
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltrasensitive single-cell proteomics workflow identifies >1000 protein groups per mammalian cell†
Yongzheng
Cong
a,
Khatereh
Motamedchaboki
b,
Santosh A.
Misal
 a,
Yiran
Liang
a,
Amanda J.
Guise
c,
Thy
Truong
a,
Romain
Huguet
b,
Edward D.
Plowey
c,
Ying
Zhu
d,
Daniel
Lopez-Ferrer
b and
Ryan T.
Kelly
*ad
a,
Yiran
Liang
a,
Amanda J.
Guise
c,
Thy
Truong
a,
Romain
Huguet
b,
Edward D.
Plowey
c,
Ying
Zhu
d,
Daniel
Lopez-Ferrer
b and
Ryan T.
Kelly
*ad
aDepartment of Chemistry and Biochemistry, Brigham Young University, Provo, UT 84602, USA. E-mail: ryan@chem.byu.edu
bThermo Fisher Scientific, San Jose, CA 95134, USA
cBiogen, Inc., Cambridge, MA 02142, USA
dEnvironmental Molecular Sciences Laboratory, Pacific Northwest National Laboratory, Richland, WA 99354, USA
First published on 17th November 2020
Abstract
We report on the combination of nanodroplet sample preparation, ultra-low-flow nanoLC, high-field asymmetric ion mobility spectrometry (FAIMS), and the latest-generation Orbitrap Eclipse Tribrid mass spectrometer for greatly improved single-cell proteome profiling. FAIMS effectively filtered out singly charged ions for more effective MS analysis of multiply charged peptides, resulting in an average of 1056 protein groups identified from single HeLa cells without MS1-level feature matching. This is 2.3 times more identifications than without FAIMS and a far greater level of proteome coverage for single mammalian cells than has been previously reported for a label-free study. Differential analysis of single microdissected motor neurons and interneurons from human spinal tissue indicated a similar level of proteome coverage, and the two subpopulations of cells were readily differentiated based on single-cell label-free quantification.
Introduction
Mass spectrometry (MS)-based proteome profiling provides insights into biological function and dysfunction that are unavailable through genomic or transcriptomic measurements.1 Extending proteomic analysis to single cells and other low-input samples sheds additional light on the roles of various cell types contributing to normal and disease processes and can yield spatial information for tissue mapping and characterization of the microenvironment.2–4 Given the absence of amplification techniques for proteins, every aspect of the analytical method must be carefully optimized to bring more protein species above detection limits and to provide a more comprehensive view of protein expression, ideally extending to thousands of proteins per cell. These optimization efforts span the entire proteomics workflow, from cell isolation and sample preparation to MS measurement and data processing, and it is the combination of these advances that has made single-cell proteomics possible.5 For example, efforts to miniaturize sample preparation to nanolitre volumes using, e.g., nanoPOTS,6 the oil-air-droplet (OAD) chip7 or the integrated proteome analysis device (iPAD),8 have effectively reduced adsorptive losses and increased sample concentrations for more efficient protein digestion for single cells and other trace samples. When coupled with fluorescence activate cell sorting and a custom autosampler, nanoPOTS has been used to analyse >150 single cells with excellent run-to-run reproducibility.9 Ultrasensitive separations have been realized by reducing total flow rates to the low-nanolitre-per-minute range using capillary electrophoresis10,11 or narrow-bore liquid chromatography (LC) with either open tubular12,13 or packed columns,14,15 providing reduced solvent contamination and improved ionization efficiency at the electrospray source.Using a combination of nanoPOTS sample preparation, nanoLC separations operated at 20 nL min−1 and the Orbitrap Eclipse Tribrid mass spectrometer, we recently identified an average of 362 and 874 protein groups from single HeLa cells15 without and with the Match Between Runs (MBR) algorithm of MaxQuant, respectively, which was the highest level of coverage reported for a label-free analysis of single mammalian cells. While this coverage is sufficient to differentiate between distinct cell types16–19 and illuminate processes involving high-abundance proteins,20 current methods are blind to expression patterns of lower abundance proteins that fall below detection limits. Tandem mass tag (TMT)-based approaches that incorporate a boosting channel21,22 can increase single-cell proteome coverage, but the quantitative accuracy is currently compromised by batch effects, ratio compression and the ‘carrier proteome effect’.23 Additional sensitivity gains achieved by further optimizing the analytical workflow are expected to improve both label-free and isobaric labelling methods.
During LC-MS analysis, tryptic peptides may be present as singly charged or multiply charged ions,24 while most contaminating species and solvent clusters are singly charged. To increase MS/MS sequencing efficiency, only multiply charged species are typically selected for fragmentation, yet the presence of these +1 species in the MS1 scan increase spectral complexity and singly charged ions may still be co-isolated for fragmentation along with selected peptides, interfering with identification. More importantly, in the case of ion trapping instruments such as the orbitrap, singly charged ions may occupy a significant portion of the trap capacity, effectively reducing sensitivity for multiply charged species and limiting those selected for fragmentation. The proportion of the ion population comprising singly charged species increases as sample size decreases, while solvent contributions remain relatively constant, so the presence of +1 ions is likely more detrimental for trace samples.
High field asymmetric ion mobility spectrometry (FAIMS)25 is a gas-phase separation technique in which an asymmetric electric field is used to disperse ions and selectively filter ion populations by varying the compensation voltage (CV). Importantly, FAIMS can selectively remove +1 ions while broadly transmitting multiply charged peptides,26,27 which should be especially beneficial for single-cell proteomic analysis. However, some signal attenuation of selected ions occurs due to imperfect transmission through FAIMS devices, so it is necessary to determine whether the benefits of FAIMS overcome any detrimental decrease in signal. Here we have evaluated the use of FAIMS for single cell proteome profiling, and incorporated the use of FAIMS with nanodroplet sample preparation, ultra-low-flow nanoLC, and the latest-generation Orbitrap Eclipse Tribrid mass spectrometer to greatly improve single-cell proteome coverage. The single cells were processed using a nanoPOTS chip, and peptides were separated using a 20 μm-i.d. home-packed nanoLC column, ionized at a chemically etched nanospray emitter and then fractionated using the FAIMS Pro interface for selective removal of singly charged species and transmission of multiply charged peptides to the Thermo Scientific Orbitrap Eclipse Tribrid MS. We identified an average of 1056 protein groups from 3912 peptides from single HeLa cells when using FAIMS and Proteome Discoverer Software 2.4 with an FDR cutoff of <0.01 at both the protein and peptide levels. This level of proteome coverage represents a respective increase of 2.3 and 2.0-fold at the protein and peptide level compared to without FAIMS, and is a far greater level of proteome coverage for single mammalian cells in a label-free study than has been previously reported.
Results and discussion
Single-cell proteome workflow
The workflow for single cell proteome profiling is shown in Fig. 1. Single cells were isolated by capillary-based micromanipulation or laser capture microdissection (LCM) and processed in ∼200 nL droplets in a nanoPOTS chip, in which the contact surface was dramatically reduced.6 Peptides were separated using a 20 μm-i.d. home-packed nanoLC column,15 ionized at a chemically etched nanospray emitter28 and then fractionated using the FAIMS Pro interface (Thermo, Waltham, MA, USA) for selective removal of singly charged species and transmission of multiply charged peptides to the Thermo Scientific Orbitrap Tribrid Eclipse mass spectrometer. Raw data were processed using Proteome Discoverer Software 2.4 (Thermo) or MaxQuant version 1.6.7.0.29 The optimized sample preparation reduced peptide losses, and when combined with ultrasensitive separation, ionization, fractionation and MS, provided dramatically increased proteome coverage.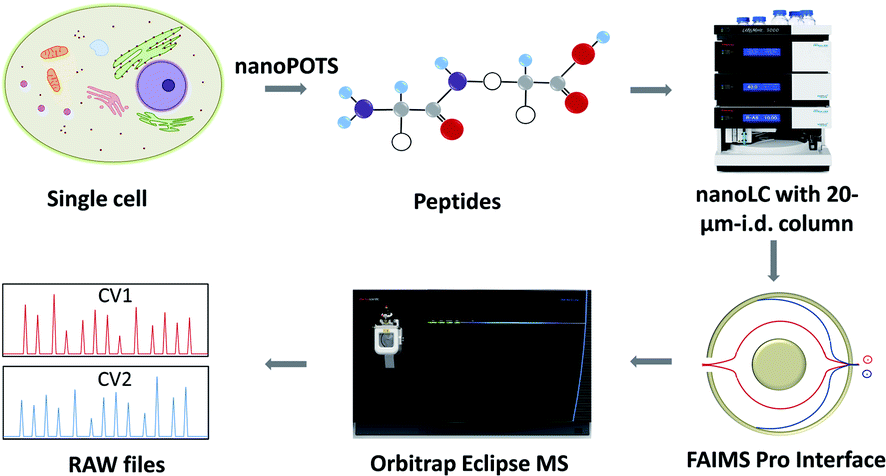 | ||
| Fig. 1 Single-cell proteomics workflow. Proteins from a single cell are extracted and digested, the resulting tryptic peptides are separated using a narrow-bore nanoLC column and ionized at an etched electrospray emitter. Singly charged ions are filtered using the FAIMS Pro interface and transmitted ions are detected using the Orbitrap Eclipse Tribrid MS. | ||
MS acquisition and FAIMS settings optimization
MS acquisition and FAIMS settings were evaluated using 0.5 ng aliquots of commercial HeLa protein digest standard, an amount equivalent to 2–3 cells. Proteome coverage increased by ∼30% when using the ion trap (IT) instead of the orbitrap (OT) for MS2 (Fig. 2A and Table S1†), which is attributed to the higher sensitivity of the ion trap. Proteome coverage was ∼12% higher when using HCD fragmentation rather than CID (Fig. 2A). We also evaluated whether scanning between 2 or 3 compensation voltage (CV) values would provide greater coverage. Under the current conditions of a 120 min LC gradient, a 2 CV method (−55 V and −70 V) provided ∼10% greater coverage than a 3 CV method (−55 V, −70 V and −85 V) (Fig. 2A). The 2 CV method with HCD fragmentation and ion trap detection was thus selected for single-cell studies. We note these settings will likely change with different sample loadings and LC gradients, etc.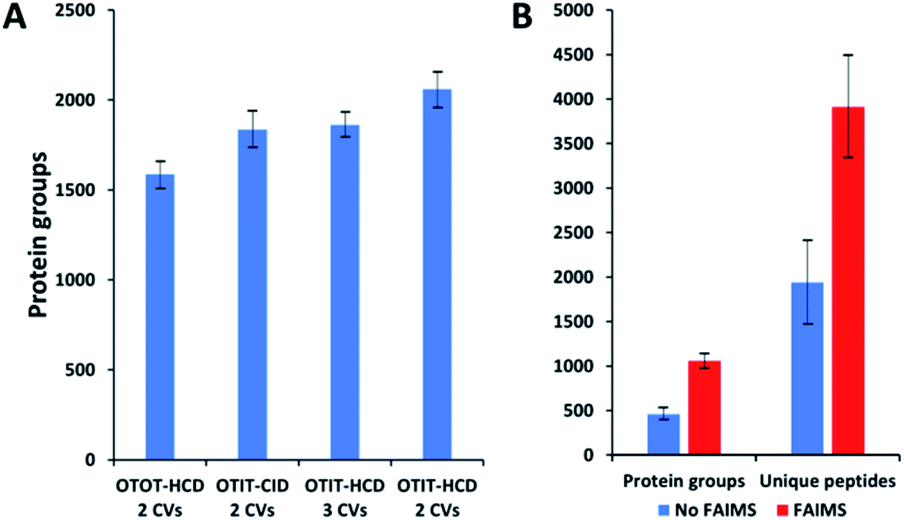 | ||
| Fig. 2 Proteome coverage for HeLa digest and single HeLa cells. (A) FAIMS method optimization using 0.5 ng aliquots of HeLa protein digest. Protein groups identified with different detection and fragmentation methods and using two or three FAIMS CVs. (B) Protein groups and unique peptides identified from single HeLa cells with and without FAIMS. Error bars indicate standard deviations based on 3 replicate measurements. | ||
Single-cell proteome profiling
Single HeLa cells were aspirated with 6 nL of supernatant, deposited into a nanoPOTS chip for sample preparation and analysed as described above. Blank samples containing an equivalent volume of cell-free supernatant were analysed in the same fashion to serve as a negative control. Mass spectra such as those shown in Fig. 3 were compared for single HeLa cells with and without the FAIMS Pro interface. Without FAIMS, spectra were primarily composed of +1 ions, whereas FAIMS effectively filtered out most singly charged species. Across all spectra, the proportion of +1 species decreased from 59% to 30% when FAIMS was employed (Fig. S1A†), and the relative abundance of those ions decreased from 75% to just 10% (Fig. S1B†). Interestingly, while the total ion abundance of ≥+4 species increased from 2% to 27%, these produced relatively few additional identifications. This is likely due to the decreased efficiency of MS/MS for such species. For example, the ∼46![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 detected +5 to +7 species only yielded 9 peptide spectrum matches such that these highly charged species may warrant exclusion in future analyses. In evaluating proteome coverage for single HeLa cells, we identified on average 1056 protein groups from 3912 peptides when using FAIMS and Proteome Discoverer Software 2.4 with an FDR cutoff of <0.01 at both the protein and peptide levels, which represents a respective increase of 2.3 and 2.0-fold compared to without FAIMS (Fig. 2B and Table S2†). We also evaluated proteome coverage using MaxQuant, which yielded an average of 683 and 1475 protein groups without and with MBR when including a matching library comprising 100 HeLa cells. While MaxQuant yielded fewer identifications without MBR relative to Proteome Discoverer, the level of proteome coverage was still 1.9 times greater than the 362 protein groups identified previously14 under identical analysis and database search conditions but without FAIMS.
000 detected +5 to +7 species only yielded 9 peptide spectrum matches such that these highly charged species may warrant exclusion in future analyses. In evaluating proteome coverage for single HeLa cells, we identified on average 1056 protein groups from 3912 peptides when using FAIMS and Proteome Discoverer Software 2.4 with an FDR cutoff of <0.01 at both the protein and peptide levels, which represents a respective increase of 2.3 and 2.0-fold compared to without FAIMS (Fig. 2B and Table S2†). We also evaluated proteome coverage using MaxQuant, which yielded an average of 683 and 1475 protein groups without and with MBR when including a matching library comprising 100 HeLa cells. While MaxQuant yielded fewer identifications without MBR relative to Proteome Discoverer, the level of proteome coverage was still 1.9 times greater than the 362 protein groups identified previously14 under identical analysis and database search conditions but without FAIMS.
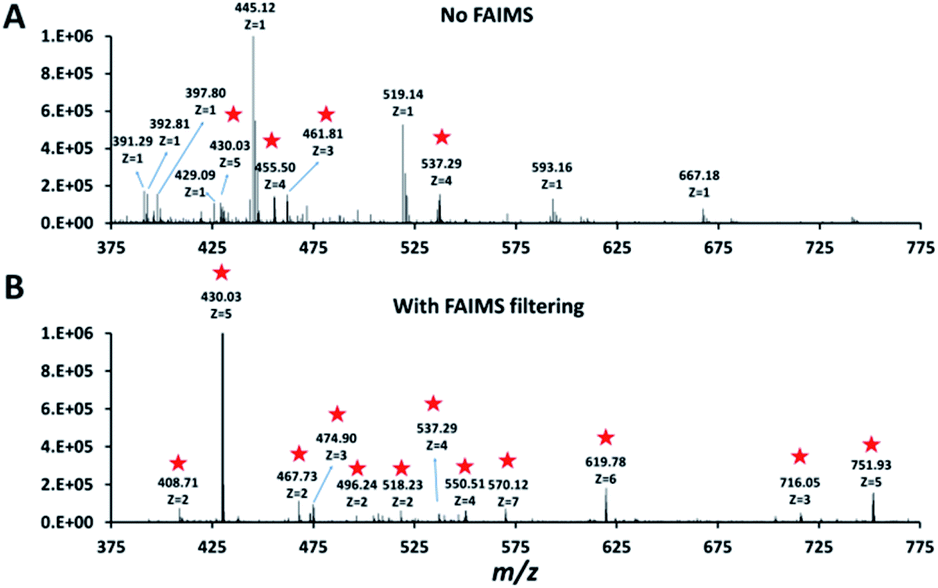 | ||
| Fig. 3 Representative mass spectra obtained without (A) and with (B) FAIMS filtering (CV −55 V). Peaks corresponding to multiply charged ions in both spectra are starred. | ||
Application to single neurons
To further explore platform performance on human post-mortem tissues and evaluate the ability of this platform to differentiate between closely related neuronal cell types, we applied our workflow to the analysis of single motor neurons (MNs) and interneurons (INs) excised by LCM from 12 μm-thick human spinal cord sections. Single-cell proteomic technologies represent an important platform for probing cell-type-specific perturbations, particularly in the context of human neurological diseases such as amyotrophic lateral sclerosis (ALS) and spinal muscular atrophy (SMA) characterized by selective vulnerability of motor neurons.30,31 An average of 1012 and 1085 protein groups were identified from single MNs (n = 3) and INs (n = 3), respectively, when applying a FDR cutoff of <0.01 and without MS1-level feature matching (Fig. 4A). The identified protein groups from the two cell types had an overlap of 77% (Fig. 4B) and were readily differentiated by principal component analysis (Fig. 4C). One motor neuron was discriminated from the remaining samples, which may reflect further subdivision into MN classes (e.g., alpha and gamma MNs). Future studies evaluating increased numbers of single cells and/or employing additional methods to selectively capture MN populations will shed additional light on the heterogeneity present among MN and IN populations. Among the 1118 quantifiable protein groups (present with ≥2 unique peptides and in ≥50% of samples), 39 were significantly differentially abundant in MNs relative to INs (p < 0.05, |fold difference| ≥ 2) (Fig. 4D and ESI Dataset II†).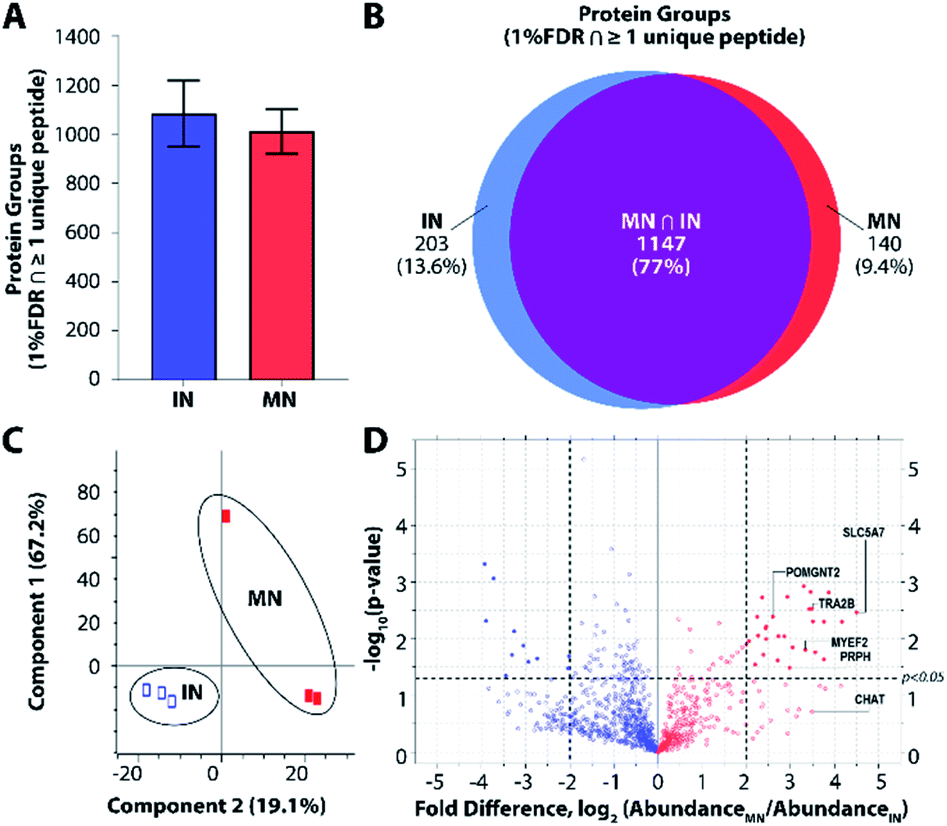 | ||
| Fig. 4 Single-cell proteomic interrogation of human spinal motor neurons and interneurons. (A) Protein groups identified from single motor neurons (MNs) and interneurons (INs). (B) Venn diagram indicating overlap of identified protein groups. (C) Principle component analysis showing differentiation of the two neuronal subtypes based on 1118 quantified features. (D) Volcano plot indicating significant differences in protein expression for quantifiable protein groups (p < 0.05, |fold difference| ≥ 2). | ||
Gene ontology (GO) enrichment analyses on the subset of enriched-in-MN proteins revealed over-representation of proteins associated with RNA processing and alternative splicing, RNA metabolism, and post-transcriptional regulation of gene expression (Fig. S3B†). Consistent with established functions of MNs and mechanisms underlying MN-mediated stimulation of muscle fibres, we identified significant MN-enrichment of the high-affinity choline transporter protein SLC5A7 that mediates choline uptake in cholinergic neurons for rapid conversion into acetylcholine by choline o-acetyltransferase (CHAT) (Fig. S3C†). CHAT itself was approximately 10 times more abundant in MNs relative to INs (ESI Dataset II†). Acetylcholine released by presynaptic MNs at the neuromuscular junction binds nicotinic receptors expressed on the post-synaptic membranes of muscle fibers.32 Furthermore, a number of proteins implicated in motor function and neuromuscular disease were represented among the subset of differentially abundant protein groups. These include RNA splicing machinery components such as TRA2B, the splicing factor that targets the survival of motor neuron (SMN) protein33,34 whose corresponding gene mutation causes SMA; MYEF2, a downstream splicing target of SMN;35 as well as multiple interactors of FUS and TARDBP (TDP43), whose corresponding gene mutations are causative for ALS (Fig. S3C†). FUS and TARDBP were detected in larger pools of MNs (5 cell pools) and in single ventral horns (not shown), but are likely below the limit of detection in single MNs. In addition to FUS and TARDBP, the intermediate filament protein peripherin (PRPH), important for axonal transport, is significantly more abundant in MNs relative to INs. Interestingly, mutations in PRPH are also associated with ALS,36 while overexpression of wild-type PRPH has been shown to result in selective motor axon degeneration in mice,37 suggesting regulation of PRPH expression is critical for motor-neuron integrity. Increased PRPH abundance in spinal motor neurons relative to interneurons is consistent with previous observations that while PRPH is predominately expressed in the peripheral nervous system, it is also expressed in subsets of central nervous system neurons containing peripheral projections (e.g., MNs), and is reported to be highly expressed in lumbar spinal MNs at the mRNA level.38 Together, these data demonstrate the ability of unbiased proteomic profiling to (1) differentiate between neuronal subpopulations at the single-cell level and (2) identify differentially-expressed proteins and pathways relevant to cell-type-specific functions of MNs in health and disease.
Conclusions
The combination of nanoPOTS sample preparation, ultra-narrow-bore LC separation, Orbitrap Eclipse Tribrid mass spectrometer and the FAIMS Pro interface provides an unprecedented label-free proteome coverage of >1000 protein groups per mammalian cell using MS/MS identification alone. While the present study focuses on label-free proteome profiling, the sensitivity gains will likely also benefit isobaric labelling workflows such as SCoPE-MS. Additional gains will likely be realized by further miniaturizing sample preparation, pushing highly efficient nanoLC to lower flow rates, and optimizing FAIMS and MS acquisition settings. This platform promises to provide insights into cellular heterogeneity and enable the characterization of tissue microenvironments through in-depth mapping of protein expression in tissues with single-cell spatial resolution.Experimental section
Material and sample preparation
Dithiothreitol (DTT), iodoacetamide (IAA), Pierce formic acid (LC-MS grade), and Pierce HeLa protein digest standard were purchased from Thermo Fisher Scientific. CHROMASOLV™ LC-MS water and acetonitrile were products of Honeywell (Charlotte, NC, USA), MS-grade trypsin and Lys-C were from Promega (Madison, WI, USA). All other chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise noted.Sample collection
HeLa cells (ATCC, Manassas, VA, USA) were cultured and isolated as described previously.14 Fresh frozen human spinal tissue (ProteoGenex, Los Angeles, CA, USA) was cryosectioned to a thickness of 12 μm and deposited onto PEN-coated microscope slides (Zeiss, Oberkochen, Germany) and fixed with 70% ethanol for 15 min. The deidentified tissue was exempt from human subjects regulations as described in US Federal Regulation 45 CFR 46. The tissue sections were stained with hematoxylin and eosin and imaged at 40× resolution using a Zeiss PALM MicroBeam system. Individual motor neurons and interneurons were selected from the ventral horn region and intermediate zone, respectively, of the spinal tissue and excised by laser capture microdissection (LCM) using the PALM MicroBeam system. The excised single neurons were collected into the nanowells of the nanoPOTS chip18 (Fig. S2†) and then prepared for analysis as described below.NanoPOTS sample processing
Samples were processed using the nanoPOTS workflow as described previously.6,15,16 Briefly, nanoliter pipetting is accomplished using an in-house-built robotic liquid handling system and a microfabricated glass chip patterned with hydrophilic nanowells arrays. The single cells were collected onto nanowells and then reagents for cell lysis, reduction, alkylation and digestion were added and incubated sequentially in a one-pot workflow as described previously6,16 to generate peptides for analysis.LC-MS analysis
Prior to separation, the sample was transferred from the storage capillary to a home-packed 75 μm-i.d. SPE column for desalting by infusing mobile phase A (MP A; 0.1% formic acid in water) at a flow rate of 1 μL min−1 for 10 min using an UltiMate 3000 RSLCnano pump (Thermo Fisher). The SPE column was then connected to an in-house slurry-packed 20 μm-i.d., 50 cm-long LC column15 with a zero-dead-volume union (Valco, Houston, TX, USA). Chromatographic media for both the SPE and LC columns were 3 μm C18 porous particles having 300 Å pores (Phenomenex, Torrance, CA, USA). The LC separation flow rate was ∼20 nL min−1, which was split from 250 nL min−1 programmed flow provided by the same UltiMate 3000 RSLCnano pump using an in-house-prepared 50 cm-long, 75 μm-i.d. column that employed the same packing material as the analytical column. A linear 100 min gradient of 8–22% mobile phase B (MP B; 0.1% formic acid in acetonitrile) was used for separation. An additional 20 min gradient of 22–45% MP B was used to elute hydrophobic peptides and the gradient was then ramped to 90% MP B over 5 min and held for 5 min to wash the column. Finally, the gradient was ramped to 2% MP B over 5 min and held for 15 min to re-equilibrate the column.A Thermo Fisher Orbitrap Eclipse Tribrid mass spectrometer with a FAIMS Pro Interface (San Jose, CA) was employed for MS analysis. For data collected without FAIMS, an electrospray potential of 2.0 kV was applied at the source for ionization, while 2200 V was applied when the FAIMS Pro interface was incorporated. The ion transfer tube was set at 150 °C for desolvation and the ion funnel RF level was 30. The orbitrap mass analyzer was used as the MS1 detector with the resolution set at 240000 (m/z 200). The resolution was set at 60000 when the orbitrap served as the MS2 detector, and the ion trap scan rate was set to rapid when the ion trap mass analyzer was used for MS2 detection. The MS1 AGC target and maximum injection time were set at 1 × 106/250 ms, the MS2 AGC target and maximum injection time were set to 1 × 105/500 ms for the orbitrap, and 3 × 104/300 ms when the ion trap was used for detection. Data-dependent acquisition mode was used to trigger precursor isolation and sequencing. Precursor ions with charges of +2 to +7 were isolated for MS2 sequencing. The MS2 isolation window was 1.6 Da, the dynamic exclusion time was set at 120 s, a mass tolerance of ±10 ppm was utilized and the signal intensity threshold was set to 8 × 103. A normalized collision energy of 30% was used for precursor fragmentation for higher energy collision-induced dissociation (HCD) mode and a normalized collision energy of 35% was used for precursor fragmentation for collision-induced dissociation (CID) mode.
Data analysis
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository39 with the dataset identifier PXD019515. Raw files were processed using Proteome Discoverer Software (version 2.4, San Jose, CA) for feature detection, database searching, and protein/peptide quantification. MS/MS spectra were searched against the UniProtKB/Swiss-Prot human database (downloaded on June 6th, 2019, containing 20353 reviewed sequences). N-terminal protein acetylation and methionine oxidation were selected as variable modifications. Carbamidomethylation of cysteine residues was set as a fixed modification. The mass tolerances of precursors and fragments were <5 and 20 ppm, respectively. The minimum peptide length was six amino acids and the maximum peptide length was 144 amino acids. The allowed missed cleavages for each peptide was 2. A second stage search was activated to identify semi-tryptic peptides. Proteins were filtered with a maximum FDR of 0.01. Both unique and razor peptides were selected for protein quantification. Other unmentioned parameters were the Proteome Discoverer default settings. Potential contaminants from culture media were filtered out using the Bos Taurus Uniprot database. MaxQuant searches performed for comparison used the same search criteria as described previously.15
Normalized abundance values for high confidence (1% protein-level FDR) master proteins determined in Proteome Discoverer Software (v2.4) (Thermo Fisher Scientific) were loaded into Perseus (v1.6.5.0),40 log2-transformed and filtered to retain proteins detected in either all three MN samples or all three IN samples. Remaining missing values (∼2% of all values) were imputed for the total matrix based on random selection from a normal distribution downshifted by 3 standard deviations (width = 0.3 standard deviations). Fold difference in abundance for individual proteins was determined by subtracting the average log2-transformed protein abundance in the IN group (n = 3) from the averaged log2-transformed protein abundance in the MN group (n = 3). Significance of differential abundance was determined by performing a two-tailed t-test and imposing a significance cutoff threshold of padj. < 0.05 and ≥2-fold differential abundance on a log2 scale.
Proteins exhibiting significant differences in single MNs vs. INs were imported into the web-based STRING (v11)41 tool for assembly of functional networks allowing a minimum interaction score cutoff of 0.4 and with the text-mining option for active interaction sources disabled. Interaction networks built in STRING were imported into Cytoscape (v3.7.2)42 to allow mapping of protein abundance data onto individual nodes. P-Values for gene ontology (GO) and pathway enrichments were calculated using a hypergeometric test (statistical background = whole genome) followed by Benjamini–Hochberg correction for multiple hypothesis testing using the STRING enrichment analysis widget.43
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under award numbers R33 CA225248 and R01 GM138931 and through a sponsored research agreement with Biogen, Inc. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or Biogen, Inc.Notes and references
- T. E. Angel, U. K. Aryal, S. M. Hengel, E. S. Baker, R. T. Kelly, E. W. Robinson and R. D. Smith, Chem. Soc. Rev., 2012, 41, 3912–3928 RSC.
- S. P. Couvillion, Y. Zhu, G. Nagy, J. N. Adkins, C. Ansong, R. S. Renslow, P. D. Piehowski, Y. M. Ibrahim, R. T. Kelly and T. O. Metz, Analyst, 2019, 144, 794–807 RSC.
- P. D. Piehowski, Y. Zhu, L. M. Bramer, K. G. Stratton, R. Zhao, D. J. Orton, R. J. Moore, J. Yuan, H. D. Mitchell, Y. Gao, B. M. Webb-Robertson, S. K. Dey, R. T. Kelly and K. E. Burnum-Johnson, Nat. Commun., 2020, 11, 8 CrossRef CAS.
- S. L. M. P. Snyder, A. Posgai and M. Atkinson, et al. , Nature, 2019, 574, 187–192 CrossRef.
- R. T. Kelly, Mol. Cell. Proteomics, 2020, 19, 1739–1748 CrossRef.
- Y. Zhu, P. D. Piehowski, R. Zhao, J. Chen, Y. Shen, R. J. Moore, A. K. Shukla, V. A. Petyuk, M. Campbell-Thompson, C. E. Mathews, R. D. Smith, W. J. Qian and R. T. Kelly, Nat. Commun., 2018, 9, 882 CrossRef.
- Z. Y. Li, M. Huang, X. K. Wang, Y. Zhu, J. S. Li, C. C. L. Wong and Q. Fang, Anal. Chem., 2018, 90, 5430–5438 CrossRef CAS.
- X. Shao, X. Wang, S. Guan, H. Lin, G. Yan, M. Gao, C. Deng and X. Zhang, Anal. Chem., 2018, 90, 14003–14010 CrossRef CAS.
- S. M. Williams, A. V. Liyu, C.-F. Tsai, R. J. Moore, D. J. Orton, W. B. Chrisler, M. J. Gaffrey, T. Liu, R. D. Smith, R. T. Kelly, L. Paša-Tolić and Y. Zhu, Anal. Chem., 2020, 92(15), 10588–10596 CrossRef CAS.
- L. Sun, G. Zhu, Y. Zhao, X. Yan, S. Mou and N. J. Dovichi, Angew. Chem., 2013, 52, 13661–13664 CrossRef CAS.
- C. Lombard-Banek, S. A. Moody and P. Nemes, Angew. Chem., Int. Ed., 2016, 55, 2454–2458 CrossRef CAS.
- S. Li, B. D. Plouffe, A. M. Belov, S. Ray, X. Wang, S. K. Murthy, B. L. Karger and A. R. Ivanov, Mol. Cell. Proteomics, 2015, 14, 1672–1683 CrossRef CAS.
- P. L. Xiang, Y. Zhu, Y. Yang, Z. T. Zhao, S. M. Williams, R. J. Moore, R. T. Kelly, R. D. Smith and S. R. Liu, Anal. Chem., 2020, 92, 4711–4715 CrossRef CAS.
- Y. Zhu, R. Zhao, P. D. Piehowski, R. J. Moore, S. Lim, V. J. Orphan, L. Pasa-Tolic, W. J. Qian, R. D. Smith and R. T. Kelly, Int. J. Mass Spectrom., 2018, 427, 4–10 CrossRef CAS.
- Y. Cong, Y. Liang, K. Motamedchaboki, R. Huguet, T. Truong, R. Zhao, Y. Shen, D. Lopez-Ferrer, Y. Zhu and R. T. Kelly, Anal. Chem., 2020, 92, 2665–2671 CrossRef CAS.
- Y. Zhu, G. Clair, W. B. Chrisler, Y. Shen, R. Zhao, A. K. Shukla, R. J. Moore, R. S. Misra, G. S. Pryhuber, R. D. Smith, C. Ansong and R. T. Kelly, Angew. Chem., Int. Ed., 2018, 57, 12370–12374 CrossRef CAS.
- M. Dou, Y. Zhu, A. Liyu, Y. Liang, J. Chen, P. D. Piehowski, K. Xu, R. Zhao, R. J. Moore, M. A. Atkinson, C. E. Mathews, W. J. Qian and R. T. Kelly, Chem. Sci., 2018, 9, 6944–6951 RSC.
- Y. Zhu, J. Podolak, R. Zhao, A. K. Shukla, R. J. Moore, G. V. Thomas and R. T. Kelly, Anal. Chem., 2018, 90, 11756–11759 CrossRef CAS.
- Y. Zhu, M. Dou, P. D. Piehowski, Y. Liang, F. Wang, R. K. Chu, W. B. Chrisler, J. N. Smith, K. C. Schwarz, Y. Shen, A. K. Shukla, R. J. Moore, R. D. Smith, W. J. Qian and R. T. Kelly, Mol. Cell. Proteomics, 2018, 17, 1864–1874 CrossRef CAS.
- Y. Zhu, M. Scheibinger, D. C. Ellwanger, J. F. Krey, D. Choi, R. T. Kelly, S. Heller and P. G. Barr-Gillespie, eLife, 2019, 8, e50777 CrossRef CAS.
- B. Budnik, E. Levy, G. Harmange and N. Slavov, Genome Biol., 2018, 19, 161 CrossRef.
- M. Dou, G. Clair, C. F. Tsai, K. Xu, W. B. Chrisler, R. L. Sontag, R. Zhao, R. J. Moore, T. Liu, L. Pasa-Tolic, R. D. Smith, T. Shi, J. N. Adkins, W. J. Qian, R. T. Kelly, C. Ansong and Y. Zhu, Anal. Chem., 2019, 91, 13119–13127 CrossRef CAS.
- T. K. Cheung, C.-Y. Lee, F. Bayer, A. McCoy, B. Kuster and C. M. Rose, Nat. Methods Search PubMed , in press.
- J. A. Loo, H. R. Udseth and R. D. Smith, Anal. Biochem., 1989, 179, 404–412 CrossRef CAS.
- R. Guevremont, J. Chromatogr. A, 2004, 1058, 3–19 CrossRef CAS.
- D. A. Barnett, B. Ells, R. Guevremont and R. W. Purves, J. Am. Soc. Mass Spectrom., 2002, 13, 1282–1291 CrossRef CAS.
- D. B. Bekker-Jensen, A. Martinez-Val, S. Steigerwald, P. Ruther, K. L. Fort, T. N. Arrey, A. Harder, A. Makarov and J. V. Olsen, Mol. Cell. Proteomics, 2020, 19, 716–729 CrossRef CAS.
- R. T. Kelly, J. S. Page, Q. Luo, R. J. Moore, D. J. Orton, K. Tang and R. D. Smith, Anal. Chem., 2006, 78, 7796–7801 CrossRef CAS.
- S. Tyanova, T. Temu and J. Cox, Nat. Protoc., 2016, 11, 2301–2319 CrossRef CAS.
- H. Fu, J. Hardy and K. E. Duff, Nat. Neurosci., 2018, 21, 1350–1358 CrossRef CAS.
- M. Bowerman, L. M. Murray, F. Scamps, B. L. Schneider, R. Kothary and C. Raoul, Eur. J. Med. Genet., 2018, 61, 685–698 CrossRef.
- C. R. Slater, Int. J. Mol. Sci., 2017, 18, 2183 CrossRef.
- Y. Hofmann, C. L. Lorson, S. Stamm, E. J. Androphy and B. Wirth, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 9618–9623 CrossRef CAS.
- Y. Hofmann and B. Wirth, Hum. Mol. Genet., 2002, 11, 2037–2049 CrossRef CAS.
- S. K. Custer, T. D. Gilson, H. Li, A. G. Todd, J. W. Astroski, H. Lin, Y. Liu and E. J. Androphy, PLoS One, 2016, 11, e0163954 CrossRef.
- F. Gros-Louis, R. Lariviere, G. Gowing, S. Laurent, W. Camu, J. P. Bouchard, V. Meininger, G. A. Rouleau and J. P. Julien, J. Biol. Chem., 2004, 279, 45951–45956 CrossRef CAS.
- J. M. Beaulieu, M. D. Nguyen and J. P. Julien, J. Cell Biol., 1999, 147, 531–544 CrossRef CAS.
- M. A. Meyer, Neurol. Int., 2014, 6, 5367 Search PubMed.
- Y. Perez-Riverol, A. Csordas, J. Bai, M. Bernal-Llinares, S. Hewapathirana, D. J. Kundu, A. Inuganti, J. Griss, G. Mayer, M. Eisenacher, E. Perez, J. Uszkoreit, J. Pfeuffer, T. Sachsenberg, S. Yilmaz, S. Tiwary, J. Cox, E. Audain, M. Walzer, A. F. Jarnuczak, T. Ternent, A. Brazma and J. A. Vizcaino, Nucleic Acids Res., 2019, 47, D442–D450 CrossRef CAS.
- S. Tyanova, T. Temu, P. Sinitcyn, A. Carlson, M. Y. Hein, T. Geiger, M. Mann and J. Cox, Nat. Methods, 2016, 13, 731–740 CrossRef CAS.
- D. Szklarczyk, A. L. Gable, D. Lyon, A. Junge, S. Wyder, J. Huerta-Cepas, M. Simonovic, N. T. Doncheva, J. H. Morris, P. Bork, L. J. Jensen and C. V. Mering, Nucleic Acids Res., 2019, 47, D607–D613 CrossRef CAS.
- P. Shannon, A. Markiel, O. Ozier, N. S. Baliga, J. T. Wang, D. Ramage, N. Amin, B. Schwikowski and T. Ideker, Genome Res., 2003, 13, 2498–2504 CrossRef CAS.
- A. Franceschini, D. Szklarczyk, S. Frankild, M. Kuhn, M. Simonovic, A. Roth, J. Lin, P. Minguez, P. Bork, C. von Mering and L. J. Jensen, Nucleic Acids Res., 2013, 41, D808–D815 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc03636f |
| This journal is © The Royal Society of Chemistry 2021 |