM), a microscopy method based on stochastic single-molecule localization by photoswitching of the fluorophore.63 This allowed the imaging of the dynamics of human histone H2B protein in living cells at ∼20 nm resolution on the 10 s timescale.
Modification of an eDHFR/TMP-tag can be used for covalent protein labeling. To generate a covalently bound protein tag, a mutant eDHFR/TMP-tag (eDHFR:L28C), where leucine 28 was replaced with cysteine, was engineered to react with electrophilic acrylamide introduced on the TMP-fluorophore conjugate (Fig. 8C).64 They also developed a chemical probe, A-TMP-ATTO655 (Fig. 8D), for covalent labeling of the eDHFR/TMP-tag with a NIR fluorophore.64 Upon treatment with A-TMP-ATTO655 of HEK293T cells transiently expressing mutant eDHFR in the nucleus and plasma membrane (Fig. 8E), the authors demonstrated selective labeling of the target proteins by confocal microscopy with a rapid labeling half-life.
FAP-tag-based systems
Szent-Gyorgyi et al. developed a fluorogen activating protein (FAP)-tag system where a chemical probe exists in a dark state but emits fluorescence when bound to a reporter protein (Fig. 9A).37 The authors screened the library of human single-chain variable fragment (scFv) antibodies (size <30 kDa) and developed FAPs binding to malachite green (MG) (Fig. 9B). MG is nonfluorescent in a buffer, but emits fluorescence in the NIR region when the chromophores are constrained upon binding to FAP. After fluorescence screening in yeast cells expressing scFv antibodies, FAP-expressing clones were obtained with enhanced fluorescence emission with MG-2p (Fig. 9C).37 The fluorogenic labeling resulted in strong NIR fluorescence in FAP-fused cell surface proteins by the MG fluorogen. Özhalici-Ünal et al. developed a NIR-based chemical probe based on a dimethylindole red (DIR) analog (Fig. 9D).65 The probes showed a high binding affinity towards specific cloned K7 scFv. Further, the authors found that the dissociation constants of these probes with K7 were in the nM range and the quantum yields were increased upon binding to K7. Moreover, the authors showed that yeast expressing FAP K7 can be fluorescently detected by the DIR probe (Fig. 9E).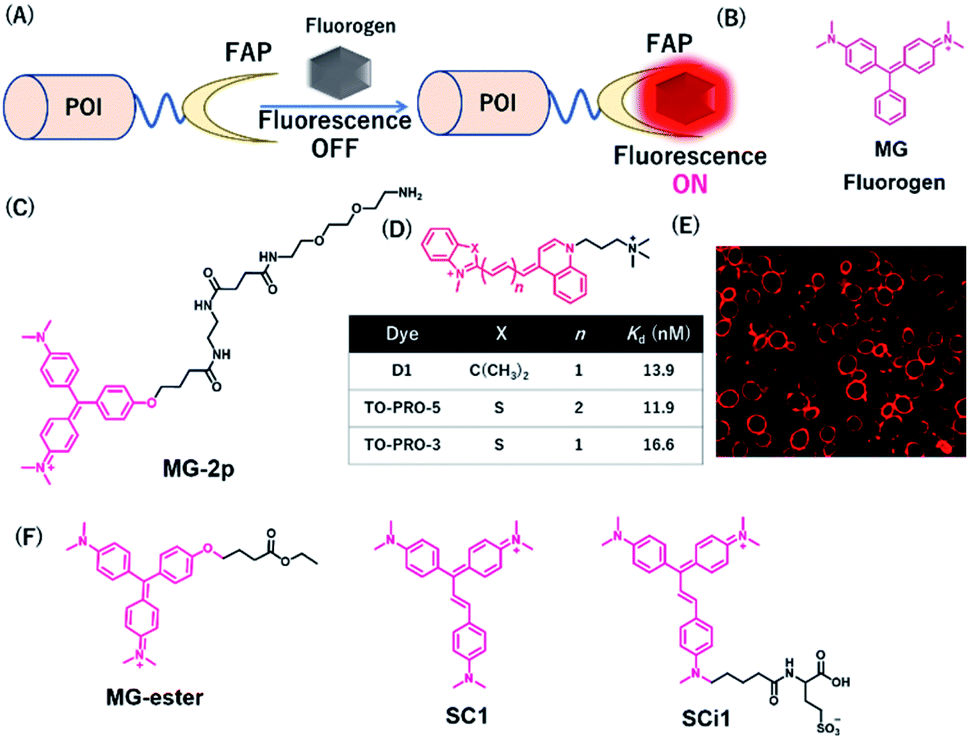 | ||
| Fig. 9 (A) Schematic representation of fluorogen activating proteins (FAPs)-based protein tag. (B) Structure of MG. (C) Structure of MG-based fluorogen for FAPs. (D) Structure of DIR-based fluorogen. (E) Fluorescence images of FAPs expressing yeast cells using D1. (F) Structure of NIR chemical probes for FAPs. POI, protein of interest. Reproduced from ref. 65 with permission from [The American Chemical Society], copyright [2008]. | ||
Using cell-permeable derivatives of MG-ester and FAPs, Yan et al. applied super-resolution imaging in fixed cells and in live cells (Fig. 9F).66 In this system, named binding and activation localization microscopy (BALM), stochastic binding of the fluorogens to FAP can be detected at the single-molecule level, resulting in subdiffraction images of actin structures.
Most FAP–fluorogen complexes emit fluorescence below the 700 nm range. Therefore, generating a fluorogen that emits >700 nm will be favorable for minimizing the effects of tissue absorption and autofluorescence for in vivo imaging. Zhang et al. developed an activatable fluorescent probe for the SC1 and SCi1 fluorogens (Fig. 9F), which emits near the 700 nm and 730 nm upon binding to FAPs, respectively.67 This probe showed an approximately thousand-fold enhancement of fluorescence emission between bound and unbound states. Furthermore, the authors demonstrated that these fluorogens can be applied to the imaging of live cells with a high signal-to-noise ratio, especially for in vivo imaging of FAP-bearing tumors in mice.
FAST-tag-based system
Plamont et al. reported a fluorescence-activating and absorption-shifting tag (FAST), a small protein tag enabling reversible fluorescence labeling of proteins with fluorogens in living cells.38 This protein tag was engineered from a variant of the monomeric small protein PYP.Recently, Li et al. described a far-red chemical probe, HPAR-3OM, targeting the far-red FAST (frFAST) tag.68 The frFAST tag was developed by a mutation of the chromophore-binding site of the original FAST tag. The probe HPAR-3OM is initially nonfluorescent in solution, but strongly emits fluorescence signal when bound to the frFAST tag with a 115 nm Stokes shift (Fig. 10). This labeling system with HPAR-3OM allowed high-contrast far-red fluorescence imaging of various fusion proteins with a frFAST tag in living cells. Further, rapid and efficient imaging of frFAST fusions was demonstrated in live zebrafish embryos and larvae and chicken embryos.
 | ||
| Fig. 10 (A) Schematic representation of a frFAST-based protein tag with chemical probe. (B) Absorption and emission spectra of HPAR-3OM in absence (black) or presence (magenta) of frFAST. (C) Fluorescence images of EGFP, mCherry, and frFAST fusions (co-incubated with HPAR-3OM) in live HeLa and U2OS cells. POI, protein of interest. frFAST, far-red fluorescence-activating and absorption-shifting tag. Reproduced from ref. 68 with permission from [Wiley-VCH GmbH], copyright [2020]. | ||
Summary and future perspectives
In this review article, we have summarized the development of NIR fluorescent probes with protein labeling ability to various imaging applications such as live cell, in vivo, single-molecule, and super-resolution imaging. NIR fluorescent probes have emerged in recent years and continue to develop as they help overcome the drawbacks of the present fluorescent probes with visible light emission. The development of SiR-based fluorescent probes with high brightness and photostability is a significant advancement in this field. For instance, NIR-based multicolor imaging studies revealed some important protein dynamics and functions in living cells. NIR-based protein tag probes can also be used to visualize protein structures in nano-dimensions by using STED or single-molecule localization microscopy. Recently, modifications of the SiR chromophore have been made to generate a cell-permeable probe for protein labeling with an improved signal-to-noise ratio. The Janelia fluorophores improved the quantum yields and modulated the fluorescence by the environment, thereby allowing fluorogenic protein labeling with high imaging contrast.In the future, further development of fluorescent probes by chemical modifications is required to render longer excitation/emission wavelengths, brightness, photostability, and efficient protein-labeling ability. NIR fluorophores presently used for imaging applications emit in the range of 650–730 nm. However, bathochromic-shifted dyes are preferred for in vivo imaging because of the improved light penetration in deep tissue.69 There is considerable scope for developing fluorescent probes that emit longer wavelengths for imaging protein function in vivo.70 More application studies with the presented labeling probes are also expected by using advanced imaging techniques. Substantial efforts in protein-labeling strategies with NIR fluorophores will elucidate the various functions of proteins and other analytes, thereby facilitating our understanding of complex cellular activities and networks.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the JSPS KAKENHI (grant numbers: JP17H06409 “Frontier Research on Chemical Communications”, JP18H03935 and 19K22255 to K. K.; JP17H02210, JP18K19402, and JP20H02879 to Y. H. and 16K01933, 20K05747 to M. M.), JSPS A3 Foresight Program, JSPS Asian CORE Program, “Asian Chemical Biology Initiative”, Japan (JSPS)-UK (RSC) Research Cooperative Program (JPJSBP120195705 to K. K.), AMED-CREST, and Toray Science Foundation (19-6008). S. I. R. would like to thank JSPS and TBRF (Tokyo Biochemical Research Foundation) for postdoctoral fellowship.Notes and references
- (a) M. Rudin and R. Weissleder, Nat. Rev. Drug Discovery, 2003, 2, 123–131 CrossRef CAS; (b) M. Schäferling, Angew. Chem., Int. Ed., 2012, 51, 3532–3554 CrossRef; (c) V. Ntziachristos, Annu. Rev. Biomed. Eng., 2006, 8, 1–33 CrossRef.
- (a) J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2006 CrossRef; (b) A. Ito, Y. Ito, S. Matsushima, D. Tsuchida, M. Ogasawara, J. Hasegawa, K. Misawa, E. Kondo, N. Kaneda and H. Nakanishi, Gastric Cancer, 2014, 17, 497–507 CrossRef CAS.
- (a) J. O. Escobedo, O. Rusin, S. Lim and R. M. Strongin, Curr. Opin. Chem. Biol., 2010, 14, 64–70 CrossRef CAS; (b) K. Kiyose, H. Kojima and T. Nagano, Chem.–Asian J., 2008, 3, 506–515 CrossRef CAS; (c) S. A. Hilderbrand and R. Weissleder, Curr. Opin. Chem. Biol., 2010, 14, 71–79 CrossRef CAS.
- (a) J. V. Frangioni, Curr. Opin. Chem. Biol., 2003, 7, 626–634 CrossRef CAS; (b) J. Huang and K. Pu, Chem. Sci., 2020 10.1039/D0SC02925D.
- (a) M. Monici, Biotechnol. Annu. Rev., 2005, 11, 227–256 CAS; (b) G. Hong, A. L. Antaris and H. Dai, Nat. Biomed. Eng., 2017, 1, 0010 CrossRef CAS.
- A. M. Smith, M. C. Mancini and S. Nie, Nat. Nanotechnol., 2009, 4, 710–711 CrossRef CAS.
- Q. Cao, N. G. Zhegalova, S. T. Wang, W. J. Akers and M. Y. Berezin, J. Biomed. Optic., 2013, 18, 101318 CrossRef.
- (a) S. Wang, B. Li and F. Zhang, ACS Cent. Sci., 2020, 6, 1302–1316 CrossRef CAS; (b) V. J. Pansare, S. Hejazi, W. J. Faenza and R. K. Prud’homme, Chem. Mater., 2012, 24, 812–827 CrossRef CAS; (c) A. K. East, M. Y. Lucero and J. Chan, Chem. Sci., 2020 10.1039/D0SC03096A.
- K. J. McHugh, L. Jing, S. Y. Severt, M. Cruz, M. Sarmadi, H. S. N. Jayawardena, C. F. Perkinson, F. Larusson, S. Rose, S. Tomasic, T. Graf, S. Y. Tzeng, J. L. Sugarman, D. Vlasic, M. Peters, N. Peterson, L. Wood, W. Tang, J. Yeom, J. Collins, P. A. Welkhoff, A. Karchin, M. Tse, M. Gao, M. G. Bawendi, R. Langer and A. Jaklenec, Sci. Transl. Med., 2019, 11, eaay7162 CrossRef CAS.
- (a) H.-W. Liu, L. Chen, C. Xu, Z. Li, H. Zhang, X.-B. Zhang and W. Tan, Chem. Soc. Rev., 2018, 47, 7140–7180 RSC; (b) J.-B. Li, H.-W. Liu, T. Fu, R. Wang, X.-B. Zhang and W. Tan, Trends Chem., 2019, 1, 224–234 CrossRef.
- (a) K. Umezawa, D. Citterio and K. Suzuki, Anal. Sci., 2014, 30, 327–349 CrossRef CAS; (b) Y. Ni and J. Wu, Org. Biomol. Chem., 2014, 12, 3774–3791 RSC; (c) F. Ding, Y. Zhan, X. Lu and Y. Sun, Chem. Sci., 2018, 9, 4370–4380 RSC.
- (a) A. Samanta, M. Vendrell, R. Das and Y.-T. Chang, Chem. Commun., 2010, 46, 7406–7408 RSC; (b) R. J. Mellanby, J. I. Scott, I. Mair, A. Fernandez, L. Saul, J. Arlt, M. Moral and M. Vendrell, Chem. Sci., 2018, 9, 7261–7270 RSC.
- K. Ilina, W. M. MacCuaig, M. Laramie, J. N. Jeouty, L. R. McNally and M. Henary, Bioconjugate Chem., 2020, 31(2), 194–213 CrossRef CAS.
- Z. Shi, X. Han, W. Hu, H. Bai, B. Peng, L. Ji, Q. Fan, L. Li and W. Huang, Chem. Soc. Rev., 2020 10.1039/D0CS00234H.
- (a) Y. Koide, Y. Urano, K. Hanaoka, T. Terai and T. Nagano, ACS Chem. Biol., 2011, 6, 600–608 CrossRef CAS; (b) M. Grzybowski, M. Taki, K. Senda, Y. Sato, T. Ariyoshi, Y. Okada, R. Kawakami, T. Imamura and S. Yamaguchi, Angew. Chem., Int. Ed., 2018, 57, 10137–10141 CrossRef CAS.
- L. Yuan, W. Lin, Y. Yang and H. Chen, J. Am. Chem. Soc., 2012, 134, 1200–1211 CrossRef CAS.
- (a) L. Yuan, W. Lin, K. Zheng, L. He and W. Huang, Chem. Soc. Rev., 2013, 42, 622–661 RSC; (b) Z. Guo, S. Park, J. Yoon and I. Shin, Chem. Soc. Rev., 2014, 43, 16–29 RSC; (c) X. Chen, F. Wang, J. Y. Hyun, T. Wei, J. Qiang, X. Ren, I. Shin and J. Yoon, Chem. Soc. Rev., 2016, 45, 2976–3016 RSC; (d) Y. V. Suseela, N. Narayanaswamy, S. Pratihara and T. Govindaraju, Chem. Soc. Rev., 2018, 47, 1098–1131 RSC.
- (a) Y. Koide, Y. Urano, K. Hanaoka, W. Piao, M. Kusakabe, N. Saito, T. Terai, T. Okabe and T. Nagano, J. Am. Chem. Soc., 2012, 134, 5029–5031 CrossRef CAS; (b) L. Wang, J. Hiblot, C. Popp, L. Xue and K. Johnsson, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202008357.
- H. Li, D. Kim, Q. Yao, H. Ge, J. Chung, J. Fan, J. Wang, X. Peng and J. Yoon, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202009796.
- (a) L. Chen, Y.-L. Lin, G. Peng and F. Li, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17966–17971 CrossRef CAS; (b) H. Li, Y. Li, Q. Yao, J. Fan, W. Sun, S. Long, K. Shao, J. Du, J. Wang and X. Peng, Chem. Sci., 2019, 10, 1619–1625 RSC; (c) H. Li, Qi. Yao, W. Sun, K. Shao, Y. Lu, J. Chung, D. Kim, J. Fan, S. Long, J. Du, Y. Li, J. Wang, J. Yoon and X. Peng, J. Am. Chem. Soc., 2020, 142, 6381–6389 CrossRef CAS.
- R. J. Iwatate, M. Kamiya, K. Umezawa, H. Kashima, M. Nakadate, R. Kojima and Y. Urano, Bioconjugate Chem., 2018, 29, 241–244 CrossRef CAS.
- (a) J. Zhang, P. Cheng and K. Pu, Bioconjugate Chem., 2019, 30, 2089–2101 CrossRef CAS; (b) M. Chiba, M. Kamiya, K. Tsuda-Sakurai, Y. Fujisawa, H. Kosakamoto, R. Kojima, M. Miura and Y. Urano, ACS Cent. Sci., 2019, 5, 1676–1681 CrossRef CAS.
- (a) R. Yan, Y. Hu, F. Liu, S. Wei, D. Fang, A. J. Shuhendler, H. Liu, H.-Y. Chen and D. Ye, J. Am. Chem. Soc., 2019, 141, 10331–10341 CrossRef CAS; (b) Y. Li, Y. Sun, J. Li, Q. Su, W. Yuan, Y. Dai, C. Han, Q. Wang, W. Feng and F. Li, J. Am. Chem. Soc., 2015, 137, 6407–6416 CrossRef CAS.
- X. Chen, D. Lee, S. Yu, G. Kim, S. Lee, Y. Cho, H. Jeong, K. T. Nam and J. Yoon, Biomaterials, 2017, 122, 130–140 CrossRef CAS.
- (a) L. O. Ofori, N. P. Withana, T. R. Prestwood, M. Verdoes, J. J. Brady, M. M. Winslow, J. Sorger and M. Bogyo, ACS Chem. Biol., 2015, 10, 1977–1988 CrossRef CAS; (b) S. L. Gibbs, Quant. Imag. Med. Surg., 2012, 2, 177–187 Search PubMed; (c) N. Kosaka, M. Ogawa, P. L. Choyke and H. Kobayashi, Future Oncol., 2009, 5, 1501–1511 CrossRef.
- (a) M. Grossi, M. Morgunova, S. Cheung, D. Scholz, E. Conroy, M. Terrile, A. Panarella, J. C. Simpson, W. M. Gallagher and D. F. O'Shea, Nat. Commun., 2016, 7, 10855 CrossRef CAS; (b) S. Benson, A. Fernandez, N. D. Barth, F. d. Moliner, M. H. Horrocks, C. S. Herrington, J. L. Abad, A. Delgado, L. Kelly, Z. Chang, Y. Feng, M. Nishiura, Y. Hori, K. Kikuchi and M. Vendrell, Angew. Chem., Int. Ed., 2019, 58, 6911–6915 CrossRef CAS; (c) E. Kozma and P. Kele, Org. Biomol. Chem., 2019, 17, 215–233 RSC.
- R. Y. Tsien, Annu. Rev. Biochem., 1998, 67, 509–544 CrossRef CAS.
- M. Fernández-Suárez and A. Y. Ting, Nat. Rev. Mol. Cell Biol., 2008, 9, 929–943 CrossRef.
- L. Wang, M. S. Frei, A. Salim and K. Johnsson, J. Am. Chem. Soc., 2019, 141, 2770–2781 CrossRef CAS.
- (a) H. M. O'Hare, K. Johnsson and A. Gautier, Curr. Opin. Struct. Biol., 2007, 17, 488–494 CrossRef; (b) S. Mizukami, S. Watanabe, Y. Akimoto and K. Kikuchi, J. Am. Chem. Soc., 2012, 134, 1623–1629 CrossRef CAS.
- A. Keppler, S. Gendreizig, T. Gronemeyer, H. Pick, H. Vogel and K. Johnsson, Nat. Biotechnol., 2003, 21, 86–89 CrossRef CAS.
- A. Gautier, A. Juillerat, C. Heinis, I. R. Corrêa, Jr., M. Kindermann, F. Beaufils and K. Johnsson, Chem. Biol., 2008, 15, 128–136 CrossRef CAS.
- G. V. Los, L. P. Encell, M. G. McDougall, D. D. Hartzell, N. Karassina, C. Zimprich, M. G. Wood, R. Learish, R. F. Ohana, M. Urh, D. Simpson, J. Mendez, K. Zimmerman, P. Otto, G. Vidugiris, J. Zhu, A. Darzins, D. H. Klaubert, R. F. Bulleit and K. V. Wood, ACS Chem. Biol., 2008, 3, 373–382 CrossRef CAS.
- Y. Hori, H. Ueno, S. Mizukami and K. Kikuchi, J. Am. Chem. Soc., 2009, 131, 16610–16611 CrossRef CAS.
- S. Mizukami, S. Watanabe, Y. Hori and K. Kikuchi, J. Am. Chem. Soc., 2009, 131, 5016–5017 CrossRef CAS.
- L. W. Miller, Y. Cai, M. P. Sheetz and V. W. Cornish, Nat. Methods, 2005, 2, 255–257 CrossRef CAS.
- C. Szent-Gyorgyi, B. F. Schmidt, Y. Creeger, G. W. Fisher, K. L. Zakel, S. Adler, J. A. J. Fitzpatrick, C. A. Woolford, Q. Yan, K. V. Vasilev, P. B. Berget, M. P. Bruchez, J. W. Jarvik and A. Waggoner, Nat. Biotechnol., 2008, 26, 235–240 CrossRef CAS.
- M.-A. Plamont, E. Billon-Denis, S. Maurin, C. Gauron, F. M. Pimenta, C. G. Specht, J. Shi, J. Quérard, B. Pan, J. Rossignol, K. Moncoq, N. Morellet, M. Volovitch, E. Lescop, Y. Chen, A. Triller, S. Vriz, T. L. Saux, L. Jullien and A. Gautier, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 497–502 CrossRef CAS.
- A. Gautier, E. Nakata, G. Lukinavičius, K.-T. Tan and K. Johnsson, J. Am. Chem. Soc., 2009, 131, 17954–17962 CrossRef CAS.
- C. Campos, M. Kamiya, S. Banala, K. Johnsson and M. González-Gaitán, Dev. Dynam., 2011, 240, 820–827 CrossRef CAS.
- X. Song, H. Bian, C. Wang, M. Hu, N. Li and Y. Xiao, Org. Biomol. Chem., 2017, 15, 8091–8101 RSC.
- Y. Koide, Y. Urano, K. Hanaoka, T. Terai and T. Nagano, J. Am. Chem. Soc., 2011, 133, 5680–5682 CrossRef CAS.
- G. Lukinaviêius, K. Umezawa, N. Olivier, A. Honigmann, G. Yang, T. Plass, V. Mueller, L. Reymond, I. R. Corrêa Jr, Z.-G. Luo, C. Schultz, E. A. Lemke, P. Heppenstall, C. Eggeling, S. Manley and K. Johnsson, Nat. Chem., 2013, 5, 132–139 CrossRef.
- J. Fölling, M. Bossi, H. Bock, R. Medda, C. A. Wurm, B. Hein, S. Jakobs, C. Eggeling and S. W. Hell, Nat. Methods, 2008, 5, 943–945 CrossRef.
- M. Heilemann, S. L. M. Schüttpelz, R. Kasper, B. Seefeldt, A. Mukherjee, P. Tinnefeld and M. Sauer, Angew. Chem., Int. Ed., 2008, 47, 6172–6176 CrossRef CAS.
- V. Westphal, S. O. Rizzoli, M. A. Lauterbach, D. Kamin, R. Jahn and S. W. Hell, Science, 2008, 320, 246–249 CrossRef CAS.
- A. N. Butkevich, H. Ta, M. Ratz, S. Stoldt, S. Jakobs, V. N. Belov and S. W. Hell, ACS Chem. Biol., 2018, 13, 475–480 CrossRef CAS.
- J. B. Grimm, B. P. English, J. Chen, J. P. Slaughter, Z. Zhang, A. Revyakin, R. Patel, J. J. Macklin, D. Normanno, R. H. Singer, T. Lionnet and L. D. Lavis, Nat. Methods, 2015, 12, 244–250 CrossRef CAS.
- N. Kosaka, M. Ogawa, P. L. Choyke, N. Karassina, C. Corona, M. McDougall, D. Lynch, C. Hoyt, R. Levenson, G. V. Los and H. Kobayashi, Bioconjugate Chem., 2009, 20, 1367–1374 CrossRef CAS.
- X. Zhou, R. Lai, J. R. Beck, H. Lia and C. I. Stains, Chem. Commun., 2016, 52, 12290–12293 RSC.
- L. Lesiak, X. Zhou, Y. Fang, J. Zhao, J. R. Becka and C. I. Stains, Org. Biomol. Chem., 2020, 18, 2459–2467 RSC.
- L. Wang, M. Tran, E. D'Este, J. Roberti, B. Koch, L. Xue and K. Johnsson, Nat. Chem., 2020, 12, 165–172 CrossRef CAS.
- J. B. Grimm, A. K. Muthusamy, Y. Liang, T. A. Brown, W. C. Lemon, R. Patel, R. Lu, J. J. Macklin, P. J. Keller, N. Ji and L. D. Lavis, Nat. Methods, 2017, 14, 987–994 CrossRef CAS.
- S. Uno, M. Kamiya, T. Yoshihara, K. Sugawara, K. Okabe, M. C. Tarhan, H. Fujita, T. Funatsu, Y. Okada, S. Tobita and Y. Urano, Nat. Chem., 2014, 6, 681–689 CrossRef CAS.
- E. Betzig, G. H. Patterson, R. Sougrat, O. W. Lindwasser, S. Olenych, J. S. Bonifacino, M. W. Davidson, J. Lippincott-Schwartz and H. F. Hess, Science, 2006, 313, 1642–1645 CrossRef CAS.
- A. Morozumi, M. Kamiya, S. Uno, K. Umezawa, R. Kojima, T. Yoshihara, S. Tobita and Y. Urano, J. Am. Chem. Soc., 2020, 142, 9625–9633 CAS.
- (a) Y. Hori and k. Kikuchi, Curr. Opin. Chem. Biol., 2013, 17, 644–650 CrossRef CAS; (b) F. Gao, T. Gao, K. Zhou and W. Zeng, Molecules, 2016, 21, 1163 CrossRef.
- S. Hirayama, Y. Hori, Z. Benedek, T. Suzuki and K. Kikuchi, Nat. Chem. Biol., 2016, 12, 853–859 CrossRef CAS.
- D. Leto and A. R. Saltiel, Nat. Rev. Mol. Cell Biol., 2012, 13, 383–396 CrossRef CAS.
- R. Sato, J. Kozuka, M. Ueda, R. Mishima, Y. Kumagai, A. Yoshimura, M. Minoshima, S. Mizukami and K. Kikuchi, J. Am. Chem. Soc., 2017, 139, 17397–17404 CrossRef CAS.
- C. Li, A. G. Tebo and A. Gautier, Int. J. Mol. Sci., 2017, 18, 1473 CrossRef.
- R. Wombacher, M. Heidbreder, S. Linde, M. P. Sheetz, M. Heilemann, V. W. Cornish and M. Sauer, Nat. Methods, 2010, 7, 717–719 CrossRef CAS.
- M. Heilemann, S. Linde, A. Mukherjee and M. Sauer, Angew. Chem., Int. Ed., 2009, 48, 6903–6908 CrossRef CAS.
- Z. Chen, C. Jing, S. S. Gallagher, M. P. Sheetz and V. W. Cornish, J. Am. Chem. Soc., 2012, 134, 13692–13699 CrossRef CAS.
- H. Özhalici-Ünal, C. L. Pow, S. A. Marks, L. D. Jesper, G. L. Silva, N. I. Shank, E. W. Jones, J. M. Burnette, P. B. Berget and B. A. Armitage, J. Am. Chem. Soc., 2008, 130, 12620–12621 CrossRef.
- Q. Yan, S. L. Schwartz, S. Maji, F. Huang, C. Szent-Gyorgyi, D. S. Lidke, K. A. Lidke and M. P. Bruchez, ChemPhysChem, 2014, 15, 687–695 CrossRef CAS.
- M. Zhang, S. K. Chakraborty, P. Sampath, J. J. Rojas, W. Hou, S. Saurabh, S. H. Thorne, M. P. Bruchez and A. S. Waggoner, J. Clin. Invest., 2015, 125, 3915–3927 CrossRef.
- C. Li, A. G. Tebo, M. Thauvin, M.-A. Plamont, M. Volovitch, X. Morin, S. Vriz and A. Gautier, Angew. Chem., Int. Ed., 2020, 59, 17917–17923 CrossRef CAS.
- S. Zhua, Q. Yang, A. L. Antarisa, J. Yue, Z. Ma, H. Wang, W. Huang, H. Wan, J. Wang, S. Diao, B. Zhang, X. Li, Y. Zhong, K. Yu, G. Hong, J. Luo, Y. Liang and H. Dai, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 962–967 CrossRef.
- M. Dai, Y. J. Reo, C. W. Song, Y. J. Yang and K. H. Ahn, Chem. Sci., 2020, 11, 8901–8911 RSC.
| This journal is © The Royal Society of Chemistry 2021 |