
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ sensors for flow reactors – a review
Jun
Li
 a,
Helena
Šimek
b,
David
Ilioae
c,
Nicole
Jung
b,
Stefan
Bräse
b,
Hans
Zappe
c,
Roland
Dittmeyer
a and
Bradley P.
Ladewig
*a
a,
Helena
Šimek
b,
David
Ilioae
c,
Nicole
Jung
b,
Stefan
Bräse
b,
Hans
Zappe
c,
Roland
Dittmeyer
a and
Bradley P.
Ladewig
*a
aInstitute for Micro Process Engineering (IMVT), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, Eggenstein-Leopoldshafen, 76344, Germany. E-mail: bradley.ladewig@kit.edu
bInstitute of Biological and Chemical Systems (IBCS-FMS), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, Eggenstein-Leopoldshafen, 76344, Germany
cGisela and Erwin Sick Laboratory for Micro-optics, Department of Microsystems Engineering, University of Freiburg, Germany
First published on 16th April 2021
Abstract
The integration of specific sensors into microfluidic reactors and devices is crucial for the optimization of controllable variables such as flow, temperature, energy input (light, microwaves etc.). In this review, we highlight the state of the art for the integration of in situ sensors.
Introduction
Microfluidic reactors have become increasingly sophisticated by the maturation of additive manufacturing1 and the enhancement of construction materials2 over the last two decades, starting from initial conceptual design to realistic, compact, automated platforms. Typical microfluidic reaction systems are constructed by microfabrication and fine-machining techniques and have served as one of the most attractive new reaction approaches in the natural sciences, involving chemistry, biochemistry, materials science and pharmaceutical industry.3–9 These systems, with dimensions of the inner channels typically in the range of 100 nanometres to several hundred micrometres, exhibit features like enhanced mass and heat transfer, improved safety, decreased reaction time, enhanced selectivity, the potential for high-throughput screening and integration of optical sensors for systematic monitoring in microfluidics. Sensors have endowed such fluidic devices with unexpectedly versatile and multifunctional utilization compared to that of conventional batch synthesis.10,11 In the meantime, the adoption of microfluidics for scientific studies is further encouraged by reported economic and eco-friendly metrics.Progress in microfluidic synthesis technology has also triggered novel demands in characterization and measurement. Calls for improving the performance of process analytical technology (PAT), which is termed as a unique system available to analyse, control, and optimize the critical product quality in a manufacturing process, have drawn considerable attention as highly sensitive and accurate analytics evolved half a century ago. Attempts to combine state-of-the-art analytical technology with microfluidic reactors for real-time measurements have promoted enhancements in flow chemistry, paving a new avenue to conduct kinetic and mechanistic studies at milliseconds to picoseconds processes.12 Traditional offline analytical methods (e.g., UV, IR, MS, NMR) are extensively used by the chemical community in both batch and flow synthesis. However, additional preparations for sampling and analysing are not well-matched with the inherent conveniences featured by flow reactors, while the integration of in situ optical sensors and other process-intensification techniques with microfluidics could be able to assemble an algorithm-modified automation platform with feedback mechanism and self-regulation,13 enabling continuous synthesis for potential industrial applications.14 These methods can further be exploited by machine-learning and artificial intelligence tools. In addition, modern data management tools be incorporated. A suitable case in this combination was shown by the Kappe group.15 They designed a real-time analytical platform for multiple synthesis of mesalazine, an active pharmaceutical ingredient (API), with three distinct steps: nitration, hydrolysis and hydrogenation, adopting four PAT tools (inline NMR, UV/vis and IR for three respective synthetic routines and online UHPLC for final quantification) as well as three inline separations. By comparison, deep-insight research might be compromised by difficulties in the capture of active intermediates or the differentiation of significant transient signals from noisy backgrounds immediately when employing comparatively time-lagged techniques. The association of process automation and flow apparatus is dedicated to undertaking automated and digital-oriented analysis comprehensively and smartly. All the aforementioned privileges contribute strong evidence of the importance of integration of real-time sensors with flow reactors for challenging academic and industrial purposes.
The combination of inline and online analytical methods with microreactors to constitute algorithm-assisted platforms was reviewed by Sans et al.,13 giving a profound overview of linking feedback algorithms and microreactor-embeddable inline/online analytics. Likewise, Baumann summarised and enumerated several cases in combining in-line purification and analysis techniques in telescoped multi-step sequences.16 However, literature outlining state-of-the-art cooperation between in situ sensors and flow reactors, which could be one of the most important issues in future flow chemistry, is rare. The following sections give an overview of recent discoveries with a particular focus on enabling in situ monitoring within microreactors for chemical transformations.
Offline, inline, online, in situ measurements: what is the best choice?
General definition
Offline measurements, the manual and interval interferences conducted by human individuals to investigate the state of a physical value of interest, have been extensively used as post-processing procedures in batch chemical synthesis for a long time. However, the accuracy of offline operations heavily relies on the professional skills of the operator. In addition to inevitable differentiation caused by the defects of analytic machines, the extra risks that can occur in manual processes may increase artificial errors and compromise results (such as incomplete reaction quenching), which may make offline measurements not as precise as other detecting operations.17Inline measurements, commonly implemented as an automatic detection method in the workflow, are specified as special integrated apparatus where detective instruments or sensors are placed within process channels or flowing materials, decreasing the need of manual interventions and increasing automation abilities in flow. Inline measurements do not require the presence of senior operators due to minor mistakes introduced, and they typically have good repeatability if precisely automated machines are employed.18
Like inline measurements, online measurements also do not require the transfer of the samples in the process. The flow stream is regularly sampled only for representatively independent samples that are of high importance for progress identification. Sometimes, physical parameters of aliquots need to be modified between sensors and analysers, conditioning testing samples (e.g., liquid or gas) in temperature, pressure, space velocity, etc.
In situ measurements, i.e., detection and analysis conducted in the primary position where active intermediate or unstable species can be monitored are considered as one of the top priorities in the characterization in chemistry and chemistry-related discipline.19 Exempting sample monitoring from manual operations and reducing exposure in the natural environment can minimize the external influence and maintain the greatest extent of the optimal working conditions. A graphical outline of the different measurements is shown in Fig. 1.
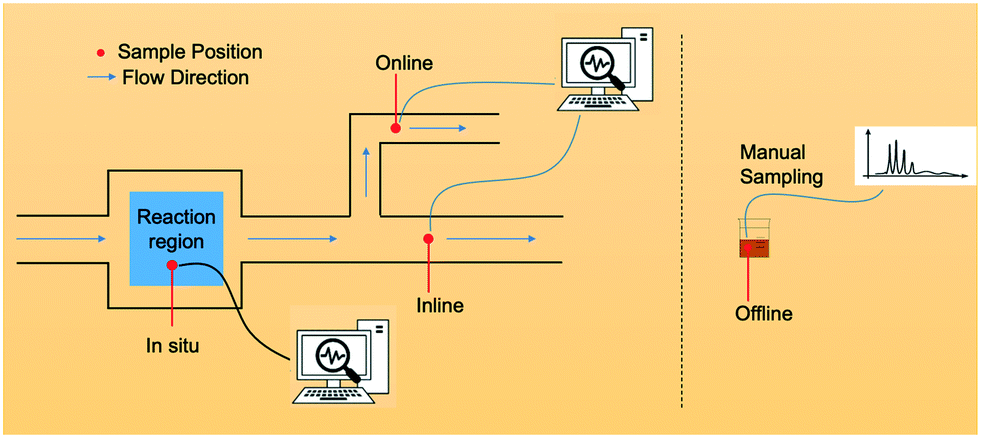 | ||
| Fig. 1 Schematic illustration of in situ, inline, online and offline measurements. | ||
Calling for innovative analytical techniques in flow manufacturing
Over the last several decades, scientists have been more inclined to batch synthesis and the use of offline measurements due to intermittent manual operations for separation and purification in the post-processing. This scenario is particularly common in organic synthesis: evaporation, distillation, crystallization and column chromatography requires a lot of effort and are time-consuming. Offline manipulations for sampling and analysing become acceptable in the case of long-term running fabrication with a comparatively short time for characterization. For instance, some photochemical transformations in flask vessels demand for response time from hours to days in some cases.10On the other hand, increased effective conversion rate, as well as improved overall yield in a short period, suggest that time-efficient flow chemistry is more constructive for industrial production. Wang and colleagues applied a photo redox-mediated Stadler–Ziegler reaction for the preparation of aryl, alkyl and diaryl sulphides to a continuous-flow process.20 The acceleration of the transformation was quite impressive, about 80% conversion in 15 s in flow versus that of 5 h in batch. Konieczynska et al. demonstrated a novel approach for symmetric anhydride production via photoinduced redox catalysis of aryl and alkyl carboxylic acids where active species-iminium ion was generated in situ. The astonishing efficiency (6.4 min residence time in flow for 97% yield vs. 85% yield after 18 h in batch synthesis) was observed when 4-tert-butylbenzoic acid was introduced in a flow reactor.21
Smart strategies such as solution-based approaches, continuous separation and distillation as well as incorporating solid-supported scavengers make multi-step organic flow synthesis practical and competitive.22 To free the process from expensive offline sampling, it is essential to integrate innovative analytical techniques capable of exempting transferring samples and manual handling. Consequently, interval-free reaction-to-analysis processes envisaged with concepts of inline, online and in situ measurements, tailored to the intrinsic nature of process control within flow reactors, contribute greatly to academia and industry.
In situ measurements for studying and precisely controlled fabrication
Many valuable concepts are introduced in flow manufacturing, indicating future requirements regarding process control. Price and co-workers summarized the scope and potential for coupling manual-free analytics with flow analysis in an excellent review where the notions of process analytical technology (PAT) and real-time release testing (RTRT) are presented.23 Correspondingly, the conceptual definitions aim to interrogate the process to guarantee high-quality production in arbitrary time intervals over the whole continuous manufacturing. Additionally, evaluating the state of flow feeds and materials produced in continuous operations as well as optimizing parameters that affect the final results, are the ultimate objective for transforming flow chemistry from cutting-edge science into realistic industrial application.The integration of in situ sensors with flow reactors to assemble an automated system is more applicable to profoundly rapid mechanistic and kinetic insights, favouring reaction optimization and repeatability.24 Inline and online measurements are available to acquire data in continuous manufacturing for process evaluation. However, the data obtained by sensors fail to reflect the real situation due to inconsistent physical parameters between the signal apparatus and reaction position. Similarly, the precision and accuracy of inline sensors and online sensors vary. As stated in a previous article, “inline strategies, when integrated to a suitable controller for automation, can qualify as an online PAT tool for flow analysis.”23 Demands for insight studying and precisely controlled fabrication encourage the marriage of in situ sensors and flow systems. The study is being advocated for the transition from online analysis tools to the monitoring of real flow manufacturing process. The monitoring and identification of transient radical intermediates at electrode–electrolyte interfaces are challenging but attainable and can be realized by the coupling of electrochemistry and in situ liquid secondary ion mass spectrometry (SIMS) for a vacuum compatible microfluidic electrochemical device, providing molecular evidence of a mechanism for the electrochemical oxidation reaction.19 Likewise, governing the nucleation and growth process is crucial to produce the controllable size and shape of Au NPs (nanoparticles), in which procedures are usually accomplished in a fleeting period (2 to 20 ms).7 The marriage of such cooperation leverages the ability to extract local information of transient species without time delay.
The ability to capture transient species during a chemical process makes insightful research accessible, helping scientists gain a better understanding of how it went through and why this happened from a micro perspective.
Integration of flow reactor and in situ optical sensors into a perfect match as art-of-process-control for chemical reaction detection
Reaction probing with in situ NMR spectroscopy
Nuclear magnetic resonance spectroscopy (NMR) is a potent, non-invasive, and by far the most information-rich analysis technique for molecular structure determination. It is nevertheless essential to improve the inherent low sensitivity of an NMR instrument for small sample volumes, which can be addressed by decreasing the diameter of the detection coil raising the possibilities for in situ measurements in microfluidic assembly as the sensitivity per amount of spins increases. The aforementioned strategies function well and show good compatibility with microfluidic NMR. Stripline-design chips, for instance, are studied as good alternatives under an unperturbed flow state for both liquid and solid samples, resulting in higher sensitivity and good resolution, as well as facilitating microfluidic NMR for fast reaction kinetic study.25,26 Other methods like adopting diamond quantum sensors27 and combing the parahydrogen-induced hyperpolarization (PHIP) tactics are also implemented into microfluidic NMR systems for liquid analytes ranging from tens of picolitres to several microliters.28 The integration of designed micro coils into microfluidic devices allows for specific optimization of materials, architectures and specific read-out.29–32Examples for exploiting in situ NMR as an analytical tool for microflow chemical synthesis are hitherto seldom. As elucidated by an earlier case, Bart et al. proposed a stripline-shape flow probe for real-time reaction kinetics study of the acylation of benzyl alcohol with acetyl chloride, tackling the incompatibility between spectral resolution and the distortion of the static magnetic field. The peak broadening and N,N-diisopropylethylamine (DIPEA) signals, as well as the absence of the 2.40 ppm peak that should be present in general NMR tests, showed strong evidence in tracing intermittent species. Further, the ability of microfluidic stripline NMR chips was extended from 1H signals into gathering 1D and 2D 1H, 13C, and heteronuclear spectra when modified stripline chips were introduced (Fig. 2).26 The inlet and outlet located on the top and bottom of the chip as well as the glued-fused silica (FS) capillaries for mass-limited NMR characterization enable a convenient microfluidic connection. However, due to the simple glue fixation of FS capillaries, the system is not appropriate to be figured as “compact” one. Therefore, the degree of leakage of these three chips is different, hence the results cannot be assessed at the same level. Some technical sealing problems seem inevitable and often challenging when trying to incorporate the fragile chips into flow system. Additionally, the maximum detection volume of three chips is 215 nL, which may not be able to meet the measurement requirements of analysing liquid volume up to several microliters.
 | ||
| Fig. 2 Schematic view of three modified stripline chips for microfluidic flow NMR spectroscopy including 1D, 2D, and heteronuclear signals. (a) 100 μm D263T borosilicate-made 165 nL volume chip with optimal resolution and sensitivity (b) 500 μm fused silica-made 145 nL volume chip, preferable for small volume samples (c) borosilicate-made with a maximum volume of 215 nL chip, similar design as (a) (adapted from ref. 26 copyright 2017 American Chemical Society). | ||
Ahmed-Omer and co-workers demonstrated the use of benchtop NMR spectroscopy for in situ monitoring of hypervalent iodine(III)-initiated cyclopropanation of styrenes. An innovative approach was developed to effectively differentiate the features between reactants and products, in which an inline solvent switching device allowing the switch from reaction to an analytical medium is incorporated for accessing spectra in a deuterium-enriched media.33 The robust and compact continuous platforms equipped with the function of evaporation, concentrating, and solvent switching are easily fabricated from commercially available elements (Fig. 3). Mass spectrometry was also combined with this system to contribute additional evidence and a multidimensional perspective to process control and real-time monitoring. The built platform is versatile, facilitating downstream processing with the strategy of online monitoring of the reaction progress over time. It also shows satisfactory adaptability of realising the interaction with MNOVA software to profile the reaction progress and record species resonance at a specific chemical shift. Since the monitoring is not accomplished in its original position, it does not strictly follow the notion of in situ NMR measurement, even though the strategy is capable of acquiring real-time data approximately.
 | ||
| Fig. 3 Robust platform for in situ reaction monitoring of a cyclopropanation reaction with Spinsolve benchtop NMR spectroscopy. (A) Vapourtec R2+ pump unit. (B) Vapourtec R4 reactor unit. (C) Flow stream input. (D) Benchtop NMR. (E) Flow stream output. (F) Glass NMR cell (adapted from ref. 33 copyright 2016 American Chemical Society). | ||
A step forward in this regard was provided by Gomez et al.,34 who assembled a novel kit with integrated planar-spiral transceiver coils for in situ NMR monitoring of UV-vis-assisted reductive dehalogenation of α-bromoacetophenone in nanolitre scale. The described process solves the plight of low photonic efficiency encountered with large-scale photochemistry and makes reduced dimension analysis feasible (see Fig. 4). The light-induced photocatalytic dehalogenation of α-bromoacetophenone and the photoconversion of o-nitrobenzaldehyde to nitrosobenzoic acid were chosen as probe reactions under a stopped-flow state, using LEDs (525 nm) and laser diode (405 nm) as the light sources, respectively. The obvious evolution of chemical progress can be visualised by the disappearance of α-bromoacetophenone at 4.93 ppm and the formation of acetophenone at 2.57 ppm as well as the chemical shift of the oxidized form of diethyl-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate at 4.32 ppm. Kinetic data from the photoconversion of o-nitrobenzaldehyde to nitrosobenzoic acid showed a good agreement with literature. These reactions can be performed in small NMR detection volumes of 25 nL and can be evenly irradiated by diverse low-power light sources with non-invasive optic fibres, enabling high photon flux. The attempts of implementing uniform UV-vis illumination in a traditional NMR device are always confronted with excessive irradiative heating, drastic decay of light strength, etc. Although the methodology presented here partially advocates a new path of problem handling to tackle difficulties listed before, the predicaments of irradiation are still to be met when the sample volume comes to a microliter scale, particularly when trying to scale up photocatalytic reactions in microfluidics.
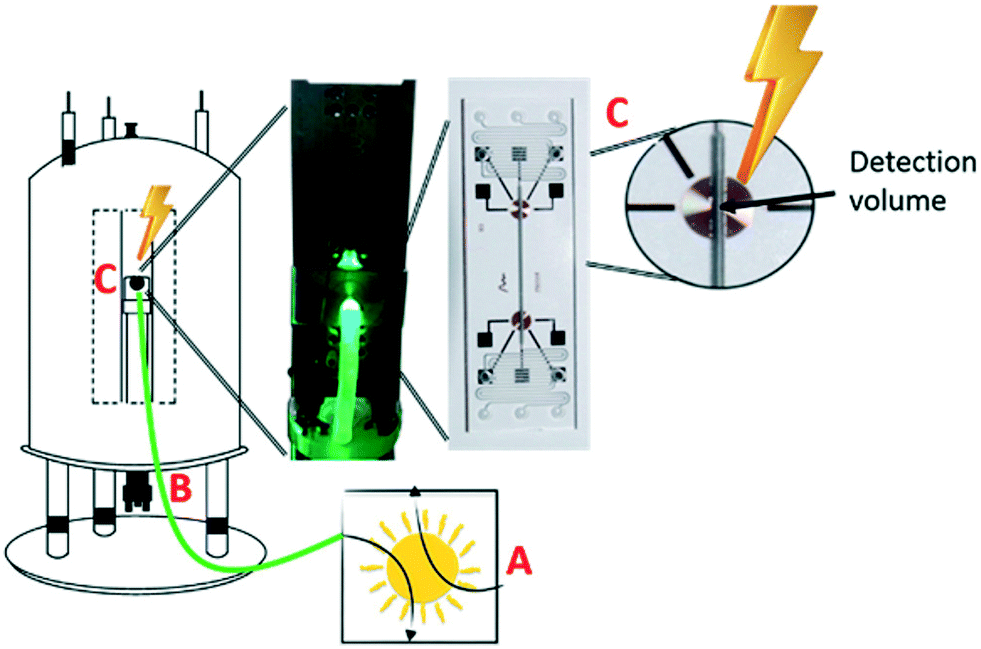 | ||
| Fig. 4 Graphic representation of the assembly of nanolitre-in situ-NMR microfluidic chips. (A) UV-vis light source. (B) Non-contact-sample optical fibre. (C) Sampling zone (adapted from ref. 34 copyright 2018 American Chemical Society). | ||
Reaction probing with in situ infrared spectroscopy
The utilization of infrared (IR) spectroscopy as a non-invasive analytical technique to obtain spectral information has been proven to be reliable and effective.36 IR spectroscopy offers an abundant capacity of spectroscopic information of interrogated chemicals due to activated molecular vibration in the range of near-IR to mid-IR. The implementation of three different FTIR techniques, diffuse reflectance (DR), attenuated total reflectance (ATR), and transmission mode, has promoted the advancement of IR detection in chemical process inspection. Furthermore, the coupling of microfluidics and mid-IR spectroscopy can be successfully applied to trace analytics and enables access to spatially-resolved spectra with a good signal-to-noise ratio (SNR). Simultaneously, the prototype of integrating FT-IR imagining with microfluidic apparatus was confirmed to be achievable, in which transmission mode results in superior sensitivity while the ATR mode contributes a preferable choice for quantitative analysis.35 Hence, it is practical to monitor a chemical process with IR detection in a flow reactor. An excellent review for combing microfluidics with FT-IR spectroscopy was provided by Perro et al., where the potentialities, strategies, and challenges are well generalized, giving a tutorial guide to constructing innovative devices for real-time IR mapping.36Examples concerning stand-alone in situ IR reaction imaging are scarce. In most cases, it is only regarded as an ancillary detection method and cannot be analysed independently to support quantitative transformations because of fragmented structural information provided. Indeed, the complexity of incorporating external analytical tools like mass spectrometry and NMR spectroscopy interferes in some cases with in situ IR implementation for chemical synthesis. There is, however, no better solution to date to circumvent these limitations.
In the first case, Gross et al.37 reported a flow system equipped with micron synchrotron IR and X-ray beams in a small cell with 2 cm length. This apparatus can associate the functions of analysing reaction transformation and observation of species evolution from vinyl ester 1, as a consequence providing strong evidence of the presence of short-lived intermediate, the allenic aldehyde 2 (Fig. 5). The yield and selectivity of products can be modified by altering the residence time of the raw feed. The results show a decreased flow rate by 50-fold to the original one (from 10 mL h−1 to 0.2 mL h−1) contributed a dramatic increase of conversion (elevated from 20% to 75%) and high chemoselectivity to give the acetal product 3 was achieved. Confirmation of the conclusion deduced from in situ IR measurement needs data from gas chromatography and NMR-spectroscopy. Additionally, the catalytically active species of Au(III) generated in the chemical process was verified by in situ X-ray microspectroscopy within the microreactor. The plot of species distribution provided insights into kinetic studies with a high spatial resolution of 15 μm. This pioneering analytic technique depicts the blueprint of in situ reaction mapping of organic synthesis, clearing the mist in the field of combining microfluidics with in situ IR measurements.
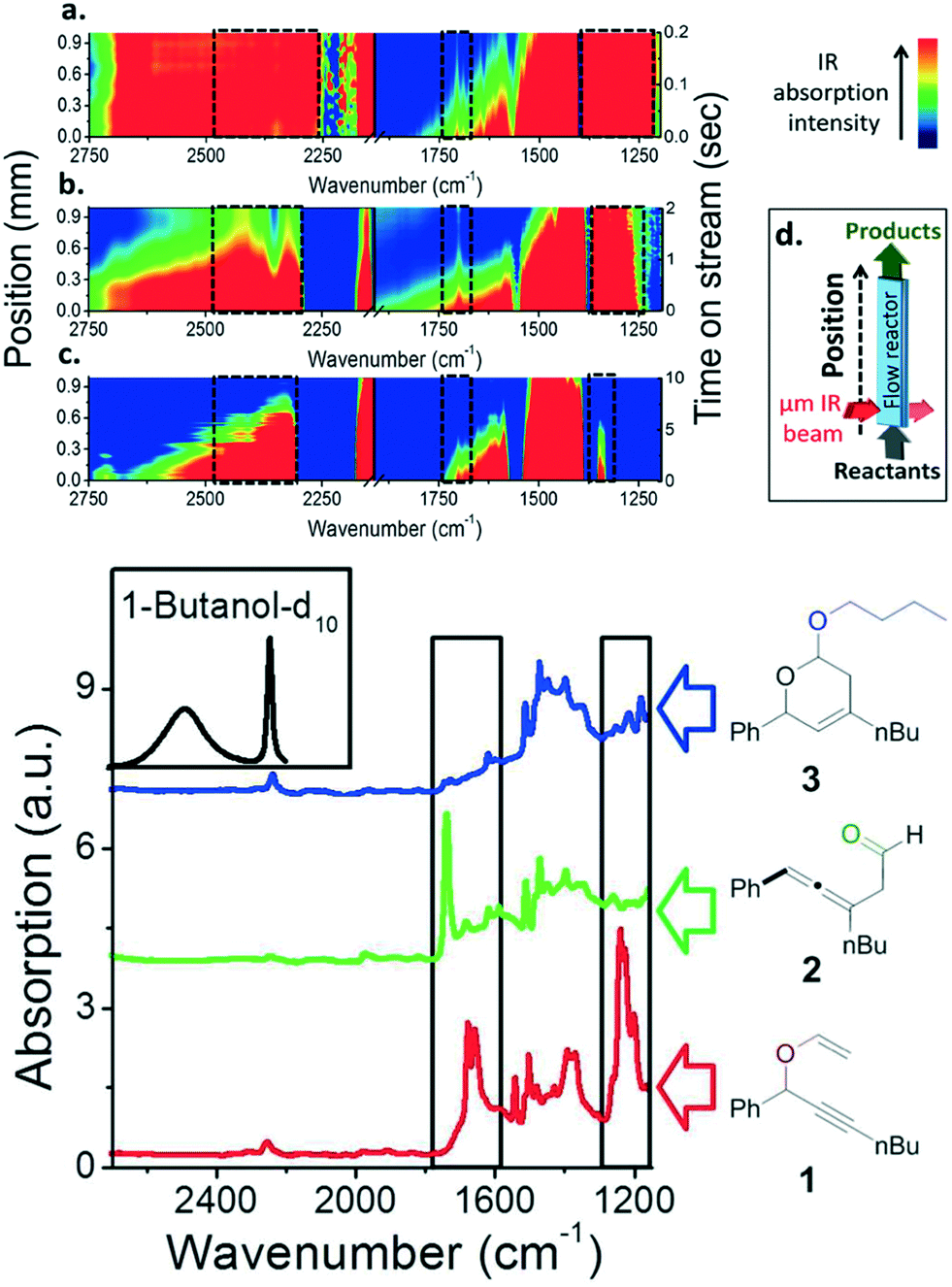 | ||
| Fig. 5 Upper: Spatially resolved IR spectroscopic imaging along the flow reactor with various flow rates, 10 (a), 1 (b), 0.2 (c) mL h−1, respectively. (d) Scheme of microreactor for in situ IR and X-ray microspectroscopy measurements. Below: FTIR spectra of reactant, vinyl ether (red), allenic aldehyde (green), and acetal (blue) (adapted from ref. 37 copyright 2014 American Chemical Society). | ||
Zhang et al.38 studied heterogeneous catalytic asymmetric hydrogenation of an α-amino ester over cinchonidine (CD)-modified palladium catalyst with several home-made in situ FTIR experimental apparatuses that enable various function-oriented characterizations (Fig. 6). The same overall shape, together with trifling distinction among three FTIR spectra depicts the evolution of hydrogenation process very well: the transmission model corroborated the formation of OH–O and NH–N hydrogenation bonding between CD and the amino ester, while DR and ATR model gave supports in monitoring the degree of interface hydrogenation. The first incorporation of diffuse reflectance infrared Fourier transform spectroscopic (DRIFTS) with a microfluidic platform offers a better option of inspecting CD-morphology on the transition-metal catalyst surface with changeable coverages, exempting from new catalyst bed preparation and reducing the waste of essential resources significantly. The described method can be used for the exploration of the morphology of a catalyst's surface in the process of heterogeneous catalysis, exploiting in situ IR analysis and microflow system.
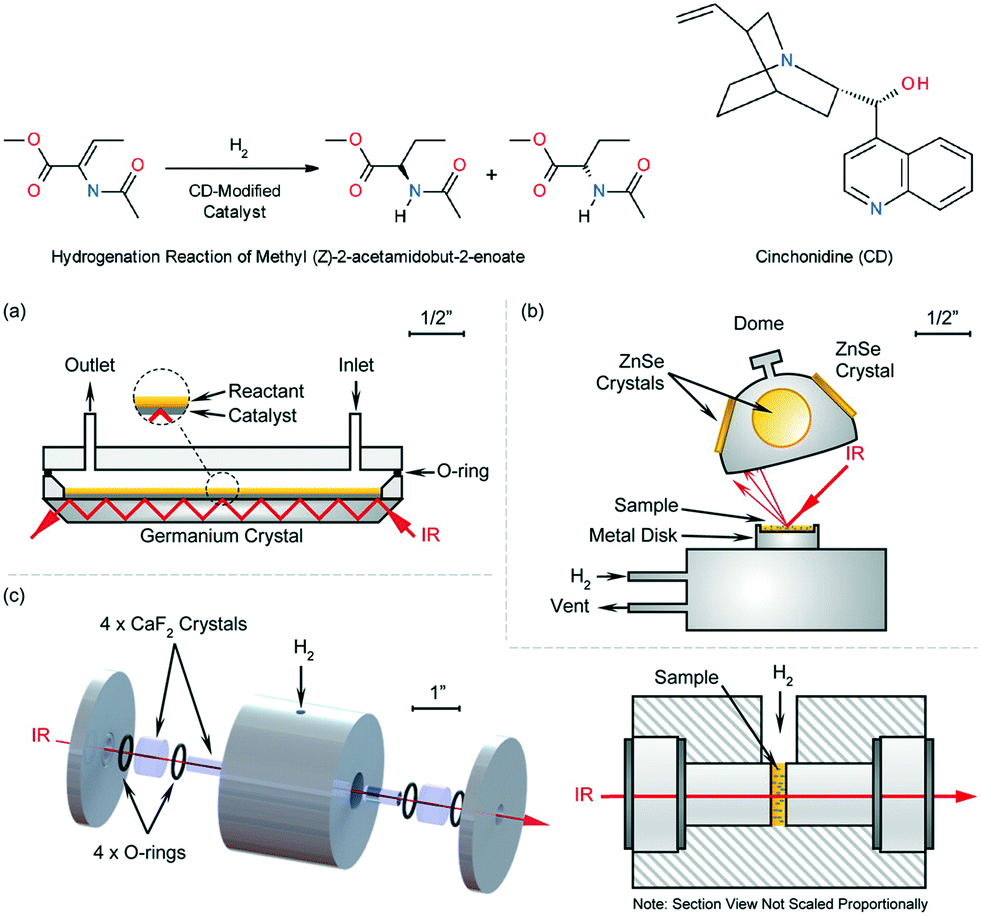 | ||
| Fig. 6 Upper: The hydrogenation of methyl (Z)-2-acetamidobut-2-enoate with a CD-doped Pd catalyst. Below: Detailed views of the in situ IR-microfluidics setup, (a) ATR infrared cell assembled by Ge crystal, (b) DRIFTS cell, (c) transmission FTIR structure (adapted from ref. 38 copyright 2016 American Chemical Society). | ||
Another case reported recently was pertinent to the improvement of a hydrosilylation reaction in a microflow system using in situ IR monitoring.39Fig. 7 shows an example of in situ IR analysis of a hydrosilylation when adopting a microfluidic reactor, achieving nearly 100% Si–H conversion of 1,1,1,3,5,5,5-heptamethyltrisiloxane (HMTS) and 1-octene with only 1 minute residence time and a very low concentration of catalyst usage (1 × 10−6 mol Pt/mol Si–H). The utilization of this analytical technique with a microreactor facilitates post-processing with the benefit of free from interval sampling procedure, preventing sampling from the reacting mixture. Another benefit is that the sample can be processed and analysed by GC before complete inactivation. The apparent disappearance of the Si–H bond during the conversion process of raw materials makes in situ IR analytics accessible to acquire real-time information. The conversion can be calculated from the reducing area of the Si–H vibration band around 915 cm−1 since the intensity of the signal is proportional to the concentration of reactants based on the Beer–Lambert law. The improved yields in the microflow system can be explained with the elimination of diffusion effects in microreactors, which cannot be mediated in batch by enhancing mixture intensity. Although results from in situ IR showed a lower yield in comparison to data from gas chromatography, the swift detection makes it an ideal tool for hydrosilylation observation. The plug-in contact measurements of ReactIR 15 for the record of IR data make it flexible and portable, but it can only reflect the state of specific local points rather than give an overview of the fluid state.
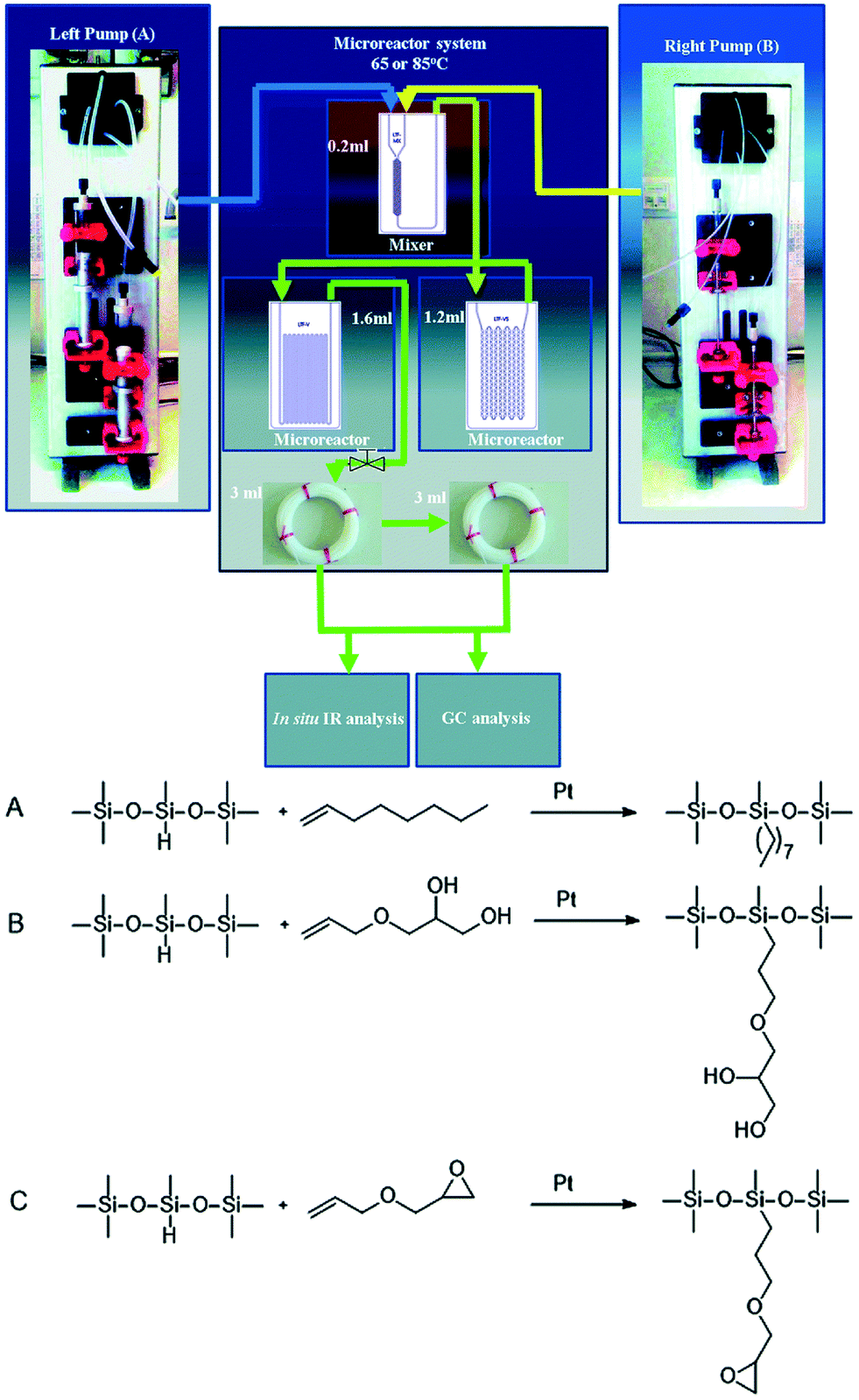 | ||
| Fig. 7 Upper: Setup of in situ IR detection in flow reactors. Below: hydrosilylation of different olefins: (A) 1-octene, (B) 3-allyloxy-1,2-propanediol and (C) allyl glycidyl ether (adapted from ref. 39 copyright 2018 The Royal Society of Chemistry). | ||
The integration of IR microspectroscopy with flow reactors for real-time monitoring of the reaction progress can be beneficial, but the requirements for differentiation between reactants and test samples as well as acquiring accurate quantitative results remain challenging.
Reaction probing with in situ Raman spectroscopy
Raman spectroscopy can compensate for some of the drawbacks that come along with IR measurements such as the sensitivity to water and inaccessibility of the low wavelength spectral range that bears structural information for hydroxyl groups and metal-connected bonds. Practising rapid and damage-free identification of test samples as well as in situ progress monitoring of chemical reactions with Raman techniques is strongly recommended in flow chemistry.40 Low scattering sensitivity confronted with surface-enhanced Raman spectroscopy (SERS) can be addressed by the addition of rough noble metal nanoparticles to accomplish several orders of magnitude increase in the analyte's Raman signals. The bridge of Raman spectroscopy and microflow setup requires developing custom-tailored flow cells and reliable probe for sensing.An initial attempt to combining in situ Raman spectroscopy for rapid condition optimization of organic synthesis was presented in 2007.41 Leadbeater et al. chose a microwave-assisted simple esterification reaction of acetic acid with butanol as a model reaction in an automated stop-flow instrument that revealed the attractive potential of quality control for organic transformations.
Another example of Raman spectroscopic studies was presented with Au–Pd bimetallic-supported TiO2 for catalytic oxidation of benzyl alcohol in silicon-glass micro-packed-bed reactors (MPBRs).42 The microreactor design and laboratory setup are shown in Fig. 8. Due to the characteristic CO stretching of benzaldehyde in the Raman spectrum at 1700 cm−1 the authors succeeded in the quantitative determination of the product and the screening different reaction parameters. External calibration was established by gas chromatography, in which the results show good accordance with the Raman measurements. An optimized reaction condition resulted in a 95% conversion of benzyl alcohol with 78% selectivity towards benzaldehyde.
 | ||
| Fig. 8 (a) Schematic illustration of MPBRs. (b) Experimental setup combining MPBRs and Raman microscope (adapted from ref. 42 copyright 2011 Elsevier). | ||
Analogously, the detection of active inorganic ion species in a chemical transformation is attainable when the challenge of small Raman cross-sections is tackled by pre-decoration of linker structure with a more powerful SERS spectrum.43 A monolayer of 4-aminothiophenol (4-ABT) was pre-decorated with silver film-coated silicon nanopillar arrays (Ag/SiNPs) within a microstructure channel for nitrite ions detection. In the presence of HCl, the nitrite ions are supposed to react with –NH3+ released by 4-ABT, contributing observation of signal changes in the spectrum for repeatable SERS detection.
More complex chemical conversions are demonstrated in a multichannel microfluidic chip fabricated from PDMS/glass with the method of moulding and plasma bonding, which approves nanolitre-scale organic synthesis for Raman imagining of dynamic flowing droplets in microchannels (Fig. 9).40 The Hantzsch syntheses of 2-aminothiazoles were chosen as model reactions, and an acquisition time of 500 ms was adopted to realize considerable SNR. Suitable indicative Raman bands characterized with apparent discrepancy between starting materials and final products were selected for spectroscopically single droplets tracing, which correspond to the dynamic view of the reaction progress. Silver nanoparticles were involved in enhancing Raman signal with the strategy of introducing downstream before arriving at acquisition points. This research paves a new path to qualitative analysis of high-throughput screening of organic reactions in flow.
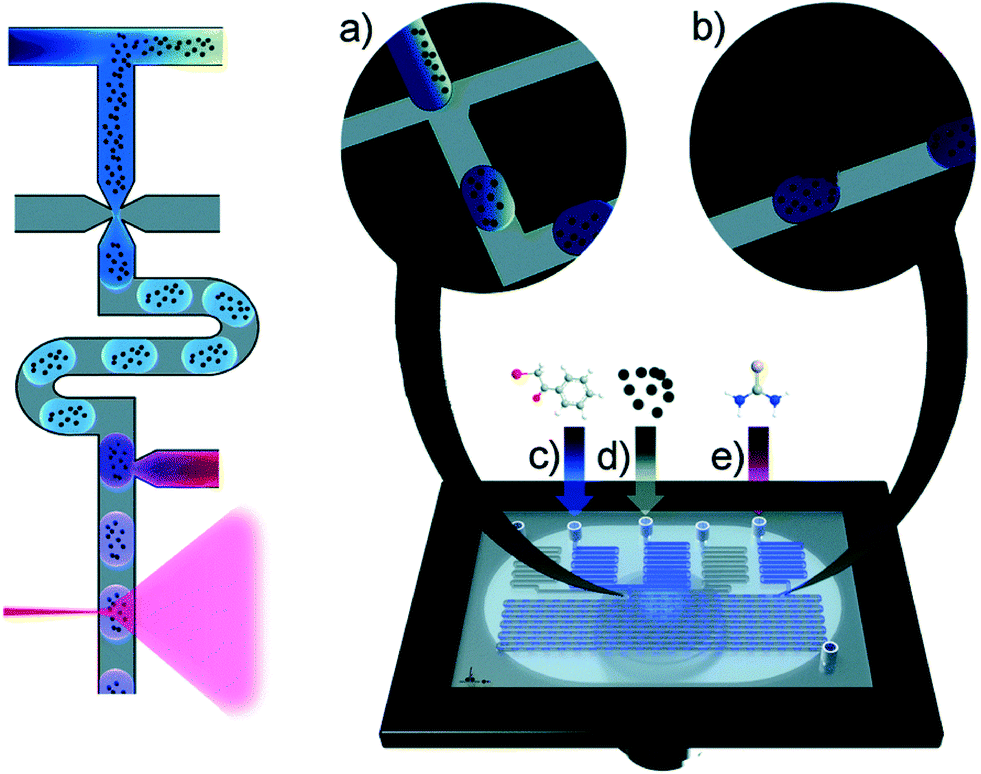 | ||
| Fig. 9 Left: Schematic diagram of in situ Raman detection. Right: multichannel microfluidic chip, the flow-focussing (a) and dispensing structure (b) within microchannel, (c) reactant 1 inlet, (d) silver suspension inlet, (e) reactant 2 inlet (adapted from ref. 40 copyright 2015 The Royal Society of Chemistry). | ||
Latterly, the first example of utilizing microfluidics with Raman spectroscopy for conducting mechanism and kinetic study of palladium-catalysed cross-coupling was elucidated by Rizkin and co-workers (Fig. 10),44 who proved that the process of PdCl2(CH3CN)2 conversion into an active Pd0L2 complex proceeds only in the interface of aqueous and organic phases. The Raman data integrated with calibration curves were used for concentration and mole fraction determination of each species. Chemical kinetics showed that neither carbopalladation nor ionic mechanisms are involved in the cross-coupling reaction while cationic and anionic deprotonation mechanisms were found to contribute. The binding of a palladium atom with ligands endows the ability of catalytic activation, which in turn promotes the chemical transformation. Such discovery supports inspirations for ubiquitous functionalization reactions in organic chemistry that resemble cross-coupling reactions.
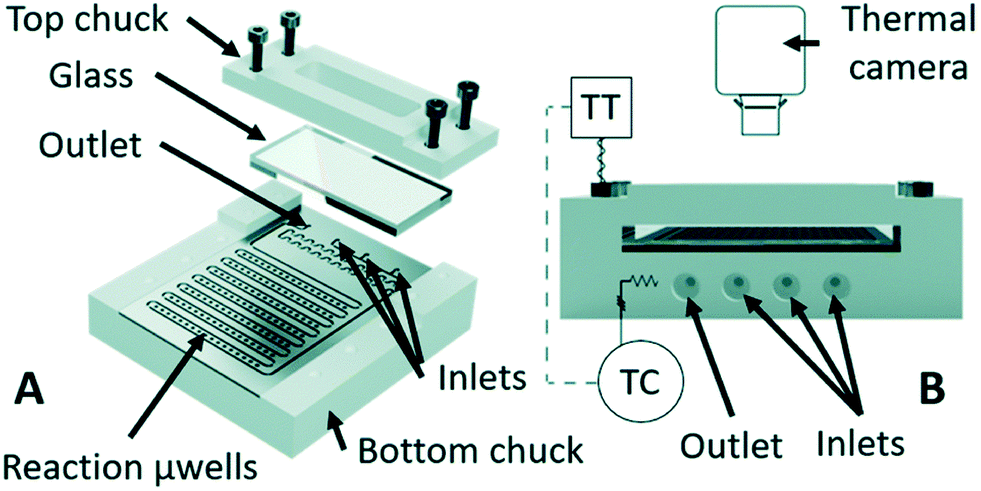 | ||
| Fig. 10 Microreactor design adopted in Pd-catalyst assisted cross-linking reaction and relative mechanism study, main (A) and back (B) view of the reactor (adapted from ref. 44 copyright 2018 The Royal Society of Chemistry). | ||
Reaction probing with in situ UV-vis spectroscopy
The first paradigm of in situ UV-vis investigation in continuous-flow was led by Jiang et al.,45 who coupled 13C MAS NMR and UV-vis spectroscopy to evaluate a catalytic methanol-to-olefins (MTO) process on silicoaluminophosphate H-SAPO-34 in a 7 mm MAS NMR rotor reactor. Evidence from in situ NMR and UV-vis spectroscopy under continuous-flow conditions discloses that dimethyl ether (DME) is the MTO's primary product at a low working temperature of 473 K and 523 K. A further increase in temperature is conducive to the simultaneous generation of smaller olefins and carbenium ions. The selectivity of propylene and ethylene can be manipulated with different catalyst working temperatures at 573 K and 623 K, respectively. Also, the resulting olefins may progressively react with the carbenium ions to generate larger aromatic deposits and larger carbenium ions, which in turn is detrimental to MTO and results in catalyst deactivation at 673 K (the formed polycyclic aromatics acting as coke deposit).Reports showed oxidative catalyst states and their reactivities are highly dependent on the contacting gas atmosphere, and some reversible changes in catalyst state are not maintainable without suitable reacting circumstances. These make directly interrogating the relationship between oxidation states of catalyst and corresponding reactivities inaccessible. The issue can be tackled with in situ UV-vis spectroscopy and continuous handling in flow operation. Bu et al. carried out another study dealing with microgram Cu-catalyst dynamics and CO-oxidation kinetics, who incorporated a pocket microreactor with in situ UV-vis and MS (see Fig. 11).46 Over a reduction to a stepwise oxidation process, the designed approach can visualize the Cu-catalyst in different valences, from metallic state to Cu(II), among which metallic Cu was found catalytically active. Deactivation occurred due to irreversible oxidation of the metallic Cu. Also, XPS is included for providing supplementary evidence as well.
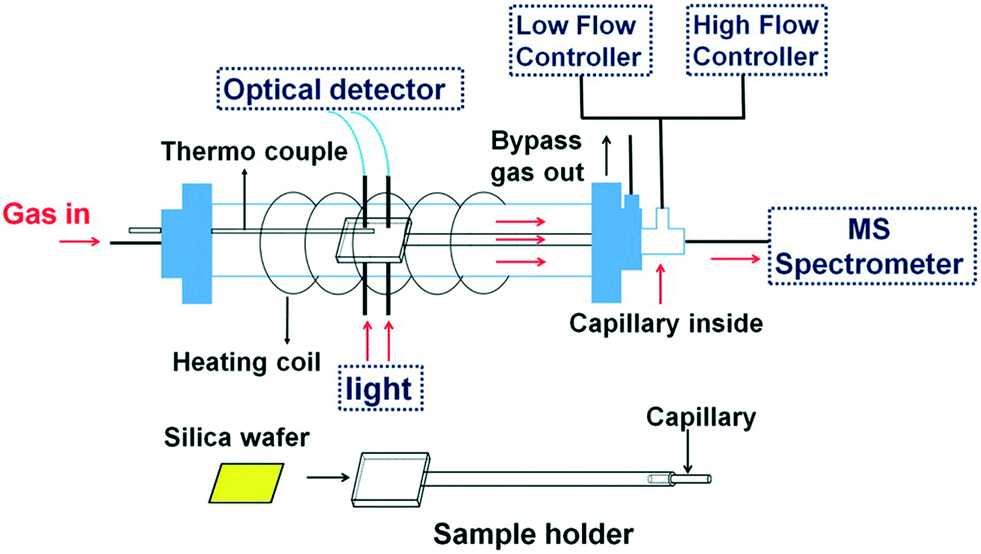 | ||
| Fig. 11 Upper: The design of microreactor integrated with optical spectroscopic apparatus and sampling capillary (adapted from ref. 46 copyright 2015 American Chemical Society). | ||
The adoption of in situ UV-vis into microfluidics reactors for organic synthesis monitoring is still far from mature. Currently, applications of UV-vis in microfluidics focus on the morphology changes on a catalyst's surface or spectral differentiations between metals in different valence states, rather than the chemical transformation itself. Besides, for reaction monitoring purpose, the chosen analytes need to possess a suitable chromophore.17
Miscellaneous
Mass spectroscopy is used in the online measurement of microfluidic analysis, however illustrative cases regarding in situ application in microfluidics are very few. Wang et al.19 confirmed the potential of in situ liquid secondary ion mass spectrometry (SIMS) to tackle the challenge in real-time detection of hypothetical short-lived radical ionic intermediates during the electrochemical conversion of ascorbic acid in a specialized microfluidic cell. The results offer clear evidence of collecting subtle information about transient radical intermediates at electrode–electrolyte interfaces.Electrochemical readout combined with other spectroscopic techniques such as NMR technique allows for the rapid dissemination of redox processes in situ.31
Other in situ optical analytics like fluorescence spectroscopy and X-ray absorption spectra (XAS) have been extensively utilized in non-interference kinetic studies of integrated microfluidic synthesis. The production of gold nanoparticles (Au-NPs),47 colloidal semiconductor nanocrystals (NCs), and quantum dots (QDs) production,48 has revealed the early stage of rapid nanostructure growth and nucleation process and corroborate the process mechanism that completes in a few milliseconds. However, literature reports concerning chemical reaction monitoring, which is the main focus of this review here, are not described yet.
Furthermore, a novel strategy in enthalpy determination of fast exothermic reactions was achieved by introducing infrared thermography in flow reactors, which enables generating time-series kinetic data.49 Coincidently, such a marriage was extended to mapping the reaction space of zirconocene polymerization catalyst and conducting kinetic study in an automated flow system.50 Real-time thermokinetic heat flux measurements in microstructured reactor via easily available Seebeck elements has also been demonstrated.51
Table 1 summarises the advantages and disadvantages of various in situ sensors coupled with flow reactors.
| In situ analytics | Advantages | Disadvantages |
|---|---|---|
| NMR | Most informative, robust scope, independent | Bulky main body, expensive, solvent-switch |
| IR | Broad scope, portable, easy to miniaturize | Water-limited, material limitation dependent |
| Raman | Broad scope, good resolution | Expensive laser required |
| UV/vis | Suitable for catalyst studies | Limited scope |
| XAS | Suitable for nano-synthesis | Limited scope |
| Infrared thermography | Can replace calorimeter | Limited scope |
| Seebeck elements | Economical, commercially available | Limited scope |
NMR has the most powerful capabilities in the assay, but the problems of bulky size, expensive instrumentation, and the need for a solvent-switch system served for the deuteration process seems to be insurmountable. IR has subordinate application scope and shows ascendency in portability, flexibility, and cost-effectiveness in microfluidic application, but some applications are restricted in water solution and low wavelength spectrum. The material challenges are confronted with inadequate wavelength coverage in the detection area of interest and high prices in some optical glasses. At the same time, additional analytical instruments are required to fulfill quantitative evaluation. Raman can compensate flaws in IR, while the costly laser is necessary for determination. The clogging of microchannels should be carefully addressed if rough metal nanoparticles are employed to achieve good resolution. A common defect associated with IR and Raman probing is the inability to use independently, and chosen chemistry in one's research should be decided by sophisticated chemists to achieve good discrimination in the obtained resultant spectra, since only partial structural information is provided. UV-vis, XAS, and infrared thermography, as well as Seebeck elements, are suitable for kinetic study in distinct fields. The adoption of these methods in chemical synthesis should be further explored.
Conclusions
The achievement of integrating in situ optical analysis with microfluidics technology has shown an impact to extract real-time information for in-depth dynamics and mechanism research, thereby reducing the gap between theoretical and realistic processes. Also, process intensification like automated optimization in microfluidic platforms was demonstrated by Jensen's group as prominent techniques to implement substantial high-yield functionalizations.52 A significant step forward is the AI-assisted organic synthesis in a robotic flow platform, revealing interesting possibilities.53 Therefore, time-lag-free acquisition of analysis that is the same initiative with the notion of RTRT becomes increasingly important when running reactions in an automated flow system. Challenges with in situ optical analysis using microfluidics, however, remain daunting. Difficulties in the miniaturization of NMR, dependence, and sensitivity of infrared, Raman, as well as UV-vis spectrometry, should not be neglected, even though spectrometers with ever-smaller footprints capable of in situ mapping was fabricated54 and field-resolved infrared spectroscopy for biological systems with a plethora of water absorptions was introduced.12With the increasing number of photochemical studies showed great success in microfluidic devices and the ease of integration of infrared sensors into microfluidic devices, further investigation may focus on in situ IR monitoring of photochemistry in flow. It should also be pointed out that the choice of model reaction is highly empirically dependent, which is now an inevitable step in this direction of research. Fortunately, the insurmountable difficulties of designing synthetic routines can be realised by progressive development of AI-assisted retrosynthetic analysis,55 which makes this accessible to the amateur in chemistry. The integration of novel technologies in flow is fascinating, but it is also a massive system engineering in the coming decades.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the DFG (Deutsche Forschungsgemeinschaft) within the Research Unit 2383 ProMiSe project.References
- A. K. Au, W. Huynh, L. F. Horowitz and A. Folch, Angew. Chem., Int. Ed., 2016, 55, 3862–3881, DOI:10.1002/anie.201504382.
- X. Hou, Y. S. Zhang, G. T.-D. Santiago, M. M. Alvarez, J. Ribas, S. J. Jonas, P. S. Weiss, A. M. Andrews, J. Aizenberg and A. Khademhosseini, Nat. Rev. Mater., 2017, 2, 17016, DOI:10.1038/natrevmats.2017.16.
- J. Wegner, S. Ceylan and A. Kirschning, Adv. Synth. Catal., 2012, 354, 17–57, DOI:10.1002/adsc.201100584.
- Y. Su, N. J. W. Straathof, V. Hessel and T. Noël, Chem. – Eur. J., 2014, 20, 10562–10589, DOI:10.1002/chem.201400283.
- A. M. Nightingale, C. L. Leong, R. A. Burnish, S.-U. Hassan, Y. Zhang, G. F. Clough, M. G. Boutelle, D. Voegeli and X. Niu, Nat. Commun., 2019, 10, 2741, DOI:10.1038/s41467-019-10401-y.
- L.-J. Pan, J.-W. Tu, H.-T. Ma, Y.-J. Yang, Z.-Q. Tian, D.-W. Pang and Z.-L. Zhang, Lab Chip, 2018, 18, 41–56, 10.1039/C7LC00800G.
- G. Tofighi, H. Lichtenberg, J. Pesek, T. L. Sheppard, W. Wang, L. Schöttner, G. Rinke, R. Dittmeyer and J.-D. Grunwaldt, React. Chem. Eng., 2017, 2, 876–884, 10.1039/C7RE00114B.
- R. Porta, M. Benaglia and A. Puglisi, Org. Process Res. Dev., 2016, 20, 2–25, DOI:10.1021/acs.oprd.5b00325.
- A. Adamo, R. L. Beingessner, M. Behnam, J. Chen, T. F. Jamison, K. F. Jensen, J.-C. M. Monbaliu, A. S. Myerson, E. M. Revalor, D. R. Snead, T. Stelzer, N. Weeranoppanant, S. Y. Wong and P. Zhang, Science, 2016, 352, 61–67, DOI:10.1126/science.aaf1337.
- D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276–10341, DOI:10.1021/acs.chemrev.5b00707.
- K. F. Jensen, AIChE J., 2017, 63, 858–869, DOI:10.1002/aic.15642.
- I. Pupeza, M. Huber, M. Trubetskov, W. Schweinberger, S. A. Hussain, C. Hofer, K. Fritsch, M. Poetzlberger, L. Vamos, E. Fill, T. Amotchkina, K. V. Kepesidis, A. Apolonski, N. Karpowicz, V. Pervak, O. Pronin, F. Fleischmann, A. Azzeer, M. Žigman and F. Krausz, Nature, 2020, 577, 52–59, DOI:10.1038/s41586-019-1850-7.
- V. Sans and L. Cronin, Chem. Soc. Rev., 2016, 45, 2032–2043, 10.1039/C5CS00793C.
- J. P. McMullen and K. F. Jensen, Annu. Rev. Anal. Chem., 2010, 3, 19–42, DOI:10.1146/annurev.anchem.111808.073718.
- P. Sagmeister, R. Lebl, I. Castillo, J. Rehrl, J. Kruisz, M. Sipek, M. Horn, S. Sacher, D. Cantillo, J. D. Williams and C. O. Kappe, Angew. Chem., Int. Ed., 2021, 60, 8139–8148, DOI:10.1002/anie.202016007.
- M. Baumann, Org. Biomol. Chem., 2018, 16, 5946–5954, 10.1039/C8OB01437J.
- D. L. Browne, S. Wright, B. J. Deadman, S. Dunnage, I. R. Baxendale, R. M. Turner and S. V. Ley, Rapid Commun. Mass Spectrom., 2012, 26, 1999–2010, DOI:10.1002/rcm.6312.
- C. Zhou, M. Keshavarz Hedayati, X. Zhu, F. Nielsen, U. Levy and A. Kristensen, ACS Sens., 2018, 3, 784–791, DOI:10.1021/acssensors.8b00030.
- Z. Wang, Y. Zhang, B. Liu, K. Wu, S. Thevuthasan, D. R. Baer, Z. Zhu, X.-Y. Yu and F. Wang, Anal. Chem., 2017, 89, 960–965, DOI:10.1021/acs.analchem.6b04189.
- X. Wang, G. D. Cuny and T. Noël, Angew. Chem., Int. Ed., 2013, 52, 7860–7864, DOI:10.1002/anie.201303483.
- M. D. Konieczynska, C. Dai and C. R. J. Stephenson, Org. Biomol. Chem., 2012, 10, 4509–4511, 10.1039/C2OB25463H.
- D. Webb and T. F. Jamison, Chem. Sci., 2010, 1, 675–680, 10.1039/C0SC00381F.
- G. A. Price, D. Mallik and M. G. Organ, J. Flow Chem., 2017, 7, 82–86, DOI:10.1556/1846.2017.00032.
- R. Chung and J. E. Hein, Top. Catal., 2017, 60, 594–608, DOI:10.1007/s11244-017-0737-9.
- J. Bart, A. J. Kolkman, A. J. Oosthoek-de Vries, K. Koch, P. J. Nieuwland, H. Janssen, J. van Bentum, K. A. M. Ampt, F. P. J. T. Rutjes, S. S. Wijmenga, H. Gardeniers and A. P. M. Kentgens, J. Am. Chem. Soc., 2009, 131, 5014–5015, DOI:10.1021/ja900389x.
- A. J. Oosthoek-de Vries, J. Bart, R. M. Tiggelaar, J. W. G. Janssen, P. J. M. van Bentum, H. J. G. E. Gardeniers and A. P. M. Kentgens, Anal. Chem., 2017, 89, 2296–2303, DOI:10.1021/acs.analchem.6b03784.
- J. Smits, J. T. Damron, P. Kehayias, A. F. McDowell, N. Mosavian, I. Fescenko, N. Ristoff, A. Laraoui, A. Jarmola and V. M. Acosta, Sci. Adv., 2019, 5, eaaw7895, DOI:10.1126/sciadv.aaw7895.
- J. Eills, W. Hale, M. Sharma, M. Rossetto, M. H. Levitt and M. Utz, J. Am. Chem. Soc., 2019, 141, 9955–9963, DOI:10.1021/jacs.9b03507.
- P. F. Silva, M. Jouda and J. G. Korvink, J. Magn. Reson., 2020, 310, 106659, DOI:10.1016/j.jmr.2019.106659.
- O. Nassar, M. Jouda, M. Rapp, D. Mager, J. G. Korvink and N. MacKinnon, Microsyst. Nanoeng., 2021, 7, 30, DOI:10.1038/s41378-021-00253-2.
- H. Davoodi, N. Nordin, L. Bordonali, J. G. Korvink, N. MacKinnon and V. Badilita, Lab Chip, 2020, 20, 3202–3212, 10.1039/d0lc00364f.
- J. G. Korvink, N. MacKinnon, V. Badilita and M. Jouda, J. Magn. Reson., 2019, 306, 112–117, DOI:10.1016/j.jmr.2019.07.012.
- B. Ahmed-Omer, E. Sliwinski, J. P. Cerroti and S. V. Ley, Org. Process Res. Dev., 2016, 20, 1603–1614, DOI:10.1021/acs.oprd.6b00177.
- M. V. Gomez, A. Juan, F. Jiménez-Márquez, A. de la Hoz and A. H. Velders, Anal. Chem., 2018, 90(3), 1542–1546, DOI:10.1021/acs.analchem.7b04114.
- K. L. A. Chan, X. Niu, A. J. de Mello and S. G. Kazarian, Lab Chip, 2010, 10, 2170–2174, 10.1039/C004246C.
- A. Perro, G. Lebourdon, S. Henry, S. Lecomte, L. Servant and S. Marre, React. Chem. Eng., 2016, 1, 577–594, 10.1039/C6RE00127K.
- E. Gross, X.-Z. Shu, S. Alayoglu, H. A. Bechtel, M. C. Martin, F. D. Toste and G. A. Somorjai, J. Am. Chem. Soc., 2014, 136, 3624–3629, DOI:10.1021/ja412740p.
- L. Zhang, M. Lohrasbi, U. Tumuluri and S. S. C. Chuang, Org. Process Res. Dev., 2016, 20, 1668–1676, DOI:10.1021/acs.oprd.6b00222.
- A. Pawlowska-Zygarowicz, R. Kukawka, H. Maciejewski and M. Smiglak, New J. Chem., 2018, 42, 15332–15339, 10.1039/C8NJ01167B.
- T. A. Meier, R. J. Beulig, E. Klinge, M. Fuss, S. Ohla and D. Belder, Chem. Commun., 2015, 51, 8588–8591, 10.1039/C4CC09595B.
- N. E. Leadbeater, R. J. Smith and T. M. Barnard, Org. Biomol. Chem., 2007, 5, 822–825, 10.1039/B615597A.
- E. Cao, M. Sankar, S. Firth, K. F. Lam, D. Bethell, D. K. Knight, G. J. Hutchings, P. F. McMillan and A. Gavriilidis, Chem. Eng. J., 2011, 167, 734–743, DOI:10.1016/j.cej.2010.08.082.
- Y. Zhao, Y.-L. Zhang, J.-A. Huang, Z. Zhang, X. Chen and W. Zhang, J. Mater. Chem. A, 2015, 3, 6408–6413, 10.1039/C4TA07076C.
- B. A. Rizkin and R. L. Hartman, React. Chem. Eng., 2018, 3, 251–257, 10.1039/C8RE00021B.
- Y. Jiang, J. Huang, V. R. Reddy Marthala, Y. S. Ooi, J. Weitkamp and M. Hunger, Microporous Mesoporous Mater., 2007, 105, 132–139, DOI:10.1016/j.micromeso.2007.05.028.
- Y. Bu, J. W. H. Niemantsverdriet and H. O. A. Fredriksson, ACS Catal., 2016, 6, 2867–2876, DOI:10.1021/acscatal.5b02861.
- K. Sai Krishna, C. V. Navin, S. Biswas, V. Singh, K. Ham, G. L. Bovenkamp, C. S. Theegala, J. T. Miller, J. J. Spivey and C. S. S. R. Kumar, J. Am. Chem. Soc., 2013, 135, 5450–5456, DOI:10.1021/ja400434c.
- I. Lignos, R. Maceiczyk and A. J. deMello, Acc. Chem. Res., 2017, 50, 1248–1257, DOI:10.1021/acs.accounts.7b00088.
- C. Zhang, J. Zhang and G. Luo, J. Flow Chem., 2020, 10, 219–226, DOI:10.1007/s41981-019-00071-8.
- B. A. Rizkin, A. S. Shkolnik, N. J. Ferraro and R. L. Hartman, Nat. Mach. Intell., 2020, 2, 200–209, DOI:10.1038/s42256-020-0166-5.
- F. Reichmann, S. Millhoff, Y. Jirmann and N. Kockmann, Chem. Eng. Technol., 2017, 40, 2144–2154, DOI:10.1002/ceat.201700419.
- A.-C. Bédard, A. Adamo, K. C. Aroh, M. G. Russell, A. A. Bedermann, J. Torosian, B. Yue, K. F. Jensen and T. F. Jamison, Science, 2018, 361, 1220–1225, DOI:10.1126/science.aat0650.
- C. W. Coley, D. A. Thomas, J. A. M. Lummiss, J. N. Jaworski, C. P. Breen, V. Schultz, T. Hart, J. S. Fishman, L. Rogers, H. Gao, R. W. Hicklin, P. P. Plehiers, J. Byington, J. S. Piotti, W. H. Green, A. J. Hart, T. F. Jamison and K. F. Jensen, Science, 2019, 365, eaax1566, DOI:10.1126/science.aax1566.
- Z. Yang, T. Albrow-Owen, H. Cui, J. Alexander-Webber, F. Gu, X. Wang, T.-C. Wu, M. Zhuge, C. Williams, P. Wang, A. V. Zayats, W. Cai, L. Dai, S. Hofmann, M. Overend, L. Tong, Q. Yang, Z. Sun and T. Hasan, Science, 2019, 365, 1017–1020, DOI:10.1126/science.aax8814.
- M. Segler, M. Preuss and M. Waller, Nature, 2018, 555, 604–610, DOI:10.1038/nature25978.
| This journal is © The Royal Society of Chemistry 2021 |