
Zeolite-mediated production of cyclic organic carbonates: reaction of CO2 with styrene oxide on zeolite Y impregnated with metal halides
Leonardo P.
Ozorio
a,
Fábio J. S.
Henrique
 b,
James W.
Comerford
c,
Michael
North
*c and
Claudio J. A.
Mota
*abd
b,
James W.
Comerford
c,
Michael
North
*c and
Claudio J. A.
Mota
*abd
aEscola de Química, Universidade Federal do Rio de Janeiro, Av. Athos da Silveira Ramos 149, CT Bl E, 21941-909, Rio de Janeiro, Brazil. E-mail: cmota@iq.ufrj.br
bInstituto de Química, Universidade Federal do Rio de Janeiro, Av. Athos da Silveira Ramos 149, CT Bl A, 21941-909, Rio de Janeiro, Brazil
cDepartment of Chemistry, Green Chemistry Centre of Excellence, The University of York, Heslington, York, YO105DD, UK. E-mail: michael.north@york.ac.uk
dINCT Energia e Ambiente, UFRJ, 21941-909, Rio de Janeiro, Brazil
First published on 2nd March 2021
Abstract
Zeolites LiY, NaY, KY and ZnY were impregnated with small amounts of the corresponding metal halides (chloride, bromide and iodide) and tested as catalysts for the carboxylation of styrene oxide with CO2. The KI/KY system was tested at 100 °C and 50 bar and yielded 100% of styrene carbonate, by 1H NMR, within 6 h, whereas KI alone required 15 h to achieve the same yield. The reaction occurs inside the zeolite pores, with the metal cations acting as Lewis acidic centers and the impregnated halides as nucleophiles. Theoretical calculations indicated that chlorides are better stabilized within the zeolite cage than iodides, explaining the reactivity order and suggesting a solvent-like behavior of the zeolite structure.
Introduction
The concept of CO2 utilization, where the captured gas is transformed into useful fuels and chemicals, is gaining importance in recent years1,2 as an interesting approach to mitigate the problems associated with the burning of fossil fuels and global warming. Urea and salicylic acid have long been produced3 from CO2. Methanol4 and organic carbonates5 are emerging as other chemicals that can be industrially produced from carbon dioxide, but improvements to the existing technologies are still necessary.Cyclic organic carbonates are important chemicals used as: polar aprotic solvents, precursors in the synthesis of polycarbonates and polyurethanes, electrolytes in lithium ion batteries and in the production of pharmaceuticals, amongst other applications.6–8 They are mostly produced by the reaction of carbon dioxide with epoxides in the presence of metal complexes and quaternary ammonium halides.5,9–11 The generally accepted mechanism for this reaction involves the interaction of the epoxide with the metal complex, which acts as Lewis acid catalyst, followed by nucleophilic attack by the halide to open the epoxide ring. The thus formed halohydrin reacts with CO2 to form a carboxylate, which after intramolecular displacement gives the cyclic organic carbonate (Scheme 1).
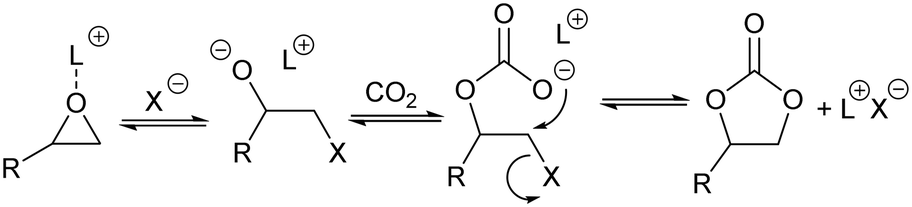 | ||
| Scheme 1 Proposed mechanism of formation of cyclic organic carbonates from the reaction of CO2 with epoxides. L+ represents a Lewis acid, usually a metal complex, and X− represents a halide, usually in the form of a quaternary ammonium salt. | ||
The use of heterogeneous catalytic systems in the reaction of CO2 with epoxides to form cyclic carbonate is receiving increased attention.12 Metal oxides, such as La2O3 and MgO, have been used in the synthesis of propylene carbonate from CO2 and propylene oxide, in the presence of DMF, at 8.0 MPa and 150 °C for 15 hours.13 The yields however, were in the range of 32 to 54% and other products were also formed. Higher catalytic activities were observed over a mixed Mg–Al oxide (Mg/Al = 5), which gave 88% of propylene carbonate in the presence of DMF as solvent.14 The authors justified the higher catalytic activity as being consequence of the cooperation between acidic and basic sites within the mixed metal oxide. However, it has been found that reaction of styrene oxide with CO2 in the presence of DMF gives styrene carbonate in 85% yield at 7.9 MPa of CO2 pressure and 150 °C for 15 hours.15 Therefore, it is not clear what role, if any, the metal oxides play in these reactions.
In recent years, the use of supported or functionalized mesoporous silicas,16–18 MOFs19 and ZIFs20–22 have appeared in the literature on the synthesis of cyclic organic carbonates. In some cases, either more severe reaction conditions or addition of a co-catalyst, usually tetraalkylammonium halides, are required to achieve high yields of products.
Among crystalline aluminosilicates, zeolite KY impregnated with caesium acetate (Cs/KX) has been used as a basic solid catalyst for the synthesis of organic carbonates from CO2 and epoxides.23 The parent KX gave poor yields of ethylene carbonate (1.8%), whereas impregnation with caesium increased the yield to 14%, at 150 °C and 36 bar initial CO2 pressure for 3 hours. The yields were, however, significantly lower than those obtained using tetraethylammonium bromide as catalyst, which gave 74% yield of ethylene carbonate under the same conditions. Amine-functionalised SAPO-34 has also been tested as a catalyst for the reaction of CO2 with epoxides.24 Nevertheless, the yields are high only with small epoxides such as epichlorohydrin and propylene oxide. The encapsulation of metal complexes in zeolites is also a reported procedure to afford cyclic organic carbonates.25,26
We have shown that zeolites may behave as solid solvents, with the ability to perform traditional nucleophilic substitution reactions, upon impregnation with metal halides.27 For instance, reaction of tert-butyl chloride over NaY zeolite impregnated with NaBr resulted in the formation of tert-butyl bromide.28 The results were interpreted as being due to the ionization of the alkyl chloride inside the pores, with subsequent nucleophilic attack by the dispersed bromide anions within the pore structure, similar to what happens in polar solvents. Nevertheless, this concept has not been widely exploited as an alternative use of zeolite as a medium to carry out ionic reaction, taking advantage of the stabilization of ions within the pores.
Considering the reaction pathway depicted in Scheme 1, we decided to investigate the use of zeolite Y impregnated with minor amounts of metal halides as potential heterogeneous catalytic systems for the synthesis of styrene carbonate from CO2 and styrene oxide (Scheme 2). It is known that metal cations are not fully coordinated within the zeolite framework, showing significant Lewis acidity,29 that could coordinate with the oxygen atom of the epoxide, whereas the impregnated halide may act as nucleophile to open the activated oxirane ring.
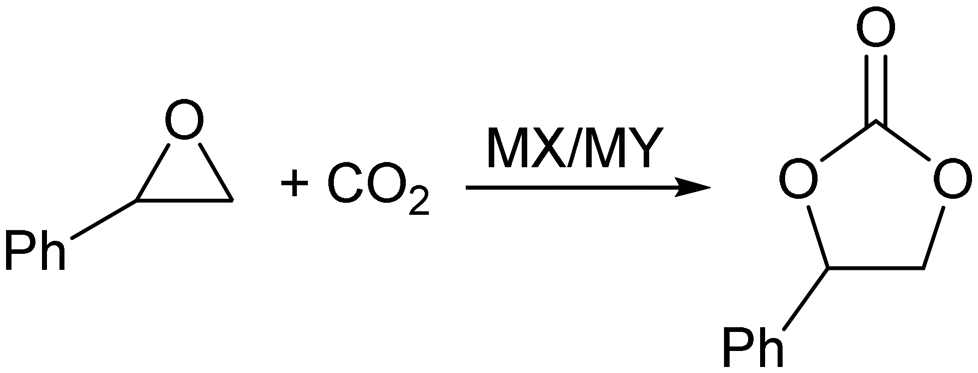 | ||
| Scheme 2 Reaction of styrene oxide with CO2 in the presence of zeolite Y impregnated with metal halide. MX stands for the metal halide and MY stands for metal-exchanged zeolite Y. | ||
Experimental part
Metal-exchanged zeolites were prepared from commercial zeolite NaY (CBV 100) obtained from Zeolyst. The NaY was initially subjected to ion exchange with metal nitrates. The general procedure involved stirring 30 g of NaY with a solution of 0.7 mol of metal nitrate in 700 mL of deionized water, at 60 °C for 24 h. Subsequently, the solid was vacuum filtered and washed with deionized water (5 × 700 mL), then dried overnight at 150 °C.Approximately 10 g batches of the exchanged zeolites were impregnated with 7.8 mmol of the metal halides by an incipient wetness procedure. The suspension was initially stirred at 60 °C for 24 h and then placed in a rotary evaporator to completely evaporate the water. The resulting powder was dried overnight at 150 °C. The final materials were named as MX/MY, where MX stands for the impregnated metal halide and MY stands for the metal-exchanged zeolite Y.
The catalysts were initially tested at atmospheric pressure and room temperature. Approximately 0.91 mL of styrene oxide and an amount of zeolite, equivalent to obtain a 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of styrene oxide to the impregnated metal halide, were placed together in the presence of CO2 for 24 h. Reactions were also carried out at 50 bar CO2 pressure and 50 or 100 °C in a stainless-steel reactor. Prior to all the reactions, the catalysts were dried overnight at 120 °C and rapidly transferred to the reaction medium. At the end of the reactions, the mixture was diluted with 15 mL of dichloromethane and centrifuged at 3500 rpm for 10 min. Subsequently, the liquid fraction was filtered through CELITE® 545 and solvent removed in vacuum. At this stage, an aliquot of the products was analysed by gas chromatography with TCD detection. The reactions that showed styrene carbonate production were then analysed by 1H NMR spectroscopy for quantification of the yield, calculated as the ratio of the 1H NMR signals of styrene carbonate and styrene oxide multiplied by 100, as no other products were detected in the NMR spectra.
:1 molar ratio of styrene oxide to the impregnated metal halide, were placed together in the presence of CO2 for 24 h. Reactions were also carried out at 50 bar CO2 pressure and 50 or 100 °C in a stainless-steel reactor. Prior to all the reactions, the catalysts were dried overnight at 120 °C and rapidly transferred to the reaction medium. At the end of the reactions, the mixture was diluted with 15 mL of dichloromethane and centrifuged at 3500 rpm for 10 min. Subsequently, the liquid fraction was filtered through CELITE® 545 and solvent removed in vacuum. At this stage, an aliquot of the products was analysed by gas chromatography with TCD detection. The reactions that showed styrene carbonate production were then analysed by 1H NMR spectroscopy for quantification of the yield, calculated as the ratio of the 1H NMR signals of styrene carbonate and styrene oxide multiplied by 100, as no other products were detected in the NMR spectra.
Calculation details
DFT calculations were done using periodic boundary conditions, at DFT level, using the codes available in the Quantum Expresso package.30 The coordinates of all atoms were optimized on the basis of the generalized gradient approximation (GGA) with the exchange and correlation functional developed by Perdew, Burke and Ernzerhof (PBE).31 The ion cores were described by Vanderbilt ultrasoft pseudopotentials.32 The electronic state was expanded in plane waves basis set with an energy cutoff of 60 Ry. Brillouin zone sampling was restricted to the Γ-point. The structure of the primitive unit cell of zeolite Y, containing one framework aluminium atom and K+ as counter ion, was considered in the calculations, totalizing 145 atoms. The cell lengths are a = b = c = 17.697 Å. For the metal halides, a cluster comprising 4 ionic units of KX (X = Cl, Br or I) was considered to describe the crystal lattice. The charge density of the ions was computed using the Bader analysis.33The embedded energy was taken as the difference between the energy of the KY zeolite embedded with the KX crystal and the sum of the energies of the isolated zeolite structure and the KX lattice, at same level of theory.
Results and discussions
Table 1 shows the styrene carbonate yields, obtained by 1H NMR spectroscopy, for the catalytic systems under different reaction conditions. The parent NaY and the metal-exchanged zeolites gave no detectable yield of styrene carbonate, indicating that the presence of the impregnated halide is necessary for the reaction to occur under the studied conditions. At room temperature and atmospheric pressure, the catalysts showed negligible production of styrene carbonate within 24 hours. However, at 50 bar and 50 °C all the impregnated catalyst systems possessed some activity for formation of styrene carbonate. The nature of the halogen atom influences the performance of the system. For instance, NaCl/NaY gave only 1% yield of styrene carbonate at 50 bar and 50 °C after 24 hours, whereas NaBr/NaY gave 2% yield. The best result in this series was observed with NaI/NaY, which produced 10% yield of styrene carbonate under the same conditions. These results are in agreement with the expected reactivity of the halides towards nucleophilic substitution in solution. The metal halide alone showed low or no conversion to the carbonate at 50 bar and 50 °C after 24 hours, except for ZnI2.| System | 25 °C; 1 bar | 50 °C; 50 bar | 100 °C; 50 bar |
|---|---|---|---|
| NaY | 0% | 0% | — |
| NaCl/NaY | 0% | 1% | — |
| NaBr/NaY | 0% | 2% | — |
| NaI/NaY | 1% | 10% | 100% |
| NaI | — | 6% | 100% |
| LiY | 0% | 0% | — |
| LiBr/LiY | 0% | 2% | — |
| LiI/LiY | 1% | 7% | — |
| KY | 0% | 0% | — |
| KBr/KY | 0% | 6% | — |
| KI/KY | 3% | 18% | 100% |
| KI | 0% | 0% | 100% |
| ZnY | 0% | 0% | — |
| ZnBr2/ZnY | 0% | 2% | — |
| ZnI2/ZnY | 1% | 34% | Polymer |
| ZnI2 | — | 38% | Polymer |
Additional experiments were carried out at 100 °C and 50 bar for 24 hours with selected zeolite systems. Quantitative yields were observed with NaI/NaY and KI/KY, whereas a polymer was observed with ZnI2/ZnY. Under these conditions, the pure metal halides also had high activity for styrene carbonate synthesis, indicating a poor discrimination amongst the various catalyst systems at higher temperature for 24 hours. On the basis of the results in Table 1, the KI/KY system was selected for further studies.
Fig. 1 shows the variation in yield of styrene carbonate with reaction time for reactions catalysed by KI/KY and for KI alone. The results show that the zeolite system is significantly more active than the pure metal halide, with the yield of styrene carbonate reaching 100% within 6 hours, whereas in the absence of zeolite the reaction took 15 hours to achieve the same result. Fig. 2 shows the effect of mixing KI and KY independently in the reaction medium. It can be seen that the impregnated system is more active, but mixing the two components during the reaction does give a significant yield of product compared to that obtained using just KI. These observations lead to two main conclusions. Firstly, the reaction takes place inside the zeolite channels and cavities, because the yield with the impregnated zeolite is higher than that obtained upon mixing the two components in the reaction medium or using just KI. Some diffusion of the metal halide into the zeolite pores can occur during styrene carbonate synthesis, which facilitates the reaction relative to use of KI in the absence of zeolite. On the other hand, KI is less reactive, probably because of solvation effects from the reaction medium and/or its low surface area. The second conclusion is linked to the diffusion of the metal halide into the zeolite pores, which suggests that small amounts of dispersed KI may be enough to catalyse the reaction, because the iodide mostly remains inside the pores.
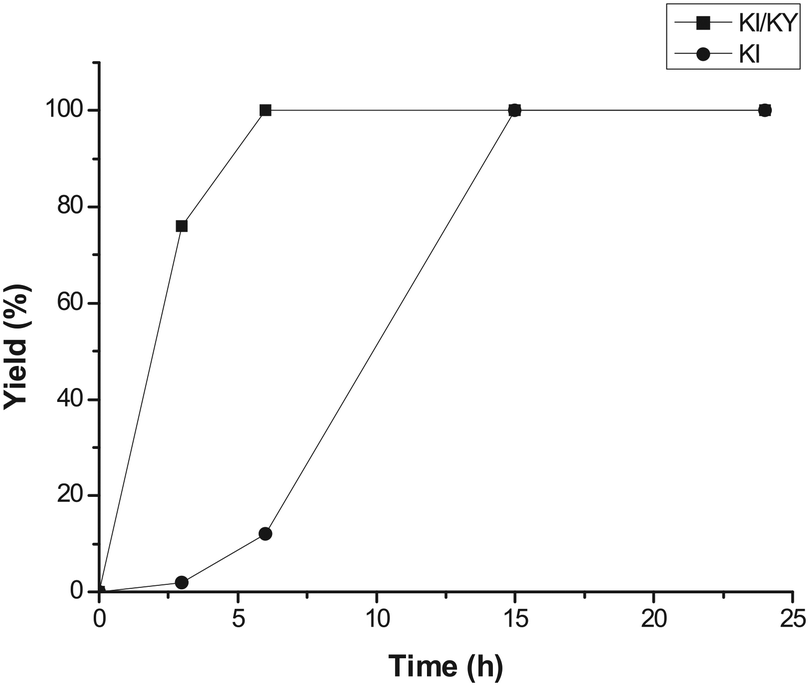 | ||
| Fig. 1 Yield of styrene carbonate against time using KI/KY and just KI, at 100 °C and 50 bar. | ||
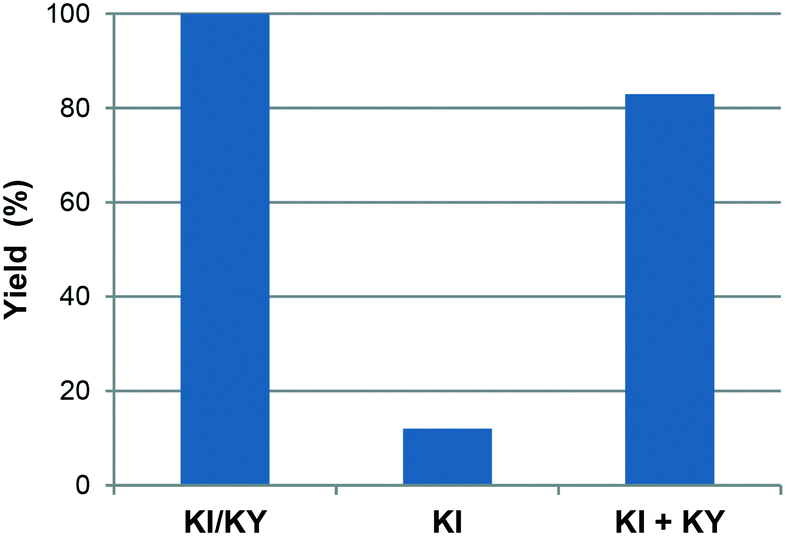 | ||
| Fig. 2 Yield of styrene carbonate using KI/KY, KI and a mixture of KI and KY during reaction at 100 °C and 50 bar for 6 h. | ||
It is worth mentioning that KI is reported34 to be a catalyst in the industrial process of ethylene carbonate production from ethylene oxide and CO2. Hence, the impregnation of this salt inside the zeolite cage can lead to significant improvement in the process with the reduction of the reaction time.
Finally, the reuse of the catalytic system was studied, but a gradual loss of activity was observed (Fig. 3). This is due to leaching of the metal halide, as shown by ICP analysis of the solution obtained at the end of the reaction. About 3% of the potassium is lost after each reaction cycle. This may be explained by the mobility of the ions from the zeolite cavity to the solution and vice versa, probably because of the high polarity of the cyclic carbonate, which may dissolve part of the salt. It must be stressed that a tiny amount of the halide was present during the reaction, as the molar ratio of the epoxide to the impregnated metal halide was about 20. Thus, any leaching of the halide would increase this molar ratio, also explaining the decrease in activity of the catalyst system. Alternatively, it might be possible to carry out the reutilization at longer times, to compensate the partial leaching or to add more halide in the reaction medium, as a type of regeneration. Nevertheless, we did not exploited these possibilities in this study.
 | ||
| Fig. 3 Reutilization of the KI/KY system in the reaction of CO2 with styrene oxide at 100 °C and 50 bar for 6 h. | ||
Table 2 shows the yields obtained for three different cyclic organic carbonates over KI/KY at 100 °C and 50 bar after 4 hours. It seems that steric effects play a significant role, because of the pore size restrictions imposed by the zeolite cavity. Thus, for bulky epoxides the reaction may not proceed properly in the zeolite system. However, from an industrial point of view, small cyclic carbonates, like ethylene and propylene carbonates, are the most relevant, which opens the possibility of exploiting the zeolite-metal halide system as a potential heterogeneous catalyst for these processes.
| Epoxide | Yield of organic carbonate (%) |
|---|---|
| Styrene oxide | 78 |
| R-Propylene oxide | 93 |
| 1,2-Epoxy-dodecane | 3 |
Table 3 compares the conditions and conversion or yield of styrene carbonate synthesis reported in the literature using other heterogeneous catalytic systems. It can be seen that the conversion of the epoxide or the yield of styrene carbonate was lower compared with the present study. In addition, in some cases, the conditions are more severe and there was solvent addition. These results highlight the potential of zeolites impregnated with metal halides as efficient heterogeneous catalytic systems for the production of cyclic organic carbonates. Zeolites have superior thermal stability to MOFs and ZIFs and are significantly cheaper than MCM-41 and other mesoporous materials, which require extensive amounts of template in the synthesis procedure. The possibility of using zeolites impregnated with minor amounts of metal halides also has the potential of suppressing the need of using tetraalkylammonium halides as co-catalysts.
| System | T (°C) | P (bar) | Conv. (%) | Yield (%) |
|---|---|---|---|---|
| a MCM-41 grafted with imidazole and 1,2-dibromoethane. Acetonitrile as solvent and 6 h reaction time; ref. 17. b 2D-MOF, 4 h; ref. 20. c 7 h; ref. 21. d Amine-functionalized SAPO-34; DMF as solvent and 5 h; ref. 24. e This work. | ||||
| MCM-41/ImBra | 140 | 40 | 84 | 81.5 |
| CdPDIA/TBABb | 80 | 20 | — | 40 |
| ZIF-8c | 100 | 7 | — | 55 |
| NH2-SAPO-34d | 85 | 6 | 77 | — |
| KI/KYe | 100 | 50 | 100 | |
In order to better understand the role of the zeolite in stabilizing the metal halide inside the pores, and to help to understand the experimental results, a theoretical study was carried out using periodic conditions to describe the zeolite system. Fig. 4 shows the calculated structures of KCl (comprising 4 ionic units) at PBE level; similar structures were found for KBr and KI, considering the same number designation for the ions. Fig. 5 shows the calculated structures of KY zeolite embedded with the KI cluster, at the same level of theory; the structures of KCl or KBr embedded inside KY zeolite were similar, considering the same number designation for the ions. Tables 4 and 5 shows the distances between the ions in the isolated metal halides and in the embedded zeolites, respectively, whereas Tables 6 and 7 report the calculated charges for the systems. Finally, Table 8 shows the embedded energy of the potassium halide (KX) clusters within the KY pore system, at PBE level of theory.
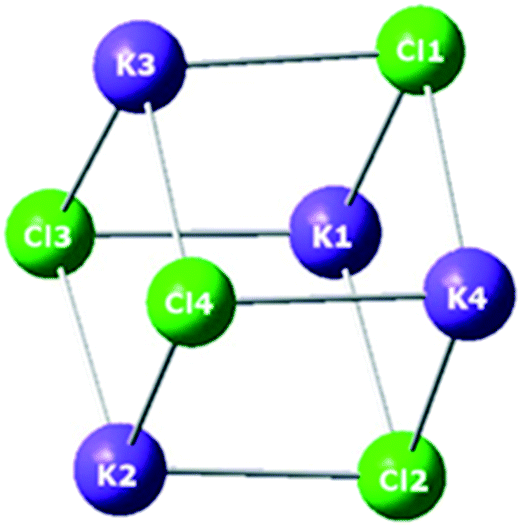 | ||
| Fig. 4 Calculated structure of KCl at PBE periodic boundary conditions. | ||
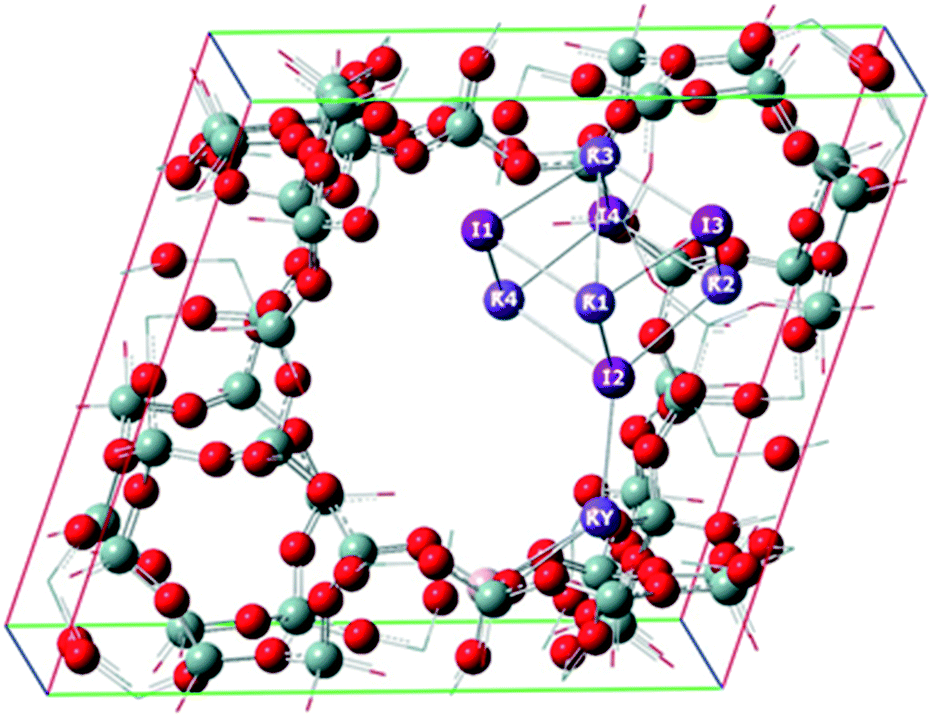 | ||
| Fig. 5 Calculated structure of KI cluster embedded inside KY zeolite, at PBE periodic boundary conditions. The structures of KCl or KBr clusters embedded in KY zeolite were similar, considering the same number designation for the ions. | ||
| KCl | KBr | KI | |||
|---|---|---|---|---|---|
| a Average distance between the ions in the embedded cluster. | |||||
| K1–Cl1 | 2.981 | K1–Br1 | 3.140 | K1–I1 | 3.387 |
| K1–Cl2 | 2.990 | K1–Br2 | 3.145 | K1–I2 | 3.389 |
| K1–Cl3 | 2.983 | K1–Br3 | 3.145 | K1–I3 | 3.393 |
| K2–Cl2 | 2.990 | K2–Br2 | 3.145 | K2–I2 | 3.387 |
| K2–Cl3 | 2.981 | K2–Br3 | 3.142 | K2–I3 | 3.390 |
| K2–Cl4 | 2.980 | K2–Br4 | 3.140 | K2–I4 | 3.387 |
| K3–Cl1 | 2.984 | K3–Br1 | 3.143 | K3–I1 | 3.396 |
| K3–Cl3 | 2.982 | K3–Br3 | 3.141 | K3–I3 | 3.391 |
| K3–Cl4 | 2.983 | K3–Br4 | 3.143 | K3–I4 | 3.395 |
| K4–Cl1 | 2.983 | K4–Br1 | 3.142 | K4–I1 | 3.390 |
| K4–Cl2 | 2.988 | K4–Br2 | 3.142 | K4–I2 | 3.386 |
| K4–Cl4 | 2.983 | K4–Br4 | 3.144 | K4–I4 | 3.392 |
| <K–Cl>a | 2.984 | <K–Br>a | 3.143 | <K–I>a | 3.390 |
| KCl/KY | KBr/KY | KI/KY | |||
|---|---|---|---|---|---|
| a Average distance between the ions in the embedded cluster. | |||||
| KY–Cl2 | 2.915 | KY–Br2 | 3.055 | KY–I2 | 3.287 |
| K1–Cl1 | 2.970 | K1–Br1 | 3.125 | K1–I1 | 3.416 |
| K1–Cl2 | 3.046 | K1–Br2 | 3.181 | K1–I2 | 3.367 |
| K1–Cl3 | 2.947 | K1–Br3 | 3.117 | K1–I3 | 3.422 |
| K2–Cl2 | 3.187 | K2–Br2 | 3.480 | K2–I2 | 3.548 |
| K2–Cl3 | 2.954 | K2–Br3 | 3.128 | K2–I3 | 3.391 |
| K2–Cl4 | 3.002 | K2–Br4 | 3.174 | K2–I4 | 3.352 |
| K3–Cl1 | 3.017 | K3–Br1 | 3.188 | K3–I1 | 3.414 |
| K3–Cl3 | 2.984 | K3–Br3 | 3.149 | K3–I3 | 3.394 |
| K3–Cl4 | 3.010 | K3–Br4 | 3.181 | K3–I4 | 3.345 |
| K4–Cl1 | 2.957 | K4–Br1 | 3.129 | K4–I1 | 3.394 |
| K4–Cl2 | 3.035 | K4–Br2 | 3.186 | K4–I2 | 3.467 |
| K4–Cl4 | 2.951 | K4–Br4 | 3.127 | K4–I4 | 3.328 |
| <K–Cl>a | 3.005 | < K–Br>a | 3.180 | <K–I>a | 3.403 |
| KCl | KBr | KI | |||
|---|---|---|---|---|---|
| a Average charge on the halogen atom. | |||||
| Cl1 | −0.855 | Br1 | −0.842 | I1 | −0.832 |
| Cl2 | −0.855 | Br2 | −0.844 | I2 | −0.831 |
| Cl3 | −0.854 | Br3 | −0.842 | I3 | −0.832 |
| Cl4 | −0.854 | Br4 | −0.843 | I4 | −0.833 |
| <Cl> | −0.854 | <Br> | −0.843 | <I> | −0.832 |
| K1 | 0.855 | K1 | 0.842 | K1 | 0.832 |
| K2 | 0.854 | K2 | 0.844 | K2 | 0.831 |
| K3 | 0.855 | K3 | 0.843 | K3 | 0.833 |
| K4 | 0.854 | K4 | 0.842 | K4 | 0.833 |
| KCl/KY | KBr/KY | KI/KY | |||
|---|---|---|---|---|---|
| a Average charge on the halogen atom. | |||||
| KY | 0.866 | KY | 0.858 | KY | 0.858 |
| Cl1 | −0.849 | Br1 | −0.836 | I1 | −0.815 |
| Cl2 | −0.846 | Br2 | −0.835 | I2 | −0.822 |
| Cl3 | −0.842 | Br3 | −0.827 | I3 | −0.807 |
| Cl4 | −0.847 | Br4 | −0.832 | I4 | −0.784 |
| <Cl> | −0.846 | <Br> | −0.833 | <I> | −0.807 |
| K1 | 0.862 | K1 | 0.851 | K1 | 0.843 |
| K2 | 0.866 | K2 | 0.861 | K2 | 0.852 |
| K3 | 0.857 | K3 | 0.846 | K3 | 0.834 |
| K4 | 0.860 | K4 | 0.850 | K4 | 0.847 |
| System | Embedded energy (kcal mol−1) |
|---|---|
| KCl/KY | −14.1 |
| KBr/KY | −11.9 |
| KI/KY | −10.8 |
It can be seen that, upon embedding the metal halide inside the zeolite Y pore, there is a slight enlargement of the distance between the ions. This may be explained by the interaction of the halide with the zeolite structure, particularly with the potassium cation associated with the framework aluminium atom. For instance, the distance between the potassium counter cation and the Cl2 ion in KCl/KY is 2.981 Å, whereas the average distance in the isolated KCl was calculated to be 2.984 Å. This trend is more pronounced for the other halides. In KBr/KY, the distance between the K+ counter ion and Br2 was 3.055 Å; shorter than the average distance of 3.143 Å in isolated KBr. The same distance in KI/KY is 3.287 Å, also shorter than the average distance between the ions in the isolated KI, computed as 3.390 Å. The stronger interaction of the lattice bromide and iodide with the K+ counter cation of the zeolite reflects the less ionic nature of the bond for these two halides compared with chloride. Hence, the bromide and iodide ions become less associated to the other potassium cations of the cluster than the chloride does. In other words, embedding the potassium halide inside the zeolite leads to a slight dissociation of the ions, due to their interaction with the zeolite structure. This trend decreases in the order I− > Br− > Cl−.
The charge distribution calculation supports the previous view. As a general trend, the average charge on the halide slightly decreases upon embedding the cluster within the zeolite pores. This is probably due to the interaction with the zeolite, which acts like a solvent to stabilize the ions. Therefore, the electrostatic interaction within the metal halide cluster slightly decreases, as a function of the larger separation between the ions.
The results of calculated embedded energy for the metal halides inside the zeolite Y cavity, at PBE level of theory, indicated that KCl is better stabilized than KBr, which in turn is more stabilized than KI. This may explain the higher reactivity of the metal iodide zeolite systems compared with metal chlorides and bromides. This behavior is similar to that observed in solution, where chlorides are better stabilized than bromides and iodides through hydrogen bonding interactions. Thus, in the present case, the zeolites act as solid solvents, providing an environment for the stabilization of ionic species and transition states. A more extensive theoretical study on the nature of the metal halide/zeolite Y systems and on the mechanistic pathway is being carried out to be reported later.
The solvent-like properties of zeolites was initially proposed by Derouane.35,36 Nevertheless, he mostly associated this behaviour to the capability of the zeolites to confine or adsorb molecules in relation to an external solution, as in a solvent extraction procedure. Our studies on the nucleophilic substitution of alkyl halides and carboxylation of styrene oxide expand the concept of zeolites as solvent, to include their performance in ionic reactions and stabilization of ionic species inside the cages.
Conclusion
Zeolite Y impregnated with metal halides are efficient catalyst systems to produce styrene carbonate from the reaction of CO2 with styrene oxide. The yield is dependent on the nature of the metal halide, with iodide giving higher yields than the corresponding bromide and chloride salts. The reaction takes place inside the zeolite cavity, but leaching of the metal halide reduces the yield of the carbonate upon reuse. The KI/KY system is significantly more active than KI alone, which is reported to be a possible catalyst in the industrial process of cyclic carbonate production.Periodic theoretical calculations, at PBE level, indicated that the KCl cluster is better stabilized within the zeolite framework than KBr and KI ones. The ions become less associated upon embedding inside the zeolite structure. The order of stabilization agrees with the reactivity of the impregnated metal halides. These data suggest that the zeolite cavities act similarly to solvents, stabilizing ionic species and transition states.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Newton Fund Cooperation between the British Council and FAPERJ for funding. They also acknowledge financial support of the Royal Society. CJAM thanks CNPq and FAPERJ for financial support and fellowships. LPO and FJSH thank CAPES for a doctoral scholarship.References
- G. Centi, E. A. Quadrelli and S. Perathoner, Energy Environ. Sci., 2013, 6, 1711–1731 RSC.
- A. Dutta, S. Farooq, I. A. Karimi and S. A. Khan, J. CO2 Util., 2017, 19, 49–57 CrossRef CAS.
- M. Aresta, A. Dibenedetto and A. Angelini, J. CO2 Util., 2013, 3–4, 65–73 CrossRef CAS.
- M. Pérez-Fortes, J. C. Schöneberger, A. Boulamanti and E. Tzimas, Appl. Energy, 2016, 161, 718–732 CrossRef.
- M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514–1539 RSC.
- L.-F. Xiao, F.-W. Li, J.-J. Peng and C.-G. Xia, J. Mol. Catal. A: Chem., 2006, 253, 265–269 CrossRef CAS.
- H. Xie, S. Li and S. Zhang, J. Mol. Catal. A: Chem., 2006, 250, 30–34 CrossRef CAS.
- V. S. Yadav, M. Prasad, J. Khan, S. S. Amritphale, M. Singh and C. B. Raju, J. Hazard. Mater., 2010, 176, 1044–1050 CrossRef CAS PubMed.
- J. W. Comerford, I. D. V. Ingram, M. North and X. Wu, Green Chem., 2015, 17, 1966–1987 RSC.
- A. Decortes, A. M. Castilla and A. W. Kleij, Angew. Chem., Int. Ed., 2010, 49, 9822–9837 CrossRef CAS PubMed.
- R. Martín and A. W. Kleij, ChemSusChem, 2011, 4, 1259–1263 CrossRef PubMed.
- A. A. Marciniak, K. Lamb, L. P. Ozório, C. J. A. Mota and M. North, Curr. Opin. Green. Sustain. Chem., 2020, 26, 100365 CrossRef.
- B. M. Bhanage, S. Fujita, Y. Ikushima and M. Arai, Appl. Catal., A, 2001, 219, 259–266 CrossRef CAS.
- H. Kawanami and Y. Ikushima, Chem. Commun., 2000, 2089–2090 RSC.
- K. Yamaguchi, K. Ebitani, T. Yoshida, H. Yoshida and K. Kaneda, J. Am. Chem. Soc., 1999, 121, 4526–4527 CrossRef CAS.
- F. Adam and M. S. Batagarawa, Appl. Catal., A, 2013, 454, 164–171 CrossRef CAS.
- J. N. Appaturi and F. Adam, Appl. Catal., B, 2013, 136–137, 150–159 CrossRef CAS.
- M. Liu, B. Liu, L. Liang, F. Wang, L. Shi and J. Sun, J. Mol. Catal. A: Chem., 2016, 418–419, 78–85 CrossRef CAS.
- B. Li, Inorg. Chem. Commun., 2018, 88, 56–59 CrossRef CAS.
- M. Zhu, D. Srinivas, S. Bhogeswararao, P. Ratnasamy and M. A. Carreon, Catal. Commun., 2013, 32, 36–40 CrossRef CAS.
- K. M. Bhin, J. Tharun, K. R. Roshan, D.-W. Kim, Y. Chung and D.-W. Park, J. CO2 Util., 2017, 17, 112–118 CrossRef CAS.
- Y.-F. Lin, K.-W. Huang, B.-T. Ko and K.-Y. A. Lin, J. CO2 Util., 2017, 22, 178–183 CrossRef CAS.
- M. Tu and R. J. Davis, J. Catal., 2001, 199, 85–91 CrossRef CAS.
- M. Ahmed and A. Sakthivel, J. CO2 Util., 2017, 22, 392–399 CrossRef CAS.
- D. Liu, G. Li, J. Liu and Y. Yi, Fuel, 2019, 244, 196–206 CrossRef CAS.
- H. He, Q. Q. Zhu, C. Zhang, Y. Yan, J. Yuan, J. Chen, C. P. Li and M. Duo, Chem. – Asian J., 2019, 14, 958–962 CrossRef CAS PubMed.
- M. Franco, N. Rosenbach, G. B. Ferreira, A. C. O. Guerra, W. B. Kover, C. C. Turci and C. J. A. Mota, J. Am. Chem. Soc., 2008, 130, 1592–1600 CrossRef CAS PubMed.
- N. Rosenbach, A. P. A. dos Santos, M. Franco and C. J. A. Mota, Chem. Phys. Lett., 2010, 485, 124–128 CrossRef CAS.
- G. Busca, Microporous Mesoporous Mater., 2017, 254, 3–16 CrossRef CAS.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. De Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- D. Vanderbilt, Phys. Rev. B, 1990, 41, 7892–7895 CrossRef PubMed.
- R. F. W. Bader, Acc. Chem. Res., 1985, 18, 9–15 CrossRef CAS.
- H. Okamoto and K. Someya, EP App., EP1630161A1, 2006.
- E. G. Derouane, C. J. Dillon, D. Bethell and S. B. Derouane-Abd Hamid, J. Catal., 1999, 187, 209–218 CrossRef CAS.
- E. G. Derouane, G. Crehan, C. J. Dillon, D. Bethell, H. He and S. B. Derouane-Abd Hamid, J. Catal., 2000, 194, 410–423 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |