
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular reactivity of thiolate-protected noble metal nanoclusters: synthesis, self-assembly, and applications
Qiaofeng
Yao
 a,
Zhennan
Wu
a,
Zhihe
Liu
ac,
Yingzheng
Lin
ac,
Xun
Yuan
*b and
Jianping
Xie
*ac
a,
Zhennan
Wu
a,
Zhihe
Liu
ac,
Yingzheng
Lin
ac,
Xun
Yuan
*b and
Jianping
Xie
*ac
aDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 4 Engineering Drive 4, Singapore 117585. E-mail: chexiej@nus.edu.sg
bCollege of Materials Science and Engineering, Qingdao University of Science and Technology, Qingdao, China 266042. E-mail: yuanxun@qust.edu.cn
cJoint School of National University of Singapore and Tianjin University, International Campus of Tianjin University, Binhai New City, Fuzhou, China 350207
First published on 23rd November 2020
Abstract
Thiolate-protected noble metal (e.g., Au and Ag) nanoclusters (NCs) are ultra-small particles with a core size of less than 3 nm. Due to the strong quantum confinement effects and diverse atomic packing modes in this ultra-small size regime, noble metal NCs exhibit numerous molecule-like optical, magnetic, and electronic properties, making them an emerging family of “metallic molecules”. Based on such molecule-like structures and properties, an individual noble metal NC behaves as a molecular entity in many chemical reactions, and exhibits structurally sensitive molecular reactivity to various ions, molecules, and other metal NCs. Although this molecular reactivity determines the application of NCs in various fields such as sensors, biomedicine, and catalysis, there is still a lack of systematic summary of the molecular interaction/reaction fundamentals of noble metal NCs at the molecular and atomic levels in the current literature. Here, we discuss the latest progress in understanding and exploiting the molecular interactions/reactions of noble metal NCs in their synthesis, self-assembly and application scenarios, based on the typical M(0)@M(I)–SR core–shell structure scheme, where M and SR are the metal atom and thiolate ligand, respectively. In particular, the continuous development of synthesis and characterization techniques has enabled noble metal NCs to be produced with molecular purity and atomically precise structural resolution. Such molecular purity and atomically precise structure, coupled with the great help of theoretical calculations, have revealed the active sites in various structural hierarchies of noble metal NCs (e.g., M(0) core, M–S interface, and SR ligand) for their molecular interactions/reactions. The anatomy of such molecular interactions/reactions of noble metal NCs in synthesis, self-assembly, and applications (e.g., sensors, biomedicine, and catalysis) constitutes another center of our discussion. The basis and practicality of the molecular interactions/reactions of noble metal NCs exemplified in this Review may increase the acceptance of metal NCs in various fields.
 Qiaofeng Yao | Qiaofeng Yao received his B.S. (2010) from the University of Science and Technology of China (USTC). He then earned his Ph.D. (2015) from the National University of Singapore (NUS), under the co-supervision of Prof. Jim Yang Lee and Prof. Jianping Xie. He is currently working as a research fellow with Prof. Jianping Xie at NUS. His current research interest focuses on developing total synthesis and self-assembly chemistry for atomically precise metal nanoclusters. |
 Zhennan Wu | Zhennan Wu obtained his Ph.D. degree (2016) from the State Key Laboratory of Supramolecular Structure and Materials at Jilin University under the supervision of Prof. Hao Zhang. Thereafter, he joined the team of Prof. Osman M. Bakr in King Abdullah University of Science and Technology (KAUST) for post-doctoral research and has been working as a research fellow with Prof. Jianping Xie in NUS since 2018. His research interests are centered on the self-assembly chemistry and optical physics of ultrasmall metal nanoclusters. |
 Zhihe Liu | Zhihe Liu received his B.S. (2015) from China University of Geosciences (CUG). Then, he obtained his M.S. (2018) from State Key Lab of Crystal Materials, Shandong University of China (SDU) under the supervision of Prof. Hong Liu. He is currently a Ph.D. student under the supervision of Prof. Jianping Xie at NUS. His research interest is in the development of conductive metal nanoclusters and their self-assembly in electrochemical applications. |
 Yingzheng Lin | Yingzheng Lin received her B.S. degree (2016) from Sun Yat-sen University, China, and M.E. degree (2019) from the Institute of Urban Environment, Chinese Academy of Sciences, China. After that, she joined Prof. Jianping Xie's group as a PhD student at the Department of Chemical and Biomolecular Engineering, NUS. She is interested in engineering metal nanoclusters for environmental applications. |
 Xun Yuan | Xun Yuan received his B.E. (2006) and M.E. (2009) degrees from Shandong University of Technology, China, and Ph.D. degree (2014) from NUS under the supervision of Prof. Jianping Xie. After 3 years of postdoctoral work at the Institute of Bioengineering and Nanotechnology (IBN), he joined Qingdao University of Science and Technology (QUST) as a professor in 2017. His research focuses on the synthesis and applications of metal nanoclusters. |
 Jianping Xie | Jianping Xie is currently a Dean's Chair Associate Professor at the Department of Chemical & Biomolecular Engineering, NUS. He received his B.S. and M.S. in Chemical Engineering from Tsinghua University, China, and his Ph.D. from the Singapore MIT Alliance (SMA) program. He joined NUS in 2010 and established the BioNanoMetals research group. His group is known for their work on engineering ultrasmall metal nanoclusters for biomedical and catalytic applications. His research interests include noble metal nanoclusters, nanomedicine, and cluster catalysis. |
1. Introduction
With the development of nanochemistry towards atomic precision, thiolate-protected noble metal (e.g., Au and Ag) nanoclusters (NCs) have attracted great interest due to their atomically tunable structures and properties.1–7 Thiolate-protected metal nanoclusters (NCs) are an emerging family of ultra-small particles (<3 nm) composed of several to several hundred metal atoms.3–5,8 They can be well described by the molecular formula [Mn(SR)m]q, where n, m and q are the number of metal atoms (M = Au or Ag) and thiolate ligands (SR), and net charge, respectively. The remarkable achievements of X-ray crystallography in the past two decades suggest a universal core–shell structure scheme for [Mn(SR)m]q NCs, in which an M(0) core is capped by a monolayer of M(I)–SR protecting motifs.9–18 Based on this molecule-like structure and the strong quantum confinement effects in this ultra-small size regime, metal NCs exhibit many molecule-like properties, such as HOMO–LUMO transition,10,12,13,19 strong luminescence,20–23 intrinsic chirality,24 and good catalytic activity.5,25–27 More intriguingly, almost all these molecule-like properties show an atomic-level dependency on the cluster structure. Such structural dependency not only provides a good means to diversify cluster properties for practical applications in various fields (e.g., biomedicine,28–34 green energy,35–37 and catalysis),38–41 but also offers an ideal platform for revealing the structure–property relationship of metal-based nanomaterials at the atomic level.40,42,43In the past few decades, the development of synthetic chemistry has allowed atomically precise [Mn(SR)m]q NCs to be widely used by the community as high-quality “metallic molecules”, thus motivating various further developments of metal NCs such as self-assembly and practical application.7,8 In these developments, an individual metal NC behaves like a molecular entity, and its practicality is inherently determined by its molecular interactions/reactions with ions, molecules, and other NCs. For example, extensive experimental and theoretical explorations have shown that the adsorption and subsequent activation of O2 on the surface of metal NCs is crucial in the aerobic oxidation reaction catalysed by metal NCs,40,43 and the development of sensors is highly dependent on chemical adsorption or reaction of analytes with metal NCs.44–47 Recent mechanistic discoveries also suggest that the growth and functionalization of metal NCs depends on their molecular interactions/reactions with reactive molecular species, including reducing agents,48–51 metal salts,52,53 SR ligands,54–57 M(I)–SR/M(II)–SR complexes,58–62 and metal NCs.63,64 Moreover, continuous developments in mass spectrometry, X-ray crystallography, X-ray absorption fine structure (XAFS) analysis, and theoretical simulations of noble metal NCs have enabled researchers to understand the pathways and dynamics of the above-mentioned reactions/interactions at the unprecedented molecular and atomic levels.8,65–72
Although in the past two decades, great efforts have been witnessed to establish the interaction/reaction chemistry of metal NCs, there is a lack of systematic summary and discussion on this topic in the current literature. Therefore, in this Review, we aim at discussing the latest developments in the basis and practicality of the molecular interactions/reactions between metal NCs and ions, molecules, and other NCs at the molecular and atomic levels. The discussion will be based on the aforementioned M(0)@M(I)–SR core–shell structure of metal NCs. The Review will start with a brief summary of the basic principles of molecular interactions/reactions between metal NCs and ions, molecules, and other NCs, focusing on the active sites of metal NCs for such interactions/reactions. Based on these fundamental insights, the use of molecular interactions/reactions of metal NCs in the synthetic chemistry (including bottom-up growth, alloying, and ligand exchange reactions) and post-synthesis development of metal NCs (including self-assembly, sensing, biomedical, and catalytic explorations) will be discussed in the following sections. This Review will be concluded with a concise summary of the main findings discussed above and our perspectives on the future development of interaction/reaction chemistry of noble metal NCs.
2. Molecular interaction/reaction fundamentals of metal NCs
The research on thiolate-protected noble metal NCs can be traced back to the 1990s, when pioneers worked together to establish the primary methods for the synthesis and characterization of sub-3 nm metal nanoparticles and NCs.73–77 These metal NCs have shown additional charm since the first successful attempt in resolving their crystal structure by X-ray crystallography in 2007, where a racemic mixture of Au102(SR)44 was identified by Jadzinsky et al.9 Another milestone achievement was then made independently by Heaven et al.11 and Zhu et al.10 in 2008, where the crystal structure of [Au25(SR)18]− was resolved. Such success in structure determination of Au NCs at the atomic resolution has also been propagated to Ag NCs, although Ag(0) is more susceptible to oxidation than Au(0). For example, the crystal structure of [Ag44(SR)30]4– has been revealed separately by Desireddy et al.12 and Yang et al.13 in 2013, while a silver analogue of [Au25(SR)18]−, i.e., [Ag25(SR)18]−, was synthesized and structurally analysed by X-ray crystallography by Joshi et al. in 2015.14 Based on the accumulated successes in the crystallization and structure determination of Au and Ag NCs, the crystal structures of numerous Au- or Ag-based alloy NCs have also been resolved with various heteroatoms of Au,13,78 Ag,79,80 Cu,17,81 Pt,82,83 Pd,83,84 Hg,85 and Cd.53,58With the increasing accessibility to the crystal structures of metal NCs with various sizes, compositions, and spatial arrangements of metal atoms or SR ligands, it has become increasingly known that the expanding structural library of thiolate-protected noble metal NCs can be in general described by a core–shell scheme, in which the M(0) core is protected by a monolayer of M(I)–SR protecting motifs.18 For example, [Au25(SR)18]− features a centred icosahedral Au13 core, which is further wrapped by six dimeric SR–[Au(I)–SR]2 motifs,10,11 while Au38(SR)24 possesses a bi-icosahedral Au23 core protected by three monomeric SR–Au(I)–SR motifs and six dimeric SR–[Au(I)–SR]2 motifs.86 It should be mentioned that such M(0)@M(I)–SR core–shell structural scheme remains valid even when the cluster size approaches the threshold value defining the molecular–metallic transition of metal nanomaterials. Recent transient and steady-state absorption spectroscopy analysis has identified Au246(SR)80 as the hitherto largest NC with a discrete and thus non-metallic electronic structure.87,88 The X-ray crystallography analysis indicates that Au246(SR)80 has an Ino-decahedron-based Au206 core.15 Depending on the varying curvature on this huge Au206 core, Au246(SR)80 possesses diverse terminal SR ligands, monomeric SR–Au(I)–SR motifs, and dimeric SR–[Au(I)–SR]2 motifs in its protecting shell. More interestingly, the mutual fit scheme of the Au(0) core and Au(I)–SR motifs not only leads to a high packing symmetry of the Au(0) core, but also results in an ordered arrangement pattern of SR ligands on the cluster surface, ultimately producing a structural complexity similar to that of proteins. The hierarchical protein-like structure of noble metal NCs indeed provides diverse active sites for their interactions/reactions with ions, molecules, and other NCs.7 Therefore, the molecular interactions/reactions of metal NCs will be summarized in terms of their active sites deciphered based on the M(0)@M(I)–SR core–shell structural scheme, as illustrated in Fig. 1.
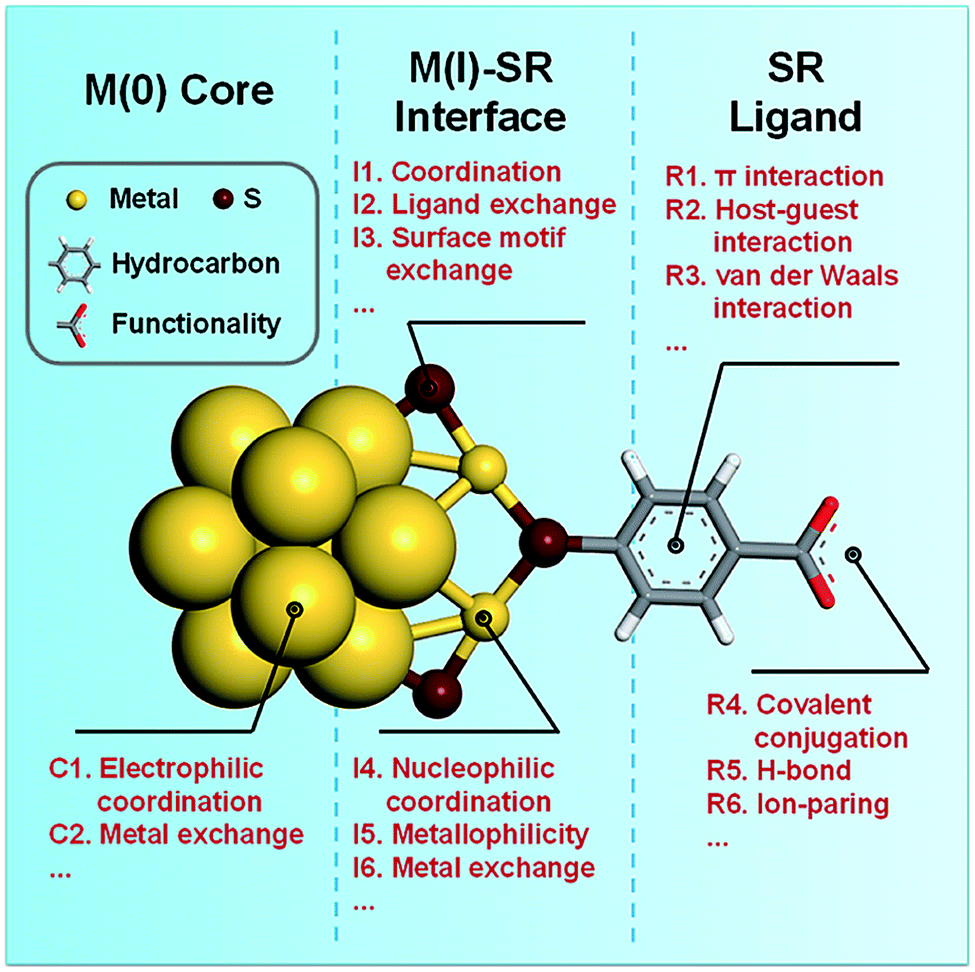 | ||
| Fig. 1 Schematic illustration of the active sites of noble metal NCs for their interactions/reactions with ions, molecules, and other NCs. The crystal structure of [Au25(SR)18]− (SR = thiolate ligand) was redrawn as a demonstrative example based on the reported crystallography data,10 in which only one SR–[Au(I)–SR]2 motif is shown for clarity. | ||
2.1. Molecular interactions/reactions based on the M(0) core
One of the most significant structural features of metal NCs is the diverse packing modes of their M(0) core, including icosahedron,10–14 decahedron,9 face-centred cubic (FCC),89 and hexagonal close packed (HCP)90 modes. The diverse structures of the M(0) core not only induce molecule-like electronic properties, but also provide various types of coordination-unsaturated M(0) sites for chemical or physical adsorption. In particular, due to the delocalization of valence electrons in the M(0) core, the M(0) core usually exhibits an electron-rich (electropositive) feature, which enhances its affinity to elements with high electronegativity (entry C1, Fig. 1). As a demonstrative example, [Au25(SR)18]− has a valence electron count of eight (N = n − m − q = 25 − 18 − 1 = 8), and these valence electrons are distributed in its Au13 core.10 The electron-rich Au13 core adopts a centred-icosahedral morphology, in which twelve facets are capped by six dimeric SR–[Au(I)–SR]2 motifs. This leaves eight uncapped triangular Au3, which have been documented as active sites for activating the –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond in the catalytic hydrogenation of ketones.91,92 For example, Zhu et al. recorded a nearly 100% stereo-selectivity of Au25(SR)18 toward the hydrogenation of bicyclic ketone 7-(phenylmethyl)-3-oxa-7-azabicyclo[3.3.1]nonan-9-one, exclusively producing the bicyclic alcohol with the as-formed –OH group adopting an equatorial orientation relative to the N-containing six-member ring (Fig. 2a).91 The authors attributed this remarkable stereo-selectivity to the preferential coordination of –CO with the electron-rich Au13 core of Au25(SR)18 NCs, followed by a partial electron transfer from the Au13 core to the O atom which activates the –CO bond. A similar –CO bond activation mechanism was also used to achieve 100% chemo-selectivity toward α,β-unsaturated alcohols in the hydrogenation reaction of α,β-unsaturated ketones or aldehydes catalysed by [Au25(SR)18]−.92
O bond in the catalytic hydrogenation of ketones.91,92 For example, Zhu et al. recorded a nearly 100% stereo-selectivity of Au25(SR)18 toward the hydrogenation of bicyclic ketone 7-(phenylmethyl)-3-oxa-7-azabicyclo[3.3.1]nonan-9-one, exclusively producing the bicyclic alcohol with the as-formed –OH group adopting an equatorial orientation relative to the N-containing six-member ring (Fig. 2a).91 The authors attributed this remarkable stereo-selectivity to the preferential coordination of –CO with the electron-rich Au13 core of Au25(SR)18 NCs, followed by a partial electron transfer from the Au13 core to the O atom which activates the –CO bond. A similar –CO bond activation mechanism was also used to achieve 100% chemo-selectivity toward α,β-unsaturated alcohols in the hydrogenation reaction of α,β-unsaturated ketones or aldehydes catalysed by [Au25(SR)18]−.92
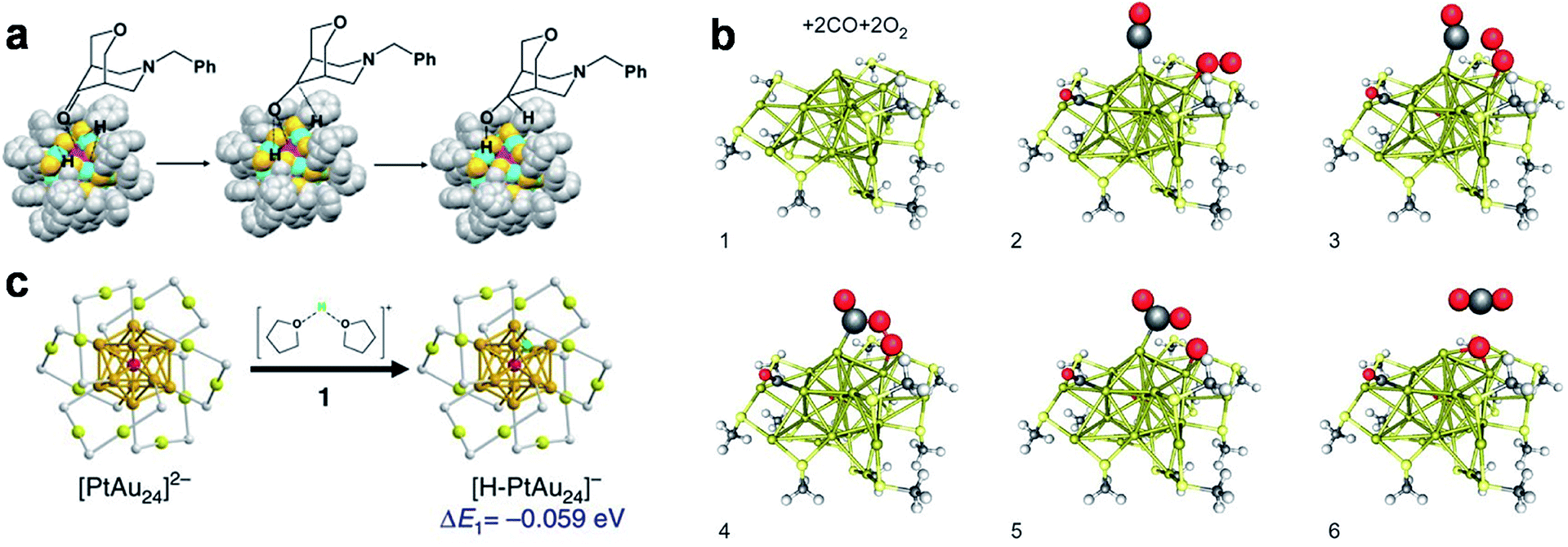 | ||
| Fig. 2 Schematic illustration of adsorption and activation of (a) –CO, (b) O2, and (c) solvated H+ on the Au13 core of [Au25(SR)18]− (a, b) or Au12Pt core of [Au24Pt(SR)18]2− (c). SR in (a) is –S-C2H4Ph (colour code: pink, core Au(0); blue, motif Au(I); golden, S; grey, C). SR in (b) is –S-CH3; two SR–[Au(I)–SR]2 motifs are removed for co-adsorption of O2 and CO (colour code: golden, Au; yellow, S; grey, C; light grey, H; red, O). The solvent molecules in (c) are tetrahydrofuran (THF) (colour code: golden, Au; red, Pt; yellow, S; green, H; light grey, C). (a) is reproduced with permission from ref. 91. Copyright 2010, Elsevier. (b) is reproduced with permission from ref. 43. Copyright 2010, Springer Nature. (c) is reproduced with permission from ref. 97. Copyright 2017, the Authors, published by Springer Nature. | ||
In addition to ketones and aldehydes, the M(0) core of metal NCs may also exhibit reaction/interaction activity toward gas molecules such as CO and O2. The exploration of the reactivity of atomically precise metal NCs toward O2 was pioneered by Zhu et al. The authors reported the aerobic oxidation of the anionic [Au25(SR)18]− into the charge-neutral Au25(SR)18.93 Based on the atomically precise structures revealed by X-ray crystallography or theoretical prediction, Lopez-Acevedo et al. carried out systematic density functional theory (DFT) calculations on a series of Au NCs (e.g., Au11(PH3)7Cl3, [Au25(SR)18]−, [Au39(PH3)14Cl6]−, Au102(SR)44, and Au144(SR)60, where PH3 denotes phosphine), and studied the activation process of O2 on Au NCs in a typical aerobic oxidation reaction of CO.43 The authors found that after partial removal of the Cl ligands or Au(I)–SR motifs, the affinity (approximated by the binding energy) of these NCs with O2 is different, which largely depends on their HOMO–LUMO gaps. As shown in Fig. 2b, removing one dimeric SR–[Au(I)–SR]2 motif from the surface of [Au25(SR)18]− can not only return one valence electron to the Au13 core, but also create an open site on the Au13 core for O2 adsorption. The additional valence electron occupies an electron state over the HOMO–LUMO gap of [Au25(SR)18]−. The electron transferred from such an electron state to one of the 2π* orbitals of O2 can activate O2 to superoxo O2−. By removing another SR–[Au(I)–SR]2 motif close to the superoxo species, a core Au site can be made available for CO adsorption. The subsequent reaction between the adsorbed CO and superoxo species provides a plausible mechanism for the aerobic oxidation of CO. In addition to the core Au site exposed by partial removal of M(I)–SR protecting motifs, the originally uncapped triangular Au3 may also be the active site of [Au25(SR)18]− for CO and O2 activation. In a recent contribution from Liu et al.,94 a triangular Au3 based self-promoting mechanism was proposed for the aerobic oxidation of CO, in which the co-adsorption of two CO and one O2 on an open triangular Au3 can facilitate the cleavage of O–O bonds conducive to the simultaneous formation of two CO2.
In addition to partially removing SR ligands or M(I)–SR ligands, the molecular reactivity of the M(0) core can also be improved by doping heteroatoms in the M(0) core. Doping Au(0) or Ag(0) cores with heteroatoms has long been employed as an effective means to enhance the stability and catalytic activity of metal NC-based catalysts.95,96 The heteroatoms usually have two functions: (1) manipulate the electronic structure of NCs, and (2) introduce additional active sites to chemically or physically adsorb substrate molecules. For example, Kwak et al. compared the electronic structure and electrocatalytic performance of [Au25(S-C6H13)18]− and Au24Pt(S-C6H13)18 through optical spectroscopy and voltammetry, where HS-C6H13 denotes 1-hexanethiol.97 They concluded that embedding the dopant Pt in the centre of the icosahedral M13 core would induce significant changes in the electron energy diagram and the redox potential of M25(SR)18 NCs. The superior electrocatalytic activity of Au24Pt(SR)18 for the hydrogen evolution reaction (HER) in tetrahydrofuran (THF) is attributed to the well-matched redox potential of [Au24Pt(SR)18]−/2− and the onset potential of the HER, indicating that Au24Pt(SR)18 can act as a charge-state-dependent electron mediator in the HER. The detailed DFT calculations by these authors further revealed that another equally important contributor to the enhanced HER catalytic activity is the preferential adsorption of H on the Pt heteroatom (Fig. 2c), although the Pt heteroatom is located at the centre of the icosahedral M13 core. In a follow-up study, the authors found that the HER catalytic activity of Au24Pt(SR)18 can be further enhanced by incorporating a proton-relaying ligand (e.g., 3-mercapto-1-propanesulfonic acid) into its protecting shell.98 It should be noted that in many other catalytic reaction systems, including oxidation99 and C–C coupling100 reactions, similar doping effects of M(0) have also been recorded.
In addition to the electropositive coordination sites, the M(0) core can also be the active site for the metal exchange reactions (entry C2, Fig. 1). For example, Wang et al. employed [Au25(SR)18]− as a template NC for the metal exchange reaction with Cu(I)–SR, Ag(I)–SR, Cd(II)–SR, and Hg(II)–SR complexes in toluene, generating [Au25−xCux(SR)18]− (x = 0–2), [Au25−xAgx(SR)18]− (x = 0–4), Au24Cd(SR)18, and Au24Hg(SR)18, respectively.58 The X-ray crystallography and DFT calculation from the same authors and other groups53,79 revealed that such metal exchange reactions do not change the M25S18 framework of [Au25(SR)18]−, in which the Cu, Ag, and Cd heteroatoms are consistently accommodated in the M13 core. These data clearly suggest that the icosahedral Au13 core of [Au25(SR)18]− provides active sites for the metal exchange reactions.
2.2. Molecular interactions/reactions at the M–SR interface
In sharp contrast to the electron-rich M(0) core, the M(I) atoms in the M(I)–SR protecting motifs are commonly electron-deficient (electronegative), owing to the electron withdrawing properties of the SR ligands coordinated to them. Such electronegative feature endows M(I) atoms with an increased affinity to electropositive molecules (entry I4, Fig. 1). For instance, in the aforementioned selective hydrogenation reaction of ketones or aldehydes catalysed by [Au25(SR)18]−, the adsorption sites of the molecular H2 are believed to be Au(I) in the SR–[Au(I)–SR]2 motifs.91,92 It has been well documented in the nanocatalysis literature that coordination-unsaturated Au atoms are active for adsorbing molecular H2.101,102 The subsequent dissociation of the molecular H2 may result in two cleaved H atoms, whose reaction with the activated –CO bond will eventually produce –CH–OH. In addition to the electron-deficiency, another important attribute that determines the reactivity of M(I) atoms in the M(I)–SR motifs is their improved accessibility compared to the core M(0) atoms. For example, Li et al. performed a detailed DFT calculation to reveal the basis of the interaction/reaction between the terminal alkynes (e.g., phenylacetylene) and [Au25(SR)18]−.103 The data suggest that the adsorption of phenylacetylene occurs on a triangular Au3 facet composed of Au(I) atoms from three different dimeric SR–[Au(I)–SR]2 motifs, because the triangular Au3 facet is less blocked and it is more accessible to both the solvent and substrate molecules. It is believed that the adsorption and activation of –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH in a similar manner can promote various reactions of –CCH (e.g., semi-hydrogenation,104 carboxylation,105 and Sonogashira cross-coupling103). It should also be noted that the motif-type Au3 facet, together with three sulfur atoms coordinated to it and a core-type Au3 facet underneath it, forms a “pocket-like cavity” on the surface of [Au25(SR)18]−, which can provide heterogeneous active sites that can accommodate various catalytic reactions (vide infra).
CH in a similar manner can promote various reactions of –CCH (e.g., semi-hydrogenation,104 carboxylation,105 and Sonogashira cross-coupling103). It should also be noted that the motif-type Au3 facet, together with three sulfur atoms coordinated to it and a core-type Au3 facet underneath it, forms a “pocket-like cavity” on the surface of [Au25(SR)18]−, which can provide heterogeneous active sites that can accommodate various catalytic reactions (vide infra).
In addition to the coordination-unsaturated metal centre, the M(I) in the M(I)–SR motifs of the metal NCs can also be active sites for metallophilicity (i.e., M(I)⋯M(I) interaction; entry I5, Fig. 1) and metal exchange reaction (entry I6, Fig. 1). For example, De Nardi et al. synthesized and crystallized Au25(S-nBu)18 NCs (HS-nBu denotes n-butanethiol).106 The X-ray crystallography analysis suggests that the Au25(S-nBu)18 NCs align themselves into a one-dimensional (1D) polymeric chain, in which the adjacent NCs are connected by Au(I)⋯Au(I) interactions with a typical bond length of 3.15 Å (Fig. 3a). The formation of extensive Au(I)⋯Au(I) bonds should be attributed to the large content of Au(I) species in the protecting shell of Au25(S-nBu)18 NCs, as well as the appropriately flexible hydrocarbon chain of the –S-nBu ligand. Similar M(I)⋯M(I) interactions have been widely observed in aggregation-induced emission (AIE)-type metal NCs, offering an important mechanism for delivering enhanced luminescence.108 The M(I) in the M(I)–SR motifs has also been identified as the active site for the metal exchange reaction (entry I6, Fig. 1). For example, Liao et al. performed a metal exchange reaction between [Au25(S-C2H4Ph)18]− and Hg2+ in the presence of excess 2-phenylethanethiol (HS-C2H4Ph), leading to the formation of Au24Hg(S-C2H4Ph)18.85 X-ray crystallography analysis showed that this metal exchange reaction occurred on the dimeric SR–[Au(I)–SR]2 motifs, in which a Au atom in the protecting motif was replaced by the Hg heteroatom. It should be noted that in a very recent contribution, Fei et al. performed a comprehensive examination on the heteroatom doping site in Au24M(SR)18 NCs (M = Pt, Pd, Cd and Hg) by combining nuclear magnetic resonance (NMR), isotope labelling and X-ray crystallography, where the preferential doping site of Hg was identified as the vertex of the icosahedral Au13 core.109 The different doping sites of Hg may be attributed to the dynamic diffusion of the Hg heteroatom in the frame of Au24Hg(SR)18.
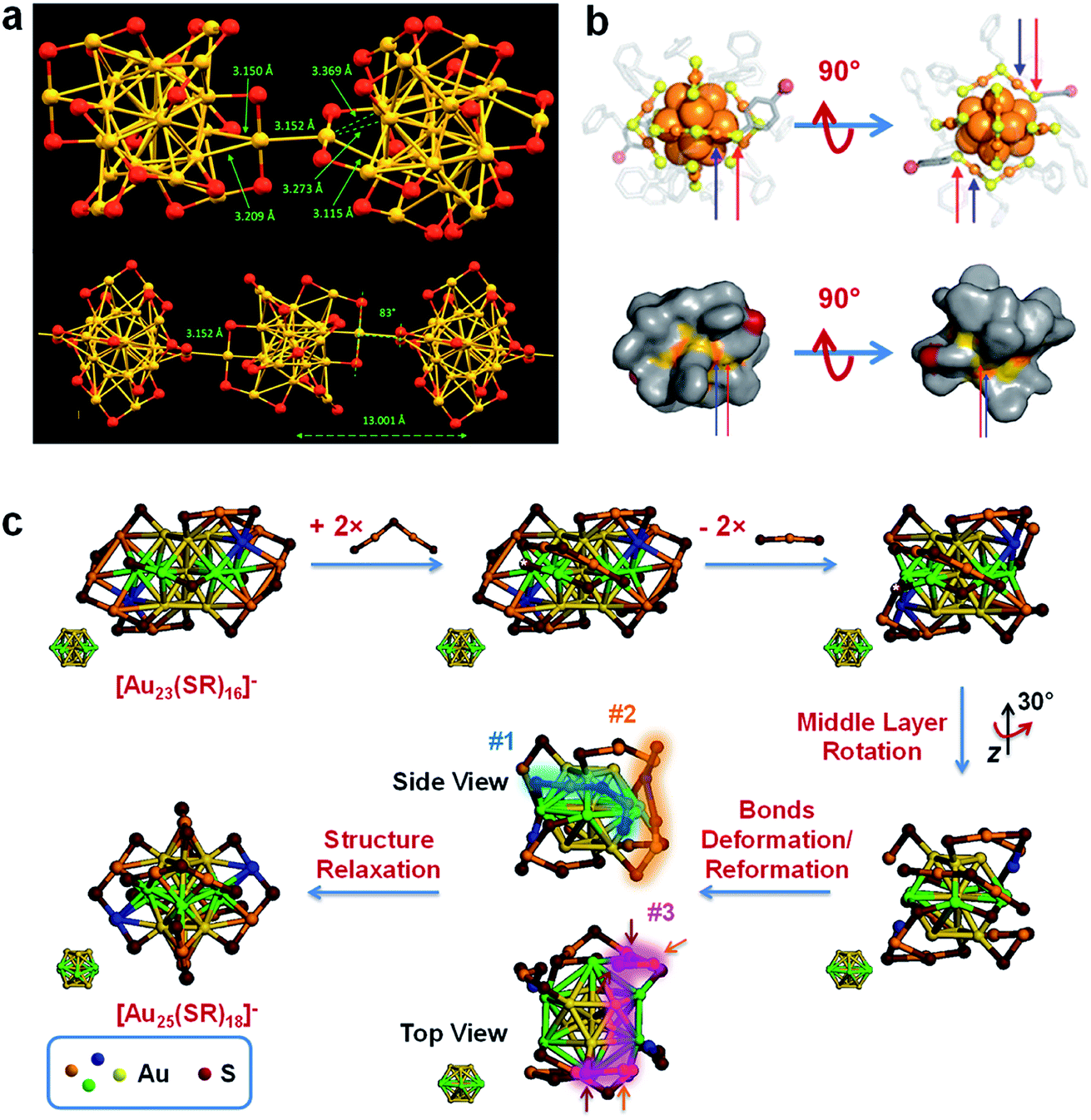 | ||
| Fig. 3 Schematic illustration of (a) inter-cluster aurophilic interaction, (b) ligand exchange reaction, and (c) surface motif exchange reaction based on the Au–S interface of Au25(SR)18 (a), [Au25(SR)18]− (b), and [Au23(SR)16]− (c). (a) shows the formation of a 1D assembly by Au25(SR)18 based on the inter-cluster aurophilic interaction (colour code: golden, Au; red, S); reproduced with permission from ref. 106. Copyright 2014, American Chemical Society. (b) illustrates the preferential ligand exchange site on [Au25(S-C2H4Ph)18]− by HS-Ph-p-Br based on its crystal structure (top panel) and solvent-accessibility surface model (bottom panel), where blue arrows indicate the solvent-exposed Au and red arrows indicate the preferential ligand exchange site (colour code: orange, Au; yellow, S; grey, C; red, Br); reproduced with permission from ref. 107. Copyright 2014, American Chemical Society. (c) shows surface motif exchange induced size conversion from [Au23(SR)16]− to [Au25(SR)18]−; the glowing belts #1–#3 indicate SR–[Au(I)–SR]2 motifs formed in the size conversion process; reproduced with permission from ref. 61. Copyright 2018, the Authors, published by Springer Nature. | ||
Interestingly, recent progress in the experimental and theoretical catalysis of metal NCs suggests that the S atom of the SR ligand can also provide additional coordination sites for the substrate molecules in the catalytic reactions (entry I1, Fig. 1). For example, Kauffman et al. evaluated the electrocatalytic activity of [Au25(SR)18]q (q = −1, 0 and +1) for CO2 reduction, CO oxidation, and O2 reduction.110 The DFT calculations indicate that the adsorption of CO2 and CO may occur on the three S atoms in the pocket-like cavity of [Au25(SR)18]q. The authors further pointed out that the charge state of [Au25(SR)18]q affected the adsorption energy of the reactants (e.g., CO2 and H+ in CO2 reduction, and CO and OH− in CO oxidation) and products (e.g., OH− in O2 reduction), which ultimately affected the catalytic activity of [Au25(SR)18]q in the above reactions.
Compared with the weak adsorption sites, the M–SR interface can mainly provide reactive sites for ligand exchange reactions (entry I2, Fig. 1), which have been widely used to customize the size, structure, surface chemistry and properties of metal NCs.7,111–113 Ligand exchange reactions largely depend on the etching reaction of the foreign ligands on the existing M–SR interface of the metal NCs. As M(I)–SR motifs have been discovered with various structures and arrangement patterns on the surface of the M(0) core, the chemical environment of SR ligands can be diverse in the metal NCs.114,115 Therefore, extensive research efforts have been made to determine the preferential reaction sites of ligand exchange reactions. For example, Heinecke et al. investigated the ligand exchange reaction of Au102(p-MBA)40 (p-MBA = para-mercaptobenzoic acid or HS-Ph-p-COOH) with two molar equivalents of para-bromobenzene thiol (p-BBT or HS-Ph-p-Br), and successfully obtained single crystals of Au102(p-MBA)40−x(p-BBT)x with partial exchange of ligands (the average value of x, xave = 1.08).116 The X-ray crystallography analysis showed that this ligand exchange reaction occurs preferentially on the Au(I) atoms in the two SR–Au(I)–SR motifs that are most accessible to the solvent. A similar observation was recorded by the same group in the ligand exchange reaction between [Au25(S-C2H4Ph)18]− and HS-Ph-p-Br, where the incoming HS-Ph-p-Br preferentially bonded to the Au(I) atoms in the protecting shell of [Au25(S-C2H4Ph)18]− that were most accessible to the solvent (Fig. 3b).107 Considering that SR–[Au(I)–SR]2 is an exclusive protecting motif in the protecting shell of [Au25(SR)18]−, there exist one core-type (bonded to the Au(0) core) and one apex-type (not bonded to the Au(0) core) –S-C2H4Ph ligands bonded to the Au(I) atom that is most accessible to the solvent. However, the X-ray crystallography data show that the core-type –S-C2H4Ph ligand is more likely to be replaced by the incoming HS-Ph-p-Br ligand, with an occupancy of 74.6%. The authors attributed the preferential exchange of the core-type SR ligands by the incoming HS-Ph-p-Br ligand to the enhanced π–π interactions of the latter with the remaining –S-C2H4Ph ligands.
Niihori et al. also observed a similar preferential exchange of the core-type –S-C2H4Ph ligands in the reaction of Au24Pd(S-C2H4Ph)18 with 1-dodecanethiol (HS-C12H25).117 The authors analysed the population distribution of the coordination isomers produced by the above ligand exchange reaction by reversed-phase high performance liquid chromatography (HPLC), and found that, at the early stage of the reaction (reaction time = 4 min), the abundance of the core-type exchanged Au24Pd(S-C2H4Ph)17(SC12H25) was 13 times higher than that of the apex-type exchanged one. In order to gain fundamental insights into the different reactivities of the core-type and apex-type SR ligands in the ligand exchange reactions, Pengo et al. monitored the ligand exchange kinetics of [Au25(S-C2H4Ph)18]− with various incoming thiols (including HS-Ph, HS-Ph-p-CH3, and HS-Ph-p-OCH3) by 1H nuclear magnetic resonance (1H-NMR) spectroscopy.118 The most important finding is that the selectivity to the core-type exchanged product is highly dependent on the electronic structure of the incoming thiols, where thiols with an electron-donating substituent at the para position have lower selectivity for the core-type exchanged products. This conclusion is consistent with recent findings of Salassa et al., where the result of the ligand exchange reaction of [Au25(SR)18]− is highly dependent on solution stability and intermolecular interaction of thiols, as well as their affinity to the Au frame.119 In addition to 1H NMR spectrometry, isothermal titration calorimetry (ITC) has also been introduced by Baksi et al. to evaluate the reactivity of metal NCs with free thiols.120
Another important reaction that occurs at the M–S interface is the surface motif exchange reaction (SME; entry I3, Fig. 1). The significant difference from conventional ligand exchange reactions is that the exchange units in SME are M(I)–SR motifs/complexes, which should be inherently attributed to the dynamic nature of M(I)–SR motifs on the surface of the M(0) core.112 In metal-cum-ligand exchange reaction, the discovery of M(I)–SR motifs/complexes as the exchanging units can be traced back to 2003, when both metal and ligand exchanges were observed in a two-phase reaction of hexanethiolate-protected Au NCs (∼1.6 nm in the toluene phase) and tiopronin-protected Ag NCs (∼1.6 nm in the aqueous phase).62 Since the metal and ligand exchange reaction can be almost prohibited under a N2 environment, the authors proposed a Au(I)–SR complex-assisted mechanism, in which the Au(I)–SR complexes formed by oxidative etching of Au NCs were identified as the “catalysts” that made possible simultaneous exchange of metal and ligand. This assertion is supported by a control experiment, which shows that adding a catalytic amount of Au(I)–SR complexes to the reaction system under N2 can restore the exchange reactions. Yao et al. recently demonstrated the versatility of SME reaction in the cluster reactions based on extensive mass spectrometry analysis.60 By monitoring the reaction between [Ag44(p-MBA)30]4− and Au(I)–(p-NTP) complexes (p-NTP = para-nitrothiophenol or HS-Ph-p-NO2) using electrospray ionization mass spectrometry (ESI-MS), the authors found that the exchange reaction mainly occurred through the following reaction (eqn (1)).
| [Ag44(p-MBA)30]4− + x[Au2(p-NTP)2Cl]− → [Ag44−xAux(p-MBA)30−2x(p-NTP)2x]4− + x[Au(p-MBA)2]− + xAgCl(x ≤ 12) | (1) |
It was also found that a similar SME reaction could effectively drive the size conversion reaction of Au NCs. An isoelectronic size conversion reaction of [Au23(SR)16]− to [Au25(SR)18]− was closely tracked by UV-vis absorption spectroscopy and ESI-MS, indicating that the isoelectronic size conversion reaction was based on the exchange of SR–[Au(I)–SR]2 units.61 Therefore, the reaction of the isoelectronic size conversion can be described by the following equation (eqn (2)).
| [Au23(SR)16]− + 2[Au2(SR′)3]− → [Au25(SR)12(SR′)6]− + 2[Au(SR)2]− | (2) |
2.3. Molecular interactions/reactions on SR ligands
Because SR ligands constitute the outermost layer of metal NCs, they largely determine the recognition chemistry and surface reaction of metal NCs. Due to the size, structure, and function diversity of the hydrocarbon tails of SR ligands, they can adapt to various covalent and non-covalent interactions/reactions.Covalent conjugation is commonly used to introduce additional functions into metal NCs for biomedical applications.121–126 Among the various covalent conjugation strategies documented in biomedical and organic chemistry, the amination reaction based on 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) is the most widely used method for surface modification of metal NCs (entry R4, Fig. 1), due to its mild operation conditions as well as abundant carboxyl or amine groups on the surface of water-soluble metal NCs.121,122,124,125 For example, Pyo et al. used EDC chemistry to conjugate folic acid (FA) with Au22(SG)18 (H-SG or GSH = glutathione, Fig. 4a),122 where the latter is the first atomically precise Au NC with strong red emission (emission wavelength, λem = 665 nm; excitation wavelength, λex = 520 nm).42 It is worth noting that, in order to effectively inhibit the inter-particle coupling of SG ligands through –NH2 and –COOH, the authors protected the –NH2 groups with benzyl chloroformate (CBz) before EDC coupling. In a separate study, Vankayala et al. made full use of EDC chemistry to activate the carboxyl group of 11-mercaptoundecanoic acid (MUA), and conjugated a nucleus-targeting peptide (TAT, peptide sequence: N-GRKKRRQRRR-C) to the Au NCs protected by MUA.127 Such conjugation allows Au NCs to have a high nucleus targeting efficacy in HeLa cells, while keeping the emission intensity of Au NCs unaffected. In addition, the chemically conjugated TAT peptide can change the zeta potential of Au NCs from negative to positive, making them also useful for gene delivery.
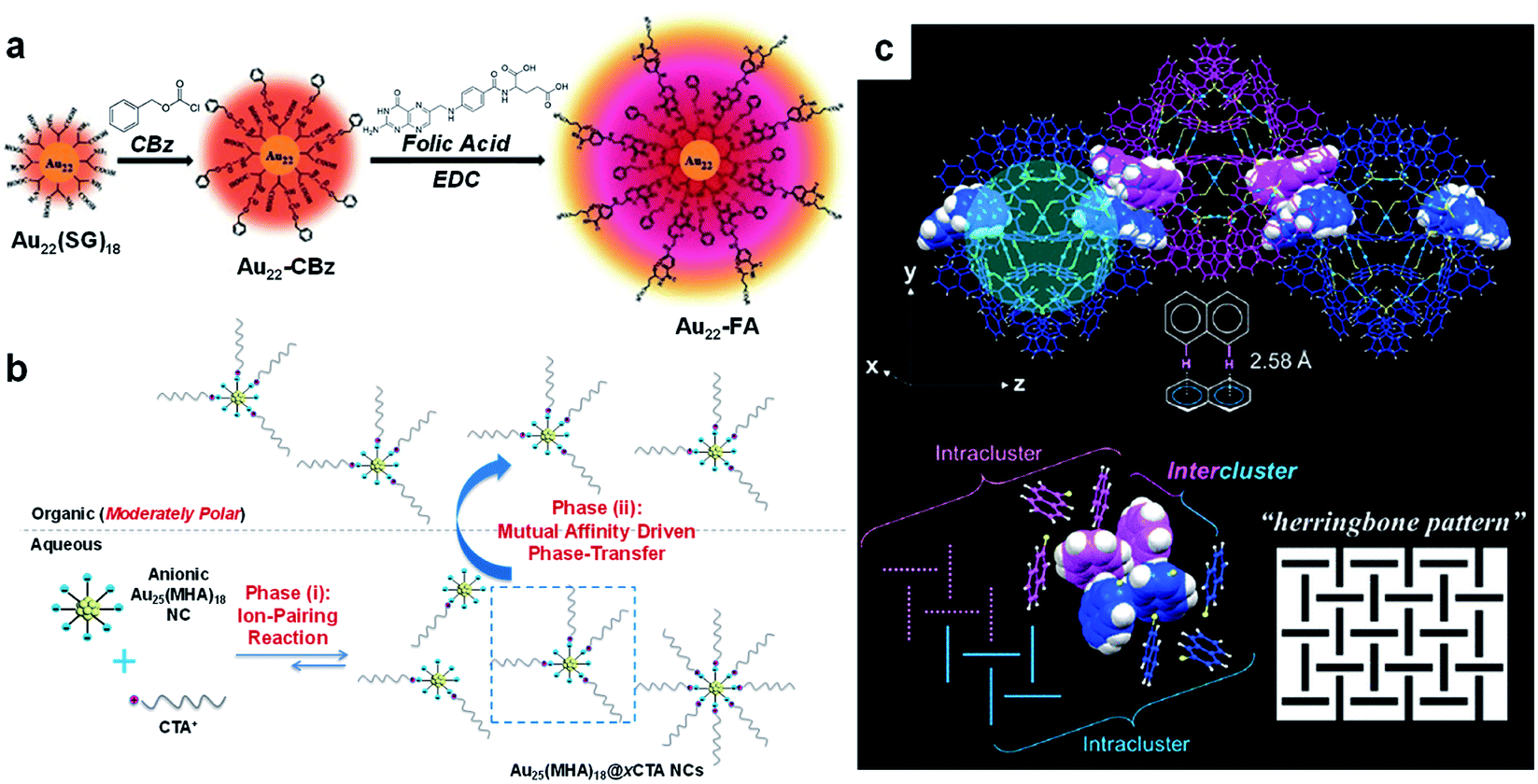 | ||
| Fig. 4 Schematic illustration of (a) 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) conjugation reaction, (b) ion-pairing reaction, and (c) CH–π interaction occurring on the SR ligands of metal NCs. (a) illustrates the EDC conjugation between folic acid (FA) and Au22(SG)18 (SG = glutathione ligand), where the –NH2 groups of SG ligands are protected by benzyl chloroformate (CBz) prior to the EDC reaction; reproduced with permission from ref. 122. Copyright 2017, Wiley-VCH. (b) illustrates the production of amphiphilic Au25(MHA)18@xCTA NCs (x = 6–9) by the phase-transfer-driven ion-pairing reaction, where MHA denotes 6-mercaptohexanoic acid and CTA denotes cetyltrimethylammonium; reproduced with permission from ref. 129. Copyright 2015, American Chemical Society. (c) exhibits inter-cluster CH–π interactions (top panel) and the “herringbone pattern” of –S-Nap ligands in the supercrystals of Au103S2(S-Nap)41 NCs, where HS-Nap denotes 2-naphthalenethiol (colour code: light blue, motif Au(I); yellow, S; light grey, H; blue/magenta, C); reproduced with permission from ref. 55. Copyright 2017, American Chemical Society. | ||
In addition to covalent conjugations, a large number of non-covalent interactions/reactions have also been documented based on the SR groups for surface modification or self-assembly of metal NCs. The carboxyl, amine or hydroxyl groups of the SR ligands are the ideal sites for the H-bonds on the surface of metal NCs (entry R5, Fig. 1). The directional H-bond not only plays a key role in determining the solvation behaviour of metal NCs, but is also effective in controlling the self-assembly and/or crystallization behaviour of metal NCs. For example, based on the well-known Brust–Schiffrin method, Desireddy et al. designed an efficient protocol to synthesize molecularly pure [Ag44(p-MBA)30]4− on a large scale, followed by its crystallization in a dimethylformamide (DMF) solution in its fully protonated form.12 The X-ray crystallography examination of the obtained single crystals showed that a strong network of H-bonds may be formed between the p-MBA ligands of adjacent NCs. Extensive H-bonds have also been found in the assemblies or supercrystals of other metal NCs protected by p-MBA ligands, among which Au102(p-MBA)44 is a good example.128 In sharp contrast, after deprotonation, the carboxyl group can be turned into an active site for electrostatic interaction (entry R6, Fig. 1). For example, Yao et al. designed a phase-transfer-driven ion-pairing reaction for facile preparation of amphiphilic metal NCs (Fig. 4b).129 The ion-pairing reaction is based on the electrostatic interaction between the –COO− group of Au25(MHA)18 (MHA = 6-mercaptohexanoic acid) and hydrophobic cations such as CTA+ (CTA+ = cetyltrimethylammonium cation). Considering the dynamics of the ion-paring reaction in the aqueous phase, different numbers of CTA+ species can be patched on the hydrophilic surface of Au25(MHA)18. A mixture of toluene and ethanol was then introduced as the organic phase, and amphiphilic Au25(MHA)18@xCTA (x = 6–9) NCs were selectively extracted from the aqueous phase. The equilibrium of the ion-pairing reaction can effectively regenerate the extracted Au25(MHA)18@xCTA species in the aqueous phase, constituting a sustained mechanism for the production of amphiphilic Au NCs.
The obvious difference from the prosperity of electrostatic interactions in the water phase is that organic-soluble metal NCs usually lack surface charges, so electrostatic interactions are less efficient in directing the self-assembly and application of metal NCs. However, because aromatic thiols are widely used in the synthesis of metal NCs in the organic phase, π interactions (e.g., π–π and CH–π interactions) have become the dominant factor in controlling the self-assembly of NCs and their applications in the organic phase (entry R1, Fig. 1). For example, Higaki et al. recently produced molecularly pure Au103S2(S-Nap)41 NCs (HS-Nap = 2-naphthalenethiol) through a ligand exchange reaction between Au99(S-Ph)42 and HS-Nap.55 The obtained Au103S2(S-Nap)41 NCs were crystallized into a monoclinic lattice by a typical vapour diffusion method. Of particular interest, the Au103S2(S-Nap)41 NCs in the supercrystals are packed through hierarchical CH–π interactions (Fig. 4c). At the intra-cluster level, the –S-Nap ligands can first form dimers through T-shaped CH–π interactions (which may also be regarded as T-shaped π–π interactions). Through similar T-shaped CH–π interactions, two pairs of such dimers can form a cyclic tetramer. The lateral C–H and naphthalene ring in the formed tetramers provide active sites for the CH–π interaction between the clusters, which binds the two tetramers from adjacent NCs together, resulting in a herringbone-like arrangement of naphthalene rings across intra- and inter-cluster levels. In addition to the CH–π interaction, it has also been found that the inter-cluster π–π interactions are important to determine the assembly behaviour of metal NCs. For example, Hossain et al. obtained single crystals of five different Au4Pt2(SR)8 NCs with different SR ligands, namely isopropanethiol (HS-CH(CH3)2), 2,4,6-trimethylbenzylmercaptane (HS-CH2Ph(CH3)3), 4-tert-butylbenzylmercaptane (HS-CH2PhtBu), and HS-C2H4Ph, respectively.130 Interestingly, only Au4Pt2(S-CH2PhtBu)8 and Au4Pt2(S-C2H4Ph)8 NCs can organize themselves into a 1D structure in their supercrystals, and the Au–Au distance between clusters is short, within 3.08 Å. The preferential 1D assembly is attributed to the strong π–π interactions and appropriately weak steric repulsion of –S-CH2PhtBu or –S-C2H4Ph ligands, thereby enabling extensive aurophilic interactions along the longitudinal orientation of the 1D structure. It should be noted that van der Waals interactions are also commonly observed in the metal NCs protected by alkyl and aryl ligands (entry R3, Fig. 1), which can facilitate their self-assembly into hierarchical architectures (see more details in Section 4.1).15,131
Host–guest interaction is an important type of supramolecular interaction, finding increasing acceptance in molecular recognition and self-assembly chemistry.132–134 A useful host–guest interaction can be established on the surface of metal NCs through an appropriate surface engineering strategy (entry R2, Fig. 1). For example, Mathew et al. demonstrated that a host–guest interaction can be established between β-cyclodextrin (CD) and the 4-tert-butylbenzenethiolate (–S-Ph-p-tBu) ligand of Au25(S-Ph-p-tBu)18 NCs.135 In this way, the authors can anchor β-CD on the surface of Au25(S-Ph-p-tBu)18, producing Au25(S-Ph-p-tBu)18@CDx (x = 1–4). It has been demonstrated that the β-CD modification on the surface of Au25(S-Ph-p-tBu)18 can not only enhance its stability against Cu2+ etching, but also change its absorption and emission spectra, providing a useful mechanism for sensing applications. Recently, similar host–guest interactions have been used to decorate the Au40(S-Adm)22 (HS-Adm = adamantanethiol) with metal organic frameworks (MOFs) constructed by γ-CD and K+ (γ-CD-MOFs).136 The as-produced Au40(S-Adm)22@γ-CD-MOF exhibits enhanced water solubility and improved catalytic activity for H2O2 reduction. In addition to modifying CDs on the surface of metal NCs, thiolated CDs can also be used as thiolate ligands in the synthesis of metal NCs. For example, per-6-thio-β-cyclodextrin (HS-β-CD) was mixed with HAuCl4, followed by NaBH4 reduction to synthesize Au NCs@β-CD.137 As evidenced by transmission electron microscopy (TEM) images, the core size of the as-synthesized Au NCs@β-CD is less than 3 nm. After being immobilized on the surface of TiO2, the TiO2–Au NCs@β-CD nanocomposite has a 2-fold enhanced photocatalytic activity (compared with pure TiO2) for the degradation of a model pollutant, methyl orange (MO), which should be attributed to the host–guest interactions between β-CD and MO molecules.
Other than CD, another marked host–guest interaction between metal NCs and fullerenes has been documented.138–140 For example, Chakraborty et al. mixed a toluene solution of C60 with a DMF solution of [Ag29(BDT)12]3− NCs (BDT = 1,3-benzenedithiol), forming [Ag29(BDT)12@(C60)x]3− (x = 1–9) adducts.139 DFT calculations suggest that the C60 is preferentially tethered in the cavity formed by the arrangement of BDT ligands, and the π–π interaction between the BDT ligand and C60 is identified as the predominant driving force for this complexation event. Very recently, the authors demonstrated that similar host–guest interactions can easily be established between C60 and [Ag25(DMBT)18]− or [Au25(S-C2H4Ph)18]− NCs (DMBT = 2,4-dimethylbenzenethiol), providing an easy way to assemble clusters into dendritic networks.140 More interestingly, by virtue of the supramolecular interaction between crown ethers and aromatic protecting ligands, co-crystallization of dibenzo-18-crown-6 and [Ag29(BDT)12(PPh3)x]3− NCs (PPh3 = triphenylphosphine) can also be achieved.141 This chemistry should be useful in engineering both physicochemical properties and self-assembly behaviour of metal NCs.
3. Molecular interactions/reactions in synthesis of metal NCs
With the continuous development of synthetic chemistry, several efficient strategies have been devised to produce atomically precise metal NCs with customizable size, composition, and structure. These methods include bottom-up growth,48,49,51 ligand exchange,111 and heteroatom alloying.142 These approaches inevitably involve the reaction of metal NCs with redox species, SR ligands, metal ions, M(I)–SR/M(II)–SR complexes, and other NCs. In this section, we will briefly discuss the molecular interactions/reactions that control the synthesis of thiolate-protected noble metal NCs.3.1. Molecular interactions/reactions in bottom-up growth of metal NCs
In a typical bottom-up growth method, metal salts are mixed with thiol ligands to generate M(I)–SR complexes, followed by a reduction-induced growth of the M(0) core.73,76 A balance between the reduction-induced growth and the SR/M(I)–SR passivation of the M(0) core leads to the formation of stable metal NCs. Therefore, the bottom-up growth of metal NCs is largely determined by the reaction of growing NCs with the reductive species, M(I)–SR complexes, or intermediate NC species.Due to its mild reduction kinetics, CO has been widely used as a reducing agent for the production of a variety of Au NCs, including Au15(SR)13,143 Au18(SR)14,143 Au22(SR)18,42 [Au25(SR)18]−,143 Au29(SR)20,144 Au38(SR)24,49 and [Au44(SR)26]2−.49 The mild and easily quenched reduction kinetics of CO can not only maintain a well-controlled environment to produce atomically precise metal NCs with desired sizes, but also provide a good means for capturing important intermediates along the growth path of metal NCs. For example, Luo et al. fine-tuned the reducing capability of CO at the optimal pH (= 11.6) for synthesis of [Au25(S-Ph-m-COOH)18]− (HS-Ph-m-COOH = meta-mercaptobenzoic acid), in an appropriately slow manner (reaction time = 72 h).48 Such a slow size evolution process allows the authors to monitor the size growth reaction of [Au25(S-Ph-m-COOH)18]− by UV-vis absorption spectroscopy and ESI-MS. Through this method, the authors identified a total of 29 Au(I)–SR complex/NC species during the entire growth process of [Au25(S-Ph-m-COOH)18]−. All these species have an even number of valence electrons (N), which indicates that the growth reaction of [Au25(SR)18]− features a step-wise 2-electron (2e−) hopping mechanism. The 2e− hopping mechanism is indeed fuelled by the reactions of CO, initially with the Au(I)–SR complexes and subsequently with the intermediate Au NC species. Based on the time-dependent abundance of individual Au(I)–SR complex/NC species, the authors can also propose detailed formation and consumption reactions for individual NC species, thus constituting a step-by-step growth pathway for [Au25(SR)18]−. For example, the formation of an important intermediate, Au18(SR)14, powered by CO reduction can be made possible by the following reaction (eqn (3)).
| Au15(SR)13 + Au3(SR)3 + CO + 4OH− → Au18(SR)14 + 2[SR]− + CO32− + 2H2O | (3) |
The molecular reaction between Au NCs and CO was further investigated by Yao et al. in seed-mediated growth of [Au25(SR)18]− to form [Au44(SR)26]2−.49 As shown in Fig. 5a, the molecularly pure [Au25(p-MBA)18]− NCs were allowed to react with Au(I)-(p-MBA) complexes and CO in an aqueous solution under highly alkaline conditions (pH = 13.0), generating atomically precise [Au44(p-MBA)26]2−. This seed-mediated growth reaction was carefully tracked by time-course UV-vis absorption spectroscopy and ESI-MS. By systematically analysing the time-dependent abundance of a total of 35 Au(I)–SR complex/NC species in the seed-mediated growth reaction, the authors identified two different size-growth pathways for [Au44(SR)26]2−: monotonic LaMer growth and volcano-shaped aggregative growth. Although the size evolution patterns can be diverse, the valence electrons of Au NCs evolve by following the universal 2e− hopping mechanism, which is in good agreement with the previous report.48 More importantly, the authors succeeded in capturing an important reaction intermediate, [Au25(p-MBA)18CO]−, in an aqueous solution of [Au25(p-MBA)18]− saturated by CO (Fig. 5b). These data clearly suggest that the size growth based on the stable cluster size can be initiated by adsorbing reductive substances (e.g., CO) on the growing NC, where [Au25(SR)18]− is a good example. The CO adsorption site is likely to be the pocket-like cavity on the surface of [Au25(SR)18]−, as discussed in the previous section.
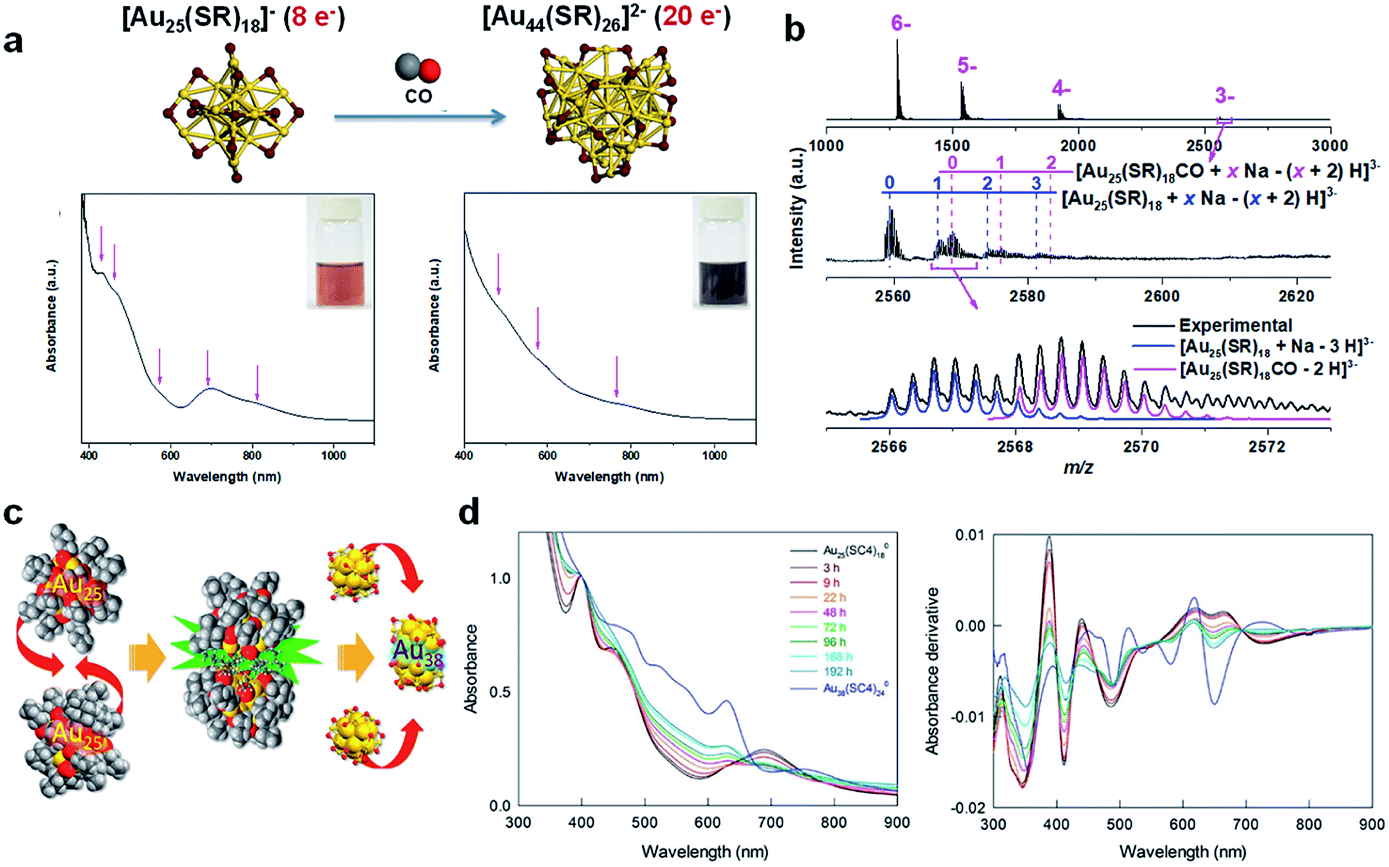 | ||
| Fig. 5 (a) Schematic illustration (top panel) and UV-vis absorption spectra (bottom panel) of size growth from [Au25(SR)18]− to [Au44(SR)26]2−via the CO-reduction method, where magenta arrows indicate the characteristic absorption of [Au25(SR)18]− and [Au44(SR)26]2− (colour code: golden, Au; purple, S). (b) ESI-MS spectra of [Au25(SR)18CO]− adducts identified in the [Au25(SR)18]− solution saturated by CO. (a) and (b) are reproduced with permission from ref. 49. Copyright 2017, the Authors, published by Springer Nature. (c) Schematic illustration of the formation of Au38(SR)24 by the fusion of two Au25(SR)18 NCs. (d) Time-dependent UV-vis absorption and the corresponding absorption-derivative spectra of the size growth of Au38(SR)24 by the fusion of two Au25(SR)18 NCs. (c) and (d) are reproduced with permission from ref. 148. Copyright 2018, American Chemical Society. | ||
Compared with the growth reaction promoted by CO, the molecular-level fundamentals of the NaBH4-driven size growth reaction of metal NCs are less investigated, although NaBH4 is widely used as a versatile reducing agent in the Brust–Schiffrin synthesis of metal NCs. Specifically, although several remarkable achievements have been made in the past decade regarding the chemical states of Au precursors prior to NaBH4 reduction in the typical Brust–Schiffrin method,145–147 little is known about the size-growth pathway of metal NCs after addition of NaBH4. This is probably due to the strong reducing ability of NaBH4, which usually provides a fast and less-controllable reduction kinetics in the size growth reaction of metal NCs. This technical challenge has been recently addressed by Chen et al.51 In contrast to feeding a large excess of NaBH4 (>10 molar equivalent of Au) into the reaction mixture in the typical Brust–Schiffrin methods, the authors significantly reduced the feeding amount of NaBH4 to the stoichiometric value according to eqn (4).
| 32/x [Au(SR)]x + 8e− = [Au25(SR)18]− + 7[Au(SR)2]− | (4) |
The low dose of NaBH4 maintains an appropriately slow reduction kinetics, so that ESI-MS can track the growth reaction of [Au25(SR)18]−. Therefore, real-time ESI-MS was used to monitor the size growth reaction powered by NaBH4, showing a 2e− hopping mechanism similar to that observed in CO reduction fuelled size growth. Given the ubiquitous observation of the 2e− hopping mechanism in various reducing systems, the root cause of this 2e− growth in Au NCs may be the inherent stability of Au NCs with even valence electrons.
In addition to reductive species prompted size growth, the size evolution of metal NCs can also be accomplished by many other cluster reactions, including inter-cluster reactions (e.g., disproportionation and comproportionation reactions) and cluster reactions with Au(I)–SR complexes (e.g., isoelectronic addition reactions). It should be mentioned that all these reactions have been observed in the size growth pathways of Au NCs induced by CO or NaBH4 reduction.48,49 For example, it is suggested that 8e− [Au25(SR)18]− can be formed through the disproportionation reaction of 6e− [Au20(SR)15]− (eqn (5)) or the comproportionation reaction between 6e− [Au21(SR)16]− and 10e− [Au29(SR)20]− (eqn (6)).48
| Disproportionation: 2[Au20(SR)15]− (N = 6) → [Au15(SR)12]− (N = 4) + [Au25(SR)18]− (N = 8) | (5) |
| Comproportionation: [Au21(SR)16]− (N = 6) + [Au29(SR)20]− (N = 10) → 2[Au25(SR)18]− (N = 8) | (6) |
In a recent contribution, Dainese et al. reported that the 14e− Au38(SR)24 was produced through comproportionation of two 7e− Au25(SR)18 NCs, in which the incubation of neutral Au25(SR)18 in toluene at 65 °C for 10–14 days can complete the size conversion (Fig. 5c).148 The slow reaction kinetics and distinctly different electronic structures of the precursor and product NCs facilitate monitoring the entire size conversion reaction by UV-vis absorption spectroscopy (Fig. 5d) and differential pulse voltammetry (DPV), which suggests a second-order rate law for such comproportionation reaction. The authors further found that the neutral core charge and paramagnetic nature of Au25(SR)18 are essential for the size conversion, which indicates that the size conversion is most likely to be accomplished by the collision and fusion of two Au25(SR)18 NCs following eqn (7).
| 2Au25(SR)18 (N = 7) → Au38(SR)24 (N = 14) + 12Au(SR) | (7) |
More recently, Suyama et al. reported a comproportionation reaction between 6e− Au24Pt(SR)18 and 8e− [Au24Pt(SR)18]2−, yielding 7e− [Au24Pt(SR)18]− (eqn (8)) with its structure unambiguously resolved by X-ray crystallography.149 It is interesting to note that this is a comproportionation reaction that changes the valence electron count while keeping the cluster size unchanged. The formation of open-shell 7e− [Au24Pt(SR)18]− is attributed to the superior adiabatic electron affinity of Au24Pt(SR)18 (3.32 eV) to that of [Au24Pt(SR)18]− (2.47 eV). The authors further proposed a dimerization mechanism assisted by the inter-cluster aurophilic interaction for this comproportionation reaction.
| Au24Pt(SR)18 (N = 6) + [Au24Pt(SR)18]2− (N = 8) → 2 [Au24Pt(SR)18]− (N = 7) | (8) |
In addition to disproportionation and comproportionation reactions that change the valence electrons of metal NCs, the size growth of metal NCs can also be achieved through isoelectronic addition reactions without changing the valence electron count. Isoelectronic addition reaction refers to the size growth reaction by adding 0e− M(I)–SR complexes to the preformed metal NCs, and a good example is to convert [Au23(SR)16]− (N = 8) to [Au25(SR)18]− (N = 8).61 The molecular-level stoichiometry (eqn (2)) and atomic-level mechanism (Fig. 3c) of this reaction have been discussed in detail in Section 2.2.
Intrinsic chirality has become one of the most attractive properties in cluster research, which should be attributed to the increasing recognition of the importance of cluster chirality in determining the biomedical and catalytic applications of metal NCs.24,150,151 The unique interactions/reactions of chiral metal NCs with chiral counterions have therefore been used for the enantioselective separation or synthesis of metal NCs. For example, Yao et al. originally developed a phase transfer method based on chiral counterions to enhance the enantiomeric excess of Au NCs protected by penicillamine.152 The Au NCs protected by racemic penicillamine (rac-Pen) were electrostatically paired with a chiral cation, (1R,2S)-N-dodecyl-N-methylephedrinium (DME+), completely transferring the rac-Pen-protected Au NCs from the aqueous phase into the chloroform phase. The phase-transferred Au NCs show circular dichroism (CD) signals in the wavelength window of 300–700 nm, while the original Au NCs in the aqueous phase are optically inactive. These data suggest that DME+ has a favourable interaction with one specific enantiomer of penicillamine-protected Au NCs, preferentially making it stable and enriched in the chloroform phase. Knoppe et al. used a similar chiral phase transfer method, and performed a DME+-induced phase transfer for the racemic mixture of Au102(SR)44.153 It should be recalled that the chirality of Au102(SR)44 intrinsically originates from the mirror arrangement of Au(I)–SR motifs on the Au79 core with a capped Marks-decahedron (MD) structure. In this way, the partial phase transfer of Au102(SR)44 NCs can produce opposite CD responses in the aqueous and organic (chloroform) phases, which clearly indicates a preferential binding habit of DME+ to one specific enantiomer of Au102(SR)44.
Yan et al. have also used the enantioselective ion pairing between metal NCs and chiral counterions in a single phase system to separate a racemic mixture of metal NCs.151N-Benzylcinchonidinium (BCD+) and N-benzylcinchoninium (BCN+) were used as chiral cations to react with a racemic mixture of [Ag28Cu12(SR)24]4−, whose chirality originated from the asymmetric packing of both the Ag28 core and Cu3(SR)6 motifs. It has been shown that incubating racemic [Ag28Cu12(SR)24]4− with BCD+ or BCN+ can selectively impart a specific enantiomer (the R enantiomer for BCD+ and the L one for BCN+) with a higher solution durability. Based on this knowledge, the authors directly synthesized [Ag28Cu12(SR)24]4− in the presence of BCD+ or BCN+, which led to the formation of a large number of optically active enantiomers. These data indicate the usefulness of the enantioselective ion-pairing reaction in the asymmetric synthesis of metal NCs.
3.2. Molecular interactions/reactions in alloying of metal NCs
It has been demonstrated that alloying can effectively enhance the stability and materials performance (e.g., luminescence154 and catalytic activity99,105,155) of metal NCs. In addition to the well-known co-reduction method, the alloying can be achieved at atomic precision through reactions of the preformed metal NCs with various heteroatom sources, including metal cations,53 M(I)–SR/M(II)–SR complexes, metal NCs, and metal surfaces. Based on the various sources of heteroatoms, the molecular interactions/reactions of metal NCs that control the alloying process are diverse.Inspired by the co-reduction method, where different metal salts are used to generate alloy NCs, metal salts or metal cations have long been used as sources of heteroatoms to react with the preformed metal NCs (i.e., parent metal NCs), aiming at producing alloy NCs while keeping the size monodispersity of the parent NCs uncompromised. The reactions between metal cations and the parent metal NCs can be accomplished by a conventional galvanic reaction. For example, several galvanic reactions have been conducted based on atomically precise [Ag25(SR)18]−.78,157 A galvanic reaction between [Ag25(SR)18]− and Au+ or Au3+ usually produces [AuAg24(SR)18]−, where the Au heteroatom is located at the centre of the icosahedral M13 core as revealed by X-ray crystallography analysis.78,157 In sharp contrast, the galvanic reaction between Pd2+ and [Ag25(SR)18]− can produce Ag4Pd4(SR)8 with a completely different M–S framework from the parent metal NCs, while a similar reaction of [Ag25(SR)18]− with Pt2+ can only result in large-sized nanoparticles with a diameter of ∼5 nm.157 These results highlight the decisive role of metal cations in the reaction with metal NCs.
In addition to the conventional galvanic reaction, the alloying reaction can also occur through the anti-galvanic mechanism, where a more reactive metal cation can be reduced by a less reactive M(0) core of the corresponding metal NCs. For example, by reacting [Au25(SR)18]− with Ag+, Hg2+, and Cd2+, a variety of bimetallic NCs, i.e., [Au25−xAgx(SR)18]q,158 Au24Hg(SR)18,85 and Au24Cd(SR)18,53 respectively, can be produced without changing the M–S framework of the parent metal NCs. In contrast, the reaction of Au44(SR)28 with Cd2+ generates Au47Cd2(SR)31, whose M–S framework is completely different from that of the parent NCs.159 It is worth noting that by deliberately controlling the solvent and time of the reaction between [Au25(SR)18]− and Ag+, Yao et al. were also able to deposit two additional Ag heteroatoms on the surface of [Au25(SR)18]− to produce Au25Ag2(SR)18, which showed improved catalytic activity for the hydrolysis of 1,3-diphenylprop-2-ynyl acetate (Fig. 6a).156 Dou et al. also adopted a similar surface addition strategy to enhance the luminescence of water-soluble Au18(SR)14 NCs.160 In this study, foreign Ag+ was introduced to bridge the Au(I)–SR motifs in the protecting shell of Au18(SR)14, leading to the formation of AuAg NCs with strong emission at 667 nm (λex = 520 nm; quantum yield, QY = ∼6.8%) via aggregation-induced emission (AIE).
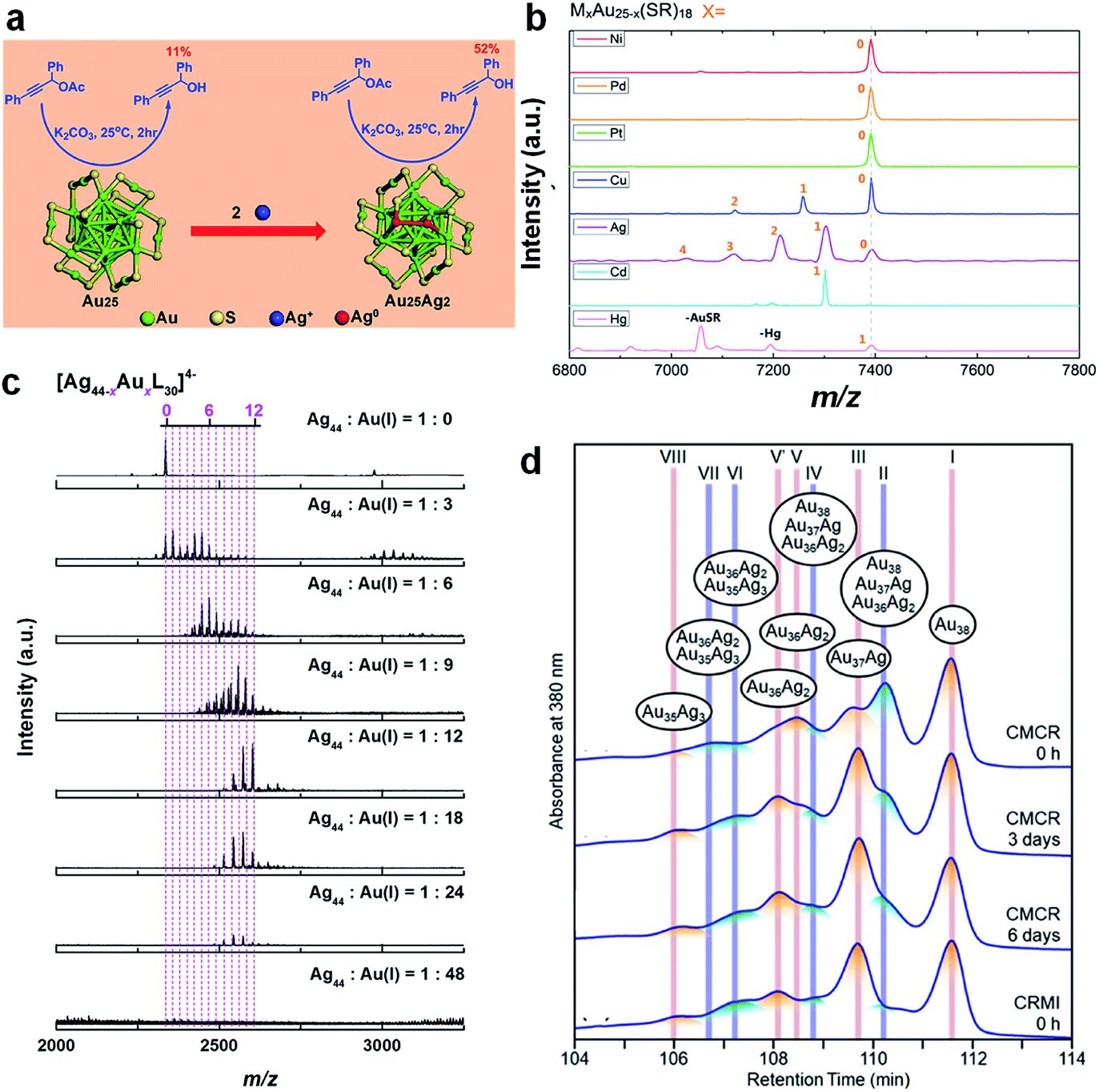 | ||
Fig. 6 (a) Schematic illustration of the formation of Au25Ag2(SR)18 by adding two additional Ag atoms to [Au25(SR)18]−; reproduced with permission from ref. 156. Copyright 2015, American Chemical Society. (b) ESI-MS spectra of Au25−xMx(SR)18 formed by reacting [Au25(SR)18]− with the corresponding M(I)–SR/M(II)–SR complexes (M = Ni, Pd, Pt, Cu, Ag, Cd, and Hg); reproduced with permission from ref. 58. Copyright 2015, American Chemical Society. (c) ESI-MS spectra of [Ag44(p-MBA)30]4− reacted with Au(I)-(p-MBA) at different feeding ratios of Ag44![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Au(I); L denotes the protecting ligand; reproduced with permission from ref. 60. Copyright 2017, the Authors, published by Springer Nature. (d) HPLC spectra of Au38−xAgx(SR)24 formed by cluster-metal complex reaction (CMCR) and co-reduction of metal ions (CRMI) at different reaction times; reproduced with permission from ref. 59. Copyright 2019, American Chemical Society. :Au(I); L denotes the protecting ligand; reproduced with permission from ref. 60. Copyright 2017, the Authors, published by Springer Nature. (d) HPLC spectra of Au38−xAgx(SR)24 formed by cluster-metal complex reaction (CMCR) and co-reduction of metal ions (CRMI) at different reaction times; reproduced with permission from ref. 59. Copyright 2019, American Chemical Society. | ||
Because M(I)–SR or M(II)–SR complexes have good stability and easy accessibility, they have attracted increasing interest as a source of heteroatoms for alloying NCs. Similar to metal cations, the reaction of M(I)–SR or M(II)–SR complexes with the parent metal NCs can produce a variety of alloy NCs, regardless of whether the original M–SR framework of the parent NCs is changed. For example, Wang et al. reported the reactions of [Au25(SR)18]− with Ag(I)–SR, Cu(I)–SR, Cd(II)–SR, and Hg(II)–SR complexes, to produce the alloys [Au25−xAgx(SR)18]−, [Au25−xCux(SR)18]−, Au24Cd(SR)18, and Au24Hg(SR)18, respectively (Fig. 6b).58 In sharp contrast, the reaction of [Au23(SR)16]− with Ag(I)–SR and Cd(II)–SR complexes can initiate alloying and size growth reactions on the parent NCs, producing [Au25−xAgx(SR)18]− (ref. 161) and Au20Cd4(SH) (SR)19,162 respectively. Due to its increasing importance in the synthetic chemistry of metal NCs, the underlying chemistry of the reaction between metal NCs and M(I)–SR or M(II)–SR complexes has been intensely pursued in the past few years. By comparing the reactions of [Au25(S-C2H4Ph)18]− with four different types of Ag(I) sources (i.e., AgNO3, Ag(I)–EDTA, Ag(I)–(S-C2H4Ph), and Ag(I)–DTZ complexes, where EDTA and DTZ are ethylenediaminetetraacetic acid and dithizone, respectively), Tian et al. found that the composition and population of the product alloy NCs were highly dependent on the chelating environment of the incoming heteroatoms.52 Yao et al. conducted detailed ESI-MS and tandem MS analysis on the reaction of [Ag44(SR)30]4− with Au(I)–SR under different feeding ratios of Ag44 to Au (Fig. 6c).60 The authors revealed that up to 12 Au heteroatoms can be incorporated into the M44S30 framework through the SME mechanism described in eqn (1). More interestingly, Zheng et al. observed the dynamic diffusion of a heteroatom (i.e., Au) inside the parent [Ag25(MHA)18]− at the intra-cluster level by using real-time ESI-MS and tandem MS.163 The authors demonstrated that [Ag25(MHA)18]− shows considerable reactivity to Au(I)–(MHA) complexes, thus producing molecularly pure [Ag24Au(MHA)18]− based on a SR–Au(I)–SR unit assisted quasi-SME mechanism. By tracking the position of the Au heteroatom in [Ag24Au(MHA)18]− using real-time tandem MS, the authors unambiguously proved the heteroatom dynamics in the alloy NCs. Specifically, after being incorporated into the protecting motif of [Ag24Au(MHA)18]−, the Au heteroatom will diffuse inward through the outer layer and eventually diffuse to the centre of the icosahedral M13 core. Such dynamics of metal atoms in the metal NCs should be taken into consideration in the further design of metal NC-based nanomaterials.
The inter-cluster reaction is a recent discovery, which provides another mechanism for the alloying of metal NCs.65 For example, Krishnadas et al. demonstrated the inter-cluster reaction between [Au25(SR)18]− and [Ag44(SR)30]4− in dichloromethane (DCM), which led to the formation of both [Au25−xAg(SR)18]− and [Ag44−xAux(SR)30]4− NCs.64,164 The alloying content (i.e., x value) of both [Au25−xAg(SR)18]− and [Ag44−xAux(SR)30]4− has been demonstrated to be highly dependent on the feeding ratio of [Au25(SR)18]− and [Ag44(SR)30]4−, and nearly molecularly pure [Ag32Au12(SR)30]4− can be produced by implementing an appropriate excess of [Ag44(SR)30]4− in the inter-cluster reaction. In a follow-up contribution, Baksi et al. investigated the kinetics of this inter-cluster reaction by time-dependent ESI-MS, mapping out the relative abundance of the individual [Ag44−xAux(SR)30]4− product (x = 1–12) in a range of m/z and reaction time.165 More intriguingly, by using trapped ion mobility mass spectrometry (TIMS), the authors also deliberately monitored the collision cross-section change of individual [Ag44−xAux(SR)30]4− during the inter-cluster reaction. The TIMS results suggest that the Au heteroatom diffused from M2(SR)5 motifs all the way into the icosahedral M12 core of [Ag44−xAux(SR)30]4− after the inter-cluster collision. These findings also highlight the usefulness of mass spectrometry in revealing the reaction kinetics and dynamics of inter-cluster reactions.166
A similar inter-cluster reaction can also occur between [Au25(SR)18]− and [Ag25(SR)18]− to produce [Au25−xAgx(SR)18]−, where the value of x can be continuously varied from 1 to 24 simply by altering the feeding ratio of parent NCs.63 ESI-MS successfully captured an important intermediate, [Au25Ag25(SR)36]2−, which indicates that the inter-cluster reaction is likely to occur through a dimerization mechanism. Combined molecular docking and DFT calculations elucidate that the dimerization is made possible most likely by an inter-cluster Ag–S bond. This mechanism is also in line with the observation of Zhang et al., where the inter-cluster reaction between Au38(SR)24 and Au38−xAgx(SR)24 can be completely terminated through physical separation of these two NCs by a dialysis membrane (molecular weight cut-off, MWCO = 1000 Da).167 Recently, Niihori et al. showed that the inter-cluster reaction might occur among a mixture of Au38−xAgx(SR)24 NCs (x = 0–3), even after the apparent abundance of Au38−xAgx(SR)24 NCs remained stable in the ESI-MS spectra.59 The authors prepared Au38−xAgx(SR)24 (x = 0–3) NCs by mixing Au38(SR)24 with Ag(I)–SR complexes in DCM for 1 min. ESI-MS analysis showed that the population abundance of Au38−xAgx(SR)24 (x = 0–3) remained unchanged within 6 days after synthesis. However, when these ESI-MS identical samples were checked by HPLC, they showed different retention profiles, which largely depended on the incubation time after synthesis (Fig. 6d). This discrepancy is attributed to the inter-cluster reaction between the newly formed Au38−xAgx(SR)24 (x = 0–3) NCs. This reaction can generate not only composition isomers, but also structural isomers in the elution column of HPLC.
In addition to the inter-cluster reaction, alloy NCs can also be produced by reacting metal NCs with bulk metals. For example, Kazan et al. obtained alloy [Au25−xAgx(SR)18]−, [Au25−xCux(SR)18]−, and Au24Cd(SR)18 by incubating [Au25(SR)18]− in the presence of bulk Ag, Cu, and Cd foil, respectively.168 The similar reaction of Au38(SR)24 with Ag and Cu foil can produce Au38−xAg(SR)24 and Au38Cu(SR)24, respectively, while incubating Au38(SR)24 with Cd foil cannot induce any alloying reaction.168
High quality bimetallic NCs fabricated by the delicate chemistry discussed above can also be employed as parent NCs for controlled production of trimetallic and tetrametallic NCs.169,170 Bootharaju et al. reacted bimetallic [Ag24Pt(SR)18]2− with Au+, giving rise to [Ag24−xPtAux(SR)18]2− (x = 1–2).171 However, a similar reaction between [Ag24Pd(SR)18]2− and Au+ could only lead to the formation of intermediate [Ag24−xPdAux(SR)18]2− (x = 1–2) with a limited lifetime, which was ultimately transformed into stable [Ag25−xAux(SR)18]2−.171 Mixing Au24M(SR)18 (M = Hg/Pd) and Ag(I)–SR complexes can inevitably produce Au24−xMAgx(SR)18.172,173 The crystal structure of Au24−xHgAgx(SR)18 has been resolved, showing that the Ag heteroatoms prefer the vertex sites of the icosahedral M13 core.172 Moreover, the as-produced trimetallic Au24−xPdAgx(SR)18 has also been utilized as a starting cluster, and its reaction with Cu–SR complexes was used to synthesize tetrametallic Au24−x−yPdAgxCuy(SR)18.173 In a recent contribution, Khatun et al. demonstrated inter-cluster reaction as another feasible approach for the production of trimetallic metal NCs, where the reaction between [Ag28M(BDT)12(PPh3)4]4− (M = Ni, Pd and Pt) and [Au25(S-C2H4Ph)18]− resulted in a mixture of [Ag28−xMAgx(BDT)12(PPh3)4]4− (x = 1–12) and [Au25−xAgx(S-C2H4Ph)18]− (x = 1–7).174 It is worth noting that the central M atom and protecting ligands of [Ag28M(BDT)12(PPh3)4]4− remain unchanged throughout the inter-cluster reaction, highlighting the decisive impact of the central M atom and BDT ligand on the cluster stability of [Ag28M(BDT)12(PPh3)4]4−.
3.3. Molecular interactions/reactions in ligand exchange of metal NCs
The ligand exchange of metal NCs represents one of the most intensively studied reactions in cluster chemistry.3,4,111,175 The ligand exchange reaction is usually induced by introducing a designed amount of foreign thiols (HSR′) into the parent [Mn(SR)m]q NCs, and recent progress in the surface engineering of metal NCs suggests that the source of foreign thiol ligands can be diverse (e.g., M(I)–SR′ complexes or [Mn(SR′)m]q NCs). Owing to the well-controlled kinetics and thermodynamics through a proper selection of the original SR and the incoming SR′ ligands, ligand exchange reactions have been widely used to fine-tune the surface chemistry of metal NCs, and they can also adjust the size and atom packing patterns of metal NCs.111As a rule of thumb, at low doses, reacting [Mn(SR)m]q NCs with the foreign HSR′ with a structure similar to that of SR tends to produce metal NCs protected by mixed ligands without changing the M–S framework of the parent NCs. For example, Murray and co-workers performed ligand exchange reactions on [Au25(S-C2H4Ph)18]− with different incoming thiol ligands, including HS-(CH2CH2O)5CH3, HS-(CH2CH2O)CH3, HS-(CH2)5CH3, and toluene-3,4-dithiol ((SH)2PhCH3), leading to the formation of mixed-ligand-protected Au25 NCs.176,177 Although the tested monodentate ligands showed very little preference for the exchange sites of the thermodynamic products (obtained at prolonged reaction time), the bidentate (SH)2PhCH3 showed preference for bridging Au(I) atoms from the adjacent SR–[Au(I)–SR]2 motifs. Similarly, monodentate (e.g., HS-Ph) and bidentate (e.g., 1,1′-binaphthyl-2,2′-dithiol or BINAS) thiols have also been used as foreign ligands to react with Au38(S-C2H4Ph)24, generating inhomogeneity in the protecting shell of Au38 NCs.57 In addition to the above-mentioned partial ligand exchange scenarios, the ligand exchange reaction can also be carried out on the surface of metal NCs till completion. The complete ligand exchange reaction usually involves native SR and incoming HSR′, which differ greatly in structure and solubility. For example, AbdulHalim et al. designed a simple method to engineer the surface properties of [Ag44(SR)30]4− NCs based on a complete ligand exchange reaction.178 [Ag44(MNBA)30]4− (MNBA = 5-mercapto-2-nitrobenzoic acid) was synthesized in an aqueous solution, followed by introduction of a DCM/ethanol (1/1 vol/vol) solution of the desired aryl thiols (e.g., HS-Ph-p-F, HS-Ph-p-NO2, and 2-naphthalenethiol). In this two-phase setup, the ligand exchange reaction can be completed within 30 s, accompanied by the total phase transfer of Ag44 NCs from the aqueous phase to the organic phase. A similar phase-transfer-assisted complete ligand exchange was also reported on Au18(SR)14 NCs, in which a complete ligand exchange from GSH to cyclohexanethiol (HS-c-C6H11) was observed.90
Another attractive feature of the ligand exchange reaction is its ability to change the M–S framework of metal NCs. By conducting ligand exchange reaction with a structurally and electronically different incoming thiol ligand at an elevated temperature, the size and structure transformation of metal NCs are likely to be evoked. In the past two decades, this ligand exchange induced size/structure transformation (LEIST) strategy has achieved great success in the synthetic chemistry of metal NCs.3,111 For example, Zeng et al. treated [Au25(S-C2H4Ph)18]− with HS-Ph-p-tBu at 80 °C for 2 h, which converted the icosahedral Au13 core based [Au25(S-C2H4Ph)18]− into the FCC Au20 core based Au28(S-Ph-p-tBu)20.179 Eswaramoorthy et al. synthesized Au279(S-Ph-p-tBu)84 by reacting Au329(S-C2H4Ph)84 with excess HS-Ph-p-tBu at 80 °C for 6 days.180 Time-course ESI-MS analysis suggests that this LEIST reaction occurs by following a three-stage mechanism: initial ligand exchange → core size conversion → complete ligand exchange. This three-stage LEIST mechanism is completely consistent with previous observations in the size transformation reaction from Au38(S-C2H4Ph)24 to Au36(S-Ph-p-tBu)24.66 It is worth noting that the LEIST reaction can be reversible since the size of the stable clusters is closely related to the structure of SR ligands. For example, Bootharaju et al. demonstrated a reversible size conversion between [Ag44(p-MBA)30]4− and [Ag25(S-PhMe2)18]− (HS-PhMe2 = 2,4-dimethylbenzenethiol) by a LEIST reaction in a two phase system (water/DCM), while a similar LEIST reaction can be performed in a one phase system (DCM) for inter-conversion between [Ag44(S-Ph-p-F)30]4− and [Ag25(S-PhMe2)18]−.67 Similar ligand exchange induced reversible size conversions have also been reported on [Ag44(S-Ph-p-F)30]4−/Ag35(SG)18 (ref. 181) and Au30(S-tBu)18/Au36(S-Ph-p-X)24 (X = H or tBu).182
In addition to the size-cum-structure transition discussed above, ligand exchange may also induce a quasi-isomerization reaction, in which the cluster size (i.e., the value of n and m in [Mn(SR)m]q) remains unchanged, while the atomic packing pattern varies. For example, Chen et al. obtained Au28(S-c-C6H11)20 by thermal treatment (at 80 °C) of Au28(S-Ph-p-tBu)20 in the presence of excess HS-c-C6H11.183 These two Au28(SR)20 NCs have a similar FCC Au20 core capped by eight bridging SR ligands. However, the Au(I)–SR motifs are different in these two quasi-isomers. Au28(S-c-C6H11)20 has two trimeric SR–[Au(I)–SR]3 and two monomeric SR–Au(I)–SR motifs, while Au28(S-Ph-p-tBu)20 has four dimeric SR–[Au(I)–SR]2 motifs. More interestingly, Au28(S-c-C6H11)20 can be converted back to Au28(S-Ph-p-tBu)20 by a similar ligand exchange process under thermal treatment (at 80 °C).183 DFT calculations suggest that the driving force for this reversible quasi-isomerization reaction is the balance between DFT energy and van der Waals contribution.
Due to the limited content, many noteworthy achievements on this topic cannot be included in this Review. We apologize for this and would like to refer interested readers to several well-written reviews published elsewhere.3,111,184,185
4. Molecular interactions/reactions in post-synthesis development of metal NCs
In the past two decades, the prosperity and development of synthetic chemistry has enabled a great variety of atomically precise metal NCs with customizable size, composition, structure, and surface chemistry, forming a library of functional building blocks for self-assembly and practical application exploration.3–5,7,8 The self-assembly and practical application exploration of metal NCs also highly depends on the interactions/reactions of metal NCs with ions, molecules, and other metal NCs, which will be discussed in this section.4.1. Molecular interactions/reactions in self-assembly of metal NCs
Self-assembly provides an alternative method to customize or enhance the materials properties and performance of metal NCs through the collective and synergistic effects of adjacent NCs in the assembly, which can be fabricated in different sizes, morphologies, symmetries, and orders.106,108,128–131,186 Since the self-assembly process of metal NCs is mainly governed by either the inter-cluster interactions/reactions or clusters' interactions/reactions with the cross-linker ions/molecules, superstructures with 1D, 2D and 3D configurations have been constructed based on metal NCs by deliberately controlling such interactions/reactions.Oligomerization can be considered as the 1D self-assembly of metal NCs. For example, Pradeep and co-workers captured a dimer of [Au25(S-C2H4Ph)18]− and [Ag25(S-PhMe2)18]− as the intermediate of inter-cluster reaction between these two NCs.63 Their computational calculations suggest that this dimerization is achieved likely through the interdigitation (e.g., van der Waals interactions) of ligands and inter-cluster Ag–S bonds. Very recently, foreign Ag+ has also been employed as a cross-linker to combine two isolated [Au25(S-C2H4Ph)18]− together and produces atomically precise Ag2Au50(S-C2H4Ph)36 (Fig. 7a), where a single Ag is accommodated in the pocket-like cavity of [Au25(S-C2H4Ph)18]−, forming a Ag–S bond with another NC.187 In addition to M–S bonds, dimerization and trimerization can also be achieved by introducing dithiolate ligands as cross-linkers.188 For example, Lahtinen et al. carried out a ligand exchange of Au102(p-MBA)44 with bidentate biphenyl-4,4′-dithiols (SH–Ph–Ph–SH), which covalently linked Au102 NCs into dimers or trimers.188
 | ||
| Fig. 7 (a) Schematic illustration of the dimerization of [Au25(SR)18]− through bridging Ag atoms to form Ag2Au50(SR)36 (colour code: green, core Au(0); cyan, motif Au(I); yellow, S; red, Ag); reproduced with permission from ref. 187. Copyright 2020, Wiley-VCH. (b) Schematic illustration of the formation of nanoribbons by inter-cluster aurophilic interactions between Au NCs: structural anatomy of [Au25(SR)18]− (i), smaller-sized Au NCs formed by surface rearrangement of [Au25(SR)18]− (ii) and their self-assembly into nanoribbons (iii) (colour code: golden, core Au(0); black, motif Au(I); light grey, S); reproduced with permission from ref. 108. Copyright 2019, Wiley-VCH. (c) Structural model of partially protonated Au102(p-MBA)44 (i); TEM image (ii), electron tomographic model (iii), and cartoon structural model (iv) of spherical capsids formed by Au102(p-MBA)44; TEM image of hexagonal close packed (HCP) layered architectures formed by Au102(p-MBA)44 (v); the arrows in (i) indicate the orientations of p-MBA ligands; the insets of (v) are the high resolution TEM image (top panel) and packing model (bottom panel) of HCP layered architectures; reproduced with permission from ref. 128 and 189. Copyright 2016 and 2017, respectively, Wiley-VCH. | ||
In addition to the aforementioned covalent bonds, metallophilic interactions have also been proven to be effective in controlling the self-assembly of metal NCs into 1D architectures. As shown in Fig. 3a, Au25(S-nBu)18 NCs can be organized into 1D nanowires in their single crystals, through inter-cluster aurophilic interactions.106 This 1D configuration also changed their magnetic response from paramagnetic (in the isolated state) to antiferromagnetic (in the assembled state). Wu et al. also demonstrated the effectiveness of aurophilic interactions in directing the 1D self-assembly of Au NCs.108 [Au25(p-MBA)18]− NCs were subjected to a cyclic dialysis treatment in water, which slightly disturbed the surface structure of NCs and enriched long Au(I)–SR motifs in their protecting shell. The abundant Au(I) sites and enhanced flexibility of such long Au(I)–SR motifs form the structural basis for the anisotropic aurophilic interactions between adjacent NCs, thereby promoting their assembly into 1D nanoribbons (Fig. 7b). It is worth noting that the extensive inter-cluster aurophilic interactions in the generated nanoribbons also provide additional radiative relaxation pathways for excited electrons, which contributes to the two-orders-of-magnitude improvement in the cluster emission. A similar aurophilic interaction is also observed in the 1D self-assembly of Au4Pt2(SR)8.130 It should be mentioned that the regular 1D arrangement is a complex undertaking, typically involving more than one type of inter-cluster interaction. For example, through the above success, the close contact between the component NCs required by significant aurophilic interactions (<3.6 Å) is made possible by anisotropic rearrangement of the hydrocarbon tails of the SR ligands, manifesting van der Waals interactions, π–π interactions, and dipole–dipole interactions among the component NCs.106,108,130
Metal NCs have also been arranged into 2D architectures by amphiphilicity,129 van der Waals interactions,131 and H-bonds.128,189 As discussed in a previous section, amphiphilicity can be introduced into Au25(MHA)18 NCs through a phase-transfer-driven ion-pairing reaction with CTA+ (Fig. 4b).129 The as-produced amphiphilic Au25(MHA)18@xCTA (x = 6–9) NCs are able to organize themselves into regularly stacked bilayers at the air–liquid interface, which is reminiscent of the formation of a lyotropic liquid crystalline phase by molecular amphiphiles. Au15 NCs protected by –S-C12H25 have also been assembled into mono-, few- and multi-layered 2D superstructures in both mono-solvent (e.g., dibenzyl ether)131 and dual-solvent (e.g., dibenzyl ether/liquid paraffin, 1/7.5 vol/vol)192 systems, largely leveraging on the van der Waals interactions of –S-C12H25 ligands between adjacent NCs. Rival et al. developed a light-triggered strategy for stacking thiolated azobenzene-protected Au25 NCs into disk-like superstructures, where light-switchable dipole–dipole interactions of azobenzene govern the self-assembly behaviour of NCs.193 Moreover, H-bonds have also been employed by Nonappa et al. for packing Au102(p-MBA)44 into 2D nanosheets, which can be further bent into spherical capsids or stacked into faceted multi-layers (Fig. 7c).128 By directly adding an aqueous solution of Au102(p-MBA)44 into methanol, the –COO− group of the p-MBA ligand can be partially protonated. This partial protonation will further induce preferential inter-cluster H-bonds within the equatorial plane of NCs according to the anisotropic distribution of p-MBA ligands on the surface of Au102(p-MBA)44 (panel (i), Fig. 7c), leading to the formation of spherical capsids with a monolayer of NCs as the shell. By changing the way of introducing the bad solvent (i.e., methanol) from direct mixing to controlled dialysis, the authors were also able to obtain regularly stacked HCP layers of Au102(p-MBA)44, reminiscent of the layered structure of graphite.
In addition to the 2D architectures, 3D supercrystals can also be produced based on the extensive H-bonds among the component metal NCs.12,190 For example, the fully protonated [Ag44(p-MBA)30]4− NCs have been crystallized into a triclinic superlattice in DMF solution (Fig. 8a).12 X-ray crystallography examination suggests that the protonated p-MBA ligands were bundled together into two different patterns, namely a double-bundle containing two p-MBA ligands and a triple-bundle involving three p-MBA ligands (Fig. 8b).12,190 These different bundle patterns of p-MBA ligands determine the inter-cluster H-bonding within and between the crystalline layers. As illustrated in Fig. 8b, NCs in the same crystalline layer are connected by the double-bundles and those in adjacent crystalline layers are connected by the triple-bundles. In this way, an individual NC is connected to other NCs through 60 H-bonds (each p-MBA contributes 2 H-bonds), thus forming a cohesive H-bond network within the NC supercrystals. More interestingly, this cohesive H-bond network endows NC supercrystals with a unique mechanical response, in which NCs in adjacent crystalline layers can rotate in opposite directions upon compression (Fig. 8c). Given that the triclinic supercrystals of [Ag44(p-MBA)30]4− are dominated by H-bonds, Yao et al. envisioned that the crystallization behaviour of [Ag44(p-MBA)30]4− might be changed by removing the directional H-bonds.191 The authors thereby deprotonated the p-MBA ligands by CsOH in a mixture of dimethyl sulfoxide (DMSO) and water, and crystallized [Ag44(p-MBA)30]4− into FCC supercrystals with well-defined octahedral morphology by entropy effects. More importantly, by changing the ionic strength and solvent polarity in the dual-solvent crystallization system, the authors were able to fine-tune the crystallization kinetics of [Ag44(p-MBA)30]4− and shape the obtained supercrystals from an octahedron to a concave octahedron (Fig. 8d).
 | ||
| Fig. 8 (a) Packing structure, (b) H-bonds, and (c) mechanical response of protonated [Ag44(p-MBA)30]4− in its triclinic supercrystals; the top panel in (b) illustrates H-bonds in a typical double-bundle of p-MBA ligands, while the bottom panel exhibits H-bonds within (horizontal direction) and between (vertical direction) the crystalline layers; V and V0 in (c) are the volume and initial volume of supercrystals, respectively; (a)–(c) are reproduced with permission from ref. 190. Copyright 2014, Springer Nature. (d) Formation diagram of the octahedral and concave-octahedral supercrystals by deprotonated [Ag44(p-MBA)30]4− in a range of Cs+ concentrations and dimethyl sulfoxide (DMSO) fractions in the crystallization solution; reproduced with permission from ref. 191. Copyright 2015, Wiley-VCH. (e) Schematic illustration of intra- (blue dashed lines) and inter-cluster (yellow dashed lines) CH–π interactions between two [Au52Cu72(S-Ph-p-CH3)55]+ NCs approaching in an edge-to-edge fashion in their supercrystals, where a triangular mosaic pattern of –S-Ph-p-CH3 ligands can be identified (colour code: pink/golden, Au/Cu; yellow, S; grey/magenta/orange, C; light grey, H); reproduced with permission from ref. 17. Copyright 2020, the Authors, published by Springer Nature. | ||
Besides H-bonds and entropy effects, π interactions (e.g., π–π and CH–π interactions) are another type of interaction most involved in metal NC crystallization research. This is most probably due to their effectiveness in inducing aesthetic and hierarchical molecular packing patterns at the intra- and inter-cluster levels, which helps to maximize the inter-cluster bindings among the supercrystals.9,15,17,55 A good example of this aesthetic and hierarchical arrangement of SR ligands is the herringbone-like arrangement of naphthalene rings through CH–π or π–π interactions in the supercrystals of Au103S2(S-Nap)41 NCs (Fig. 4c), which has been detailed in Section 2.3.55 In addition to Au103S2(S-Nap)41 NCs, similar highly symmetric and aesthetic ligand patterns packed by CH–π and/or π–π interactions have been increasingly observed in large-sized metal NCs, including Au52Cu72(S-Ph-p-CH3)55 (ref. 17) and Au246(S-Ph-p-CH3)80.15 As shown in Fig. 8e, in the triclinic supercrystal of Au52Cu72(S-Ph-p-CH3)55 NCs, a triangular mosaic pattern of HS-Ph-p-CH3 packed through the intra- and inter-cluster CH–π interactions (blue and yellow dashed lines, respectively) has been identified. More interestingly, the intra-cluster CH–π interactions all come from methyl H, while the inter-cluster ones are uniformly composed of phenyl ring H. It should be noted that besides CH–π and π–π interactions, the inter-cluster van der Waals interactions also help to stabilize the supercrystals of metal NCs.15 Similar to the DNA-assisted crystallization of metal nanoparticles,194–196 the crystallization of thiolate-protected noble metal NCs seems to occur via maximizing the attractive ligand interactions (e.g., van der Waals, CH–π, and π–π interactions) between adjacent NCs.15
Other than the template-free self-assembly, the arrangement of metal NCs can also be templated by larger-sized nanomaterials. For example, Chakraborty et al. assembled [Ag44(p-MBA)30]4− around p-MBA protected Au nanorods (Au NRs) with a length of ∼30 nm and a width of ∼10 nm.186 The preferential anchoring of [Ag44(p-MBA)30]4− on the <110> facets of Au NRs via H-bonds was suggested to trigger the self-assembly of [Ag44(p-MBA)30]4−, ultimately giving rise to a core-in-cage octahedral architecture of Au NR@Ag44 NCs. In sharp contrast to Au NR directed self-assembly of [Ag44(p-MBA)30]4− NCs, the self-assembly behaviour of Te nanowires (NWs) can be modulated by [Ag44(p-MBA)30]4− NCs.197 By modifying the surface of Te NWs with an optimized amount of [Ag44(p-MBA)30]4− NCs, Te NWs could be packed into 81°-crossed bilayers. As indicated by the authors' computational study, the near-orthogonal bilayer arrangement of Te NWs is driven by maximizing H-bonds of [Ag44(p-MBA)30]4− NCs from the neighbouring Te NWs.
4.2. Molecular interactions/reactions in sensing applications of metal NCs
Due to their unique molecular structures and environmentally sensitive physicochemical properties (e.g., strong luminescence), metal NCs have gained marked acceptance in sensing applications.30,198 The development of sensors relies heavily on the interactions/reactions of analytes with the metal NCs, which may cause measurable changes in the properties of the clusters. Based on the reactivity of metal NCs to selected ions, small molecules, and biomolecules, metal NCs have long been used as active probes for detecting and monitoring these species in the biomedical and environmental fields.30,199By using metal NCs as optical probes, various cations (e.g., Ag+,200 Hg2+,46,47,201,202 Cu2+,203–206 Pb2+,202,207–209 Pt2+,208,210 Au3+,208,211 Al3+,212 and As3+)213 and anions (e.g., CN− (ref. 45) and S2−)214 can be detected. Among the above-mentioned metal cations, Hg2+ has attracted great attention due to its high toxicity and diverse interactions with metal NCs. For example, Xie et al. first demonstrated the metallophilic interactions between Hg2+ and Au(I) in a red-emitting Au NC protected by bovine serum albumin (BSA), and developed a luminescent “turn-off” sensor for Hg2+ detection.46 The strong and unique Hg2+⋯Au(I) interactions make the Au NC-based luminescent probes highly selective (against 15 other cations, including Cd2+, Pb2+, and Fe3+) and sensitive (with a limit of detection (LOD) of 0.5 nM). Similar metallophilic interactions have also been documented between Hg2+ and Ag(I) provided by Ag12(SG)10 (Fig. 9a–c), whose blue emission centred at 435 nm (λex = 350 nm) can be sensitively quenched by Hg2+.47 By using Ag12(SG)10 at a concentration of 1 nM, it can provide a LOD of 0.1 nM with a good selectivity against interfering cations (e.g., Cd2+, Pb2+, and Fe3+) (Fig. 9d). In addition to the metallophilic interaction with M(I), Hg2+ can also be detected by forming Hg–S bonds and ion pairs with the SR ligands of metal NCs.208,215
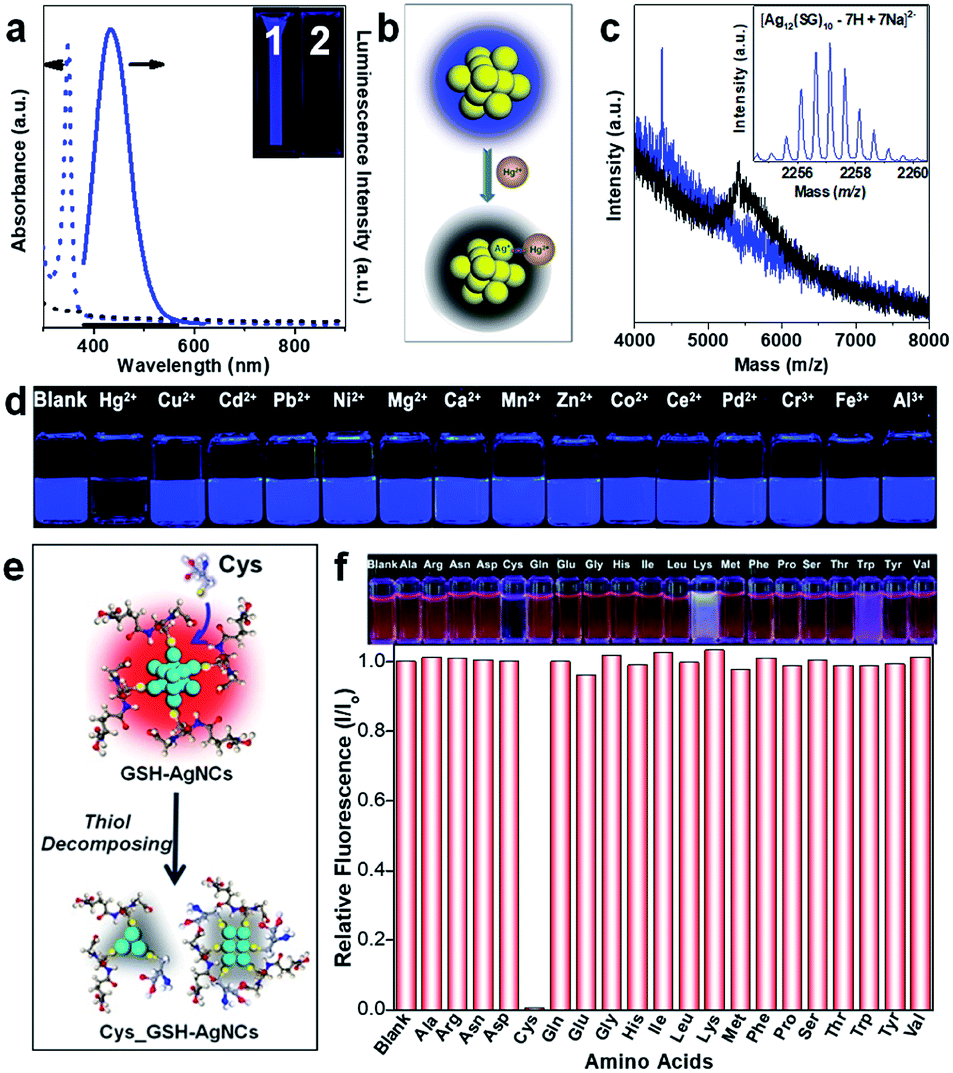 | ||
| Fig. 9 (a) UV-vis absorption (dotted lines) and emission (solid lines, λex = 350 nm) spectra of Ag12(SG)10 in the presence (black lines) and absence (blue lines) of Hg2+; insets are digital photos of Ag12(SG)10 in the presence (#2) and absence (#1) of Hg2+ under UV light; (b) schematic illustration of the metallophilic interaction between Hg2+ and Ag12(SG)10 that can quench cluster luminescence; (c) matrix-assisted laser desorption/ionization (MALDI) mass spectra of Ag12(SG)10 in the presence (black line) and absence (blue line) of Hg2+; (d) digital photos of Ag12(SG)10 in the presence of varied metal cations under UV light (365 nm); (a)–(d) are reproduced with permission from ref. 47. Copyright 2012, Royal Society of Chemistry. (e) Schematic illustration of the detection mechanism of cysteine (Cys) by Ag16(SG)9 NCs; (f) relative emission intensity and digital photos of Ag16(SG)9 NCs in the presence of 20 natural amino acids; reproduced with permission from ref. 216. Copyright 2012, American Chemical Society. | ||
Besides the above-mentioned interactions, another notable reaction that can be used for cation detection is the redox reaction between metal NCs and cations. For example, Wu et al. found that an aqueous solution of [Au25(SG)18]− had an increase in luminescence of 3.4 fold (λem = 670 nm; λex = 514 nm) upon the addition of Ag+. This luminescence enhancement is mainly attributed to the oxidation of [Au25(SG)18]− by Ag+.200 A prototype “turn-on” luminescent sensor for Ag+ detection can be developed based on this mechanism, delivering a LOD of 200 nM and good selectivity against 19 other cations, including Cd2+, Hg2+, and Cu2+. It should be mentioned that redox reaction could also occur between [Au25(SG)18]− and Au3+, leading to decomposition of the former into Au(I)–SG complexes.211 The fast decomposition kinetics (within minutes) may provide a good means for Au3+ detection. Similarly, chemical reduction of Cu2+ by Au15(SG)13 has been registered by George et al., based on which the authors devised a free-standing Au15(SG)13-coated chitosan film for sensitive Cu2+ detection.209
In contrast to the diverse interactions/reactions suitable for cation detection, the documented successes of anion detection mainly depend on the core etching reaction of the metal NCs induced by the reactive anions. For example, CN− can exert a strong etching effect on the Au(0) core of Au NCs through the well-known Elsner reaction (eqn (9)).
| 4Au + 8CN− + 2H2O + O2 → 4[Au(CN)2]− + 4OH− | (9) |
Based on this reaction, Lu et al. developed a luminescent sensor using BSA-protected Au NCs, which emit bright red light at 640 nm (λex not specified).45 After adding CN−, this red emission can be quickly and effectively quenched at the optimized pH of 12.0. A reliable selectivity was demonstrated against 15 interfering anions (e.g., SCN−, EDTA2−, and F−) and 16 cations (e.g., Mg2+, Ba2+, and Al3+), while the LOD was determined to be 200 nM. A similar strong core etching effect has been documented for S2− toward Ag NCs, based on which a “turn-off” luminescent sensor has been developed by using Ag12(SG)10 as a luminescent probe.214
In addition to anions, the core etching reaction can also be induced by small molecules such as thiols. For example, Yuan et al. developed a “turn-off” luminescent sensor for sensitive detection of cysteine (Cys), which is a naturally occurring thiol-containing amino acid.216 The metal NCs used are mainly Ag16(SG)9 NCs, which emit strong red light at 647 nm (λex = 489 nm). Upon reacting with Cys (Fig. 9e), Ag16(SG)9 NCs decomposed into smaller NCs or Ag(I)–SR complexes such as Ag8(SG)4(Cys)2, Ag6(SG)3(Cys)3, Ag3(SG)2(Cys)1, and Ag3(SG)3, thereby reducing red emission and delivering a LOD of 3 nM. Due to the lack of thiol group in the other 19 naturally occurring amino acids, a good selectivity of Cys has been achieved (Fig. 9f). The most interesting finding is the good selectivity of Cys against the thiol-containing tri-peptide GSH. This should be attributed to the size effects of the analytes, where the large GSH cannot penetrate the protecting shell of Ag16(SG)9, thus inhibiting its etching reaction with the Ag(0) core. Other than the core etching reaction, the detection of small molecules can also be achieved by the redox reaction with metal NCs. H2O2 is a common oxidative molecular species detected by this method. For example, Guan et al. developed a Zn(OH)2-assisted co-precipitation method to separate BSA-protected Au NCs from free BSA and other interfering impurities.217 The isolated Au NCs feature an enhanced emission at 615 nm (λex not specified), and the oxidative quenching of this emission provides a good mechanism for H2O2 detection. Very recently, Chen et al. reported the protic-solvent-sensitive emission in 3D superatom complex inorganic frameworks (SCIFs), which are periodically cross-linked [Au1Ag22(S-Adm)12]3+ by SbF6−.218 It was found that these SCIFs show a switchable emission at ∼650 nm (λex = 365 nm), which can be “turned on” or “turned off” in the presence or absence of protic solvents (e.g., water, methanol, and ethanol), respectively. This protic solvent selectivity is attributed to the coordination of solvent molecules with Au/Ag atoms or the formation of H-bonds between F atoms and solvent molecules. It should be noted that a similar solvent- or gas-molecule-sensitive luminescence has been widely recorded in Ag(I) NC-based metal–organic frameworks (MOFs), which are usually bridged by N-containing heterocyclic cross-linkers.219–221
The detection of large biomolecules (e.g., proteins) is another important topic in the sensor development of metal NCs. This detection largely depends on the specific bio-recognition and bio-conjugation interactions.222,223 For example, Chen et al. designed a luminescent probe for glutathione S-transferase (GST)-tagged protein based on a specific GSH–GST interaction.222 GSH-protected Au NCs with orange emission at 610 nm (λex = 395 nm) were used as luminescent probes, which can specifically bind to GST or GST-tagged proteins (e.g., clumping factor A or ClfA for short). It is worth noting that this binding between the GSH ligand and GST can induce negligible changes in cluster luminescence, thus providing a convenient method to stain GST or GST-tagged ClfA. In this way, GST or GST-tagged ClfA can be identified by the naked eye in a mixture of proteins or in cell lysates. In addition to directly using SR ligands as recognition moieties, foreign recognition moieties can also be conjugated to metal NCs to facilitate the selective detection of target biomolecules. For example, Kurdekar et al. recently developed an immunoassay based on luminescent Au NCs for sensitive detection of human immunodeficiency virus (HIV)-1 p24 antigen, which is an important biomarker for the early stage of HIV infection.223 Streptavidin was conjugated to GSH-protected Au NCs (λem = 615 nm; λex = 365 nm) by EDC chemistry. Then, the specific interaction between biotin and streptavidin was used to further couple biotinylated antibodies on the surface of Au NCs, thereby providing specific binding sites to HIV-1 p24 antigen. By forming an antibody–antigen–antibody sandwich complex with the capture antibody, the luminescent Au NCs can be immobilized in the immunoassay wells, thereby providing a means for quantitative detection of HIV-1 p24 antigen. The established immunoassay can deliver a LOD of 5 pg mL−1, which is 3-fold higher than that of the traditional enzyme-linked immunoassay. In addition, this assay has a good selectivity against hepatitis B virus (HBV)-positive and hepatitis C virus (HCV)-positive plasma samples.
4.3. Molecular interactions/reactions in biomedical applications of metal NCs
Due to their small size, strong luminescence, good biocompatibility and efficient renal clearance, metal NCs have potential applications in diverse sectors of biomedicine, like bioimaging,224,225 cancer therapy226 and antimicrobial activity.126,227 The most commonly used molecular reaction of metal NCs in these biomedical applications is covalent conjugation reaction that confers additional functional moieties to NCs, including active targeting moieties122,124,228,229 and therapeutic moieties.125,126,229 For example, since the overexpression of folate receptors has been observed on the surface of many cancer cells, Pyo et al. conjugated folic acid (FA) to Au22(SG)18 NCs through EDC chemistry, which can enhance the luminescence of Au NCs (QY = 42%, ∼6.5-fold higher than that of the original Au22(SG)18) and improve the targeting efficacy toward folate-receptor positive KB and HeLa cells.122 Similarly, by using EDC chemistry, Chen et al. co-conjugated FA and Au29–43(SG)27–37 to a pH-responsive amphiphilic copolymer, poly(DBAM-co-NAS-co-HEMA) or PDNH for short, where DBAM, NAS, and HEMA denote 4-n-dodecyloxybenzalacetal, N-succinimidylmethacrylate, and 2-hydroxyethyl methacrylate, respectively. After loading the anti-cancer drug paclitaxel (PTX), the Au NC-PDNH composite can simultaneously provide active targeting ability, luminescent imaging activity, and therapeutic effect in female athymic nude mice bearing HeLa tumors.229 In addition to FA, peptides and proteins have also been conjugated to luminescent Au NCs, thereby enhancing their ability to target cancer cells for both imaging and killing purposes.124,127 For example, streptavidin was conjugated to GSH-protected Au23 NCs, which emitted strong red light (λem = 685 nm), via EDC chemistry.124 The as-obtained Au23 NC–streptavidin conjugate exhibits enhanced activity for imaging biotin-rich human hepatoma (HepG2) cells, by virtue of the specific streptavidin–biotin interaction.Chemical and physical antimicrobial components have also been conjugated to Au or Ag NCs to evoke collective and synergistic antimicrobial mechanisms.125,126 For example, with good awareness of the bactericidal activity of Ag-based nanomaterials, Zheng et al. combined Ag16(SG)9 NCs with a commercial antibiotic, daptomycin, through EDC chemistry.125 The resulting composite (D-Ag NCs for short) appears in the form of a conjugated network of daptomycin and Ag NCs, with a typical hydrodynamic diameter of about 200 nm. It is further evident that such a conjugated network can provide a high localized concentration of both Ag NCs and daptomycin, which synergistically causes more serious damage to the membrane of bacterial cells. The damaged cell membrane in turn enhances the uptake of D-Ag NCs by bacteria, where Ag NCs can induce a high localized concentration of reactive oxygen species (ROS). Such a high concentration of ROS has been proved to be effective at destroying DNA, contributing to the enhanced antibacterial activity against Gram-positive model bacteria Staphylococcus aureus (S. aureus). In a follow-up study, the authors conjugated [Au25(Cystm)x(MHA)18−x]− NCs (Cystm = cysteamine; x = 1–4) with Ho3+-decorated graphene oxides (Ho-GOs) by a similar EDC approach.126 Au NCs are an emerging family of nanoscale bactericides,230 while Ho-GOs can serve as sharp edges with their orientation controllable by an external magnetic field. The aligned high density of sharp edges provides a physical means to destroy the bacterial membrane, which combined with the high oxidative stress induced by Au NCs can provide a synergistic mechanism to kill both Gram-positive (e.g., S. aureus) and Gram-negative (e.g., Escherichia coli or E. coli for short) bacteria. In addition to Au NCs, luminescent Ag NCs have also been conjugated to graphene sheets, which could be potentially used as antimicrobial and bioimaging agents.231
4.4. Molecular interactions/reactions in catalytic application of metal NCs
Due to the protein-like hierarchical structure, thiolate-protected noble metal NCs can provide a variety of coordination sites and a well-structured reaction environment for substrate molecules, thereby sustaining remarkable catalytic activity and selectivity toward various reactions, including oxidation, hydrogenation, and C–C coupling reactions.5,7,26,232–237 Unlike their larger counterparts, metal nanoparticles (>3 nm), metal NCs can be synthesized with molecular purity and their structures can be probed at the unprecedented Å resolution.8 The molecular level monodispersity and atomically precise structure make metal NCs particularly suitable for use as model catalysts, thus revealing the fundamentals of nanocatalysis at the molecular and atomic levels.Among the various oxidation reactions, the aerobic oxidation of CO has received special attention due to its fundamental and practical importance.238,239 From DFT calculations on three thiolate-protected (i.e., [Au25(SR)18]−, Au102(SR)44, and Au144(SR)60) and two phosphine-protected (i.e., Au11(PH3)7Cl3 and [Au39(PH3)14Cl6]−) Au NCs, Lopez-Acevedo et al. found that O2 and CO can be co-adsorbed on the exposed Au(0) sites after the partial removal of the protecting ligands or motifs.43 Afterwards, the activated O2 can react with CO via a Langmuir–Hinshelwood (LH) mechanism to generate CO2 through a peroxyformate intermediate. Interestingly, Liu et al. also performed DFT calculations on a series of Aun NCs (n = 8–34) and identified a triangular Au3 facet as the active site for aerobic CO oxidation.94 The authors proposed a novel tri-molecular LH mechanism, in which co-adsorption of one O2 molecule and two CO molecules on the Au3 facet facilitates the cleavage of the O–O bond through an OCOO* intermediate. It should be noted that adsorption of up to three molecules of O2 by a single [Au25(SR)18]− has been experimentally evidenced by Bhat et al. based on ESI-MS analysis, strongly corroborating the ability of [Au25(SR)18]− to adsorb and activate molecular O2 in various aerobic oxidation reactions including CO oxidation.240 Since metal NCs are usually immobilized on the oxide supports in practical applications, the support effects have also been intensely evaluated. For example, Wu et al. proved the Mars and van Krevelen (MvK) mechanism for CO oxidation by the 18O isotope labelling experiments, in which the lattice O atoms of the CeO2 support also participated in the aerobic oxidation of CO catalysed by CeO2-supported [Au25(S-C2H4Ph)18]−.241 The authors also found that the partial removal of SR ligands at the interface of [Au25(SR)18]− and CeO2 is beneficial to increase the catalytic activity, thus confirming the theoretical prediction of Lopez-Acevedo et al.43 In addition to support effects, in terms of the catalytic activity of metal NCs in the aerobic oxidation of CO, their size,43,242 heteroatom doping,155 ligand,241 and pre-treatment243 effects have been well studied.
Selective alkene oxidation is another oxidation reaction of current interest. For example, Zhu et al. reported a selective oxidation of styrene to benzaldehyde over styrene epoxide and acetophenone in the presence of supported (on SiO2 or Ca10(PO4)6(OH)2) or unsupported [Au25(S-C2H4Ph)18]−, Au38(S-C2H4Ph)24, and Au144(S-C2H4Ph)60, where the activation of O2 is considered to be the key step.244 This finding was further supported by the distinctly improved conversion of styrene (up to ∼10-fold increase) when the more reactive tert-butyl hydroperoxide (BuOOH) was used as the oxidant or co-oxidant. A detailed mechanism of selective oxidation of styrene has been proposed based on the known crystal structure of [Au25(S-C2H4Ph)18]− (Fig. 10), in which O2 is adsorbed on the electropositive Au13 core while the activation of the CC bond occurs on the electronegative Au(I) in the protecting shell. Then, the oxidation proceeds through a cyclic transition state F (Fig. 10), where the preferential dissociation pathway of this transition state contributes to the selectivity of the overall oxidation reaction. In order to correlate the electronic structure of Au25 with catalytic selectivity, Liu et al. performed XAFS and ultraviolet photoemission spectroscopy analysis on [Au25(S-C2H4Ph)18]− and [Au25(PPh3)10(S-C12H25)5Cl2]2+.245 It was found that [Au25(PPh3)10(S-C12H25)5Cl2]2+ has a more electropositive Au core, which yields a higher selectivity for more oxidized products (i.e., benzaldehyde). It should be pointed out that based on DFT calculations, a slightly different mechanism has been proposed for the selective production of benzaldehyde, which involves a metastable four-membered ring CCOO* intermediate.246 In addition to Au NCs, Ag NCs and alloy NCs have also been used as catalysts for the selective oxidation of styrene, where both catalytic activity and selectivity are proven to be tunable through size and composition engineering of metal NCs.247
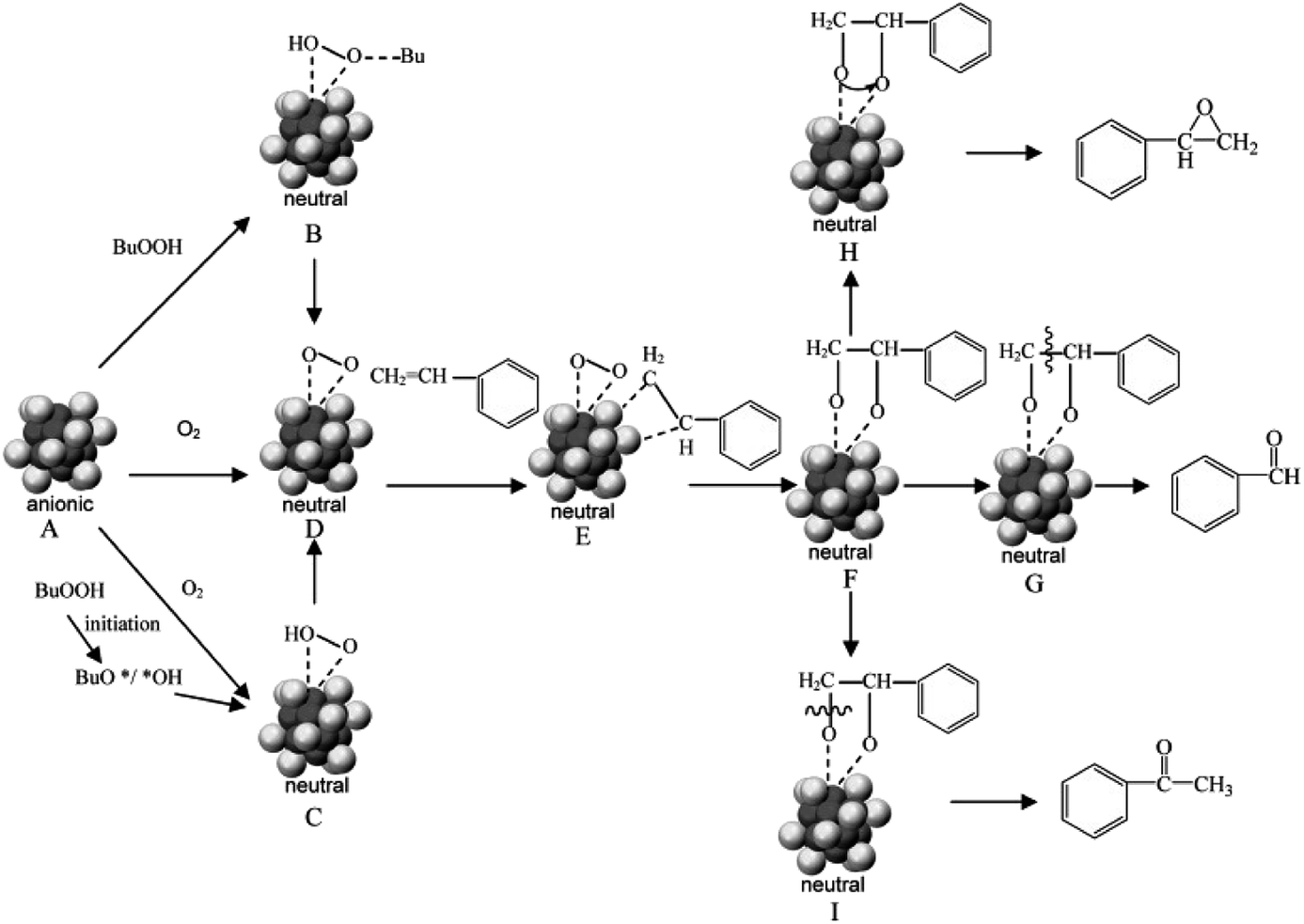 | ||
| Fig. 10 Proposed catalytic mechanism of styrene oxidation by [Au25(SR)18]− in the presence of varied oxidants: BuOOH, O2, and a mixture of them. Colour code: grey, core Au(0); light grey, motif Au(I); SR ligands are omitted for clarity. Reproduced with permission from ref. 244. Copyright 2010, Wiley-VCH. | ||
Other than oxidation reactions, metal NCs also show good catalytic activity and selectivity to reduction and hydrogenation reactions. For example, Zhu et al. measured the effectiveness of both supported (on Fe2O3, TiO2, and SiO2) and unsupported [Au25(S-C2H4Ph)18]− in the selective hydrogenation of α,β-unsaturated ketones, where the hydrogenation of benzalacetone under one atmosphere of H2 at 273 K can provide 100% selectivity to the unsaturated alcohols.92 The authors attributed the extremely high selectivity to the unique activation pathway of H2 and –CO group determined by the Au(0)@Au(I)–SR core–shell structure of [Au25(S-C2H4Ph)18]−. It is suggested that –CO adsorption occurs in the pocket-like cavity of [Au25(S-C2H4Ph)18]−, where the electronegative O is coordinated to the electropositive Au13 core. The electronegative Au(I) in the protecting shell of [Au25(S-C2H4Ph)18]− is considered to be the adsorption site of H2. Afterwards, the addition reaction between the activated –CO and H–H can generate α,β-unsaturated alcohols. In a separate study, Ouyang et al. carried out insightful DFT calculations for the selective hydrogenation of benzalacetone, and mapped out the reaction pathways at the atomic level.248 The authors proposed a mechanism for H2 activation assisted by α,β-unsaturated ketones, in which benzalacetone is co-adsorbed in the pocket-like cavity of [Au25(SR)18]− to facilitate the heterolytic cleavage of H2. Through the transition state Auδ+⋯Hδ−⋯Hδ+⋯Oδ−Cδ+, this heterolytic cleavage can occur preferentially on the –CO group, which is beneficial to the high selectivity of the unsaturated alcohol products.
Considering the unsatisfactory conversion of aldehydes (e.g., 4-nitrobenzaldehyde) by the supported [Au25(S-C2H4Ph)18]− in their hydrogenation reaction at low temperature (e.g., 13.4% at 323 K), Li et al. introduced transition metal cations (e.g., Cu+, Cu2+, Co2+, and Cr3+) as promoters to increase the catalytic activity of [Au25(S-C2H4Ph)18]− in the presence of a base (e.g., ammonia and pyridine).249 It has been proved that the addition of metal cations can effectively increase the conversion of 4-nitrobenzaldehyde to 90.8% (using Co2+ as a promoter), while the 100% selectivity to 4-nitrobenzyl alcohol remains unchanged. Based on the results of matrix-assisted laser desorption/ionization (MALDI) MS, ESI-MS, and DFT calculations, the authors proposed a partial decomposition mechanism for the activation of H2, in which [Au–SR]x modules (x = 1–4) dissociated from [Au25(S-C2H4Ph)18]− to provide active sites for the adsorption and activation of H2.
The aforementioned partial decomposition mechanism well corroborates the hydride-assisted Au(I)–SR motif desorption–re-adsorption mechanism for the hydrogenation of 4-nitrophenenol.250 Nasarudin et al. used UV-vis absorption spectroscopy and ESI-MS to track the NaBH4 hydrogenation of 4-nitrophenenol in the presence of [Au25(p-MBA)18]−.250 It was found that the addition of NaBH4 to the reaction mixture may cause partial desorption of the SR ligands or SR–[Au(I)–SR]x motifs from [Au25(p-MBA)18]−, resulting in a series of smaller-sized Au NCs, among which [Au23(p-MBA)16]− is the most abundant. Since the adsorption of hydrides on cluster species was widely observed in the early stage of the catalytic reaction, the authors concluded that the hydride adsorption might promote the partial desorption of SR or SR–[Au(I)–SR]x, which provided additional active sites for the hydrogenation reaction of 4-nitrophenol. More interestingly, the smaller-sized Au NCs (e.g., [Au23(p-MBA)16]−) can grow back to larger ones (e.g., Au26(p-MBA)19) by re-adsorbing SR or SR–[Au(I)–SR]x in the final stage of the catalytic reaction, constituting a desorption–re-adsorption mechanism for the selective hydrogenation reaction of 4-nitrophenol. In another contribution, the authors also found that the hydrogenation pathway of 4-nitrophenol could be tunable based on the hierarchical structure of [Au25(p-MBA)18]−.251 Leveraging on time-dependent UV-vis absorption spectroscopy, ESI-MS, and DFT calculations, the authors constructed a tri-molecular LH mechanism for semi-hydrogenation of 4-nitrophenol to 4,4′-dihydroxyazobenzene. The co-adsorption of two molecules of 4-nitrophenol and one molecule of BH4− in the pocket-like cavity of [Au25(p-MBA)18]− facilitates the hydrogenation and subsequent coupling of 4-nitrophenol, giving rise to 4,4′-dihydroxyazobenzene particularly in its cis configuration. The formation of the semi-hydrogenation product 4,4′-dihydroxyazobenzene is in sharp contrast to the exclusive production of 4-aminophenol by conventional nano-gold catalysts.
Additionally, the molecular purity of metal NCs may provide unprecedented opportunities to understand the kinetics of catalytic reduction at the molecular and single-cluster level. For example, Zhang et al. were able to monitor the catalytic reduction kinetics of different-sized Au NCs in real time through single-molecule fluorescence microscopy.252 The model reaction used in this study is the catalytic reduction of non-fluorescent resazurin to fluorescent resorufin through hydroxylamine, which makes possible a precise tracking of the formation and desorption of the reduction products on a single cluster by single-molecule fluorescence microscopy. The systematic kinetics analysis performed in this way shows that the catalytic reactions on [Au25(MPA)18]− and Au18(MPA)14 follow a non-competitive LH mechanism, where MPA denotes 3-mercaptopropionic acid. In contrast, the catalytic reduction on Au15(MPA)13 occurs through a competitive LH mechanism. This apparent difference in the catalytic mechanism of Au NCs with different sizes is attributed to the size-sensitive electronic structure of Au NCs, which determines their affinity to substrate and product molecules.
The diverse and delicate molecular interactions/reactions of metal NCs also make them useful for catalysing C–C coupling reactions. It is well known that the coupling reaction between p-iodoanisole (p-MeOPhI) and phenylacetylene (PhCCH) can occur through either cross-coupling or homo-coupling pathways, resulting in three different products, namely diphenyldiacetylene, 4,4′-dimethoxy-1,1′-biphenyl (DMBP), and 1-methoxy-4-(2-phenylethynyl)benzene (MPEB). Interestingly, by doping various heteroatoms (e.g., Ag, Au, and Pt) in [Au25(S-C2H4Ph)18]−, the obtained alloy NCs (supported on TiO2) showed distinctly different catalytic selectivity to the aforementioned three coupling products.100 Specifically, [Au25−xCux(S-C2H4Ph)18]− (x = 0–6) has high selectivity to DMBP (Ullmann homo-coupling product), while [Au25(S-C2H4Ph)18]− and Au24Pt(S-C2H4Ph)18 tend to form MPEB (Sonogashira cross-coupling product). In contrast, [Au25−xAgx(S-C2H4Ph)18]− (x = 0–5) shows comparable selectivity to MPEB (55.4%) and DMBP (44.6%). With the help of DFT calculations, the authors elucidate that two substrate molecules (i.e., p-MeOPhI or PhCCH) can be co-adsorbed on the motif-type Au3 facet of the pocket-like cavity of Au25−xMx(SR)18 NCs, while the core composition of Au25−xMx(SR)18 NCs determines the selective adsorption of p-MeOPhI/PhCCH or p-MeOPhI/p-MeOPhI, which ultimately dictates the pathway and final product of the C–C coupling reactions. Besides doping effects, the same group has also evaluated the ligand effects of [Au25(SR)18]− on the selectivity and activity of Ullmann coupling reaction.253 In addition to Ullmann and Sonogashira coupling, metal NCs have also been shown to be active in catalysing the Suzuki cross-coupling reaction.254 In particular, Abroshan et al. reported the high catalytic activity of TiO2-supported [Au25(S-C2H4Ph)18]− in Suzuki cross-coupling reaction in the presence of an imidazolium-based ionic liquid.254 The conversion of iodoanisole could be varied from 4% to 90% (or even higher) in its cross-coupling reaction with phenylboronic acid, when an optimized amount of BMIM·X (BMIM = 1-butyl-3-methylimidazolium; X = Br, Cl or BF4) was introduced into the reaction system. The combined results of MALDI-MS and theoretical simulation show that the adsorption of BMIM+ cations on the SR–[Au(I)–SR]2 motifs of [Au25(S-C2H4Ph)18]− may lead to a successive dissociation of Au–SR modules, generating active sites on the remaining [Au25−x(S-C2H4Ph)18−x]− (x = 1–4) NCs to activate iodoanisole.
5. Conclusions
In summary, owing to their protein-like hierarchical structure, thiolate-protected noble metal NCs possess a great diversity of active sites, making possible their interactions with ions, molecules, and other metal NCs. Such interactions include physical/chemical adsorption, metallophilicity, van der Waals interactions, π interactions, host–guest interactions, electrostatic interactions, H-bonds, and covalent bonds, depending on the chemical nature of the incoming species and their interacting sites on metal NCs. The fundamentals of the above interactions inherently determine the reactivity and selectivity of metal NCs in their formation (e.g., size growth, metal exchange, ligand exchange, and motif exchange), self-assembly (e.g., 1D, 2D, and 3D self-assembly) and applications (e.g., sensors, biomedicine, and catalysis). It is well known that the remarkable reactivity and selectivity of metal NCs are a combined result of their well-organized atomic or molecular patterns at different hierarchical levels, of which the most notable example is the pocket-like cavity of [Au25(SR)18]− consisting of the core Au(0), motif Au(I) atoms, and SR ligands. In return, the atomically precise structure and structure-sensitive properties of metal NCs provide a good platform to probe and understand their molecular interactions/reactions with ions, molecules, and other metal NCs at the molecular and atomic levels. In the past two decades, remarkable achievements have been made in this regard through a combination of experimental and theoretical studies.Although exciting achievements have been documented in understanding and implementing the molecular interactions/reactions of metal NCs, there are still challenges that require collaborative efforts of experimental and theoretical chemists. For example, customizing the composition and structure of metal NCs atom by atom is a necessary prerequisite for atomic-level control of their molecular reactivity. Although many effective methods have been developed to customize the cluster size while keeping their size monodispersity unaffected, a similar atomically precise control of the alloying/ligand-exchange sites of noble metal NCs still remains a grand synthetic challenge.7,8,142,170 In addition, the increasingly known dynamics of metal atoms and/or SR ligands in each NC imposes additional difficulties to precisely control the alloying/ligand-exchange sites in metal NCs.163,255–257 It should be noted that similar metal and ligand dynamics have also been demonstrated at the inter-cluster level,59,249,250 which suggests that the stability and integrity of noble metal NCs in practical applications should be carefully evaluated in the corresponding surroundings.
In addition to thiolates, many other organic ligands have been exploited to synthesize atomically precise metal NCs, due to their diverse binding modes and binding strength with metal centres. Such organic ligands of current interest include phosphines,258–260 hydrides,261–264 alkynyls,265–268 N-heterocyclic carbenes,269–271 carboxylates,219,272,273 and solvent molecules.219,272–275 In particular, recent findings have shown that organic ligands with relatively low binding affinity to Ag (e.g., CF3COO− and CH3CN) can be replaced by bridging ligands (e.g., 4,4′-bypyridine), further cross-linking mixed-ligand-protected Ag NCs into a 3D network reminiscent of MOF structures.219 Such Ag NC-based MOFs have enhanced stability and unique interactions with various gas and/or solvent molecules, making them particularly useful in sensing applications. This readily suggests that customizing organic ligands is an effective method that can not only tailor the metal–ligand interface, but also adjust the packing patterns of metal NCs in their assemblies. Therefore, continuous efforts should be devoted to engineering the metal–ligand interface by discovering and customizing organic ligands for metal NC fabrication.
In addition to the structure and composition customization at the intra-cluster level, engineering the packing patterns and symmetry of the metal NCs in their assemblies or supercrystals is an equally important means to regulate the molecular interactions/reactions of metal NCs. As discussed in Section 4.1, metal NCs have been assembled into 1D, 2D, and 3D superstructures with different orders, symmetries, and morphologies. However, due to its relatively short development history and sensitive dependence on high-quality building blocks, the self-assembly study (especially the exploration of crystallization) of noble metal NCs is still in its infancy. In particular, the governing principles are less well established for the self-assembly or crystallization chemistry of noble metal NCs, compared with those for the self-assembly or crystallization studies of molecular compounds or larger nanoparticles.276–278 Such knowledge gap should be narrowed or eliminated in the near future.
To sum up, we have discussed recent progress in understanding the fundamentals and practicality of the interactions/reactions of metal NCs with ions, molecules, and other NCs. Since the synthesis, self-assembly, and practical application of metal NCs are inherently determined by their molecular reactivity, the underlying and applied chemistry discussed in this Review can not only provide guiding principles for the synthesis of atomically precise noble metal NCs, but can also increase their acceptance in diverse sectors of post-synthesis developments (e.g., self-assembly and practical application).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the Ministry of Education, Singapore (under Research Grant R-279-000-580-112 and R-279-000-538-114), the Taishan Scholar Foundation of Shandong Province (under Grant tsqn201812074) and Young Talents Joint Fund of Shandong Province (under Grant ZR2019YQ07), China.References
- M.-C. Daniel and D. Astruc, Chem. Rev., 2003, 104, 293–346 CrossRef.
- P.-C. Chen, M. Liu, J. S. Du, B. Meckes, S. Wang, H. Lin, V. P. Dravid, C. Wolverton and C. A. Mirkin, Science, 2019, 363, 959–964 CrossRef CAS.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS.
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2019, 120, 526–622 CrossRef.
- A. Fernando, K. L. D. M. Weerawardene, N. V. Karimova and C. M. Aikens, Chem. Rev., 2015, 115, 6112–6216 CrossRef CAS.
- Q. Yao, X. Yuan, T. Chen, D. T. Leong and J. Xie, Adv. Mater., 2018, 30, 1802751 CrossRef.
- Q. Yao, T. Chen, X. Yuan and J. Xie, Acc. Chem. Res., 2018, 51, 1338–1348 CrossRef CAS.
- P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and R. D. Kornberg, Science, 2007, 318, 430–433 CrossRef CAS.
- M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2008, 130, 5883–5885 CrossRef CAS.
- M. W. Heaven, A. Dass, P. S. White, K. M. Holt and R. W. Murray, J. Am. Chem. Soc., 2008, 130, 3754–3755 CrossRef CAS.
- A. Desireddy, B. E. Conn, J. Guo, B. Yoon, R. N. Barnett, B. M. Monahan, K. Kirschbaum, W. P. Griffith, R. L. Whetten, U. Landman and T. P. Bigioni, Nature, 2013, 501, 399–402 CrossRef CAS.
- H. Yang, Y. Wang, H. Huang, L. Gell, L. Lehtovaara, S. Malola, H. Häkkinen and N. Zheng, Nat. Commun., 2013, 4, 2422 CrossRef.
- C. P. Joshi, M. S. Bootharaju, M. J. Alhilaly and O. M. Bakr, J. Am. Chem. Soc., 2015, 137, 11578–11581 CrossRef CAS.
- C. Zeng, Y. Chen, K. Kirschbaum, K. J. Lambright and R. Jin, Science, 2016, 354, 1580–1584 CrossRef CAS.
- H. Yang, Y. Wang, X. Chen, X. Zhao, L. Gu, H. Huang, J. Yan, C. Xu, G. Li, J. Wu, A. J. Edwards, B. Dittrich, Z. Tang, D. Wang, L. Lehtovaara, H. Häkkinen and N. Zheng, Nat. Commun., 2016, 7, 12809 CrossRef CAS.
- Y. Song, Y. Li, H. Li, F. Ke, J. Xiang, C. Zhou, P. Li, M. Zhu and R. Jin, Nat. Commun., 2020, 11, 478 CrossRef CAS.
- H. Häkkinen, M. Walter and H. Grönbeck, J. Phys. Chem. B, 2006, 110, 9927–9931 CrossRef.
- S. Chen, R. S. Ingram, M. J. Hostetler, J. J. Pietron, R. W. Murray, T. G. Schaaff, J. T. Khoury, M. M. Alvarez and R. L. Whetten, Science, 1998, 280, 2098–2101 CrossRef CAS.
- Z. Wu, Q. Yao, O. J. H. Chai, N. Ding, W. Xu, S. Zang and J. Xie, Angew. Chem., Int. Ed., 2020, 59, 9934–9939 CrossRef CAS.
- K. Pyo, V. D. Thanthirige, K. Kwak, P. Pandurangan, G. Ramakrishna and D. Lee, J. Am. Chem. Soc., 2015, 137, 8244–8250 CrossRef CAS.
- T. Wang, D. Wang, J. W. Padelford, J. Jiang and G. Wang, J. Am. Chem. Soc., 2016, 138, 6380–6383 CrossRef CAS.
- S. Choi, R. M. Dickson and J. Yu, Chem. Soc. Rev., 2012, 41, 1867–1891 RSC.
- S. Knoppe and T. Bürgi, Acc. Chem. Res., 2014, 47, 1318–1326 CrossRef CAS.
- S. Yamazoe, K. Koyasu and T. Tsukuda, Acc. Chem. Res., 2013, 47, 816–824 CrossRef.
- T. Higaki, Y. Li, S. Zhao, Q. Li, S. Li, X.-S. Du, S. Yang, J. Chai and R. Jin, Angew. Chem., Int. Ed., 2019, 58, 8291–8302 CrossRef CAS.
- W. Chen and S. Chen, Angew. Chem., Int. Ed., 2009, 48, 4386–4389 CrossRef CAS.
- M. Yu and J. Zheng, ACS Nano, 2015, 9, 6655–6674 CrossRef CAS.
- H. Zhu, J. Li, J. Wang and E. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 36831–36838 CrossRef CAS.
- L. Shang, J. Xu and G. U. Nienhaus, Nano Today, 2019, 28, 100767 CrossRef.
- L. Shang, F. Stockmar, N. Azadfar and G. U. Nienhaus, Angew. Chem., Int. Ed., 2013, 52, 11154–11157 CrossRef CAS.
- J. Zhang, Y. Fu, C. V. Conroy, Z. Tang, G. Li, R. Y. Zhao and G. Wang, J. Phys. Chem. C, 2012, 116, 26561–26569 CrossRef CAS.
- B. Du, M. Yu and J. Zheng, Nat. Rev. Mater., 2018, 3, 358–374 CrossRef.
- B. Du, X. Jiang, A. Das, Q. Zhou, M. Yu, R. Jin and J. Zheng, Nat. Nanotechnol., 2017, 12, 1096–1102 CrossRef CAS.
- K. G. Stamplecoskie and P. V. Kamat, J. Am. Chem. Soc., 2014, 136, 11093–11099 CrossRef CAS.
- Y.-S. Chen and P. V. Kamat, J. Am. Chem. Soc., 2014, 136, 6075–6082 CrossRef CAS.
- H. Zhang, H. Liu, Z. Tian, D. Lu, Y. Yu, S. Cestellos-Blanco, K. K. Sakimoto and P. Yang, Nat. Nanotechnol., 2018, 13, 900–905 CrossRef CAS.
- W. Kurashige, R. Hayashi, K. Wakamatsu, Y. Kataoka, S. Hossain, A. Iwase, A. Kudo, S. Yamazoe and Y. Negishi, ACS Appl. Energy Mater., 2019, 2, 4175–4187 CrossRef CAS.
- F. Liu, Y. Liu, Q. Yao, Y. Wang, X. Fang, C. Shen, F. Li, M. Huang, Z. Wang, W. Sand and J. Xie, Environ. Sci. Technol., 2020, 54, 5913–5921 CrossRef CAS.
- G. Zhang, R. Wang and G. Li, Chin. Chem. Lett., 2018, 29, 687–693 CrossRef CAS.
- C. Liu, X. Ren, F. Lin, X. Fu, X. Lin, T. Li, K. Sun and J. Huang, Angew. Chem., Int. Ed., 2019, 58, 11335–11339 CrossRef CAS.
- Y. Yu, Z. Luo, D. M. Chevrier, D. T. Leong, P. Zhang, D.-e. Jiang and J. Xie, J. Am. Chem. Soc., 2014, 136, 1246–1249 CrossRef CAS.
- O. Lopez-Acevedo, K. A. Kacprzak, J. Akola and H. Häkkinen, Nat. Chem., 2010, 2, 329–334 CrossRef CAS.
- Y. Chen, L. Li, L. Gong, T. Zhou and J. Liu, Adv. Funct. Mater., 2019, 29, 1806945 CrossRef.
- Y. Liu, K. Ai, X. Cheng, L. Huo and L. Lu, Adv. Funct. Mater., 2010, 20, 951–956 CrossRef CAS.
- J. Xie, Y. Zheng and J. Y. Ying, Chem. Commun., 2010, 46, 961–963 RSC.
- X. Yuan, T. J. Yeow, Q. Zhang, J. Y. Lee and J. Xie, Nanoscale, 2012, 4, 1968–1971 RSC.
- Z. Luo, V. Nachammai, B. Zhang, N. Yan, D. T. Leong, D.-e. Jiang and J. Xie, J. Am. Chem. Soc., 2014, 136, 10577–10580 CrossRef CAS.
- Q. Yao, X. Yuan, V. Fung, Y. Yu, D. T. Leong, D.-e. Jiang and J. Xie, Nat. Commun., 2017, 8, 927 CrossRef.
- B. M. Barngrover and C. M. Aikens, J. Phys. Chem. Lett., 2011, 2, 990–994 CrossRef CAS.
- T. Chen, V. Fung, Q. Yao, Z. Luo, D.-e. Jiang and J. Xie, J. Am. Chem. Soc., 2018, 140, 11370–11377 CrossRef CAS.
- S. Tian, C. Yao, L. Liao, N. Xia and Z. Wu, Chem. Commun., 2015, 51, 11773–11776 RSC.
- C. Yao, Y.-j. Lin, J. Yuan, L. Liao, M. Zhu, L.-h. Weng, J. Yang and Z. Wu, J. Am. Chem. Soc., 2015, 137, 15350–15353 CrossRef CAS.
- M. Rambukwella, L. Sementa, A. Fortunelli and A. Dass, J. Phys. Chem. C, 2017, 121, 14929–14935 CrossRef CAS.
- T. Higaki, C. Liu, M. Zhou, T.-Y. Luo, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2017, 139, 9994–10001 CrossRef CAS.
- C. Peng, M. Yu and J. Zheng, Nano Lett., 2019, 20, 1378–1382 CrossRef.
- S. Knoppe, A. C. Dharmaratne, E. Schreiner, A. Dass and T. Bürgi, J. Am. Chem. Soc., 2010, 132, 16783–16789 CrossRef CAS.
- S. Wang, Y. Song, S. Jin, X. Liu, J. Zhang, Y. Pei, X. Meng, M. Chen, P. Li and M. Zhu, J. Am. Chem. Soc., 2015, 137, 4018–4021 CrossRef CAS.
- Y. Niihori, S. Hashimoto, Y. Koyama, S. Hossain, W. Kurashige and Y. Negishi, J. Phys. Chem. C, 2019, 123, 13324–13329 CrossRef CAS.
- Q. Yao, Y. Feng, V. Fung, Y. Yu, D.-e. Jiang, J. Yang and J. Xie, Nat. Commun., 2017, 8, 1555 CrossRef.
- Q. Yao, V. Fung, C. Sun, S. Huang, T. Chen, D.-e. Jiang, J. Y. Lee and J. Xie, Nat. Commun., 2018, 9, 1979 CrossRef.
- Y. Song, T. Huang and R. W. Murray, J. Am. Chem. Soc., 2003, 125, 11694–11701 CrossRef CAS.
- K. R. Krishnadas, A. Baksi, A. Ghosh, G. Natarajan and T. Pradeep, Nat. Commun., 2016, 7, 13447 CrossRef CAS.
- K. R. Krishnadas, A. Ghosh, A. Baksi, I. Chakraborty, G. Natarajan and T. Pradeep, J. Am. Chem. Soc., 2016, 138, 140–148 CrossRef CAS.
- K. R. Krishnadas, A. Baksi, A. Ghosh, G. Natarajan, A. Som and T. Pradeep, Acc. Chem. Res., 2017, 50, 1988–1996 CrossRef CAS.
- C. Zeng, C. Liu, Y. Pei and R. Jin, ACS Nano, 2013, 7, 6138–6145 CrossRef CAS.
- M. S. Bootharaju, C. P. Joshi, M. J. Alhilaly and O. M. Bakr, Chem. Mater., 2016, 28, 3292–3297 CrossRef CAS.
- D. M. Chevrier, B. E. Conn, B. Li, D.-e. Jiang, T. P. Bigioni, A. Chatt and P. Zhang, ACS Nano, 2020, 14, 8433–8441 CrossRef CAS.
- D. M. Chevrier, L. Raich, C. Rovira, A. Das, Z. Luo, Q. Yao, A. Chatt, J. Xie, R. Jin, J. Akola and P. Zhang, J. Am. Chem. Soc., 2018, 140, 15430–15436 CrossRef CAS.
- P. Zhang, J. Phys. Chem. C, 2014, 118, 25291–25299 CrossRef CAS.
- S. Yamazoe and T. Tsukuda, Bull. Chem. Soc. Jpn., 2019, 92, 193–204 CrossRef CAS.
- S. Yamazoe, S. Takano, W. Kurashige, T. Yokoyama, K. Nitta, Y. Negishi and T. Tsukuda, Nat. Commun., 2016, 7, 10414 CrossRef CAS.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802 RSC.
- R. H. Terrill, T. A. Postlethwaite, C.-h. Chen, C.-D. Poon, A. Terzis, A. Chen, J. E. Hutchison, M. R. Clark, G. Wignall, J. D. Londono, R. Superfine, M. Falvo, C. S. Johnson Jr, E. T. Samulski and R. W. Murray, J. Am. Chem. Soc., 1995, 117, 12537–12548 CrossRef CAS.
- R. S. Ingram, M. J. Hostetler and R. W. Murray, J. Am. Chem. Soc., 1997, 119, 9175–9178 CrossRef CAS.
- R. L. Whetten, J. T. Khoury, M. M. Alvarez, S. Murthy, I. Vezmar, Z. L. Wang, P. W. Stephens, C. L. Cleveland, W. D. Luedtke and U. Landman, Adv. Mater., 1996, 8, 428–433 CrossRef CAS.
- T. G. Schaaff, M. N. Shafigullin, J. T. Khoury, I. Vezmar, R. L. Whetten, W. G. Cullen, P. N. First, C. Gutiérrez-Wing, J. Ascensio and M. J. Jose-Yacamán, J. Phys. Chem. B, 1997, 101, 7885–7891 CrossRef CAS.
- M. S. Bootharaju, C. P. Joshi, M. R. Parida, O. F. Mohammed and O. M. Bakr, Angew. Chem., Int. Ed., 2015, 55, 922–926 CrossRef.
- C. Kumara, C. M. Aikens and A. Dass, J. Phys. Chem. Lett., 2014, 5, 461–466 CrossRef CAS.
- C. Kumara, K. J. Gagnon and A. Dass, J. Phys. Chem. Lett., 2015, 6, 1223–1228 CrossRef CAS.
- H. Yang, Y. Wang, J. Yan, X. Chen, X. Zhang, H. Häkkinen and N. Zheng, J. Am. Chem. Soc., 2014, 136, 7197–7200 CrossRef CAS.
- S. Tian, L. Liao, J. Yuan, C. Yao, J. Chen, J. Yang and Z. Wu, Chem. Commun., 2016, 52, 9873–9876 RSC.
- J. Yan, H. Su, H. Yang, S. Malola, S. Lin, H. Häkkinen and N. Zheng, J. Am. Chem. Soc., 2015, 137, 11880–11883 CrossRef CAS.
- M. A. Tofanelli, T. W. Ni, B. D. Phillips and C. J. Ackerson, Inorg. Chem., 2016, 55, 999–1001 CrossRef CAS.
- L. Liao, S. Zhou, Y. Dai, L. Liu, C. Yao, C. Fu, J. Yang and Z. Wu, J. Am. Chem. Soc., 2015, 137, 9511–9514 CrossRef CAS.
- H. Qian, W. T. Eckenhoff, Y. Zhu, T. Pintauer and R. Jin, J. Am. Chem. Soc., 2010, 132, 8280–8281 CrossRef CAS.
- T. Higaki, M. Zhou, K. J. Lambright, K. Kirschbaum, M. Y. Sfeir and R. Jin, J. Am. Chem. Soc., 2018, 140, 5691–5695 CrossRef CAS.
- N. A. Sakthivel, M. Stener, L. Sementa, A. Fortunelli, G. Ramakrishna and A. Dass, J. Phys. Chem. Lett., 2018, 9, 1295–1300 CrossRef CAS.
- D. Crasto, S. Malola, G. Brosofsky, A. Dass and H. Häkkinen, J. Am. Chem. Soc., 2014, 136, 5000–5005 CrossRef CAS.
- S. Chen, S. Wang, J. Zhong, Y. Song, J. Zhang, H. Sheng, Y. Pei and M. Zhu, Angew. Chem., Int. Ed., 2015, 54, 3145–3149 CrossRef CAS.
- Y. Zhu, Z. Wu, C. Gayathri, H. Qian, R. R. Gil and R. Jin, J. Catal., 2010, 271, 155–160 CrossRef CAS.
- Y. Zhu, H. Qian, B. A. Drake and R. Jin, Angew. Chem., Int. Ed., 2010, 49, 1295–1298 CrossRef CAS.
- M. Zhu, W. T. Eckenhoff, T. Pintauer and R. Jin, J. Phys. Chem. C, 2008, 112, 14221–14224 CrossRef CAS.
- C. Liu, Y. Tan, S. Lin, H. Li, X. Wu, L. Li, Y. Pei and X. C. Zeng, J. Am. Chem. Soc., 2013, 135, 2583–2595 CrossRef CAS.
- S. Xie, H. Tsunoyama, W. Kurashige, Y. Negishi and T. Tsukuda, ACS Catal., 2012, 2, 1519–1523 CrossRef CAS.
- Y. Negishi, K. Igarashi, K. Munakata, W. Ohgake and K. Nobusada, Chem. Commun., 2012, 48, 660–662 RSC.
- K. Kwak, W. Choi, Q. Tang, M. Kim, Y. Lee, D.-e. Jiang and D. Lee, Nat. Commun., 2017, 8, 14723 CrossRef.
- K. Kwak, W. Choi, Q. Tang, D.-e. Jiang and D. Lee, J. Mater. Chem. A, 2018, 6, 19495–19501 RSC.
- Y. Lu, C. Zhang, X. Li, A. R. Frojd, W. Xing, A. Z. Clayborne and W. Chen, Nano Energy, 2018, 50, 316–322 CrossRef CAS.
- Z. Li, X. Yang, C. Liu, J. Wang and G. Li, Prog. Nat. Sci., 2016, 26, 477–482 CrossRef CAS.
- L. Andrews, Chem. Soc. Rev., 2004, 33, 123–132 RSC.
- C. Mohr, H. Hofmeister, J. Radnik and P. Claus, J. Am. Chem. Soc., 2003, 125, 1905–1911 CrossRef CAS.
- G. Li, D.-e. Jiang, C. Liu, C. Yu and R. Jin, J. Catal., 2013, 306, 177–183 CrossRef CAS.
- G. Li and R. Jin, J. Am. Chem. Soc., 2014, 136, 11347–11354 CrossRef CAS.
- Y. Liu, X. Chai, X. Cai, M. Chen, R. Jin, W. Ding and Y. Zhu, Angew. Chem., Int. Ed., 2018, 57, 9775–9779 CrossRef CAS.
- M. De Nardi, S. Antonello, D.-e. Jiang, F. Pan, K. Rissanen, M. Ruzzi, A. Venzo, A. Zoleo and F. Maran, ACS Nano, 2014, 8, 8505–8512 CrossRef CAS.
- T. W. Ni, M. A. Tofanelli, B. D. Phillips and C. J. Ackerson, Inorg. Chem., 2014, 53, 6500–6502 CrossRef CAS.
- Z. Wu, Y. Du, J. Liu, Q. Yao, T. Chen, Y. Cao, H. Zhang and J. Xie, Angew. Chem., Int. Ed., 2019, 58, 8139–8144 CrossRef CAS.
- W. Fei, S. Antonello, T. Dainese, A. Dolmella, M. Lahtinen, K. Rissanen, A. Venzo and F. Maran, J. Am. Chem. Soc., 2019, 141, 16033–16045 CrossRef CAS.
- D. R. Kauffman, D. Alfonso, C. Matranga, P. Ohodnicki, X. Deng, R. C. Siva, C. Zeng and R. Jin, Chem. Sci., 2014, 5, 3151–3157 RSC.
- C. Zeng, Y. Chen, A. Das and R. Jin, J. Phys. Chem. Lett., 2015, 6, 2976–2986 CrossRef CAS.
- K. R. Krishnadas, G. Natarajan, A. Baksi, A. Ghosh, E. Khatun and T. Pradeep, Langmuir, 2019, 35, 11243–11254 CrossRef CAS.
- E. Khatun, A. Ghosh, P. Chakraborty, P. Singh, M. Bodiuzzaman, P. Ganesan, G. Nataranjan, J. Ghosh, S. K. Pal and T. Pradeep, Nanoscale, 2018, 10, 20033–20042 RSC.
- P. Liu, R. Qin, G. Fu and N. Zheng, J. Am. Chem. Soc., 2017, 139, 2122–2131 CrossRef CAS.
- H. Häkkinen, Nat. Chem., 2012, 4, 443–455 CrossRef.
- C. L. Heinecke, T. W. Ni, S. Malola, V. Mäkinen, O. A. Wong, H. Häkkinen and C. J. Ackerson, J. Am. Chem. Soc., 2012, 134, 13316–13322 CrossRef CAS.
- Y. Niihori, Y. Kikuchi, A. Kato, M. Matsuzaki and Y. Negishi, ACS Nano, 2015, 9, 9347–9356 CrossRef CAS.
- P. Pengo, C. Bazzo, M. Boccalon and L. Pasquato, Chem. Commun., 2015, 51, 3204–3207 RSC.
- G. Salassa, A. Sels, F. Mancin and T. Bürgi, ACS Nano, 2017, 11, 12609–12614 CrossRef CAS.
- A. Baksi, M. S. Bootharaju, P. K. Chhotaray, P. Chakraborty, B. Mondal, S. Bhat, R. Gardas and T. Pradeep, J. Phys. Chem. C, 2017, 121, 26483–26492 CrossRef.
- C.-A. J. Lin, T.-Y. Yang, C.-H. Lee, S. H. Huang, R. A. Sperling, M. Zanella, J. K. Li, J.-L. Shen, H.-H. Wang, H.-I. Yeh, W. J. Parak and W. H. Chang, ACS Nano, 2009, 3, 395–401 CrossRef CAS.
- K. Pyo, N. H. Ly, S. Y. Yoon, Y. Shen, S. Y. Choi, S. Y. Lee, S.-W. Joo and D. Lee, Adv. Healthcare Mater., 2017, 6, 1700203 CrossRef.
- K. Pyo, V. D. Thanthirige, S. Y. Yoon, G. Ramakrishna and D. Lee, Nanoscale, 2016, 8, 20008–20016 RSC.
- M. A. H. Muhammed, P. K. Verma, S. K. Pal, R. C. A. Kumar, S. Paul, R. V. Omkumar and T. Pradeep, Chem. Eur. J., 2009, 15, 10110–10120 CrossRef CAS.
- K. Zheng, M. I. Setyawati, T.-P. Lim, D. T. Leong and J. Xie, ACS Nano, 2016, 10, 7934–7942 CrossRef CAS.
- K. Zheng, K. Li, T.-H. Chang, J. Xie and P.-Y. Chen, Adv. Funct. Mater., 2019, 29, 1904603 CrossRef CAS.
- R. Vankayala, C.-L. Kuo, K. Nuthalapati, C.-S. Chiang and K. C. Hwang, Adv. Funct. Mater., 2015, 25, 5934–5945 CrossRef CAS.
- Nonappa, T. Lahtinen, J. S. Haataja, T.-R. Tero, H. Häkkinen and O. Ikkala, Angew. Chem., Int. Ed., 2016, 55, 16035–16038 CrossRef CAS.
- Q. Yao, X. Yuan, Y. Yu, Y. Yu, J. Xie and J. Y. Lee, J. Am. Chem. Soc., 2015, 137, 2128–2136 CrossRef CAS.
- S. Hossain, Y. Imai, Y. Motohashi, Z. Chen, D. Suzuki, T. Suzuki, Y. Kataoka, M. Hirata, T. Ono, W. Kurashige, T. Kawawaki, T. Yamamoto and Y. Negishi, Mater. Horizons, 2020, 7, 796–803 RSC.
- Z. Wu, J. Liu, Y. Li, Z. Cheng, T. Li, H. Zhang, Z. Lu and B. Yang, ACS Nano, 2015, 9, 6315–6323 CrossRef CAS.
- D. J. Cram and J. M. Cram, Science, 1974, 183, 803–809 CrossRef CAS.
- Y. Wu, R. Shi, Y.-L. Wu, J. M. Holcroft, Z. Liu, M. Frasconi, M. R. Wasielewski, H. Li and J. F. Stoddart, J. Am. Chem. Soc., 2015, 137, 4111–4118 CrossRef CAS.
- M. A. Moussawi, N. Leclerc-Laronze, S. Floquet, P. A. Abramov, M. N. Sokolov, S. Cordier, A. Ponchel, E. Monflier, H. Bricout, D. Landy, M. Haouas, J. Marrot and E. Cadot, J. Am. Chem. Soc., 2017, 139, 12793–12803 CrossRef CAS.
- A. Mathew, G. Natarajan, L. Lehtovaara, H. Häkkinen, R. M. Kumar, V. Subramanian, A. Jaleel and T. Pradeep, ACS Nano, 2013, 8, 139–152 CrossRef.
- Y. Zhao, S. Zhuang, L. Liao, C. Wang, N. Xia, Z. Gan, W. Gu, J. Li, H. Deng and Z. Wu, J. Am. Chem. Soc., 2019, 142, 973–977 CrossRef.
- H. Zhu, N. Goswami, Q. Yao, T. Chen, Y. Liu, Q. Xu, D. Chen, J. Lu and J. Xie, J. Mater. Chem. A, 2018, 6, 1102–1108 RSC.
- P. Chakraborty, A. Nag, A. Chakraborty and T. Pradeep, Acc. Chem. Res., 2019, 52, 2–11 CrossRef CAS.
- P. Chakraborty, A. Nag, G. Paramasivam, G. Natarajan and T. Pradeep, ACS Nano, 2018, 12, 2415–2425 CrossRef CAS.
- P. Chakraborty, A. Nag, B. Mondal, E. Khatun, G. Paramasivam and T. Pradeep, J. Phys. Chem. C, 2020, 124, 14891–14900 CrossRef CAS.
- P. Chakraborty, A. Nag, K. S. Sugi, T. Ahuja, B. Varghese and T. Pradeep, ACS Mater. Lett., 2019, 1, 534–540 CrossRef CAS.
- S. Hossain, Y. Niihori, L. V. Nair, B. Kumar, W. Kurashige and Y. Negishi, Acc. Chem. Res., 2018, 51, 3114–3124 CrossRef CAS.
- Y. Yu, X. Chen, Q. Yao, Y. Yu, N. Yan and J. Xie, Chem. Mater., 2013, 25, 946–952 CrossRef CAS.
- Y. Yu, Q. Yao, K. Cheng, X. Yuan, Z. Luo and J. Xie, Part. Part. Syst. Char., 2014, 31, 652–656 CrossRef CAS.
- P. J. G. Goulet and R. B. Lennox, J. Am. Chem. Soc., 2010, 132, 9582–9584 CrossRef CAS.
- Y. Li, O. Zaluzhna, B. Xu, Y. Gao, J. M. Modest and Y. J. Tong, J. Am. Chem. Soc., 2011, 133, 2092–2095 CrossRef CAS.
- Y. Li, O. Zaluzhna and Y. J. Tong, Langmuir, 2011, 27, 7366–7370 CrossRef CAS.
- T. Dainese, S. Antonello, S. Bogialli, W. Fei, A. Venzo and F. Maran, ACS Nano, 2018, 12, 7057–7066 CrossRef CAS.
- M. Suyama, S. Takano, T. Nakamura and T. Tsukuda, J. Am. Chem. Soc., 2019, 141, 14048–14051 CrossRef CAS.
- Y. Zhu, H. Wang, K. Wan, J. Guo, C. He, Y. Yu, L. Zhao, Y. Zhang, J. Lv, L. Shi, R. Jin, X. Zhang, X. Shi and Z. Tang, Angew. Chem., Int. Ed., 2018, 57, 9059–9063 CrossRef CAS.
- J. Yan, H.-F. Su, H. Yang, C. Hu, S. Malola, S.-C. Lin, B. K. Teo, H. Häkkinen and N. Zheng, J. Am. Chem. Soc., 2016, 138, 12751–12754 CrossRef CAS.
- H. Yao, T. Fukui and K. Kimura, J. Phys. Chem. C, 2008, 112, 16281–16285 CrossRef CAS.
- S. Knoppe, O. A. Wong, S. Malola, H. Häkkinen, T. Bürgi, T. Verbiest and C. J. Ackerson, J. Am. Chem. Soc., 2014, 136, 4129–4132 CrossRef CAS.
- M. van der Linden, A. J. van Bunningen, L. Amidani, M. Bransen, H. Elnaggar, P. Glatzel, A. Meijerink and F. M. F. de Groot, ACS Nano, 2018, 12, 12751–12760 CrossRef CAS.
- W. Li, C. Liu, H. Abroshan, Q. Ge, X. Yang, H. Xu and G. Li, J. Phys. Chem. C, 2016, 120, 10261–10267 CrossRef CAS.
- C. Yao, J. Chen, M.-B. Li, L. Liu, J. Yang and Z. Wu, Nano Lett., 2015, 15, 1281–1287 CrossRef CAS.
- X. Liu, J. Yuan, J. Chen, J. Yang and Z. Wu, Part. Part. Syst. Char., 2019, 36, 1900003 CrossRef.
- J.-P. Choi, C. A. Fields-Zinna, R. L. Stiles, R. Balasubramanian, A. D. Douglas, M. C. Crowe and R. W. Murray, J. Phys. Chem. C, 2010, 114, 15890–15896 CrossRef CAS.
- S. Zhuang, D. Chen, L. Liao, Y. Zhao, N. Xia, W. Zhang, C. Wang, J. Yang and Z. Wu, Angew. Chem., Int. Ed., 2020, 59, 3073–3077 CrossRef CAS.
- X. Dou, X. Yuan, Y. Yu, Z. Luo, Q. Yao, D. T. Leong and J. Xie, Nanoscale, 2014, 6, 157–161 RSC.
- Q. Li, S. Wang, K. Kirschbaum, K. J. Lambright, A. Das and R. Jin, Chem. Commun., 2016, 52, 5194–5197 RSC.
- M. Zhu, P. Wang, N. Yan, X. Chai, L. He, Y. Zhao, N. Xia, C. Yao, J. Li, H. Deng, Y. Zhu, Y. Pei and Z. Wu, Angew. Chem., Int. Ed., 2018, 57, 4500–4504 CrossRef CAS.
- K. Zheng, V. Fung, X. Yuan, D.-e. Jiang and J. Xie, J. Am. Chem. Soc., 2019, 141, 18977–18983 CrossRef CAS.
- K. R. Krishnadas, A. Baksi, A. Ghosh, G. Natarajan and T. Pradeep, ACS Nano, 2017, 11, 6015–6023 CrossRef CAS.
- A. Baksi, E. K. Schneider, P. Weis, K. R. Krishnadas, D. Ghosh, H. Hahn, T. Pradeep and M. M. Kappes, J. Phys. Chem. C, 2019, 123, 28477–28485 CrossRef CAS.
- P. Chakraborty and T. Pradeep, NPG Asia Mater., 2019, 11, 48 CrossRef.
- B. Zhang, G. Salassa and T. Bürgi, Chem. Commun., 2016, 52, 9205–9207 RSC.
- R. Kazan, U. Müller and T. Bürgi, Nanoscale, 2019, 11, 2938–2945 RSC.
- Z. Gan, N. Xia and Z. Wu, Acc. Chem. Res., 2018, 51, 2774–2783 CrossRef CAS.
- A. Ghosh, O. F. Mohammed and O. M. Bakr, Acc. Chem. Res., 2018, 51, 3094–3103 CrossRef CAS.
- M. Bootharaju, L. Sinatra and O. M. Bakr, Nanoscale, 2016, 8, 17333–17339 RSC.
- N. Yan, L. Liao, J. Yuan, Y.-j. Lin, L.-h. Weng, J. Yang and Z. Wu, Chem. Mater., 2016, 28, 8240–8247 CrossRef CAS.
- S. Sharma, S. Yamazoe, T. Ono, W. Kurashige, Y. Niihori, K. Nobusada, T. Tsukuda and Y. Negishi, Dalton Trans., 2016, 45, 18064–18068 RSC.
- E. Khatun, P. Chakraborty, B. R. Jacob, G. Paramasivam, M. Bodiuzzaman, W. A. Dar and T. Pradeep, Chem. Mater., 2019, 32, 611–619 CrossRef.
- E. S. Shibu, M. A. H. Muhammed, T. Tsukuda and T. Pradeep, J. Phys. Chem. C, 2008, 112, 12168–12176 CrossRef CAS.
- A. Dass, A. Stevenson, G. R. Dubay, J. B. Tracy and R. W. Murray, J. Am. Chem. Soc., 2008, 130, 5940–5946 CrossRef CAS.
- C. A. Fields-Zinna, J. F. Parker and R. W. Murray, J. Am. Chem. Soc., 2010, 132, 17193–17198 CrossRef CAS.
- L. G. AbdulHalim, N. Kothalawala, L. Sinatra, A. Dass and O. M. Bakr, J. Am. Chem. Soc., 2014, 136, 15865–15868 CrossRef CAS.
- C. Zeng, T. Li, A. Das, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2013, 135, 10011–10013 CrossRef CAS.
- S. K. Eswaramoorthy, N. A. Sakthivel and A. Dass, J. Phys. Chem. C, 2019, 123, 9634–9639 CrossRef CAS.
- M. S. Bootharaju, V. M. Burlakov, T. M. D. Besong, C. P. Joshi, L. G. AbdulHalim, D. Black, R. Whetten, A. Goriely and O. M. Bakr, Chem. Mater., 2015, 27, 4289–4297 CrossRef CAS.
- A. Dass, T. C. Jones, S. Theivendran, L. Sementa and A. Fortunelli, J. Phys. Chem. C, 2017, 121, 14914–14919 CrossRef CAS.
- Y. Chen, C. Liu, Q. Tang, C. Zeng, T. Higaki, A. Das, D.-e. Jiang, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2016, 138, 1482–1485 CrossRef CAS.
- X. Kang and M. Zhu, Chem. Mater., 2019, 31, 9939–9969 CrossRef CAS.
- Y. Niihori, S. Hossain, S. Sharma, B. Kumar, W. Kurashige and Y. Negishi, Chem. Rec., 2017, 17, 473–484 CrossRef CAS.
- A. Chakraborty, A. C. Fernandez, A. Som, B. Mondal, G. Natarajan, G. Paramasivam, T. Lahtinen, H. Häkkinen, Nonappa and T. Pradeep, Angew. Chem., Int. Ed., 2018, 57, 6522–6526 CrossRef CAS.
- X. Liu, G. Saranya, X. Huang, X. Cheng, R. Wang, M. Chen, C. Zhang, T. Li and Y. Zhu, Angew. Chem., Int. Ed., 2020, 59, 13941–13946 CrossRef CAS.
- T. Lahtinen, E. Hulkko, K. Sokolowska, T.-R. Tero, V. Saarnio, J. Lindgren, M. Pettersson, H. Häkkinen and L. Lehtovaara, Nanoscale, 2016, 8, 18665–18674 RSC.
- Nonappa and O. Ikkala, Adv. Funct. Mater., 2017, 28, 1704328 CrossRef.
- B. Yoon, W. D. Luedtke, R. N. Barnett, J. Gao, A. Desireddy, B. E. Conn, T. Bigioni and U. Landman, Nat. Mater., 2014, 13, 807–811 CrossRef CAS.
- Q. Yao, Y. Yu, X. Yuan, Y. Yu, D. Zhao, J. Xie and J. Y. Lee, Angew. Chem., Int. Ed., 2015, 54, 184–189 CrossRef CAS.
- Z. Wu, C. Dong, Y. Li, H. Hao, H. Zhang, Z. Lu and B. Yang, Angew. Chem., Int. Ed., 2013, 52, 9952–9955 CrossRef CAS.
- J. V. Rival, Nonappa and E. S. Shibu, ACS Appl. Mater. Interfaces, 2020, 12, 14569–14577 CrossRef CAS.
- R. J. Macfarlane, B. Lee, M. R. Jones, N. Harris, G. C. Schatz and C. A. Mirkin, Science, 2011, 334, 204–208 CrossRef CAS.
- S. Y. Park, A. K. R. Lytton-Jean, B. Lee, S. Weigand, G. C. Schatz and C. A. Mirkin, Nature, 2008, 451, 553–556 CrossRef CAS.
- D. Nykypanchuk, M. M. Maye, D. van der Lelie and O. Gang, Nature, 2008, 451, 549–552 CrossRef CAS.
- A. Som, I. Chakraborty, T. A. Maark, S. Bhat and T. Pradeep, Adv. Mater., 2016, 28, 2827–2833 CrossRef CAS.
- L.-Y. Chen, C.-W. Wang, Z. Yuan and H.-T. Chang, Anal. Chem., 2015, 87, 216–229 CrossRef CAS.
- A. Mathew and T. Pradeep, Part. Part. Syst. Char., 2014, 31, 1017–1053 CrossRef CAS.
- Z. Wu, M. Wang, J. Yang, X. Zheng, W. Cai, G. Meng, H. Qian, H. Wang and R. Jin, Small, 2012, 8, 2028–2035 CrossRef CAS.
- L. Shang, L. Yang, F. Stockmar, R. Popescu, V. Trouillet, M. Bruns, D. Gerthsen and G. U. Nienhaus, Nanoscale, 2012, 4, 4155–4160 RSC.
- M. S. Bootharaju and T. Pradeep, Langmuir, 2011, 27, 8134–8143 CrossRef CAS.
- G. Zhang, Y. Li, J. Xu, C. Zhang, S. Shuang, C. Dong and M. M. F. Choi, Sens. Actuators B Chem., 2013, 183, 583–588 CrossRef CAS.
- Y.-T. Su, G.-Y. Lan, W.-Y. Chen and H.-T. Chang, Anal. Chem., 2010, 82, 8566–8572 CrossRef CAS.
- E. S. Shibu and T. Pradeep, Chem. Mater., 2011, 23, 989–999 CrossRef CAS.
- H. Liu, X. Zhang, X. Wu, L. Jiang, C. Burda and J.-J. Zhu, Chem. Commun., 2011, 47, 4237–4239 RSC.
- N. Goswami, A. Giri, M. S. Bootharaju, P. L. Xavier, T. Pradeep and S. K. Pal, Anal. Chem., 2011, 83, 9676–9680 CrossRef CAS.
- I. Chakraborty, T. Udayabhaskararao and T. Pradeep, J. Hazard. Mater., 2012, 211–212, 396–403 CrossRef CAS.
- A. George, E. S. Shibu, S. M. Maliyekkal, M. S. Bootharaju and T. Pradeep, ACS Appl. Mater. Interfaces, 2012, 4, 639–644 CrossRef CAS.
- Y. Guo, Z. Wang, H. Shao and X. Jiang, Analyst, 2012, 137, 301–304 RSC.
- M. A. Habeeb Muhammed and T. Pradeep, Chem. Phys. Lett., 2007, 449, 186–190 CrossRef CAS.
- J. Shen, Z. Wang, D. Sun, C. Xia, S. Yuan, P. Sun and X. Xin, ACS Appl. Mater. Interfaces, 2018, 10, 3955–3963 CrossRef CAS.
- S. Roy, G. Palui and A. Banerjee, Nanoscale, 2012, 4, 2734–2740 RSC.
- T. Zhou, M. Rong, Z. Cai, C. J. Yang and X. Chen, Nanoscale, 2012, 4, 4103–4106 RSC.
- C.-C. Huang, Z. Yang, K.-H. Lee and H.-T. Chang, Angew. Chem., Int. Ed., 2007, 46, 6824–6828 CrossRef CAS.
- X. Yuan, Y. Tay, X. Dou, Z. Luo, D. T. Leong and J. Xie, Anal. Chem., 2012, 85, 1913–1919 CrossRef.
- G. Guan, S.-Y. Zhang, Y. Cai, S. Liu, M. S. Bharathi, M. Low, Y. Yu, J. Xie, Y. Zheng, Y.-W. Zhang and M. Han, Chem. Commun., 2014, 50, 5703–5705 RSC.
- S. Chen, W. Du, C. Qin, D. Liu, L. Tang, Y. Liu, S. Wang and M. Zhu, Angew. Chem., Int. Ed., 2020, 59, 7542–7547 CrossRef CAS.
- R.-W. Huang, Y.-S. Wei, X.-Y. Dong, X.-H. Wu, C.-X. Du, S.-Q. Zang and T. C. W. Mak, Nat. Chem., 2017, 9, 689 CrossRef CAS.
- X.-H. Wu, P. Luo, Z. Wei, Y.-Y. Li, R.-W. Huang, X.-Y. Dong, K. Li, S.-Q. Zang and B. Z. Tang, Adv. Sci., 2019, 6, 1801304 CrossRef.
- M. Cao, R. Pang, Q.-Y. Wang, Z. Han, Z.-Y. Wang, X.-Y. Dong, S.-F. Li, S.-Q. Zang and T. C. W. Mak, J. Am. Chem. Soc., 2019, 141, 14505–14509 CrossRef CAS.
- C.-T. Chen, W.-J. Chen, C.-Z. Liu, L.-Y. Chang and Y.-C. Chen, Chem. Commun., 2009, 7515–7517 RSC.
- A. D. Kurdekar, L. A. Avinash Chunduri, C. S. Manohar, M. K. Haleyurgirisetty, I. K. Hewlett and K. Venkataramaniah, Sci. Adv., 2018, 4, eaar6280 CrossRef CAS.
- Nonappa, Beilstein J. Nanotechnol., 2020, 11, 533–546 CrossRef CAS.
- X. Jiang, B. Du, Y. Huang and J. Zheng, Nano Today, 2018, 21, 106–125 CrossRef CAS.
- N. Goswami, Z. Luo, X. Yuan, D. T. Leong and J. Xie, Mater. Horizons, 2017, 4, 817–831 RSC.
- K. Zheng, M. I. Setyawati, D. T. Leong and J. Xie, ACS Nano, 2017, 11, 6904–6910 CrossRef CAS.
- C. Zhang, C. Li, Y. Liu, J. Zhang, C. Bao, S. Liang, Q. Wang, Y. Yang, H. Fu, K. Wang and D. Cui, Adv. Funct. Mater., 2015, 25, 1314–1325 CrossRef CAS.
- D. Chen, Z. Luo, N. Li, J. Y. Lee, J. Xie and J. Lu, Adv. Funct. Mater., 2013, 23, 4324–4331 CrossRef CAS.
- Y. Li, J. Zhen, Q. Tian, C. Shen, L. Zhang, K. Yang and L. Shang, J. Colloid Interface Sci., 2020, 569, 235–243 CrossRef CAS.
- A. Chandrasekar and T. Pradeep, J. Phys. Chem. C, 2012, 116, 14057–14061 CrossRef CAS.
- Y. Yun, H. Sheng, K. Bao, L. Xu, Y. Zhang, D. Astruc and M. Zhu, J. Am. Chem. Soc., 2020, 142, 4126–4130 CrossRef CAS.
- J. Chai, S. Yang, Y. Lv, H. Chong, H. Yu and M. Zhu, Angew. Chem., Int. Ed., 2019, 58, 15671–15674 CrossRef CAS.
- J. Zhao, Q. Li, S. Zhuang, Y. Song, D. J. Morris, M. Zhou, Z. Wu, P. Zhang and R. Jin, J. Phys. Chem. Lett., 2018, 9, 7173–7179 CrossRef CAS.
- M. Urushizaki, H. Kitazawa, S. Takano, R. Takahata, S. Yamazoe and T. Tsukuda, J. Phys. Chem. C, 2015, 119, 27483–27488 CrossRef CAS.
- T. Yoskamtorn, S. Yamazoe, R. Takahata, J.-i. Nishigaki, A. Thivasasith, J. Limtrakul and T. Tsukuda, ACS Catal., 2014, 4, 3696–3700 CrossRef CAS.
- Z. Li, C. Liu, H. Abroshan, D. R. Kauffman and G. Li, ACS Catal., 2017, 7, 3368–3374 CrossRef CAS.
- M. Haruta, T. Kobayashi, H. Sano and N. Yamada, Chem. Lett., 1987, 16, 405–408 CrossRef.
- A. Haruta, Chem. Rec., 2003, 3, 75–87 CrossRef.
- S. Bhat, R. P. Narayanan, A. Baksi, P. Chakraborty, G. Paramasivam, R. R. J. Methikkalam, A. Nag, G. Natarajan and T. Pradeep, J. Phys. Chem. C, 2018, 122, 19455–19462 CrossRef CAS.
- Z. Wu, D.-e. Jiang, A. K. P. Mann, D. R. Mullins, Z.-A. Qiao, L. F. Allard, C. Zeng, R. Jin and S. H. Overbury, J. Am. Chem. Soc., 2014, 136, 6111–6122 CrossRef CAS.
- Y. Gao, N. Shao, Y. Pei, Z. Chen and X. C. Zeng, ACS Nano, 2011, 5, 7818–7829 CrossRef CAS.
- X. Nie, H. Qian, Q. Ge, H. Xu and R. Jin, ACS Nano, 2012, 6, 6014–6022 CrossRef CAS.
- Y. Zhu, H. Qian and R. Jin, Chem.–Eur. J., 2010, 16, 11455–11462 CrossRef CAS.
- J. Liu, K. S. Krishna, Y. B. Losovyj, S. Chattopadhyay, N. Lozova, J. T. Miller, J. J. Spivey and C. S. S. R. Kumar, Chem.–Eur. J., 2013, 19, 10201–10208 CrossRef CAS.
- S. Lin and Y. Pei, J. Phys. Chem. C, 2014, 118, 20346–20356 CrossRef CAS.
- J. Chai, H. Chong, S. Wang, S. Yang, M. Wu and M. Zhu, RSC Adv., 2016, 6, 111399–111405 RSC.
- R. Ouyang and D.-e. Jiang, ACS Catal., 2015, 5, 6624–6629 CrossRef CAS.
- G. Li, H. Abroshan, Y. Chen, R. Jin and H. J. Kim, J. Am. Chem. Soc., 2015, 137, 14295–14304 CrossRef CAS.
- R. R. Nasaruddin, Q. Yao, T. Chen, M. J. Hülsey, N. Yan and J. Xie, Nanoscale, 2018, 10, 23113–23121 RSC.
- R. R. Nasaruddin, T. Chen, N. Yan and J. Xie, Coord. Chem. Rev., 2018, 368, 60–79 CrossRef CAS.
- Y. Zhang, P. Song, T. Chen, X. Liu, T. Chen, Z. Wu, Y. Wang, J. Xie and W. Xu, Proc. Natl. Acad. Sci., 2018, 115, 10588–10593 CrossRef CAS.
- G. Li, H. Abroshan, C. Liu, S. Zhuo, Z. Li, Y. Xie, H. J. Kim, N. L. Rosi and R. Jin, ACS Nano, 2016, 10, 7998–8005 CrossRef CAS.
- H. Abroshan, G. Li, J. Lin, H. J. Kim and R. Jin, J. Catal., 2016, 337, 72–79 CrossRef CAS.
- S. Malola and H. Häkkinen, J. Am. Chem. Soc., 2019, 141, 6006–6012 CrossRef CAS.
- B. Zhang and T. Bürgi, J. Phys. Chem. C, 2016, 120, 4660–4666 CrossRef CAS.
- K. Salorinne, S. Malola, O. A. Wong, C. D. Rithner, X. Chen, C. J. Ackerson and H. Häkkinen, Nat. Commun., 2016, 7, 10401 CrossRef CAS.
- G. Schmid, R. Pfeil, R. Boese, F. Bandermann, S. Meyer, G. H. M. Calis and J. W. A. van der Velden, Chem. Ber., 1981, 114, 3634–3642 CrossRef CAS.
- L. G. AbdulHalim, M. S. Bootharaju, Q. Tang, S. del Gobbo, R. G. AbdulHalim, M. Eddaoudi, D.-e. Jiang and O. M. Bakr, J. Am. Chem. Soc., 2015, 137, 11970–11975 CrossRef CAS.
- S.-S. Zhang, L. Feng, R. D. Senanayake, C. M. Aikens, X.-P. Wang, Q.-Q. Zhao, C.-H. Tung and D. Sun, Chem. Sci., 2018, 9, 1251–1258 RSC.
- S. Takano, S. Hasegawa, M. Suyama and T. Tsukuda, Acc. Chem. Res., 2018, 51, 3074–3083 CrossRef CAS.
- S. Takano, S. Ito and T. Tsukuda, J. Am. Chem. Soc., 2019, 141, 15994–16002 CrossRef CAS.
- S. Takano, H. Hirai, S. Muramatsu and T. Tsukuda, J. Am. Chem. Soc., 2018, 140, 12314–12317 CrossRef CAS.
- Q. Tang, Y. Lee, D.-Y. Li, W. Choi, C. W. Liu, D. Lee and D.-e. Jiang, J. Am. Chem. Soc., 2017, 139, 9728–9736 CrossRef CAS.
- Z. Lei, X.-K. Wan, S.-F. Yuan, Z.-J. Guan and Q.-M. Wang, Acc. Chem. Res., 2018, 51, 2465–2474 CrossRef CAS.
- X.-S. Han, X. Luan, H.-F. Su, J.-J. Li, S.-F. Yuan, Z. Lei, Y. Pei and Q.-M. Wang, Angew. Chem., Int. Ed., 2020, 59, 2309–2312 CrossRef CAS.
- Z.-J. Guan, F. Hu, J.-J. Li, Z.-R. Wen, Y.-M. Lin and Q.-M. Wang, J. Am. Chem. Soc., 2020, 142, 2995–3001 CrossRef CAS.
- Y. Wang, X.-K. Wan, L. Ren, H. Su, G. Li, S. Malola, S. Lin, Z. Tang, H. Häkkinen, B. K. Teo, Q.-M. Wang and N. Zheng, J. Am. Chem. Soc., 2016, 138, 3278–3281 CrossRef CAS.
- M. R. Narouz, K. M. Osten, P. J. Unsworth, R. W. Y. Man, K. Salorinne, S. Takano, R. Tomihara, S. Kaappa, S. Malola, C.-T. Dinh, J. D. Padmos, K. Ayoo, P. J. Garrett, M. Nambo, J. H. Horton, E. H. Sargent, H. Häkkinen, T. Tsukuda and C. M. Crudden, Nat. Chem., 2019, 11, 419–425 CrossRef CAS.
- M. R. Narouz, S. Takano, P. A. Lummis, T. I. Levchenko, A. Nazemi, S. Kaappa, S. Malola, G. Yousefalizadeh, L. A. Calhoun, K. G. Stamplecoskie, H. Häkkinen, T. Tsukuda and C. M. Crudden, J. Am. Chem. Soc., 2019, 141, 14997–15002 CrossRef CAS.
- H. Shen, G. Deng, S. Kaappa, T. Tan, Y.-Z. Han, S. Malola, S.-C. Lin, B. K. Teo, H. Häkkinen and N. Zheng, Angew. Chem., Int. Ed., 2019, 58, 17731–17735 CrossRef CAS.
- Z. Wang, H.-F. Su, C.-H. Tung, D. Sun and L.-S. Zheng, Nat. Commun., 2018, 9, 4407 CrossRef.
- M. J. Alhilaly, R.-W. Huang, R. Naphade, B. Alamer, M. N. Hedhili, A.-H. Emwas, P. Maity, J. Yin, A. Shkurenko, O. F. Mohammed, M. Eddaoudi and O. M. Bakr, J. Am. Chem. Soc., 2019, 141, 9585–9592 CrossRef CAS.
- Z. Wang, H.-F. Su, M. Kurmoo, C.-H. Tung, D. Sun and L.-S. Zheng, Nat. Commun., 2018, 9, 2094 CrossRef.
- Z.-Y. Wang, M.-Q. Wang, Y.-L. Li, P. Luo, T.-T. Jia, R.-W. Huang, S.-Q. Zang and T. C. W. Mak, J. Am. Chem. Soc., 2018, 140, 1069–1076 CrossRef CAS.
- M. A. Boles, M. Engel and D. V. Talapin, Chem. Rev., 2016, 116, 11220–11289 CrossRef CAS.
- A. Hernandez-Garcia, D. J. Kraft, F. J. JanssenAnne, H. H. BomansPaul, A. J. M. SommerdijkNico, M. E. Thies-WeesieDominique, M. E. Favretto, R. Brock, F. A. de Wolf, M. W. T. Werten, P. van der Schoot, M. C. Stuart and R. de Vries, Nat. Nanotechnol., 2014, 9, 698–702 CrossRef CAS.
- A. Walther and A. H. E. Müller, Chem. Rev., 2013, 113, 5194–5261 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |