
CO2 adsorption mechanisms on MOFs: a case study of open metal sites, ultra-microporosity and flexible framework
Renata Avena
Maia
 *ab,
Benoît
Louis
b,
Wanlin
Gao
c and
Qiang
Wang
c
*ab,
Benoît
Louis
b,
Wanlin
Gao
c and
Qiang
Wang
c
aUniversité de Strasbourg, CMC UMR 7140, 4 rue Blaise Pascal, 67081, Strasbourg, France. E-mail: avenamaia@unistra.fr
bUniversité de Strasbourg, ICPEES UMR 7515, 25 rue Becquerel, 67087, Strasbourg Cedex 2, France
cCollege of Environmental Science and Engineering, Beijing Forestry University, 35 Qinghua East Road, Haidian District, Beijing 100083, P. R. China
First published on 28th April 2021
Abstract
The hazardous emission of CO2 in the atmosphere is a concerning topic when addressing climate change issues. The mitigation of this greenhouse gas has attracted great attention in the realm of a sustainable economy, especially regarding the design of novel CO2 capture technologies. Metal–organic frameworks (MOFs), a class of hybrid porous materials, stand out as efficient carbon capture and sequestration (CCS) materials, which have a CO2 capacity of similar range or superior to the standard employed adsorbents, such as zeolite 13X and activated carbon. Taking into account the promising future of MOFs as CO2 adsorbents, it is therefore of utter importance to understand the CO2 adsorption mechanisms in these porous materials, which can contribute to further improvement of their performance. In this review, we explore the CO2 adsorption mechanisms of diverse MOFs, namely MOF-74-Mg, HKUST-1, SIFSIX-3-M (M = Fe, Co, Ni, Cu, Zn), and ZIF-8. These materials show interesting features as open metal sites, ultra-microporosity, and flexible framework, which are present in the majority of MOFs used for this application. Studies regarding their preferential adsorption sites, water stability, CO2–MOF complex configuration, CO2 adsorption dynamics, bonding angle, decomposition mechanism, and swing effects were addressed in this contribution.
 Renata Avena Maia | Renata obtained her PhD in Materials Chemistry (2019) from the Federal University of Rio de Janeiro (UFRJ, Brazil) on the topic of design and applications of covalent organic frameworks (COFs), mainly focusing on heterogeneous catalysis and CO2 capture. Currently, Renata is a postdoctoral fellow at the University of Strasbourg in France, where she investigates green methodologies for the synthesis of metal–organic frameworks. In 2021, Renata was selected as member of the steering committee of the French Group of zeolites, an association which aims to promote fundamental and applied research related to the science of zeolites and microporous materials. |
 Benoît Louis | Benoît Louis graduated from the University of Strasbourg (1998) and completed his PhD at the Swiss Federal Institute of Technology (EPFL, 2002). He is currently CNRS Research Director at the Institute of Chemical and Processes for Energy, Environment and Health (UMR 7515). Since twenty years, Benoît is focused on designing zeolites, mainly for acid-catalysed reactions. The originality of his research relies on the combination of molecular and microscopic design of microporous solids along with a suitable macro-scale (shaping, coating) for a use in real reactors. His current research interests encompass heterogeneous catalysis, zeolites and porous materials, biomass valorisation, alkane activation chemistry, CO2 capture and conversion and more generally C1 chemistry. He was awarded by the Guy Ourisson Prize (University of Strasbourg, 2015). In 2013, he was the first laureate of the Young Scientist Award in acid–base catalysis (Tokyo) and awarded by the French Chemical Society SCF Catalysis Division Prize. He also received the CNRS Bronze medal in 2009. |
 Wanlin Gao | Wanlin Gao is currently a PhD candidate in the College of Environmental Science and Engineering, Beijing Forestry University in China under the supervision of Prof. Qiang Wang. She worked on the fabrication and application of MgO based CO2 adsorbents for sorption enhanced water gas shift reaction and her current research interest is focused on the deployment of in situ and operando analysis for CO2 capture mechanism study. |
 Qiang Wang | Professor Qiang Wang received BSc (2003) and MSc (2005) from Harbin Institute of Technology in China, and PhD (2009) from POSTECH in South Korea. In 2009–2011, he worked as Research Fellow in the Institute of Chemical and Engineering Sciences under A*STAR, Singapore. In 2011–2012, he worked as postdoctoral associate in the Department of Chemistry, University of Oxford. From 2012, he holds a full professor position in the College of Environmental Science and Engineering, Beijing Forestry University. His research interests are heterogeneous catalysis and materials chemistry. He was selected into the National Overseas High-level Talent Introduction Youth Project, National Excellent Youth, New Century Excellent Talents, Excellent young scholars in the special technology of national environmental protection awarded by the Ministry of Environmental Protection of P. R. China, Beijing Youth Award and Science and Technology Rising Star. |
Introduction
Metal–organic frameworks (MOFs) have been regarded as an undoubtedly promising class of porous materials for a breadth of applications. The straightforward synthesis of these materials makes them eligible candidates for scale-up methodologies, which in turn resulted in their ready availability as far as one click away. Under the tradename of Basolite, Sigma-Aldrich markets the well-known MIL-53(Al), HKUST-1, ZIF-8, and MOF-177, among others, which is an indicative of the global commercial demand for these materials with a variety of specific functionalities, mainly controlled by pore size aperture, framework flexibility, and open metal sites. In general, MOFs can be synthesized by the mixture of a metallic salt with an organic linker by the usual solvothermal method, which requires an appropriate solvent and a given reaction time. The resulting material unveils an extended structure based on the premises of coordination chemistry, which can be, in some cases, modulated by variation of the reaction parameters and/or the ratio of the starting materials. Hence, such discrete molecules, when placed in the same reaction vessel, can either form the simplest or the most complex topology, and the beauty of this chemistry relies on unravelling the reaction parameters and characterization techniques that enables one to better decipher the full potential of these materials.One particular application of MOFs is CO2 capture, the latter has attracted much scientific attention due to the current climate issues related to the hazardous CO2 emissions in the atmosphere, which have been addressed in some comprehensive reviews.1–12 Hence, the mitigation of excessive CO2 emissions, using advanced capture technologies, is mandatory to avoid, or at least delay, the alarming greenhouse effects. Nowadays, MOF materials have displayed excellent results towards CO2 capture, it is therefore of paramount importance to understand the CO2 adsorption mechanisms in these porous MOFs-based materials, which can contribute to further performance improvements and lead to new design strategies. From this perspective, this review focuses on the different CO2 adsorption mechanisms occurring in MOFs MOF-74-Mg, HKUST-1, SIFSIX-3-M (M = Fe, Co, Ni, Cu, Zn) and ZIF-8. These MOFs were chosen due to their diverse structural architectures towards CO2 capture: open metal sites, ultra-microporosity, and flexible framework, features which are present in the majority of MOFs used for this application. By scrutinizing a relevant material of each category, this contribution aims to summarize their particular interactions with CO2, e.g. bonding angle, adsorption sites, molecular dynamics, water stability, decomposition mechanism, and swing effects. The chosen MOFs also display distinguished CO2 uptake values, which are in the range or superior when comparing to well-established adsorbents, as zeolite 13X and activated carbon materials (Table 1).13 Although the latter are still less expensive than MOFs, the main drawback of zeolite 13X and activated carbons is the high energy requirement for regeneration posterior to CO2 capture (Table 1), driving an obvious reason for the quest of new CO2 capture materials. Particularly, MOFs provide an overall advantage of synthesis by design, as the judicious choice of their building blocks can tailor the physicochemical properties of the targeted adsorbent. Although a limited number of MOFs were studied for regeneration processes, they bear overall lower energy costs when compared to traditional sorbents,9 elevating MOFs to a platform worth pursuing regarding CO2 capture.
| Material | Averagepore size (nm) | Pore volume (cm3 g−1) | BET surface area (m2 g−1) | Adsorption mechanism | CO2 uptake (mmol g−1) at 313 K and 0.15 bar | CO2 uptake (mmol g−1) at 298 K and 1 bar | CO2 uptake (mmol g−1) at high pressure | Advantages | Disadvantages |
|---|---|---|---|---|---|---|---|---|---|
| a Temperature varied in a temperature-programmed desorption (TPD) analysis. b Estimated visually from the CO2 isotherms at 298 K. c At 318 K. d At 298 K. e At 273 K. f At 33 bar and 278 K. g At 40 bar and 303 K. h At 300 bar and 313 K. i At 30 bar and 298 K. j Estimated visually from the CO2 isotherm at 35 bar and 323 K. k At 40 bar. l At 20 bar and 298 K. | |||||||||
| MOF-74-Mg | 1.014 | 0.385–0.68214 | 882–141614 | Physisorption (open metal site) | 5.2815 | 8–9.914,16,17 | 15.7f,18 | High CO2 uptake in dry conditions | Water sensitive |
| HKUST-1 | 0.5; 0.919 | 0.80–0.8220 | 1270–223020,21 | 1.6a,22 | 4.5;23 11.6e,20 | 9.1g,24; 9.7h,21 | |||
| SIFSIX-3-Fe | 0.3–0.425 | 0.167–0.19725 | 35826 | Physisorption (size exclusion separation/molecular sieving)7 | ~ 2.6b,25 | 2.825 | — | Water stable | Few studies for high-pressure CO2 adsorption |
| SIFSIX-3-Zn | 25027 | 2.3c,5,27 | 2.825 | — | |||||
| SIFSIX-3-Co | 22328 | ~ 2.6b,25 | 2.925 | — | |||||
| SIFSIX-3-Ni | 36828 | 2.2c,5,29 | 2.625 | 3.2i,30 | |||||
| SIFSIX-3-Cu | 30031 | 2.4c,5,31 | 2.625 | — | |||||
| ZIF-8 | 0.3; 1.232 | 0.66332 | 194732 | Physisorption (flexible framework) | < 0.133 | 2.434 | ~5.9j,35 | Flexible framework | Structure collapse under humid CO2 conditions |
| Zeolite 13X | 1.0–2.236,37 | 0.21–0.3836,37 | 160–87636,37 | Physisorption | 3.2a,22 | 3.738 | ~7b,k,39 | High thermal stability | High energy requirement for regeneration; water sensitive |
| Activated carbon | 0.740 | 1.040 | 371–231840,41 | Physisorption | 1.9d,42 | 1.8–5.640,41,43 | 19-21l,44 | Low acid/base reactivity | High energy requirement for regeneration |
MOF-74-Mg
Currently taken as the benchmark in CO2 adsorption regarding MOF materials, MOF-74-Mg (CPO-27-Mg, Mg2(dobdc))45 is known for its high CO2 uptake in dry conditions and its remarkable selectivity.7 Extensive studies have demonstrated that the dominant interaction mechanism between CO2 molecules and MOF-74-Mg is through an open Mg2+ site, generated upon the thermal activation of the material under vacuum. As an overview of this section, it will be addressed (i) the structural configuration of the CO2 molecule on the MOF-74-Mg;(ii) the temperature-related molecular dynamic of CO2, and (iii) the decrease of CO2 uptake on humid conditions, as the water dissociation causes hydroxyl-poisoning that impairs CO2 sorption by both high temperature and moisture exposure. Finally, (iv) the presence of methanol/isopropanol was shown to either accelerate/slow down the water poisoning, since methanol can complex with the metal centre and is small enough to still allow water molecules to approach its vicinity, whereas isopropyl alcohol sterically hinders this approximation and slows down water dissociation by 40%, when compared to humid alcohol-free conditions.Wu et al.46 employed neutron diffraction and density functional theory (DFT) calculations to identify the primary binding sites in MOF-74-Mg. By loading sufficiently low amounts of CO2 on the activated MOF-74-Mg to ensure a monolayer formation, the neutron diffraction data categorically showed that CO2 molecules were preferentially adsorbed on Mg2+ sites (Fig. 1a) and the adsorbed CO2 molecule possessed a ∼160.5° O–C–O bond angle. Generally, the theoretical O–C–O bond angles are in the range of 175–179°, which are quite distant from the one found in the neutron diffraction experiment. To better understand this unusual bending for the linear CO2 molecule, further DFT calculations highlighted that the CO2 molecule interacts with the open metal site in an end-on orientation (one of the oxygen atoms in the CO2 is fixed, while the rest of the molecule is orientationally free). Thus, the freedom attributed to the “end-part” of the CO2 molecule allows an orientational disorder of a certain degree, which was therefore observed in the neutron diffraction data as an apparent larger bond bending angle. Additionally, since the bond angle from the diffraction data can be assumed as a statistical measurement of the structural components, this angle value can be strongly biased in terms of these disorder features and could not be taken as an accurate bond angle value. This study thus provided a better understanding of an unusual bond angle of CO2, which was found with the integrated combination of theoretical calculations and experimental data.
 | ||
| Fig. 1 a) CO2 preferential binding site at magnesium cations, by real space Fourier difference scattering length density (yellow) superimposed on the MOF-74-Mg structure (reprinted with permission from J. Phys. Lett. 2010, 1, 1946–1951. Copyright (2010) American Chemical Society). b) Distances (in angstroms) and angles (in degrees) of the adsorbent-(MOF-74-Mg) complexes for CO, N2, and CO2, from left to right (reprinted with permission from J. Phys. Chem. C., 2010, 114, 11185–11191. Copyright (2010) American Chemical Society). | ||
Focused on the interaction angle of adsorbent-(MOF-74-Mg), Valenzano et al.47 investigated the adsorption of CO, N2, and CO2 on MOF-74-Mg by spectroscopic and computational studies based on the B3LYP functional. The theoretical data showed that the CO2-framework complex is rather angular in contrast to CO and N2, which form a complex with the Mg2+ site in a linear fashion (Fig. 1b). The carbon dioxide in the Mg2+–(O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO) complex is tilted at a 129° angle, which was hypothesized as a lateral interaction between the CO2 molecule and the carboxylate oxygen of the 2,5-dihydroxydeteraphtalate linker, mainly due to the quadrupole moment of the former. The standard adsorption enthalpy of the gas molecules on MOF-74-Mg was obtained by variable-temperature infrared (VTIR) technique, and the CO2 adsorption enthalpy was found to be −47 kJ mol−1, whilst N2 and CO gave values of −21 kJ mol−1 and −29 kJ mol−1, respectively. The difference in the adsorption enthalpy was highlighted as an energetic tool to enable gas separation: the stronger interaction between MOF-74-Mg and CO2 could facilitate a greater affinity towards CO2, and thus enhance selectivity versus other molecules with weaker adsorbent–framework interactions.
CO) complex is tilted at a 129° angle, which was hypothesized as a lateral interaction between the CO2 molecule and the carboxylate oxygen of the 2,5-dihydroxydeteraphtalate linker, mainly due to the quadrupole moment of the former. The standard adsorption enthalpy of the gas molecules on MOF-74-Mg was obtained by variable-temperature infrared (VTIR) technique, and the CO2 adsorption enthalpy was found to be −47 kJ mol−1, whilst N2 and CO gave values of −21 kJ mol−1 and −29 kJ mol−1, respectively. The difference in the adsorption enthalpy was highlighted as an energetic tool to enable gas separation: the stronger interaction between MOF-74-Mg and CO2 could facilitate a greater affinity towards CO2, and thus enhance selectivity versus other molecules with weaker adsorbent–framework interactions.
Focused on the end-on complexation of CO2–(MOF-74-Mg), but aware of the importance of dynamic CO2 studies in a wide temperature range, Reimer's group48 reported an in situ13C NMR experiment where 13C-enriched CO2 was adsorbed on MOF-74-Mg in a flame-sealed glass tube, at temperatures from 12 K up to 400 K. Physisorption could be easily detected by the substantial decrease of the CO2 motional freedom in the 13C spectra. A uniaxial rotation angle θ was also observed, which was in agreement with a CO2–(MOF-74-Mg) complex reported by Valenzano et al.,47 as well as with an the orientationally free CO2 configuration pointed out by Wu et al.46 Their simulations demonstrated that the rotation angle θ was in the range of 56–69°, which varies within this range depending on the temperature and CO2 concentration loaded in the sample (Fig. 2). For example, when a concentration of 0.5 CO2/Mg2+ was employed, it was observed that the rotation angle θ was 56°, 59°, 61° and 63° at 375 K, 300 K, 250 K, and 200 K, respectively. These results are rather surprising, since one could imagine that, a priori, the CO2 motion would occupy a wider rotational cone with increasing thermal energy subjected to the system. In a follow-up study, Reimer's group49 was able to correlate the decrease in the rotation angle θ with the increase of temperature. Canonical Monte Carlo simulations demonstrated that a motion along the z-direction was responsible for a larger tilt of the carbon dioxide regarding the x-y plane, resulting in a decrease in the equivalent rotation angle θ. Furthermore, the latter study also contributed to a better understanding of the CO2 molecule dynamics, which could differentiate the modes of: (i) fluctuation of the CO2 molecule near the minimum-energy configuration (bounded to the same metal site, localized motion) and (ii) hops of the CO2 molecules between different Mg2+ sites (non-localized hopping motion). An illustrative scheme of those modes can be visualized at the original publication at Angew. Chem. Int. Ed. 2013, 52, 4410–4413. Indeed, by comparing the simulated and experimental 13C NMR chemical shift anisotropy patterns, it was evident that the localized motion is the dominant mode at 100 K, followed by a transition behaviour at 150 K containing localized motions with rare events of CO2 hopping. Finally, at temperatures higher than 200 K the non-localized hopping motion becomes the major mode and the localized motion has no relevant contribution to the system. Thus, these studies applying in situ13C NMR to CO2 adsorption on the MOF-74-Mg provided insightful results towards the in-depth understanding of its adsorption molecular dynamics, which have been widely reported for other MOFs50–54 and summarized in a perspective by Bernin & Hedin.55
 | ||
| Fig. 2 Representation of the uniaxial rotation of the CO2 molecule complexed in an end-on orientation with the Mg2+ open site of MOF-74-Mg as a function of the θ angle (reprinted with permission from J. Am. Chem. Soc. 2012, 134, 14341–14344. Copyright (2012) American Chemical Society). | ||
Given that the CO2 capture performance of MOF-74-Mg drastically decreases under humid conditions, efforts have been devoted to understanding the moisture effect on the adsorption capacity of the MOF-74-M (Mg, Zn, Ni and Co) series, as reported by Zuluaga et al.56 In this study, they investigated deeper their first findings57,58 on water dissociation (D2O → D + OD) in MOF-74-M by infrared spectroscopy and ab initio calculations. The materials exhibited a significant reduction in CO2 uptake after water exposure as MOF-74-Zn > MOF-74-Mg > MOF-74-Ni > MOF-74-Co, and particularly poor performance with increasing temperatures from 100 °C to 200 °C. Initially, they demonstrated that neither the water nor the temperature was solely responsible for this behaviour, since the exposure of MOF-74-Zn in anhydrous conditions at 300 °C or in humid conditions at 100 °C did not induce any change in the crystalline structure of the MOF. Only with the combination of both factors, namely humid conditions at 300 °C, the crystalline structure was completely destroyed. These results, along with the detection of the dissociated water (OD) peak at 970 cm−1 in the infrared spectra, revealed that those factors are intertwined. Theoretical calculations showed that the dissociation reaction D2O → D + OD proceeds by (i) adsorption on the metal centre, followed by (ii) D donation to the nearest hydroxyl group of a linker, thus leaving the OD connected to the metal centre. Therefore, the decrease in CO2 adsorption capacity on the series of MOF-74-M could not be attributed to the sole presence of water per se, but indeed to the hydroxyl poisoning effect that occurs by water dissociation, especially at higher temperatures.
Additionally, the tendency of the metal ability towards catalysing the water dissociation reaction was explained by a linear relationship between the catalytic activity versus the calculated radii of the metal atoms in the MOF-74-M structure, which showed that a smaller M radius enabled higher water dissociation. Thus, this correlation was interpreted as: the smaller the M atom, the closer the D atom of the complexed water molecule (D–OD) is to the hydroxyl group of the linker, thus facilitating the water dissociation due to its closer interaction with the linker. Thus, firstly, the D⋯OHlinker interaction causes elongation of the M–Ocarboxylate(linker) bond, which leads to the water dissociation effect, and thus finally results in the collapse of the structure. The authors were also able to correlate the framework stability with the electronegativity of the metal atoms, where a higher metal electronegativity led to a less stable framework. Consequently, MOF-74-Mg presented itself as the most stable MOF among the studied scope, followed by MOF-74-Zn, MOF-74-Co, and MOF-74-Ni, respectively. A decrease in CO2 adsorption capacity and loss of crystallinity upon exposure to humid conditions at temperatures higher than 150 °C are subsequent (and different) events: first, the water complexes with the metal site and its dissociation takes place to poison the MOF with OD and impairs CO2 adsorption (driven by the smaller metal radius), followed by a possible collapse of the structure (which depends highly on the electronegativity of the metal site).
To further explore the water–MOF relationship, Fuentes-Fernandez et al.59 recently demonstrated that exposing MOF-74-Zn to an alcohol/water mixture could either speed up or reduce the water dissociation reaction, depending on the nature of the alcohol employed. Based on the greater affinity towards moisture, MOF-74-Zn was chosen for this study, although a priori their results could be extrapolated to other members of its series (MOF-74-M). The results led to a very interesting alternative to the CO2 capture field, since this methodology could potentially slow down the degree of –OH poisoning for this class of MOFs. It was found that methanol speeded up the dissociation reaction by hydrogen bonding interactions, while isopropyl alcohol led to the opposite effect, mainly due to its steric hindrance. The proposed mechanism for the higher rate of dissociation induced by methanol (Fig. 3a) starts with a preferential binding to the metal site, driving the water molecule to bind to the oxygen atom (hydroxyl group) from the linker. The Me–OH bond becomes weaker, especially when a D2O molecule (associated with the linker) is close. This proximity favours the dissociation of water, leaving the D bounded to the oxygen atom of the linker, and the –OD part binds to its neighbouring alcohol molecule, resulting in the MeOD molecule and the poisoned MMOF–OH. Additionally, the formation of water clusters was found to diminish the energetic barrier of the water molecule dissociation, mainly due to H-bonding. In stark contrast, isopropyl alcohol slows down this dissociation. Its bulkier size, when compared to methanol, prevents a favourable approximation to the water molecule bonded to the linker (Fig. 3b), thus preventing water cluster formation. Those combined effects of isopropyl alcohol slowed down the dissociation reaction of water by approximately 40% in relation to the exposure to pure D2O. This study showed promising results towards controlled poisoning of MOF-74-M adsorbents and hopefully it could be further applied to the lifetime extension of moisture-sensitive MOFs.
 | ||
| Fig. 3 Schematic mechanisms of a) methanol speeding up the water dissociation due to spatial proximity of D2O, thus enabling water cluster formation; and b) isopropanol slowing down the same reaction by means of steric hindrance (reprinted with permission from Fuentes-Fernandez, E. M. A., Jensen, S., Tan, K., Zuluaga, S., Wang, H., Li, J., Thonhauser, T., Chabal, Y. J. App. Sci. 2018, 8, 270. Copyright 2018 by the authors. Licensee MDPI, Basel, Switzerland, https://www.mdpi.com/2076-3417/8/2/270). | ||
HKUST-1
As another example of a robust MOF containing open metal sites,60 HKUST-1 (Cu3(BTC)2, Cu–BTC) has been thoroughly investigated for its CO2 capture properties,10,11 either in its pristine conditions or after chemical modifications.20,61–63 First described by Chui et al.64 as the product of the solvothermal synthesis between cupric nitrate and trimesic acid, HKUST-1 is composed of dimeric cupric tetracarboxylate units, where water molecules are coordinated on the axial positions of the paddlewheel units. Further activation of this material, mainly by thermal treatment under vacuum, provides available open Cu2+ binding sites due to the elimination of these water molecules, which is visually observable by its shift in colour from turquoise to dark blue.64–66 Besides the Cu2+ open site, this section also addresses other reported adsorption sites of HKUST-1, whether it is related to structural defects or small cages present in this material. Furthermore, a similar rotational motion of the MOF-complexed CO2, that was studied for MOF-74-Mg, was also present in HKUST-1. Regarding its moisture stability, the water presence was demonstrated by PXRD studies and detected by the 311 plane reflection intensity, which regulates either the degree of hydration or the CO2 loading capacity, as this plane is related to the Cu2+ available binding site. The degradation of this MOF was also evidenced by the presence of both moisture and temperature, and ligand displacement was indicated as the degradation mechanism, which was reported as being initiated by water dissociation triggered by the H2O–MOF complex.In a spectroscopic study, Bordiga et al.65 investigated the CO2 interaction with the metallic Cu2+ center by loading 9 mbar of CO2 at 150 K in a HKUST-1 sample, and progressively decreasing the equilibrium pressure to 0 with an increasing temperature up to 300 K. At low coverage, the Cu2+ interaction with the carbon dioxide was evidenced by the red shift of the CO2 gas phase frequency from 2349 cm−1 to 2333 cm−1, which differed from the bare Cu2+ cation interaction with CO2 (which bears a typical blue shift). Interestingly, the interaction with 13CO2 for HKUST-1 was also detected, owing to the presence of ca. 1% of its natural isotope. Thus, it was evidenced that there were two slightly different Cu2+ adsorption sites present in HKUST-1, mainly attributed to (i) Cu2+ present on the external surface and/or related to structural defects and (ii) Cu2+ present in the inner framework of the MOF. Accordingly, a XAFS study34 also demonstrated that the CO2 physisorption occurs on the CO2 molecule binding with the Cu2+ centre of HKUST-1.
To further explore the CO2 interaction with the HKUST-1 framework, Wu et al.46 executed neutron diffraction experiments combined with theoretical calculations for HKUST-1, via the same methodology as they had performed for MOF-74-Mg (vide supra). Upon activation of HKUST-1 at 150 °C for 24 h, two main adsorption sites were observed upon analysis of the Fourier difference maps (Fig. 4). As expected, the preferential adsorption site is Cu2+, which is first occupied by CO2 molecules at low loadings due to the strong electrostatic interaction between Cu2+ and the quadrupole moment of CO2 (Fig. 4, left). When the CO2 concentration was increased, a second adsorption site was detected: the CO2 molecule occupied the window opening of the small octahedral cage (Fig. 4, right). The secondary adsorption site was attributed to stronger van der Waals interactions, since the small cage enables the interaction of a single CO2 molecule with multiple surfaces, generating an enhanced adsorption potential. Those multiple adsorption sites were also reported by Grajciar et al.,67 by performing theoretical and experimental studies on CO2 adsorption over this material.
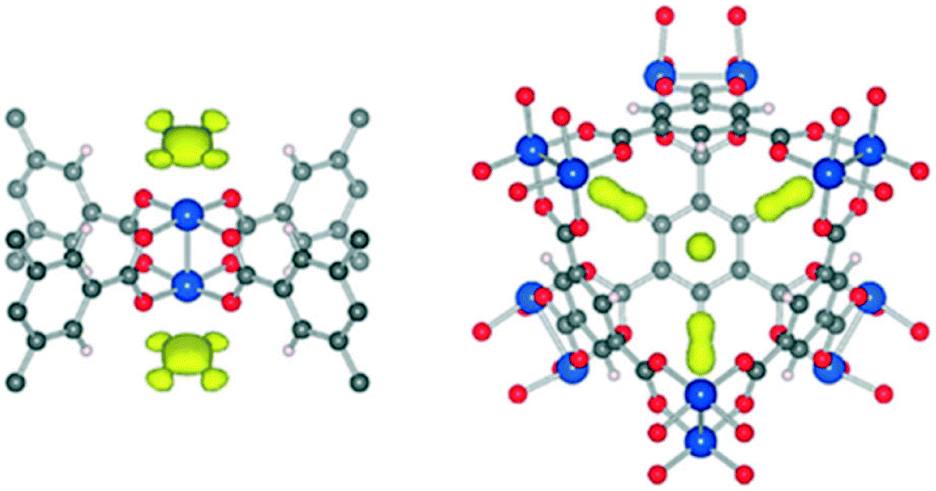 | ||
| Fig. 4 Fourier difference maps of CO2 adsorption on HKUST-1 and its main adsorption sites: (left) the Cu2+ site and (right) the triangular-shaped window opening of the small octahedral cage (reprinted with permission from J. Phys. Lett. 2010, 1, 1946–1951. Copyright (2010) American Chemical Society). | ||
Based on molecular simulations, Gutiérrez-Sevillano et al.19 suggested five possible adsorption sites in HKUST-1 (Fig. 5a), described as L1, L2, L3, Tw and Lw. T1 is a small 5 Å cavity that surrounds the larger central cavities L2 and L3 of 9 Å, whereas only the latter bears the copper sites pointed into the pore. TW is the triangular-shaped aperture that connects T1 and L3, while LW represents the windows connecting L3 and L2. Their results demonstrated that the preferential adsorption site for CO2 in HKUST-1 is T1 for all studied loadings that varied from 4 to 192 molecules per unit cell. The preferential attraction was also attributed to the quadrupole moment of CO2 combined with the molecular size of CO2, which led to its preferential adsorption in the small tetrahedral cage T1.
 | ||
| Fig. 5 a) The simulated adsorption sites of HKUST-1: L1, L2, L3, Tw and Lw (reprinted with permission from J. Phys. Chem. C. 2013, 117, 11357–11366 copyright (2013) American Chemical Society). b) Proposed rotational disorder (red arrows) of adsorbed CO2 (oxygen, blue; carbon, grey) molecules on the Cu2+ sites (green) (reprinted from Wong-Ng, W., Levin, I., Kaduk, J. A., Espinal, L., Wu, H. J. Alloys Compd. 2016, 656, 200–205, CO2 capture and positional disorder in Cu3(1,3,5-benzenetricarboxylate)2: an in situ laboratory X-ray powder diffraction study. Copyright (2016) with permission from Elsevier). | ||
In 2016, an in situ PXRD study regarding CO2 adsorption on HKUST-1 was reported by Wong-Ng et al.68 by locating CO2 molecules inside the framework. Through the investigation of the 311 reflection intensity variation upon dehydration and further CO2 adsorption, it was clear that the removal of water from the MOF structure led to an increase in this diffraction peak, which progressively diminished upon CO2 exposure. This indicated the emptiness of the pores/binding sites upon dehydration (activation) were subsequently occupied by CO2 molecules. Additionally, the lack of well-defined difference in electron-density peaks for the “free” oxygen of adsorbed CO2 (Cu2+⋯OCO), assuming an end-on configuration, was attributed to a rotational motion of CO2 (Fig. 5b), which was reported to hold an interaction Cu–O–C angle of ca. 110°. A similar Cu–O–C interaction angle and a flexible CO2-framework interaction were also reported by Supronowicz et al.69 In their study regarding the interaction of small gases with an unsaturated Cu2+ centre of HKUST-1, the geometry of CO2 was practically linear and the interaction angle of Cu–(OCO) was estimated at 120°.
The collapse of the structural integrity of HKUST-1 towards water exposure has been thoroughly studied, mainly in order to understand its dissociation mechanism and in view of the potential applications of this copper-based MOF. Particularly, the interest on this topic is of great importance for the CO2 capture from flue gas stream, as a post-combustion coal-fired power plant containing up to 5–7% of water.70 Hence, the effect of water adsorption on the structure of HKUST-1 was investigated by solid-state NMR spectroscopy by Gul-E-Noor et al.66 By controlling the exposure time and water amounts, the stability of the HKUST-1 framework remained seemingly the same when exposed to low amounts of water (0.5 mol equivalent with respect to the copper atom) even after 60 days, which was evidenced by an unchanged 1H MAS spectrum profile. However, with increasing water content (0.75, 1.0, 1.5, 2.0 and 5.0 mol equivalent), different decomposition pathways were assumed, since there was no apparent correlation with the water content and the change in the spectra profiles up to 60 days of exposure. For example, when 1.0 mol equivalent of water reacted with HKUST-1, the 1H MAS NMR spectrum kept the same profile until 12 days of exposure, whereas the 1H MAS NMR spectrum of the sample with exposure to 5.0 mol equivalent of water presented minimal changes over the course of the 60 days of experiment. To further investigate the change in the chemical environment of the framework structure, the 13C NMR spectra was acquired for the samples with 1.0 and 2.0 mol of water adsorbed after 21 and 12 days, respectively (see Fig. 5 at the original publication at Phys. Chem. Chem. Phys. 2011, 13, 7783–7788). For the sample treated with 1.0 mol equivalent of water, an extension in signal ranges was evidenced for both types of carbon, the aromatic and carboxylate ones. Besides, narrow resonances around 130–140 ppm and 180 ppm were observed, which are typical of free ligand trimesic acid in the absence of the paramagnetic environment of Cu2+, which considerably affects the magnetic properties of its surroundings due to the hyperfine interactions of the copper unpaired electron with its neighbourhood. Apparently, for the sample with water content of 2.0 mmol equivalent, the major carbon chemical shifts were attributed to the diamagnetic free ligand, indicating the absence of interaction between the linker and paramagnetic copper atom, which clearly suggested the decomposition of the pristine chemical structure.
The moisture stability of HKUST-1 was also investigated by Fan's group,71 which focused on analysing the samples after water sorption experiments with an exposure time of 90 hours. By comparing the PXRD patterns of the parent material and the HKUST-1 after exposure to a relative humidity of 90% at 25 °C, the results showed that the diffraction pattern was retained for pristine sample, but a discrete increase of the amorphous background could be detected. For higher humidity contents, the crystalline structure remained at 50 °C and relative humidity of 70%, but it totally collapsed when the humidity was increased to 90%. In a follow-up study,72 the HKUST-1 samples, exposed to water vapor experiments up to 90% of relative humidity at different temperatures, were investigated by small-angle X-ray scattering (SAXS), infrared spectroscopy, and differential scanning calorimetry (DSC). SAXS experiments confirmed the decrease in crystallinity for the sample exposed to water vapor at 25 °C and a total loss of structure ordering at 50 °C. Since the thermal stability of HKUST-1 reached up to 260 °C, the authors claim that the collapse of the structure cannot be attributed to the temperature alone, adding that the presence of water plays a central role regarding the long-range order. By further analysing the infrared spectra of the sample at 50 °C, they observed the presence of free carboxylate groups by the appearance of the CO and C–O carboxylate stretching modes. Additionally, a characteristic sharp endothermic peak around 310–340 °C was found in the DSC analysis, which was absent in the other samples. The peak correlates with the DSC curve of pure trimesic acid ligand, demonstrating that in a humid environment at 50 °C the water coordinates with the metal centre and causes the displacement of the ligands from the framework.
To understand the loss of HKUST-1 crystalline structure with the presence of water, Xue et al.73 investigated the initial stages of the hydrolytic mechanism of HKUST-1 by ab initio molecular dynamics simulations and DFT calculations. The proposed mechanism involves a three-step process, including water adsorption, ligand displacement, and water dissociation, which eventually leads to the collapse of the HKUST-1 framework in the presence of water. They demonstrated that the water dissociation occurs first at the Cu2+ center by an acid–base Lewis process whereas the water donates its free electron pair to the metallic cation. The process of water adsorption was also linked to the elongation of the Cu–Ocarboxylate bonds, especially when only one water molecule is adsorbed on one side of the Cu–Cu dimer, although this effect was discrete when two molecules of water are present and are adsorbed on each side of the Cu–Cu node cluster. For the ligand displacement step, three situations were investigated, based on the presence of one, two, and three or more water molecules. For the one and two water molecules situation, the water adsorption takes place, followed by a shortening of the Cu–OH2 bond and the subsequent breaking of the Cu–Ocarboxylate bond. Although one bond of the linker is broken, it is still not enough to collapse the structure. The bonds of at least two ligands at one side of the dimer would need to be broken due to the symmetric nature of HKUST-1. The following water dissociation while bonded to the Cu2+ centre would be difficult to occur due to the high free energy of the activation barrier. However, when three or more molecules were placed near the MOF, it led to two ligands fully disconnected from the metallic centre, leading to probable destruction of the crystalline nature of HKUST-1. This study corroborates well with the experimental findings of water exposure and structure collapse,66,71,72 which places this theoretical study as a relevant contribution towards the full understanding of this hydrolytic mechanism.
The acidic behaviour of water towards the decomposition of the framework was evidenced by Álvarez et al.74 through the investigation of the stability of HKUST-1 towards water and ethanol. Exposure to both solvents separately for 9 h showed its higher stability in ethanol, as its structure crystallinity decreased when exposed to water, according to PXRD diffractograms. Additionally, the morphology of the samples was also evaluated by SEM: while the sample exposed to ethanol preserved the double-sided pyramidal shape of the pristine material, the one exposed to water showed a majority of needle-shaped crystals. The theoretical calculations demonstrated a strong stability dependence to the acidity of the proton of the solvent, as the Brønsted–Lowry acidity of water is 80 times higher than the acidity of ethanol, given the logarithmic nature of the scale. Thus, when coordinated to the metallic centre of HKUST-1, the water molecule is more prone than an ethanol molecule to donate its proton to an adjacent oxygen atom of the linker. Thus, this proton exchange facilitates the breaking of the carboxylate(linker)–copper bond, leading to a partial loss of crystallinity, as observed by PXRD. Although a complete structure degradation was not observed, the authors proposed two possible phases for the water-degraded framework diffractogram: (i) a one-dimensional chain of Cu:BTC coordination polymer and (ii) a layered structure of [Cu2OH(BTC)(H2O)].
SIFSIX-3-M (M = Fe, Co, Ni, Cu, Zn)
The series SIFSIX-3-M are a class of hybrid ultra-microporous materials with pore size smaller than 0.7 nm. The SIFSIX family differs from other MOFs by the employment of both organic (pyrazine, pyz) and inorganic ligands (SiF62−) in their structure. The combination of these two factors enhances its high selectivity towards CO2, once the ultra-microporosity approaches the kinetic diameter of CO2 and the inorganic ligands contribute to a stronger electrostatic interaction with polarizable guests.75 Regarding their topology, while the organic ligand pyrazine grants an extended 2D framework in the x-y plane, the inorganic anions SiF62− function as pillars, which provide the 3D extension of the latter through bonding of F–Si–F to the M site in the z axis (Fig. 6a and b). For the SIFSIX-3-M family, the metal M2+ has already been described as Fe, Co, Ni, Cu, Zn, and the general formula for this MOF can be taken as [M(pyz)2SiF6]n.25 | ||
| Fig. 6 a) Top view SIFSIX-3-M framework representation and b) an up-close view of the pore whereas the equatorial fluorine atoms point inwards, both exemplified by the variant SIFSIX-3-Zn. Atom colors: F = cyan, Zn = purple, Si = yellow, H = white, C = gray, N = blue (reprinted with permission from Cryst. Growth Des. 2016, 16, 3890–3897. Copyright (2016) American Chemical Society). | ||
Recent advances on the insights of CO2 adsorption on the SIFSIX-3-M (Fe, Co, Ni, Cu, Zn) were experimentally and theoretically explored by Forrest et al.25 Regarding the CO2 sorption isotherms at 298 K for the SIFSIX-3-M family, the results found for the 0.10 atm region pointed out interesting results. Indeed, the MOFs that presented relatively larger pore sizes, namely SIFSIX-3-Fe and SIFSIX-3-Zn, exhibited a lower CO2 uptake, which was attributed to a non-optimal interaction between the CO2 molecule and the MOF (Table 2).
| Material | S BET (m2 g−1) | Pore Sizea,25 (Å) | Exp. CO2 uptakeref (mmol g−1) | Exp. Qstref (kJ mol−1) |
|---|---|---|---|---|
| a The pore size was considered as taking the diagonal F⋯F distance across the channel of the crystal structure as the pore size, minus the van der Waals radius of each F atom (Fig. 6b). | ||||
| SIFSIX-3-Fe | 358 (ref. 26) | 3.84 | 2.43/2.83 (ref. 25) | 42 (ref. 25) |
| SIFSIX-3-Zn | 250 (ref. 27) | 3.84 | 2.43/2.64 (ref. 27) | 45 (ref. 27) |
| SIFSIX-3-Co | 223 (ref. 28) | 3.77 | 2.51/2.79 (ref. 28) | 47 (ref. 28) |
| SIFSIX-3-Ni | 368 (ref. 28) | 3.55 | 2.68/2.88 (ref. 28) | 51 (ref. 28) |
| SIFSIX-3-Cu | 300 (ref. 31) | 3.54 | 2.46/2.58 (ref. 31) | 54 (ref. 31) |
Even though the Co and Ni homologues displayed higher CO2 uptake capacity than SIFSIX-3-Cu at 1.0 atm, the latter exhibited particularly a greater uptake in the 0.001 atm pressure range (Fig. 7a). This feature was attributed to its smallest pore size within the SIFSIX-3-M family, thus ensuring a higher CO2 affinity. This explanation was also in agreement with the experimental isosteric heats of adsorption, described as SIFSIX-3-Cu > SIFSIX-3-Ni > SIFSIX-3-Co > SIFSIX-3-Zn > SIFSIX-3-Fe (Table 2). Importantly, the heat of adsorption values were basically constant throughout the selected interval of CO2 uptake (Fig. 7b), indicating that only one type of adsorption site is present in those materials. Further investigations by periodic DFT calculations employing radial distribution functions showed that the preferential position for the CO2 molecule is usually at the centre of the pore. SIFSIX-3-Cu displayed a peculiar behaviour, whereas the CO2 molecule within this material is much closer to the SiF62− pillars than in other analogues. This was attributed to the smaller pore size of this material, which makes this particular F⋯CO2 distance inevitably closer. Furthermore, its particular shorter a/b cell lattice parameters and elongated c lattice were linked to the Jahn–Teller effect, resulting in a smaller average distance between the CO2 molecule and the MOF pillars in SIFSIX-3-Cu due to its d9 open shell configuration.
 | ||
| Fig. 7 a) CO2 adsorption isotherms of SIFSIX-3-M at 298 K for pressures up to 0.1 atm and b) isosteric heat of adsorption for SIFSIX-3-M (reprinted with permission from Cryst. Growth Des. 2019, 19, 3732–3743. Copyright (2019) American Chemical Society). | ||
SIFSIX-3-Cu was first reported by Shekhah et al.31 in 2014. The rational reason for employing the copper cation lied promptly on the consequences of the said Jahn–Teller effect, as they observed the direct enhancement of CO2 adsorption by pore contraction within the SIFSIX family. As expected, SIFSIX-3-Cu presented a smaller unit cell than its Zn analogue and based on the higher affinity towards CO2 of SIFSIX-3-Cu at lower pressures, the authors were able to target the direct air capture (DAC) of CO2. The DAC is a particularly challenging but useful application, since it requests an extremely CO2-philic material at low CO2 concentration. For the CO2 adsorption experiments, the steeper adsorption branch of the CO2 isotherm of SIFSIX-3-Cu in a series of experiments at low CO2 pressures (400 ppm – 5%) showed higher CO2 affinity for this hybrid ultra-microporous material. Compared with other adsorbents, it adsorbed 86.2 cm3(STP) cm−3 of CO2 at 0.01 bar, versus 55 cm3(STP) cm−3 and 28 cm3(STP) cm−3 regarding SIFSIX-3-Zn and MOF-74-Mg, respectively. Likewise, the breakthrough experiments for SIFSIX-3-Cu were performed using 1000 ppm of diluted CO2 in N2 under dry and humid conditions (relative humidity of 74%), whereas the latter did not interfere with the CO2 adsorption properties. The stability towards humidity highlights an important advantage of employing a saturated metal site-type material as an adsorbent, once it is not deactivated by complexation of the water molecule on the open metal site, as observed in MOF-74-Mg and HKUST-1. Remarkably, the CO2 selectivity under breakthrough conditions induced a 50% enhancement for SIFSIX-3-Cu with respect to SIFSIX-3-Zn. This extreme CO2-philic behaviour of the copper homologue was attributed to a higher charge density due to smaller pores in SIFSIX-3-Cu, whereas the fluorine atoms can interact in a closer fashion with the guest molecule. Besides, the adsorption kinetics favoured a rapid CO2 adsorption compared with the N2 molecule. Finally, the substitution of Zn by Cu in the SIFSIX structure also resulted in an increase of 20% in the isosteric heat of adsorption, from 45 kJ mol−1 to 54 kJ mol−1 (Table 2), which did not interfere with the full regeneration of the adsorbent up to 4 cycles after evacuation or under a N2 flow.
Regarding the effect of pore size and chemical composition upon CO2 adsorption on SIFSIX-3-M, Ziaee et al.75 employed classical grand canonical Monte Carlo (GCMC), classical molecular dynamics (MD) and DFT to investigate those features on SIFSIX-3-Cu, SIFSIX-3-Ni and SIFSIX-3-Zn. Firstly, from a structural point of view, they studied the specific position of the equatorial fluorine atoms. By employing DFT-2D optimization, they concluded that fluorine atoms form a θ = 45° angle with respect to the a lattice in all studied materials (Fig. 6b). This angle remains unchanged even when the pore is loaded with CO2 molecules, in which the authors attributed this orientation to the highest degree of interaction between the electronegative fluorine and the electropositive carbon from carbon dioxide. Accordingly, they also found that the smaller pore size, ruled ultimately by the cell parameter dimensions, granted higher CO2 affinity. Thus, the pore size hierarchy SIFSIX-3-Zn > SIFSIX-3-Ni > SIFSIX-3-Cu dictates both, (i) the distance between the fluorine atoms and the carbon of CO2 and (ii) the CO2 affinity, which was also confirmed by the same trend of the isosteric heat of adsorption. The effect of pore size was also explored by Eddaoudi and co-workers,76 which correlated the latter (related to the F⋯F distance, Fig. 6b) with the strength of CO2 adsorption: SIFSIX-3-Zn (Qst = 45 kJ mol−1, F⋯F dist. = 6.784 Å), SIFSIX-3-Ni (Qst = 47 kJ mol−1, F⋯F dist. = 6.694 Å) and SIFSIX-3-Cu (Qst = 54 kJ mol−1, F⋯F dist. = 6.483 Å). Notably, Ziaee et al.75 attributed two main forces to the CO2–SIFSIX-3M complex, (i) an electrostatic attraction between the electronegative equatorial fluorine of SiF62− and the electropositive carbon of CO2 and (ii) the repulsion between the fluorine atoms and the electronegative oxygen atoms of CO2. Thus, the optimized CO2 position was evidenced by its presence in the centre of the pore (Fig. 8), whereas the attraction and repulsion interactions are maximized and minimized, respectively, leading to the most stable host–guest structure. Additionally, the CO2 orientation within the material is calculated when this molecule lies vertically at the pore centre, with the carbon atom equidistant to the four equatorial fluorine atoms. It is worth mentioning that this interaction is notably stronger for SIFSIX-3-Cu, which holds the smallest a and b cell parameters. Furthermore, simulated CO2 isotherms also showed excellent uptake at lower pressures for SIFSIX-3-Cu, SIFSIX-3-Ni and SIFSIX-3-Zn, highlighting this family as promising candidates towards DAC applications, which were experimentally investigated by other groups.25,27,28,31
 | ||
| Fig. 8 Optimized position of the CO2 molecule at the centre of the pore of the SIFSIX-3-M structure (reprinted with permission from Cryst. Growth Des. 2016, 16, 3890–3897. Copyright (2016) American Chemical Society). | ||
In 2015, SIFSIX-3-Co and SIFSIX-3-Ni materials were reported for the first time by Elsaidi et al.,28 which exhibited high CO2 affinity for both dry and wet flue gas, taking advantage of the water-resistant feature of those saturated metal sites MOFs. The CO2 uptakes for SIFSIX-3-Co and SIFSIX-3-Ni were 62.6 cm3 g−1 and 64.5 cm3 g−1 at 298 K and at 1 atm, respectively, with their isosteric heat of adsorption summarized in Table 2. Their N2 uptakes were remarkably low, exhibiting values of ca. 5 cm3 g−1 for the copper and the nickel variants. The higher selectivity towards CO2 was mainly attributed, especially in this case, to the pore size of the MOFs. Since they possess pore sizes slightly above the kinetic diameter of CO2 (3.3 Å12) and comparable to the kinetic diameter of N2 (3.64 Å12), their relatively higher CO2 uptake over N2 was correlated with the pore size tuning derived from the metal centre substitution in the SIFSIX-3-M, indicating that even slightly structural changes can impact their adsorption properties. Furthermore, column breakthrough experiments for SIFSIX-3-M (Ni, Co, Zn, Cu) were performed under a 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :85 CO2/N2 flow, as it represents the usual flue gas composition. As expected, a superior affinity of both materials towards CO2 rather than N2 was observed, given the much shorter breakthrough time of the latter. Importantly, when a humid gas stream was employed (50% humidity) instead of the dry one, the breakthrough times were basically unchanged, demonstrating experimentally the remarkable water stability of these SIFSIX-3-M homologues. Their metal saturated nature renders them an advantage of inert property to moisture, when comparing to the open metal site MOFs that can be easily coordinated by water, e.g. MOF-74-Mg and HKUST-1. To further understand the CO2 adsorption mechanism in the frameworks of SIFSIX-Co and Ni, in situ PXRD results suggested that the CO2 molecule was located at the centre of the squared-channels, as later evidenced for other SIFSIX-3-M homologues.25,75 Additionally, modelling studies in SIFSIX-3-M (Zn, Cu, Ni, Co) showed that there is a correlation between the strength of interaction of SiF62−⋯CO2 and the lattice parameters a and b, whereas the shorter the lattices a and b, the stronger the SiF62−⋯CO2 interaction.
:85 CO2/N2 flow, as it represents the usual flue gas composition. As expected, a superior affinity of both materials towards CO2 rather than N2 was observed, given the much shorter breakthrough time of the latter. Importantly, when a humid gas stream was employed (50% humidity) instead of the dry one, the breakthrough times were basically unchanged, demonstrating experimentally the remarkable water stability of these SIFSIX-3-M homologues. Their metal saturated nature renders them an advantage of inert property to moisture, when comparing to the open metal site MOFs that can be easily coordinated by water, e.g. MOF-74-Mg and HKUST-1. To further understand the CO2 adsorption mechanism in the frameworks of SIFSIX-Co and Ni, in situ PXRD results suggested that the CO2 molecule was located at the centre of the squared-channels, as later evidenced for other SIFSIX-3-M homologues.25,75 Additionally, modelling studies in SIFSIX-3-M (Zn, Cu, Ni, Co) showed that there is a correlation between the strength of interaction of SiF62−⋯CO2 and the lattice parameters a and b, whereas the shorter the lattices a and b, the stronger the SiF62−⋯CO2 interaction.
A recent contribution from Desveaux et al.77 investigated the CO2 adsorption process at a molecular level for SIFSIX-3-Zn with the combination of single-crystal XRD and solid-state NMR studies. They reported the crystal structure of the CO2-loaded SIFSIX-3-Zn for the first time, in which the pore size was 3.72 Å, slightly smaller than the guest-free homologue (3.84 Å25). Solid-state NMR was employed to explore the mobility of adsorbed CO2 on the MOF, since the SCXRD measurement at 110 K may underestimate the CO2 dynamic behavior due to the low-temperature influence on the guest motion. Thus, the NMR measurements performed at 223 K and 293 K were applied to describe the short-range structure changes of CO2 on SIFSIX-3-Zn, and spectral line-shapes could be simulated concerning the anisotropic surroundings of host–guest interactions. The spectra of the as-made SIFSIX-3-Zn (Fig. 9a) is broader than the activated one, evidenced by the greater coupling constant of 16.0 MHz, in comparison with 13.5 MHz. This was attributed to an enhancement of the local spherical symmetry around the Zn nucleus, due to the removal of the methanol used to synthesize SIFSIX-3-Zn. Additionally, the alike coupling constants of the activated and CO2-loaded material indicated that the Zn local structure remains unchanged upon adsorption. To directly investigate the interaction between CO2 and the MOF, a 19F → 13C CP-MAS experiment was performed (Fig. 9b). Since the CO2 peak at 124 ppm was only observable at longer contact times >4 ms (the peak at 146 ppm is attributed to the pyr ligands of the SIFSIX-3-Zn), the CO2 molecule is relatively far from the fluorine atoms, which is compatible with single-crystal XRD obtained for this CO2 loaded MOF. Furthermore, the rotational-echo double-resonance 13C{1H} REDOR was employed to verify the interaction between the protons of the pyr linker and the CO2 molecule. The analysis showed that the pyr protons also contributed to CO2 adsorption, for a relatively strong interaction between 13C–1H was detected due to the closer proximity of the protons to the carbon atom of CO2.
 | ||
| Fig. 9 a) Experimental and simulated 67Zn solid-state NMR spectra of SIFSIX-3-Zn; b) 19F → 13C CP/MAS ssNMR spectra of the CO2 loaded SIFSIX-3-Zn (reprinted with permission from J. Phys. Chem. C. 2019, 123, 17798–17807. Copyright (2019) American Chemical Society). | ||
ZIF-8
Besides the hybrid ultra-microporous materials, another MOF subgroup of relevance is the zeolitic-imidazolate frameworks (ZIFs). By taking advantage of the zeolite topology, this emerging class is designed by replacing the T-atoms of the aluminosilicate with transition metals and the oxygen atoms with imidazolate linkers to fabricate the so-called ZIFs.78 Formed by reaction between ZnNO3·6H2O and 2-methylimidazole in either DMF, methanol, or water, ZIF-8 bears its zinc atoms bridging the imidazolate likers to form a sodalite structure (isostructural to ZIF-9479 and SIM-180) that has windows of 3.4 Å diameter, which interconnects cages of 11.6 Å.32 Particularly, ZIF-8 is a flexible MOF, as it was demonstrated to bear a “swing effect” or “window opening” upon a continuous adsorption-induced deformation of N2 and other guests. This results in a high-pressure phase (HP) which differs from the ambient pressure one (AP).81–83 In particular, ZIF-8 is one of the most studied materials from the ZIF class, mainly due to its commercial availability, high BET surface areas (1300–1800 m2 g−1), thermal stability up to ca. 670 K, and its high resistance towards water, alkaline solutions, and organic solvents.34 Regarding the CO2 interaction features, this material takes advantage of its small pores comparable to the kinetic diameter of carbon dioxide and its hydrophobic framework, whereas the methyl groups in the imidazolate fragment offer a high ratio for the CO2/water selectivity.Liu et al.84 reported CO2 adsorption on ZIF-8 and its amino-functionalized analogues, ZIF-8-NH2 and ZIF-8-(NH2)2via grand canonical Monte Carlo simulations and DFT calculations. By calculating the CO2 adsorption isotherms in the range of 0 to 30 bar at 298 K, they found that at low pressures (below 12 bar), the CO2 capacity follows the trend: ZIF-8-(NH2)2 > ZIF-8-NH2 > ZIF-8, which could be attributed to the basicity of the NH2 groups and a higher quadrupole-π electron interaction in the amino-functionalized ZIFs. By bearing NH2 groups in their structure, the ZIF-8-NH2 and ZIF-8-(NH2)2 exhibit an enhanced interaction with the CO2 molecule due to their basic feature, favouring a stronger Lewis acid–base interaction. As an additional effect, the electron donor characteristic of the NH2 groups increased the electronic density of the π electrons in the imidazolate scaffold. This effect enhanced the quadrupole interaction between the CO2 molecule and the imidazolate ring, thus resulting in a higher quadrupole-π electron interaction for the amino-decorated ZIFs. To explore the framework integrity upon CO2 adsorption in the bare scaffold of ZIF-8, Du et al.34 carried out adsorption–desorption CO2 isotherms at 298 K and 103 kPa, exhibiting a CO2 uptake of 10.4 cm3 g−1 for ZIF-8. The XANES analysis showed that the oxidation state of the zinc atom did not change during the process of CO2 adsorption, revealing the redox stability of the Zn2+-based framework. Additionally, EXAFS experiments were performed to determine its structural parameters. The results showed no changes in the coordination number of ZIF-8, indicating that there is no change in the short-range local structure around the zinc atoms in this material during the CO2 adsorption.
In an in-depth molecular approach, Hu et al.85 investigated CO2 adsorption on ZIF-8 scaffold at high pressures via in situ FTIR spectroscopy. By means of tracking the CO2 insertion within the pores through analysing the CO2 overtone/combination bands,86 the authors suggested the existence of two different CO2 species upon adsorption, one that stays outside of the framework and another that is inserted in ZIF-8. Additionally, a pressure-regulated mechanism was found to be responsible for a cyclic CO2 loading–unloading process. Initially, the spectrum of CO2 on ZIF-8 at 0.47 GPa (Fig. 10a, bottom) is similar to the profile of pure CO2 spectrum. When the CO2 pressure is increased (Fig. 10a), a new peak at a higher wavenumber appears, associated with the CO2 trapped inside the pores. For the reverse process of decompression, it is possible to observe that this doublet merges back into a singlet at the pressure of 0.47 GPa, indicating that the carbon dioxide molecules located inside are removed from the ZIF-8 structure. This process was reported to endure several cycles reversibly, and always presented the peak splitting at 0.4–0.8 GPa. Furthermore, by analysing the absorption intensity, it was possible to estimate that approximately 68% of loaded CO2 was adsorbed by the ZIF-8 upon compression, whilst a minimum of 45% still remained trapped in the framework upon decompression. Regarding the mobility of CO2 molecules, Chmelik78 investigated the diffusion of small probe molecules in ZIF-8 by IR micro-imaging. It was found that an enhanced diffusivity in ZIF-8 was associated with loadings of 3 molecules of carbon dioxide per cage, with saturation loadings reaching up to 16–18 molecules per cage at 198 K. This surprising mobility of CO2 molecules was associated with a phenomenon that acts upon systems with large cages and small windows, resulting in an increase of molecular mobility. This behaviour was suggested to occur due to the intermolecular repulsive forces among the guests due to the narrow windows, pushing these molecules into the next and further cavity. Typically, these repulsion forces lower the energy barrier for this “push” to another cavity, thus facilitating this process even at lower loadings for certain guests.
 | ||
| Fig. 10 IR spectra of CO2 adsorption on ZIF-8 at room temperature for the a) compression and b) decompression modes (reprinted with permission from J. Am. Chem. Soc. 2013, 135, 9287–9290. Copyright (2013) American Chemical Society). | ||
Tian et al.87 studied the CO2 adsorption on ZIF-8 films by employing temperature-programmed desorption (TPD). By using both surface and a bulk-responsive technique, it was possible to differentiate the adsorption behaviour that occurs specifically at the surface and in the totality of the film, respectively. As shown in Fig. 11c, three different temperatures were observed related to specific events, β1 (105 K), α1 (116 K), and α2 (ca. 175 K). The β1 peak was assigned to the desorption of CO2 multilayer, since further saturation of this peak was not possible in their experiments. The α1 and the α2 peaks were attributed to CO2 molecules within the pores of ZIF-8 and the ones caught in diffusion traps of the ZIF film. These diffusion traps among the nanoparticles would hinder the CO2 mobility into and out of the film, whereas the CO2 molecules located at the pores move quite easily due to an open channel system between connected cages. Hence, one would expect a lower temperature for the latter, and it is suggested that α2 > α1, where α is the temperature for the occurrence of these events. To confirm their hypothesis, the authors attempted to diminish the diffusion traps by heating up the as-prepared film (Fig. 11a) at 500 K, which caused particles agglomeration and led to a more uniform film (Fig. 11b). After this procedure, the improved film was submitted to the same TPD experiment. The α2 peak greatly diminished and the peak α1 was sharpened, confirming the aforementioned attributions for those α peaks.
 | ||
| Fig. 11 SEM micrographs of ZIF-8 film a) as-prepared and b) after annealing at 500 K for 30 min and c) TPD spectra of CO2 desorption (reprinted with permission from J. Phys. Chem. C. 2015, 119, 15248–15253. Copyright (2015) American Chemical Society). | ||
Regarding CO2 separations in humid conditions, ZIF-8 presents a hydrophobic behaviour due to its methyl groups that hinder the diffusion of water in its pores,88 which has been widely investigated towards the enhancement of methane adsorption under hydrate formation conditions.89 For instance, Liu et al.90 investigated the use of ZIF-8 for CO2 adsorption under humid conditions of 30 wt% and 77 wt%. They reported an increase of CO2 adsorption in the aqueous ZIF-8 system, reaching up 39% of CO2 adsorption enhancement for 77 wt% humidity conditions. However, such feature was related to an irreversible collapse of the structure of ZIF-8, caused by a combination of both water and acidic media. The change in the structure was attributed to the chemical reaction between carbonate ions, formed upon CO2 interaction with water, and the metal node of ZIF-8 to form a stable zinc carbonate and 2-methylimidazole ligand (Fig. 12). The irreversible nature of this transformation was attributed to the poor solubility of the zinc carbonate in water, thus impairing an equilibrium reaction and leading to a permanent collapse of ZIF-8 framework. The formation of these species was confirmed by spectroscopic techniques, such as FT-IR and Raman. It was clear that the characteristic bands related to the ZIF-8 in the infrared spectrum were replaced by the zinc carbonate ones, and the –NH stretching modes appeared as the imidazolate linkers were protonated in the process of structure dissociation. Likewise, this –NH band could be assessed by Raman, which showed the typical peak at 3169 cm−1 for the water exposed samples. Additionally, the PXRD patterns of pristine and humid samples were compared to confirm a complete destruction of ZIF-8 structure after contact with water at longer exposure times. This change was also followed by SEM analysis, indicating the formation of bigger aggregates that differed from ZIF-8 pristine morphology. By performing these CO2 adsorption studies in glycol instead of water, no structure collapse could be observed, thus suggesting that the combination of water and CO2 unravelled the undesired reaction that led to ZIF-8 permanent structure decomposition and, subsequently, loss of porosity.
 | ||
| Fig. 12 Permanent collapse of ZIF-8 structure upon CO2 adsorption in aqueous media (reprinted with permission from J. Phys. Chem. C. 2016, 120, 13287–13294. Copyright (2016) American Chemical Society). | ||
Cryogenic temperature studies of CO2 adsorption were studied by Russell et al.,91 in which they investigated the CO2 adsorption from 133 K to 237 K. The CO2 isotherms (Fig. 13a) showed two steps: one at lower specific loadings (from ca. 2 mmol g−1 to 10 mmol g−1) related to the gas adsorption, and a saturation step starting at approximately 12 mmol g−1, corresponding to the saturated vapor pressure for CO2 at the measured temperature. On the contrary to other systems, the sub-step related to the phase transition was not present in those isotherms. As reported in other studies, ZIF-8 exhibits additional substeps related to the HP → AP phase transition when exposed to Ar,92 O2,93 N2,94 CO (ref. 94) and Xe.95 However, the absence of this step is not a confirmation that there is no phase transition upon CO2 adsorption, as theoretical simulations have shown that, for this guest, such transitions could occur around room temperature and high pressures.82 Additionally, it was also shown that the phase transition steps regarding Xe adsorption on ZIF-8 disappeared at temperatures below 145 K.95 The isosteric heat of adsorption (Fig. 13b) shows a coverage dependence, whereas it has a value of 24.2 kJ mol−1 up to 9 mmol g−1, reaching a maximum value of 28.8 kJ mol−1 when the coverage is 11.5 mmol g−1. This distinct isosteric heat maximum coincided with the reported value of the latent heat of CO2 sublimation that occurs when CO2 saturation takes place. For coverages measured after vapor pressure saturation, the isosteric heat returns to a value of ca. 26 kJ mol−1. This coverage-dependence of the adsorption heat was comparable to exfoliated graphite, whereas the isosteric heat of adsorption increases towards the bulk heat of transition as the pressure increases and the saturated vapor pressure is reached.96,97
 | ||
| Fig. 13 a) CO2 adsorption isotherms on ZIF-8 at a range of cryogenic temperatures and b) the isosteric heat of adsorption as a function of the surface coverage (reprinted from Russel, B. A., Migone, A. D. Microporous Mesoporous Mat. 2017, 246, 178–185. Low-temperature adsorption study of CO2 in ZIF-8, copyright (2017), with permission from Elsevier). | ||
Conclusions
In summary, four types of MOFs were reviewed in this study, as their preferential adsorption sites, water stability, CO2–MOF complex configuration, CO2 adsorption dynamics, and swing effects were described by experimental and theoretical data (diffraction studies, spectroscopic techniques, theoretical calculations, solid-state NMR, and water vapor experiments). The high CO2 uptake of MOF-74-Mg and HKUST-1 is granted by their CO2–OMS interaction, which can be disturbed by the presence of water molecules, which compete for the metal node interaction. The flexible ZIF-8, however, is stable to the sole presence of water, but the combination of water + CO2 destroys the structure by the formation of a stable zinc carbonate, also impairing its use for practical adsorption conditions under a humid CO2 flow. Preparation of MOFs that are water stable is of utter importance, as, realistically, drying of the CO2 flow would be impractical and expensive. The SIFSIX-3 series represent a set of materials that are stable to humid conditions, and even though they do not possess the highest CO2 adsorption uptakes, its moisture stability grants them recyclability potential and the possibility to enhance drastically these uptake values by their recuperation and reuse in several cycles. Finally, it is expected that this review may encourage further studies devoted to CO2 adsorption mechanisms applied to other classes of MOFs and porous solids in general, aiming to better comprehend their particularities, and to further fine-tune the design of high capacity and water stable CO2 capture materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the Chinese Science Council and the French Ministry for Foreign Affairs for funding their Cai Yuanpei project. RAM and BL are grateful to University of Strasbourg Institute for Advanced Study (USIAS) for its support. The authors declare no conflict of interest.References
- M. I. Nandasiri, S. R. Jambovane, B. P. McGrail, H. T. Schaef and S. K. Nune, Coord. Chem. Rev., 2016, 311, 38–52 CrossRef CAS.
- R. Ben-Mansour, M. A. Habib, O. E. Bamidele, M. Basha, N. A. A. Qasem, A. Peedikakkal, T. Laoui and M. Ali, Appl. Energy, 2016, 161, 225–255 CrossRef CAS.
- J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- M. Alhamami, H. Doan and C. H. Cheng, Materials, 2014, 7, 3198–3250 CrossRef PubMed.
- H. Li, K. Wang, Y. Sun, C. T. Lollar, J. Li and H. C. Zhou, Mater. Today, 2018, 21, 108–121 CrossRef CAS.
- M. Younas, M. Rezakazemi, M. Daud, M. B. Wazir, S. Ahmad, N. Ullah and S. Ramakrishna, Prog. Energy Combust. Sci., 2020, 80, 100849 CrossRef.
- C. Petit, Curr. Opin. Chem. Eng., 2018, 20, 132–142 CrossRef.
- A. Alonso, J. Moral-Vico, A. A. Markeb, M. Busquets-Fité, D. Komilis, V. Puntes, A. Sánchez and X. Font, Sci. Total Environ., 2017, 595, 51–62 CrossRef CAS PubMed.
- C. A. Trickett, A. Helal, B. A. Al-Maythalony, Z. H. Yamani, K. E. Cordova and O. M. Yaghi, Nat. Rev. Mater., 2017, 2, 1–16 Search PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- J. Yu, L. H. Xie, J. R. Li, Y. Ma, J. M. Seminario and P. B. Balbuena, Chem. Rev., 2017, 117, 9674–9754 CrossRef CAS PubMed.
- J. Liu, P. K. Thallapally, B. P. McGrail, D. R. Brown and J. Liu, Chem. Soc. Rev., 2012, 41, 2308–2322 RSC.
- D. K. Yoo, H. C. Woo and S. H. Jhung, Coord. Chem. Rev., 2020, 422, 213477 CrossRef CAS.
- X. Wu, Z. Bao, B. Yuan, J. Wang, Y. Sun, H. Luo and S. Deng, Microporous Mesoporous Mater., 2013, 180, 114–122 CrossRef CAS.
- J. A. Mason, K. Sumida, Z. R. Herm, R. Krishna and J. R. Long, Energy Environ. Sci., 2011, 4, 3030 RSC.
- R. Ben-Mansour and N. A. A. Qasem, Energy Convers. Manage., 2018, 156, 10–24 CrossRef CAS.
- Z. Bao, L. Yu, Q. Ren, X. Lu and S. Deng, J. Colloid Interface Sci., 2011, 353, 549–556 CrossRef CAS PubMed.
- P. D. C. Dietzel, V. Besikiotis and R. Blom, J. Mater. Chem., 2009, 19, 7362–7370 RSC.
- J. J. Gutiérrez-Sevillano, J. M. Vicent-Luna, D. Dubbeldam and S. Calero, J. Phys. Chem. C, 2013, 117, 11357–11366 CrossRef.
- X. Yan, S. Komarneni, Z. Zhang and Z. Yan, Microporous Mesoporous Mater., 2014, 183, 69–73 CrossRef CAS.
- J. Moellmer, A. Moeller, F. Dreisbach, R. Glaeser and R. Staudt, Microporous Mesoporous Mater., 2011, 138, 140–148 CrossRef CAS.
- A. Kumar, D. G. Madden, M. Lusi, K.-J. Chen, E. A. Daniels, T. Curtin, J. J. Perry and M. J. Zaworotko, Angew. Chem., Int. Ed., 2015, 54, 14372–14377 CrossRef CAS PubMed.
- P. Aprea, D. Caputo, N. Gargiulo, F. Iucolano and F. Pepe, J. Chem. Eng. Data, 2010, 55, 3655–3661 CrossRef CAS.
- L. Hamon, E. Jolimaître and G. D. Pirngruber, Ind. Eng. Chem. Res., 2010, 49, 7497–7503 CrossRef CAS.
- K. A. Forrest, T. Pham, S. K. Elsaidi, M. H. Mohamed, P. K. Thallapally, M. J. Zaworotko and B. Space, Cryst. Growth Des., 2019, 19, 3732–3743 CrossRef CAS.
- S. K. Elsaidi, M. H. Mohamed, C. M. Simon, E. Braun, T. Pham, K. A. Forrest, W. Xu, D. Banerjee, B. Space, M. J. Zaworotko and P. K. Thallapally, Chem. Sci., 2017, 8, 2373–2380 RSC.
- P. Nugent, E. G. Giannopoulou, S. D. Burd, O. Elemento, E. G. Giannopoulou, K. Forrest, T. Pham, S. Ma, B. Space, L. Wojtas, M. Eddaoudi and M. J. Zaworotko, Nature, 2013, 495, 80–84 CrossRef CAS PubMed.
- S. K. Elsaidi, M. H. Mohamed, H. T. Schaef, A. Kumar, M. Lusi, T. Pham, K. A. Forrest, B. Space, W. Xu, G. J. Halder, J. Liu, M. J. Zaworotko and P. K. Thallapally, Chem. Commun., 2015, 51, 15530–15533 RSC.
- O. Shekhah, Y. Belmabkhout, K. Adil, P. M. Bhatt, A. J. Cairns and M. Eddaoudi, Chem. Commun., 2015, 51, 13595–13598 RSC.
- M. Khraisheh, F. Almomani and G. Walker, Sci. Rep., 2020, 10, 1–12 CrossRef PubMed.
- O. Shekhah, Y. Belmabkhout, Z. Chen, V. Guillerm, A. Cairns, K. Adil and M. Eddaoudi, Nat. Commun., 2014, 5, 4228 CrossRef CAS PubMed.
- K. S. Park, Z. Ni, A. P. Cote, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS PubMed.
- D. G. Madden, H. S. Scott, A. Kumar, K.-J. Chen, R. Sanii, A. Bajpai, M. Lusi, T. Curtin, J. J. Perry and M. J. Zaworotko, Philos. Trans. R. Soc., A, 2017, 375, 20160025 CrossRef PubMed.
- M. Du, L. Li, M. Li and R. Si, RSC Adv., 2016, 6, 62705–62716 RSC.
- M. Etxeberria-Benavides, T. Johnson, S. Cao, B. Zornoza, J. Coronas, J. Sanchez-Lainez, A. Sabetghadam, X. Liu, E. Andres-Garcia, F. Kapteijn, J. Gascon and O. David, Sep. Purif. Technol., 2020, 237, 116347 CrossRef CAS.
- Z. Zhang, W. Zhang, X. Chen, Q. Xia and Z. Li, Sep. Sci. Technol., 2010, 45, 710–719 CrossRef CAS.
- C. Chen, D.-W. Park and W.-S. Ahn, Appl. Surf. Sci., 2014, 292, 63–67 CrossRef CAS.
- J.-S. Lee, J.-H. Kim, J.-T. Kim, J.-K. Suh, J.-M. Lee and C.-H. Lee, J. Chem. Eng. Data, 2002, 47, 1237–1242 CrossRef CAS.
- S. Cavenati, C. A. Grande and A. E. Rodrigues, J. Chem. Eng. Data, 2004, 49, 1095–1101 CrossRef CAS.
- S.-Y. Lee and S.-J. Park, J. Colloid Interface Sci., 2013, 389, 230–235 CrossRef CAS PubMed.
- N. A. Rashidi and S. Yusup, J. CO2 Util., 2016, 13, 1–16 CrossRef CAS.
- D. Li, J. Zhou, Z. Zhang, L. Li, Y. Tian, Y. Lu, Y. Qiao, J. Li and L. Wen, Carbon, 2017, 114, 496–503 CrossRef CAS.
- J. Silvestre-Albero, A. Wahby, A. Sepúlveda-Escribano, M. Martínez-Escandell, K. Kaneko and F. Rodríguez-Reinoso, Chem. Commun., 2011, 47, 6840 RSC.
- M. Sevilla, W. Sangchoom, N. Balahmar, A. B. Fuertes and R. Mokaya, ACS Sustainable Chem. Eng., 2016, 4, 4710–4716 CrossRef CAS.
- P. D. C. Dietzel, R. Blom and H. Fjellvåg, Eur. J. Inorg. Chem., 2008, 2008, 3624–3632 CrossRef.
- H. Wu, J. M. Simmons, G. Srinivas, W. Zhou and T. Yildirim, J. Phys. Chem. Lett., 2010, 1, 1946–1951 CrossRef CAS.
- L. Valenzano, B. Civalleri, S. Chavan, G. T. Palomino, C. O. Areán and S. Bordiga, J. Phys. Chem. C, 2010, 114, 11185–11191 CrossRef CAS.
- X. Kong, E. Scott, W. Ding, J. A. Mason, J. R. Long and J. A. Reimer, J. Am. Chem. Soc., 2012, 134, 14341–14344 CrossRef CAS PubMed.
- L.-C. Lin, J. Kim, X. Kong, E. Scott, T. M. McDonald, J. R. Long, J. A. Reimer and B. Smit, Angew. Chem., Int. Ed., 2013, 52, 4410–4413 CrossRef CAS PubMed.
- B. E. G. Lucier, H. Chan, Y. Zhang and Y. Huang, Eur. J. Inorg. Chem., 2016, 2016, 2017–2024 CrossRef CAS.
- W. D. Wang, B. E. G. Lucier, V. V. Terskikh, W. Wang and Y. Huang, J. Phys. Chem. Lett., 2014, 5, 3360–3365 CrossRef CAS PubMed.
- Y. Lu, B. E. G. Lucier, Y. Zhang, P. Ren, A. Zheng and Y. Huang, Phys. Chem. Chem. Phys., 2017, 19, 6130–6141 RSC.
- S. Chen, B. E. G. Lucier, P. D. Boyle and Y. Huang, Chem. Mater., 2016, 28, 5829–5846 CrossRef CAS.
- S. Bin Baek and H. C. Lee, Bull. Korean Chem. Soc., 2016, 37, 588–591 CrossRef.
- D. Bernin and N. Hedin, Curr. Opin. Colloid Interface Sci., 2018, 33, 53–62 CrossRef CAS.
- S. Zuluaga, E. M. A. Fuentes-Fernandez, K. Tan, F. Xu, J. Li, Y. J. Chabal and T. Thonhauser, J. Mater. Chem. A, 2016, 4, 5176–5183 RSC.
- K. Tan, S. Zuluaga, Q. Gong, P. Canepa, H. Wang, J. Li, Y. J. Chabal and T. Thonhauser, Chem. Mater., 2014, 26, 6886–6895 CrossRef CAS.
- K. Tan, N. Nijem, Y. Gao, S. Zuluaga, J. Li, T. Thonhauser and Y. J. Chabal, CrystEngComm, 2015, 17, 247–260 RSC.
- E. Fuentes-Fernandez, S. Jensen, K. Tan, S. Zuluaga, H. Wang, J. Li, T. Thonhauser and Y. Chabal, Appl. Sci., 2018, 8, 270 CrossRef.
- E. Haldoupis, J. Borycz, H. Shi, K. D. Vogiatzis, P. Bai, W. L. Queen, L. Gagliardi and J. I. Siepmann, J. Phys. Chem. C, 2015, 119, 16058–16071 CrossRef CAS.
- M. Mohamedali, A. Henni and H. Ibrahim, Microporous Mesoporous Mater., 2019, 284, 98–110 CrossRef CAS.
- C. Y. Chuah, W. Li, S. A. S. C. Samarasinghe, G. S. M. D. P. Sethunga and T.-H. Bae, Microporous Mesoporous Mater., 2019, 290, 109680 CrossRef.
- J. Cortés-Súarez, V. Celis-Arias, H. I. Beltrán, A. Tejeda-Cruz, I. A. Ibarra, J. E. Romero-Ibarra, E. Sánchez-González and S. Loera-Serna, ACS Omega, 2019, 4, 5275–5282 CrossRef PubMed.
- S. S. Y. Chui, S. M. F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS PubMed.
- S. Bordiga, L. Regli, F. Bonino, E. Groppo, C. Lamberti, B. Xiao, P. S. Wheatley, R. E. Morris and A. Zecchina, Phys. Chem. Chem. Phys., 2007, 9, 2676 RSC.
- F. Gul-E-Noor, B. Jee, A. Pöppl, M. Hartmann, D. Himsl and M. Bertmer, Phys. Chem. Chem. Phys., 2011, 13, 7783 RSC.
- L. Grajciar, A. D. Wiersum, P. L. Llewellyn, J.-S. Chang and P. Nachtigall, J. Phys. Chem. C, 2011, 115, 17925–17933 CrossRef CAS.
- W. Wong-Ng, I. Levin, J. A. Kaduk, L. Espinal and H. Wu, J. Alloys Compd., 2016, 656, 200–205 CrossRef CAS.
- B. Supronowicz, A. Mavrandonakis and T. Heine, J. Phys. Chem. C, 2013, 117, 14570–14578 CrossRef CAS.
- E. J. Granite and H. W. Pennline, Ind. Eng. Chem. Res., 2002, 41, 5470–5476 CrossRef CAS.
- N. Al-Janabi, P. Hill, L. Torrente-Murciano, A. Garforth, P. Gorgojo, F. Siperstein and X. Fan, Chem. Eng. J., 2015, 281, 669–677 CrossRef CAS.
- N. Al-Janabi, A. Alfutimie, F. R. Siperstein and X. Fan, Front. Chem. Sci. Eng., 2016, 10, 103–107 CrossRef CAS.
- W. Xue, Z. Zhang, H. Huang, C. Zhong and D. Mei, J. Phys. Chem. C, 2020, 124, 1991–2001 CrossRef CAS.
- J. R. Álvarez, E. Sánchez-González, E. Pérez, E. Schneider-Revueltas, A. Martínez, A. Tejeda-Cruz, A. Islas-Jácome, E. González-Zamora and I. A. Ibarra, Dalton Trans., 2017, 46, 9192–9200 RSC.
- A. Ziaee, D. Chovan, M. Lusi, J. J. Perry, M. J. Zaworotko and S. A. M. Tofail, Cryst. Growth Des., 2016, 16, 3890–3897 CrossRef CAS.
- P. M. Bhatt, Y. Belmabkhout, A. Cadiau, K. Adil, O. Shekhah, A. Shkurenko, L. J. Barbour and M. Eddaoudi, J. Am. Chem. Soc., 2016, 138, 9301–9307 CrossRef CAS PubMed.
- B. E. Desveaux, Y. T. A. Wong, B. E. G. Lucier, V. V. Terskikh, P. D. Boyle, S. Jiang and Y. Huang, J. Phys. Chem. C, 2019, 123, 17798–17807 CrossRef CAS.
- C. Chmelik, Microporous Mesoporous Mater., 2015, 216, 138–145 CrossRef CAS.
- T. Johnson, M. M. Łozińska, A. F. Orsi, P. A. Wright, S. Hindocha and S. Poulston, Green Chem., 2019, 21, 5665–5670 RSC.
- A. M. Marti, M. Van and K. J. Balkus, J. Porous Mater., 2014, 21, 889–902 CrossRef CAS.
- F. Coudert, ChemPhysChem, 2017, 18, 2732–2738 CrossRef CAS PubMed.
- T. Chokbunpiam, S. Fritzsche, C. Chmelik, J. Caro, W. Janke and S. Hannongbua, Chem. Phys. Lett., 2016, 648, 178–181 CrossRef CAS.
- L. Zhang, Z. Hu and J. Jiang, J. Am. Chem. Soc., 2013, 135, 3722–3728 CrossRef CAS PubMed.
- D. Liu, Y. Wu, Q. Xia, Z. Li and H. Xi, Adsorption, 2013, 19, 25–37 CrossRef CAS.
- Y. Hu, Z. Liu, J. Xu, Y. Huang and Y. Song, J. Am. Chem. Soc., 2013, 135, 9287–9290 CrossRef CAS PubMed.
- M. Santoro, F. Gorelli, J. Haines, O. Cambon, C. Levelut and G. Garbarino, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 7689–7692 CrossRef CAS PubMed.
- F. Tian, A. M. Mosier, A. Park, E. R. Webster, A. M. Cerro, R. S. Shine and L. Benz, J. Phys. Chem. C, 2015, 119, 15248–15253 CrossRef CAS.
- G. Ortiz, H. Nouali, C. Marichal, G. Chaplais and J. Patarin, Phys. Chem. Chem. Phys., 2013, 15, 4888 RSC.
- L. Mu, B. Liu, H. Liu, Y. Yang, C. Sun and G. Chen, J. Mater. Chem., 2012, 22, 12246 RSC.
- H. Liu, P. Guo, T. Regueira, Z. Wang, J. Du and G. Chen, J. Phys. Chem. C, 2016, 120, 13287–13294 CrossRef CAS.
- B. Russell and A. Migone, Microporous Mesoporous Mater., 2017, 246, 178–185 CrossRef CAS.
- H. Tanaka, S. Ohsaki, S. Hiraide, D. Yamamoto, S. Watanabe and M. T. Miyahara, J. Phys. Chem. C, 2014, 118, 8445–8454 CrossRef CAS.
- B. Russell, J. Villaroel, K. Sapag and A. D. Migone, J. Phys. Chem. C, 2014, 118, 28603–28608 CrossRef CAS.
- C. O. Ania, E. García-Pérez, M. Haro, J. J. Gutiérrez-Sevillano, T. Valdés-Solís, J. B. Parra and S. Calero, J. Phys. Chem. Lett., 2012, 3, 1159–1164 CrossRef CAS PubMed.
- D. H. Gallaba, A. G. Albesa and A. D. Migone, J. Phys. Chem. C, 2016, 120, 16649–16657 CrossRef CAS.
- V. Krungleviciute, A. D. Migone, M. Yudasaka and S. Iijima, J. Phys. Chem. C, 2012, 116, 306–310 CrossRef CAS.
- M. Bienfait, P. Zeppenfeld, N. Dupont-Pavlovsky, M. Muris, M. R. Johnson, T. Wilson, M. DePies and O. E. Vilches, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 035410 CrossRef.
| This journal is © The Royal Society of Chemistry 2021 |