
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImproved continuous synthesis of UiO-66 enabling outstanding production rates†
Angelos
Polyzoidis
 *,
Steffen
Reichle
,
Maud
Schwarzer
,
Calogero Giancarlo
Piscopo
,
Stefan
Löbbecke
and
Dusan
Boskovic
*,
Steffen
Reichle
,
Maud
Schwarzer
,
Calogero Giancarlo
Piscopo
,
Stefan
Löbbecke
and
Dusan
Boskovic
Fraunhofer Institute for Chemical Technology ICT, Joseph-von-Fraunhofer Str. 7, 76327 Pfinztal, Germany. E-mail: angelos.polyzoidis@ict.fraunhofer.de
First published on 25th January 2021
Abstract
As the applicability of MOFs progresses, the demand for production of large quantities in a safe and reproducible manner is constantly growing. UiO-66 is considered to be one of the most stable MOFs and for this reason it is attractive for a wide range of applications. This work describes the upscaled continuous synthesis of UiO-66 and its derivatives by implementation of a simple milli-structured reactor. High quality material can be produced within 8 min of reaction under unprecedented reproducibility and Space time yields up to 7162 kg m−3 d−1. The optimization of the reaction was carried out in a smaller reactor where the effects of concentration, crystallization agent (HCl) and temperature were studied and the best conditions were applied in a bigger reactor capable of delivering up to 200 g d−1 in just 40 mL of reactor volume. Finally, UiO-66 derivatives were also synthesized while the reactor and process stability were tested through a number of long runs.
Introduction
After more than 20 years of research, MOFs1,2 have now surpassed the stage of basic research and are in the process of being used on an industrial scale. The introduction of these materials opened a new chapter of research for a wide range of applications.3–7 Already, in their first ten years of their existence, several popular MOFs had been created, such as the MIL,8 UiO,9 IRMOFs,10 and ZIFs11 families that the research focused on. Among them, due to its high stability, UiO-66 has been tested in a broad spectrum of applications.12,13The more these materials are tested in commercially relevant applications, the more important it becomes to find a controlled and reproducible way for their synthesis on a pilot and industrial scale. Usually after the initial results obtained with the powder form efforts are given in shaping14–16 and coating17 the MOFs in order to allow easier handling.
Studies have already emerged that stress the importance of reducing production costs18 as well as adopting processes that lead to production in a more repeatable and reliable way.19 To this extent the demand for production of large quantities of UiO-66 in a safe and reproducible manner is constantly growing.20 Unsurprisingly, like in most MOFs, UiO-66 was firstly prepared under discontinuous conditions and this reaction has been extensively studied and optimized.21
Focus on the development of continuous synthetic pathways has also been invested, leading to very interesting results. In 2013, UiO-66 was first synthesized continuously in a microreactor in a study that served mainly as a feasibility demonstration.22 Other studies exploited the advantages of flow chemistry for controlling the size of the particles,23 or simply achieving higher production rates.24 Rubio-Martinez et al.24 using a milli-structured reactor (internal diameter 1 mm) and a reaction time of only 10 min achieved a productivity of about 1.7 g h−1 which corresponds to a space–time yield (STY) of 672 kg m−3 d−1 – the highest by that time.
In an attempt to further intensify the production of the MOF flow chemistry was combined with various techniques such as spray drying,25 ultrasound26 and microwave irradiation.27 While spray drying reduced the reaction time to just over one minute and ultrasound ensured a higher product concentration without the danger of clogging, the reported space–time yields reached only the value of 428 kg m−3 d−1. The use of microwave radiation on the other hand showed more promising results. A capillary reactor with an internal diameter of 4.35 mm placed in a microwave oven realized the synthesis of UiO-66 with a space–time yield as high as 4899 kg m−3 d−1 and a productivity of about 9.8 g h−1.27
In a previous work28 the UiO-66 continuous synthesis using a T-type micromixer has been presented, highlighting the consequences that intensified flow reaction conditions induce to its textural properties. In the present work, the already reported method is consistently developed, allowing the production of UiO-66 and derivatives with higher surface areas and outstanding production rates, using a larger reactor. Special attention was given in understanding the effect of reaction parameters and finding the optimal reaction conditions that maintain good product characteristics while allowing an effective and trouble-free production.
Experimental
Chemicals and solvents were analytical grade, except methanol which was technical grade, purchased from various commercial suppliers and used without further purification. See ESI† for more details. For the purpose of this paper reaction ratios correspond to Zr![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :linker:DMF:HCl:H2O.
:linker:DMF:HCl:H2O.
Analytical methods
Powder diffraction measurements were performed on a Bragg–Brentano diffractometer, D8 Advance from Bruker AXS, equipped with copper tube, 2.5° Soller collimators, 0.3 mm divergence slit, anti-scatter screen, flip-stick stage and silicon strip detector (LynxEye) with 3° 2θ detector opening. The diffraction patterns were monitored between 5° and 60° 2θ with 0.02° 2θ step width and 1 s counting time per step. The STOE WinXPOW software (version 1.2, STOE & Cie GmbH) was used to display the powder diffractograms. The calculation of the powder diffractogram of UiO-66 was based on information from the literature.29TGA experiments were carried out on a TGA Q5000 TA Instruments device. Samples were heated under air at a heating rate of 10 K min−1 up to 700 °C.
Nitrogen adsorption isotherms were measured at −196 °C using a Quantachrome Autosorb iQ surface area analyser. Samples – if not stated otherwise – were degassed in vacuum for 12 h at 200 °C prior to the analysis. BET pressure range varied between 3.0 × 10−2 and 7.0 × 10−2 bar for most of the samples (ESI†).
Reactor design
The two reactors used consisted of a T-type mixer (inner diameter 1.25 mm, ETFE, Upchurch Scientific), which was connected to a helically wound PFA tube (Bola), that acted as the reaction zone. The helical shape of the reactor body was chosen in order to avoid “U” points. These areas are potentially adverse for the long-term operation of the reactor since solid particles can be trapped there, eventually leading to clogging. The length of each tube was 5 m, while the inner diameter of the so-called small reactor was about 1.6 mm and that of the so-called large reactor about 3.2 mm (Fig. S1†). The volume of the small reactor was thus 10 mL and that of the large reactor 40 mL. For the synthesis, the reactors were dipped into an oil bath. The reactant solutions were conveyed by means of two syringe pumps (S1610, Sykam GmbH) to the micromixer, passed through the reactor and the product formed was recovered by filtration.For the segmented flow synthesis (large reactor only), a second T-type mixer was placed after the first one, in which the reaction mixture was segmented with nitrogen. The flow rate of nitrogen was adjusted using a mass flow controller (Analyt-MTC). Fig. S2† shows a schematic representation of the experimental setup.
UiO-66 small scale synthesis
A 0.35 M solution of ZrOCl2·8H2O and a 0.35 M solution of BDC in DMF were used as precursors. In the metal precursor 10 eq. of HCl (37%) were added as a modulator. The flow rate of each pump was set at 0.6 mL min−1 and the reactor was operated for 60 min at 110 °C. The molar ratio of the reaction was Zr:BDC:DMF:HCl:H2O 1:1:63:10:34. The formed solid was separated continuously using membrane filtration and was washed with a small amount of fresh methanol while being on the filter. Afterwards the solid was suspended overnight in methanol, recovered by filtration and dried at room temperature.
UiO-66 large scale synthesis
A 0.35 M solution of ZrOCl2·8H2O and a 0.35 M solution of BDC in DMF were used as precursors. In the metal precursor 10 eq. of HCl (37%) were added as a crystallization agent. The flow rate of each pump was set at 1.9 mL min−1 while the flow rate of the N2 at 0.25 mL min−1 and the reactor was operated for 15 min at 110 °C. The molar ratio of the reaction was Zr:BDC:DMF:HCl:H2O 1:1:63:10:34. The formed solid was separated continuously using membrane filtration and the same washing procedure was followed like in the small-scale synthesis.
Results and discussion
Effect of HCl
The starting point for the improvement of the continuous synthesis of UiO-66 was the results of the previous work conducted on this topic.28 The increase of the amount of crystallization agent (HCl) was chosen as means for increasing the reaction yield and thus productivity. To study the effect of hydrochloric acid, reactions were carried out in which the proportion of HCl in the reaction mixture was increased to 10 eq. The concentration of the precursor solutions was left constant at 0.26 mol L−1. Reaction temperature was adjusted between 70 and 110 °C, while the residence time was either 8 or 24 min Table 1.:1:98:2:7 and 1:1:89:10:34
| HCl | Time | Temp. | Yield | BET | STY | Prod. |
|---|---|---|---|---|---|---|
| eq. | min | °C | % | m2 g−1 | kg m−3 d−1 | g d−1 |
| 2 | 24 | 90 | 36 | 1368 | 523 | 5.3 |
| 110 | 64 | 1534 | 1103 | 11.1 | ||
| 10 | 24 | 70 | — | — | — | — |
| 90 | 81 | 1545 | 1399 | 14.1 | ||
| 110 | 77 | 1471 | 1409 | 14.2 | ||
| 8 | 90 | — | — | — | — | |
| 110 | 67 | 1602 | 3509 | 35.3 |
Compared to the reactions carried out with 2 eq. HCl, significantly higher yields are found. An increase in the acid concentration thus leads to an acceleration of the reaction and correspondingly larger amounts of product. Furthermore, the influence of the acid seems to outweigh the temperature, since at a residence time of 24 min no significant differences in the yields for the reaction at 90 °C and 110 °C can be observed. This observation is in line with similar observations of HCl catalysing the formation reaction of UiO-66.30,31 Finally, using HCl as a promotor seems to lead in the incorporation of Cl− in the final product.
After establishing that the increase in the acid concentration leads to an accelerated reaction progress, it was investigated whether a temperature of 70 °C is sufficient for a synthesis. However, this did not lead to product formation.
In order to increase the productivity of the process, reactions were also carried out with a shorter residence time. Here it was found that even the temperature of 90 °C was no longer sufficient for the formation of a product. At 110 °C, however, and despite a slight yield decrease to 67%, the productivity could be significantly increased to 35.3 g d−1 (corresponds to a STY of 3509 kg m−3 d−1) due to the lower process time.
Aiming to exclude that the accelerating effect of the hydrochloric acid is not caused by the water contained therein, a control experiment was carried out at 90 °C and a residence time of 24 min, in which only the 34 eq. water were added. The product obtained had a markedly deteriorated quality (BET area: 227 m2 g−1), suggesting that the use of an acidic crystallization agent is essential for obtaining good product quality within the given time (Fig. S6†). A possible explanation for this is that in absence of the acidic medium the hydrolysis of the zirconyl chloride is not suppressed and condensed zirconium-hydroxide-clusters can be formed.32 Turbidity of the zirconium chloride solution, which is an evidence for the formation of insoluble species like Zr(OH)4, has indeed been observed. This mechanism is responsible for a reduced zirconium availability, since the hydroxide cannot be converted into the crystalline MOF. Therefore, the UiO-66 accounts only for a portion of the solid product, which hence shows significantly reduced crystallinity and porosity. Crystallographic comparison of the performed reactions can be seen in Fig. 1, while Table 1 summarizes the results of the reactions.
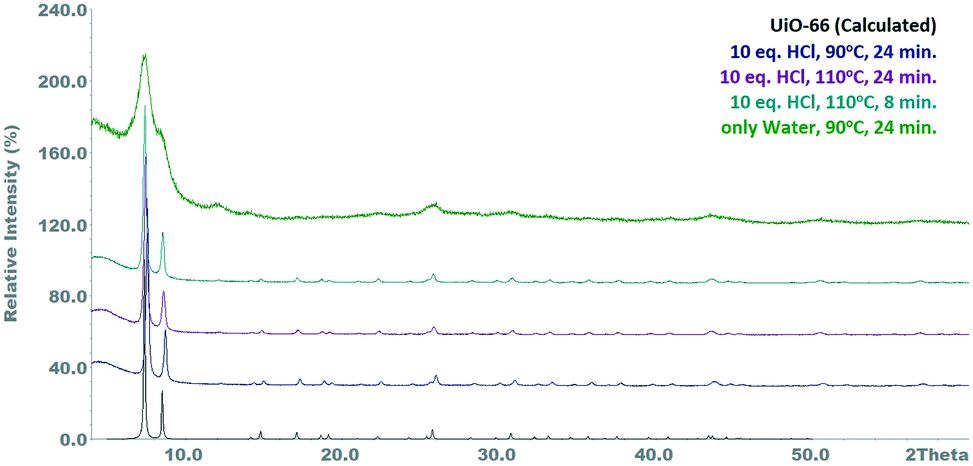 | ||
| Fig. 1 Powder diffractograms of samples synthesized with a concentration of 0.26 M. | ||
An additional reaction with 20 eq. HCl was also performed to see if the reaction can be further accelerated. The results showed that the yield did increase a bit more, but not considerably to justify any significant additional effect (Fig. 2).
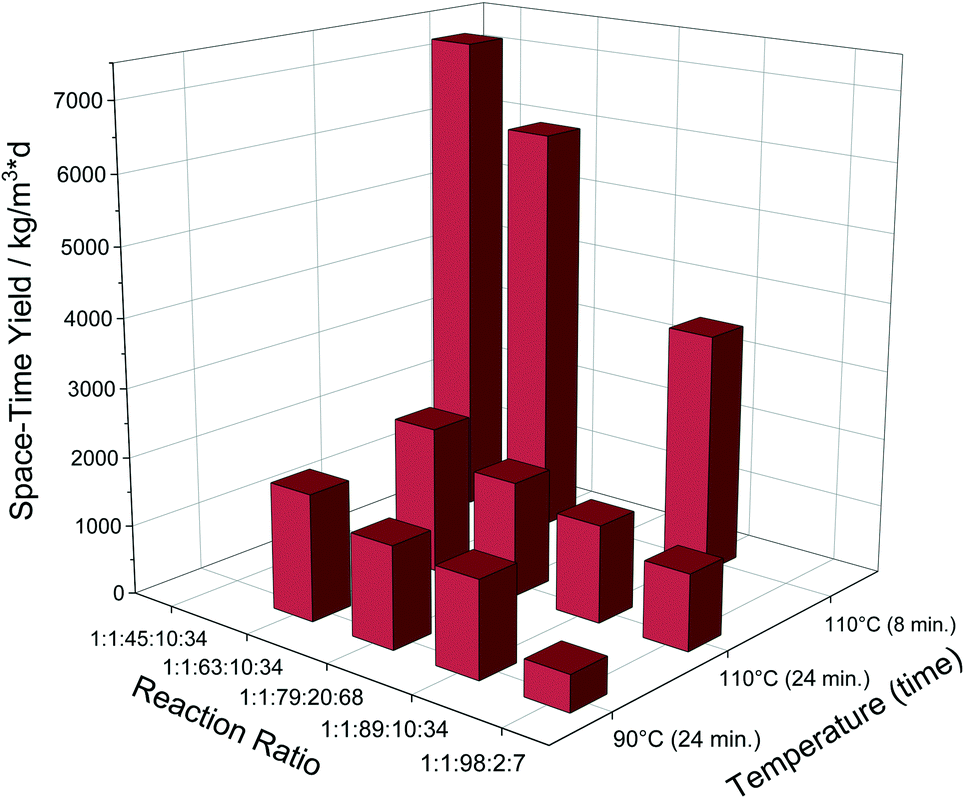 | ||
| Fig. 2 Overview of the achieved STY depending on reaction parameters. Reaction ratio corresponds to Zr:linker:DMF:HCl:H2O. | ||
Effect of concentration
Previous research on MOFs has shown that the product formation can also be accelerated by a higher precursor concentration.33 It was investigated whether this is the case also in the continuous synthesis of UiO-66 and whether the product quality is also retained. Due to the low solubility of the terephthalic acid in DMF, however, the concentration could only be increased to 0.35 mol L−1 (Table 2 – ESI†) – should no change in the yield occur, this would still correspond to a productivity increase by a factor of 1.3.:1:63:10:34 and 1:1:45:10:34
| Flow rate | Time | Temp. | Yield | BET | STY | Prod. |
|---|---|---|---|---|---|---|
| min | °C | % | m2 g−1 | kg m−3 d−1 | g d−1 | |
| Equal | 24 | 90 | 79 | 1638 | 1835 | 18.4 |
| 110 | 88 | 1684 | 2128 | 21.4 | ||
| 8 | 110 | 82 | 1678 | 5982 | 60.1 | |
| Different | 8 | 110 | 74 | 1690 | 7162 | 72 |
Concerning their textural properties, the products of this set of reactions exhibit surfaces in the range of 1650 m2 g−1 which are higher than the equivalents at lower concentration (Fig. S5†). As shown also in literature highly defective UiO-66 is expected to exhibit BET areas in this range.34
With regard to the yields, a significant increase from 67% to 82% can be found, especially for the reaction carried out within 8 minutes, supporting the initial assumption that the increased concentration promotes the progress of the reaction. With these reaction conditions the productivity of the process was almost doubled, reaching 60.1 g d−1 – a value that corresponds to a STY of 5982 kg m−3 d−1.
The final strategy for increasing the concentration and hence the productivity in the small reactor at equimolar reagent ratios was the variation of the pumping rates of the precursor solutions. So far, both solutions were dosed into the reactor at the same pump rate. Since the terephthalic acid solution concentration is limited by its solubility, varying the pump rates would allow the further increase (to 0.7 mol L−1) of the concentration of the zirconyl chloride octahydrate solution while keeping the reaction ratio and time unchanged. Pumping rates of 0.4 mL min−1 (Zr solution) and 0.8 mL min−1 (H2BDC solution) were selected, maintaining the residence time at 8 min.
Compared to the reaction with analogous reaction parameters, but with the same pumping rates, the quality of the product has remained excellent having a BET area of 1690 m2 g−1 and retaining its crystallinity (Fig. S7†).
As expected, the productivity increased further by a factor of 1.2, reaching 72 g d−1 of dry MOF. The corresponding 7162 kg m−3 d−1 STY is the highest value reported for dry UiO-66, to the best of our knowledge.35Fig. 2 summarizes all the achieved STY values.
Comparison with batch
In order to demonstrate the advantages of continuous UiO-66 synthesis using a millireactor, a batch synthesis of UiO-66 was performed using the best reaction parameters identified so far. The conditions were chosen to be as similar as possible to those of the continuous synthesis, i.e. similar reaction volume (50 mL), no slow heating of the reactants after mixing. For this purpose, each of the reactant solutions were prepared and separately heated at 110 °C. After the temperature was reached, the terephthalic acid solution was added to the zirconyl chloride solution under vigorous stirring and the reaction mixture reacted for 8 minutes before it was hot filtered off and worked up.In contrast to the continuous synthesis, this reaction method resulted in the formation of a large amount of a gel-like precipitate immediately after the mixing suggesting that the reactivity of the educts was different as already found by Taddei et al.36 Furthermore, analysis of the resulting product revealed a mainly amorphous nature and a correspondingly low porosity (BET area: 225 m2 g−1 – Fig. S6†). After a reaction time of 3 h an improvement of the crystallinity can be seen, which is still poor (Fig. 3).
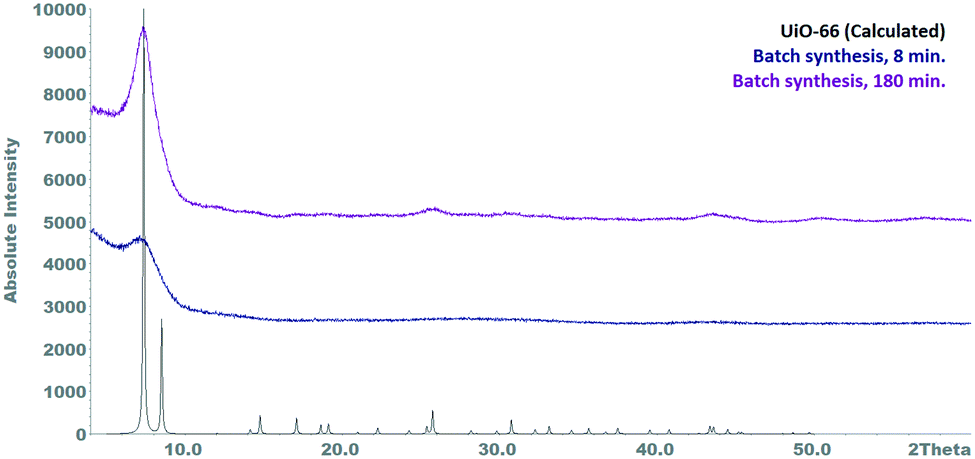 | ||
| Fig. 3 Powder diffractograms of batch samples synthesized under the best continuous conditions. | ||
Upscaled synthesis
Once the reaction was optimised on the small reactor a further increase of the productivity could only be achieved by scaling the volume of the reactor. Using tubing with inner diameter of 3.2 mm (instead of 1.6 mm) the volume of the reactor and thus the productivity could be increased by a 300% (Fig. S1†).Experience gained from the small reactor was applied directly for the study of the new reactor system. It was immediately observed that the influence of gravity resulted in some solid collecting at the bottom of the reactor tube that was transported only marginally with the rest of the reaction mixture. Such behaviour could not be observed in the small reactor in any of the reactions. It can be assumed that the radial velocity distribution, which was greatly broadened in the large reactor, led to this. Due to gravitational forces, the solid sinks into the lower part of the tube, where the flow speeds are too low for complete transport of the particles.
Additionally, in the reaction with different pump rates some solid forming on the walls of the reactor was observed. Such a phenomenon could eventually lead to clogging and is better avoided. It was believed that the two problems were connected to each other deriving from the poor solid transport near the reactor wall.
One way to ensure complete transport of the solid formed consists in a segmentation of the reaction mixture by an additional gas phase. In order to ensure a stable process in the large reactor the segmentation was realised via a second T-type mixer dosing nitrogen with the use of a mass flow controller (Fig. S2†). In order not to reduce the effective volume of the reactor too much, the amount of nitrogen used for the segmentation was regulated to be as low as possible but still allow stable segmentation.
An overview of the results acquired with the large reactor is found in Table 3 (and ESI†). It can be seen by the first entry that the reaction performed without segmentation yielded almost 80% product exceeded the expected fourfold productivity. Surprisingly, the product characteristics where slightly different after this reaction. The defectiveness of the MOF changed and an additional 0.5 linker molecule was calculated (Fig. S14†),37 affecting the BET area accordingly. The introduction of segmentation with nitrogen results in a final increase of the productivity by a factor of three, assures trouble-free operation and brings the MOF textural properties at levels comparable to those of a small reactor.
:1:63:10:34 (Segm: Segmentation with gas)
| Segm. | Temp. | Time | Yield | BET | STY | Prod. |
|---|---|---|---|---|---|---|
| °C | min | % | m2 g−1 | kg m−3 d−1 | g d−1 | |
| No | 110 | 8 | 76 | 1342 | 6850 | 271 |
| Yes | 110 | 8 | 80 | 1617 | 4542 | 180 |
| 24 | 78 | 1653 | 1196 | 47.4 |
After establishing the upscaled continuous synthesis of UiO-66 and inspired by literature31 the robustness of the process was examined with the synthesis of UiO-66 derivatives. As linker molecules 2-aminoterephthalic acid and 2,5-dihydroxyterephthalic acid were used. The MOF products are accordingly designated as UiO-66-NH2 and UiO-66-(OH)2.
By applying the optimized process parameters, the production of the UiO-66 derivatives was successful (Fig. S3 and S9†). A slight adjustment had to be made in the case of UiO-66-(OH)2 since the concentration of the reactants had to be reduced (0.15 M – molar ratio 1:1:151:10:34) due to the low solubility of the organic linker. For a residence time of 8 min in the large reactor, a space–time yield of 5202 kg m−3 d−1 could be achieved for UiO-66-NH2, comparable to UiO-66, which underlines the suitability of the parameters for the synthesis of this product. Due to the higher mass of the linker, productivity in this case is slightly increased to 205 g d−1.
Long term operation
In order to test the suitability of the developed synthesis route in the large reactor for a longer period of time, three experiments were carried out producing UiO-66 for about 4 to 4.5 h runtime each. The same reaction conditions as described in the large-scale synthesis with segmentation (see Experimental) were used, differing only in the filtration that carried out through a larger cellulose filter (diameter 110 mm). The filter was changed after every hour because the filtration rate deteriorated due to the large amount of solid material collected on it. The change was performed without stopping operating by simply switching between vacuum filtration setups.The main aim of the long-term tests was to determine whether the reactor could withstand a longer test run without showing clogging or other problems. In fact, no changes in the reactor could be observed or detected in any of the reactions performed. The temporal and spatial course of the reaction progress remained constant, no accumulations of solid matter within the experimental period could be found and pressure remained stable suggesting that no clogging is occurring, even at the narrowest points (T-type mixer). The optimized synthesis parameters are therefore indeed suitable for a longer production of the MOF.
Table 4 shows the results and product characteristics of the 3 longer runs. As expected no differences can be observed on the results compared to the ones from the shorter runs. A fact that stands out though is the excellent reproduction of the results among each other. The reaction yield based on the results of the three attempts was calculated at 80.3 ± 2.1%, STY was found 4712 ± 99 kg m−3 d−1and finally productivity was 186.6 ± 3.9 g d−1. Even more astonishing is the fact that the product quality follows the same trend, showing remarkable reproducibility – BET area of the three samples was found 1695 ± 22 m2 g−1 (Fig. S4 and S10†).
:1:63:10:34
| Run | Product | Yield | BET | STY | Prod. |
|---|---|---|---|---|---|
| Mass (g) | % | m2 g−1 | kg m−3 d−1 | g d−1 | |
| Conti-1 | 55.4 | 78 | 1725 | 4577 | 181.2 |
| Conti-2 | 57.4 | 83 | 1674 | 4809 | 190.4 |
| Conti-3 | 55 | 80 | 1685 | 4751 | 188.1 |
Long term operation confirmed that the advantages of continuous synthesis are being realised for the synthesis of UiO-66 and derivatives.
SEM images of various samples synthesized under different conditions and using both reactors revealed small spherical crystals with sized of few nm (Fig. 4). The same shape was also found for the derivatives (Fig. S17†).
 | ||
| Fig. 4 SEM images of various samples. Reaction conditions: 10 eq. HCl, 110 °C and 8 min residence time same for all samples. A: Small reactor – 0.26 M concentration (molar ratio: 1:1:89:10:34) B: Small reactor – 0.35 M concentration (molar ratio: 1:1:63:10:34) C: Big reactor – 0.35 M concentration – segmented flow (molar ratio: 1:1:63:10:34) D: Sample after long term operation – same conditions as C. | ||
Conclusions
In this study the continuous synthesis of UiO-66 was further developed and optimized towards higher production rates. In depth analysis of reaction parameters provided useful knowledge for the successful transfer of the production to a larger reactor. The high STY reached with this approach promotes quantitative production, allowing these materials to become readily available for industrial exploitation.UiO-66 and derivatives can be now synthesized at increased production rates while maintaining excellent product quality. Moreover, the advantages of the continuous synthetic approach have been demonstrated both in the process itself and in the characteristics of the product with a profound reproducibility of the results. The simple, cheap and straightforward method can be further upscaled without any risk realising a space effective method for the production of MOFs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Federal ministry of education and research under Grant No. 13N14195.References
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276 CrossRef CAS.
- H.-C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- A. J. Tansell, C. L. Jones and T. L. Easun, Chem. Cent. J., 2017, 11, 100 CrossRef CAS PubMed.
- H. Wang, Q.-L. Zhu, R. Zou and Q. Xu, Chem, 2017, 2, 52 CAS.
- A. Kirchon, L. Feng, H. F. Drake, E. A. Joseph and H.-C. Zhou, Chem. Soc. Rev., 2018, 47, 8611 RSC.
- S. Yuan, L. Feng, K. Wang, J. Pang, M. Bosch, C. Lollar, Y. Sun, J. Qin, X. Yang, P. Zhang, Q. Wang, L. Zou, Y. Zhang, L. Zhang, Y. Fang, J. Li and H.-C. Zhou, Adv. Mater., 2018, 30, e1704303 CrossRef PubMed.
- C. Livage, C. Egger and G. Férey, Chem. Mater., 1999, 11, 1546 CrossRef CAS.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850 CrossRef PubMed.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469 CrossRef CAS PubMed.
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186 CrossRef CAS PubMed.
- A. Dhakshinamoorthy, A. Santiago-Portillo, A. M. Asiri and H. Garcia, ChemCatChem, 2019, 11, 899 CrossRef CAS.
- J. Hajek, C. Caratelli, R. Demuynck, K. de Wispelaere, L. Vanduyfhuys, M. Waroquier and V. van Speybroeck, Chem. Sci., 2018, 9, 2723 RSC.
- Y. Khabzina, J. Dhainaut, M. Ahlhelm, H.-J. Richter, H. Reinsch, N. Stock and D. Farrusseng, Ind. Eng. Chem. Res., 2018, 57, 8200 CrossRef CAS.
- D. W. Lee, T. Didriksen, U. Olsbye, R. Blom and C. A. Grande, Sep. Purif. Technol., 2020, 235, 116182 CrossRef CAS.
- H. H. de Lima, C. T. da Silva, V. L. Kupfer, J. C. de Rinaldi, E. S. Kioshima, D. Mandelli, M. R. Guilherme and A. W. Rinaldi, Carbohydr. Polym., 2021, 251, 116977 CrossRef CAS PubMed.
- H. Reinsch, D. Fröhlich, S. Waitschat, S. Chavan, K.-P. Lillerud, S. Henninger and N. Stock, React. Chem. Eng., 2018, 3, 365 RSC.
- U. Ryu, S. Jee, P. C. Rao, J. Shin, C. Ko, M. Yoon, K. S. Park and K. M. Choi, Coord. Chem. Rev., 2021, 426, 213544 CrossRef CAS PubMed.
- D. DeSantis, J. A. Mason, B. D. James, C. Houchins, J. R. Long and M. Veenstra, Energy Fuels, 2017, 31, 2024 CrossRef CAS.
- J. Winarta, B. Shan, S. M. Mcintyre, L. Ye, C. Wang, J. Liu and B. Mu, Cryst. Growth Des., 2020, 20, 1347 CrossRef CAS.
- F. Ragon, P. Horcajada, H. Chevreau, Y. K. Hwang, U.-H. Lee, S. R. Miller, T. Devic, J.-S. Chang and C. Serre, Inorg. Chem., 2014, 53, 2491 CrossRef CAS PubMed.
- M. Faustini, J. Kim, G.-Y. Jeong, J. Y. Kim, H. R. Moon, W.-S. Ahn and D.-P. Kim, J. Am. Chem. Soc., 2013, 135, 14619 CrossRef CAS PubMed.
- S. Tai, W. Zhang, J. Zhang, G. Luo, Y. Jia, M. Deng and Y. Ling, Microporous Mesoporous Mater., 2016, 220, 148 CrossRef CAS.
- M. Rubio-Martinez, M. P. Batten, A. Polyzos, K.-C. Carey, J. I. Mardel, K.-S. Lim and M. R. Hill, Sci. Rep., 2014, 4, 5443 CrossRef CAS PubMed.
- L. Garzón-Tovar, M. Cano-Sarabia, A. Carné-Sánchez, C. Carbonell, I. Imaz and D. Maspoch, React. Chem. Eng., 2016, 1, 533 RSC.
- S. Waitschat, M. T. Wharmby and N. Stock, Dalton Trans., 2015, 44, 11235 RSC.
- M. Taddei, D. A. Steitz, J. A. van Bokhoven and M. Ranocchiari, Chem. – Eur. J., 2016, 22, 3245 CrossRef CAS PubMed.
- A. Polyzoidis, M. Schwarzer, S. Loebbecke and C. G. Piscopo, Mater. Lett., 2017, 197, 213 CrossRef CAS.
- L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. H. Nilsen, S. Jakobsen, K. P. Lillerud and C. Lamberti, Chem. Mater., 2011, 23, 1700 CrossRef CAS.
- M. G. Goesten, M. F. de Lange, A. I. Olivos-Suarez, A. V. Bavykina, P. Serra-Crespo, C. Krywka, F. M. Bickelhaupt, F. Kapteijn and J. Gascon, Nat. Commun., 2016, 7, 11832 CrossRef CAS PubMed.
- M. J. Katz, Z. J. Brown, Y. J. Colón, P. W. Siu, K. A. Scheidt, R. Q. Snurr, J. T. Hupp and O. K. Farha, Chem. Commun., 2013, 49, 9449 RSC.
- A. Clearfield, J. Mater. Res., 1990, 5, 161 CrossRef CAS.
- A. Polyzoidis, T. Altenburg, M. Schwarzer, S. Loebbecke and S. Kaskel, Chem. Eng. J., 2016, 283, 971 CrossRef CAS.
- H. Wu, Y. S. Chua, V. Krungleviciute, M. Tyagi, P. Chen, T. Yildirim and W. Zhou, J. Am. Chem. Soc., 2013, 135, 10525 CrossRef CAS PubMed.
- M. Taddei, N. Casati, D. A. Steitz, K. C. Dümbgen, J. A. van Bokhoven and M. Ranocchiari, CrystEngComm, 2017, 19, 3206 RSC.
- M. Taddei, J. A. van Bokhoven and M. Ranocchiari, Inorg. Chem., 2020, 59, 7860 CrossRef CAS PubMed.
- G. C. Shearer, S. Chavan, S. Bordiga, S. Svelle, U. Olsbye and K. P. Lillerud, Chem. Mater., 2016, 28, 3749 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0re00464b |
| This journal is © The Royal Society of Chemistry 2021 |