
Co–Nx catalyst: an effective catalyst for the transformation of nitro compounds into azo compounds
Renjie
Huang
,
Yanxin
Wang
,
XiXi
Liu
,
Peng
Zhou
,
Shiwei
Jin
 * and
Zehui
Zhang
*
* and
Zehui
Zhang
*
Key Laboratory of Catalysis and Materials Sciences of the Ministry of Education, South-Central University for Nationalities, Wuhan, 430074, P. R. China. E-mail: jinsw@mail.scuec.edu.cn; zehuizh@mail.ustc.edu.cn; Fax: +86 27 67842572; Tel: +86 27 67842572
First published on 12th September 2020
Abstract
The selective hydrogenation of nitro compounds into their corresponding azo compounds is challenging in organic synthesis. In this study, we have developed a facile and efficient catalytic system for the transformation of nitro compounds into azo compounds under mild conditions over a heterogeneous cobalt catalyst (Co–Nx/C-800-AT). The Co–Nx/C-800-AT catalyst was found to be highly active for the reductive-coupling of nitro compounds into azo compounds in ethanol at 110 °C and 2.5 bar H2 pressure with the addition of NaOH as an additive, affording structurally-diverse azo compounds with satisfactory yields (70.4–98.3%). The developed method demonstrated several advantages such as high catalytic activity and high selectivity with tolerance to other functional groups and high stability.
Introduction
Aromatic azo compounds are high-value materials, which have been widely used in the field of organic dyes, food additives, indicators, and drugs.1–3 Traditional methodologies for the synthesis of azo compounds mainly include the azo-coupling reactions with electron-rich aromatic compounds by the formation of diazonium salts,4 the Mills reaction (the reaction between aromatic nitroso derivatives and anilines),5,6 and the Wallach reaction (acid-catalyzed rearrangement of azoxybenzenes to p-hydroxyazobenzenes).7 However, these traditional methods suffer from several drawbacks such as the generation of highly toxic waste, low atom efficiency, and high production cost by the use of excess oxidizing or reducing agents. Therefore, it is very urgent to develop efficient and sustainable protocols for the synthesis of these compounds.In the current reported methods, the reduction of nitro compounds and the oxidation of anilines are the two most common methods for the synthesis of azo compounds. Obviously, the reductive method is much more preferable, because of nitro compounds being readily available and valuable starting materials, while anilines should be obtained by the hydrogenation of nitroarenes in advance for the oxidative methods.8 For example, a two-step method was adopted by Corma and co-workers for the synthesis of azobenzene from nitrobenzene, in which the first step was the hydrogenation of nitrobenzene into aniline at 9 atm H2 and the second step was the oxidation of aniline into azobenzene at 5 atm of O2 for the transformation of nitrobenzene into azobenzene.9 Hence, the direct reductive coupling of readily available nitroarenes is much more sustainable and economical for the synthesis of azo compounds by avoiding intermediate separation and purifications steps, and minimizing energy and solvents.
In recent years, some new methods have been reported for the reduction of nitro aromatics into azo compounds with H2 or other hydrogen donors such as CO and organic silicane.10–14 Organic silicane is a strong reducing reagent, and hence the reduction of nitro compounds can be performed under relative mild conditions. For example, Sakai et al. reported an InX3/Et3SiH reduction system for the production of azo compounds with yields of 41–96% at 60 °C.11 This method is constrained by the use of expensive organic silicane, low atom efficiency by the release of silicon oxides and difficulty in the product separation and product purification. Meanwhile, CO-mediated hydrogenation reactions have also received some attention.15,16 Cao and co-workers developed a method for the reductive coupling of nitro compounds into azo compounds by using CO as the deoxygenative reagent at a high temperature of 150 °C based on a mesostructured ceria supported gold (Au/meso-CeO2) catalyst.17 Compared with the deoxygenative reagents CO and organic silicane, H2 is undoubtedly the most attractive and popular reducing agent as H2O is the only oxidation product with high atom efficiency, thereby leading to a number of catalytic hydrogenative coupling procedures for nitroarenes.18–20 The current methods for the reductive coupling of nitro compounds into azo compounds have been mainly performed over noble metal catalysts such as Pd, Pt and Au catalysts,18–21 but the high cost of noble metal catalysts limits their practical application. Therefore, the development of non-noble metal catalytic systems for the efficient and selective transformation of nitro compounds into azo-compounds is highly desirable.
In our previous work, we have prepared cobalt/nitrogen-doped carbon catalysts and identified that Co–Nx was effective for the hydrogenation of nitro compounds into anilines.20 As part of our continuing investigation into reduction chemistry with the Co–Nx catalyst, we wondered whether Co–Nx would also be active for the synthesis of azo compounds via the reductive coupling of nitro compounds.
Experimental
Materials
All of the solvents were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). All of the chemicals were purchased from Aladdin Chemicals Co. Ltd. (Beijing, China). The catalyst (abbreviated as Co–Nx/C-800-AT) was prepared and described in our previous work.20 Briefly, the preparation of Co–Nx/C-800-AT includes the pyrolysis of the cobalt phthalocyanine/silica composite at 800 °C under an N2 atmosphere, and the subsequent treatment with 10 wt% HF to simultaneously remove Co nanoparticles and silica.General procedure
Reactions were performed in a 50 mL autoclave, which was equipped with a magnetic stirrer and a temperature controller. In a typical run, ethanol (10 mL), nitrobenzene (1 mmol), the Co–Nx/C-800-AT catalyst (40 mg), and NaOH (0.5 mmol) were added to the reactor. Then, the autoclave was purged with H2 five times to completely remove the air. After being sealed, the autoclave was charged with 2.5 bar H2 at room temperature. Then, the reaction mixture was stirred at 110 °C for 3 h with mechanical stirring at 1000 rpm. Afterwards, the autoclave was cooled to room temperature and depressurized. Phenyl ether was added as an internal standard, and the reaction mixture was diluted with ethanol. The products in the reaction mixture were identified by comparison of their retention times with those of the reference chemicals, and further confirmed and quantified by GC-MS (Agilent 7890A GC/5973 MS, HP-5 column).Analytical methods
Analysis of the products was performed by gas chromatography (GC) on an Agilent 7890A instrument with a cross-linked capillary HP-5 column (30 m × 0.32 mm × 0.4 mm) equipped with a flame ionization detector. The operating conditions were as follows: the flow rate of the N2 carrier gas was 40 mL min−1 and the injection port temperature was 300 °C. The GC oven temperature program was as follows: 10 °C min−1 ramp from 50 °C to 280 °C, and the detector temperature was set to 300 °C. The content of each compound was determined based on the internal standard.Recycling experiments
After reaction, the Co–Nx/C-800-AT catalyst was separated from the reaction mixture by centrifugation, and the spent catalyst was exhaustively washed with water and ethanol, respectively. The spent catalyst was used for the next run under the same conditions. Other cycles were repeated with the same procedure.Results and discussion
Optimal reaction conditions for the reductive-coupling of nitrobenzene
The well-defined Co–Nx/C-800-AT catalyst was prepared and characterized as described in our previous work. In our previous work, it showed high catalytic activity in the hydrogenation of nitro compounds into amines.20 In this study, we tried to transform nitro compounds directly into azo compounds, which have also been of great importance in the fields of dyes and pharmaceuticals. Firstly, the reductive coupling of nitrobenzene was used as the model reaction over the Co–Nx/C-800-AT catalyst. In our previous work, the hydrogenation of nitrobenzene was proved to occur via a direct way, involving the consecutive hydrogenation of nitrobenzene into nitrosoarene (PhNO), the hydrogenation of nitrosoarene into phenylhydroxylamine (PhNHOH), and the final hydrogenation of phenylhydroxylamine to aniline. According to ref. 22 and 23, a base was used as the additive to promote the transformation of nitroarenes into azo compounds. Therefore, the hydrogenation of nitrobenzene over the Co–Nx/C-800-AT catalyst was firstly performed by the use of different bases in ethanol, and the results are summarized in Table 1.|
|
|||||
|---|---|---|---|---|---|
| Entry | Base | Conversion (%) | Selectivity (%) | ||
| 1 | 2 | 3 | |||
| a Reaction conditions: nitrobenzene (1 mmol), Co–Nx/C-800-AT catalyst (40 mg), ethanol (10 mL), 90 °C, H2 (10 bar), base (0.5 mmol), 2 h. | |||||
| 1 | NaHCO3 | 47.0 | >99 | 0 | 0 |
| 2 | Na2CO3 | 54.7 | 45.7 | 0 | 0 |
| 3 | K2CO3 | 83.1 | 96.4 | 3.6 | 0 |
| 4 | Cs2CO3 | 100 | 53.4 | 33.8 | 12.8 |
| 5 | NaOH | 100 | 35.0 | 43.1 | 21.9 |
| 6 | Triethylamine | 100 | >99 | 0 | 0 |
| 7 | Pyridine | 100 | >99 | 0 | 0 |
It was noted that the base showed a great effect both on the nitrobenzene conversion and the product distribution. Full nitrobenzene conversion was achieved within 2 h at 90 °C, when the reactions were performed using organic bases (Table 1, entries 6 and 7) or strong inorganic bases NaOH and Cs2CO3 (Table 1, entries 4 & 5). The organic bases all dissolved in ethanol. These are Lewis bases, which are helpful to promote the H2 heterolytic dissociation with the Co sites to generate hydrogen species (H+ and H−), in which H+ was freely bonded with the organic base, and H− was bonded on the Co sites (Co–H−). Unlike the organic bases, the inorganic bases cannot dissolve in ethanol, and these inorganic bases should first react with ethanol to generate organic bases such as EtONa. Therefore, the inorganic base with strong basicity should generate a much stronger organic base. Hence, the conversion of nitrobenzene increased with the increase of the basicity of the inorganic bases (Table 1, entries 1–4).
Interestingly, it was noted that the selectivity to the products was greatly affected by the base. Under weakly basic conditions, aniline is the only product or the major product (Table 1, entries 1 and 2 & 6 and 7). Azobenzene was the major product when the hydrogenation of nitrobenzene was performed by the use of strong bases such as NaOH and Cs2CO3 (Table 1, entries 4 & 5). In addition, azoxybenzene as the intermediate of the transformation of nitrobenzene into azobenzene was also observed in these cases (Table 1, entries 4, 5 & 8). These results suggested that the bases with strong basicity greatly promoted the transformation of nitrobenzene into azobenzene, which was also reported by other researchers.22,23 Considering the fact that t-BuOK is sensitive to moisture in the air and expensive, NaOH was selected as an appropriate base for the reductive coupling of nitrobenzene into azobenzene.
With the aim of improving the selectivity to azobenzene, the solvent effect on this transformation was also investigated under similar conditions (Table 2). A comparison of water and commonly used organic solvents showed that anhydrous ethanol is the best solvent for the generation of azobenzene in this system, which resulted in a high nitrobenzene conversion and the highest azobenzene selectivity of 43.1% (Table 2, entry 8). Generally, the reaction in the non-protic solvents such as toluene, ethyl acetate and hexane produced much lower nitrobenzene conversions (Table 2, entries 2–6), and the reason should be that NaOH cannot dissolve in these solvents and react with them to generate the organic base.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Base | Conversion (%) | Selectivity (%) | ||
| 1 | 2 | 3 | |||
| a Reaction conditions: nitrobenzene (1 mmol), Co–Nx/C-800-AT catalyst (40 mg), solvent (10 mL), 90 °C, H2 (10 bar), NaOH (0.5 mmol), 2 h. b Sodium isopropoxide was used. | |||||
| 1 | H2O | 22.1 | 20.0 | 9.5 | 0 |
| 2 | THF | 23.8 | >99 | 0 | 0 |
| 3 | CH3CN | 35.9 | >99 | 0 | 0 |
| 4 | Toluene | 45.6 | >99 | 0 | 0 |
| 5 | Ethyl acetate | 39.4 | >99 | 0 | 0 |
| 6 | Hexane | 28.2 | >99 | 0 | 0 |
| 7 | Methanol | 100 | 73.8 | 12.8 | 13.4 |
| 8 | EtOH | 100 | 35.0 | 43.1 | 21.9 |
| 9 | Isopropanol | 67.3 | 56.0 | 15.8 | 28.2 |
| 10b | EtOH | 90.8 | 48.2 | 30.2 | 21.6 |
The reaction in protic solvents such as methanol, ethanol and isopropanol produced high conversions of nitrobenzene (Table 2, entries 7–9). Particularly, the conversion of nitrobenzene increased in the order isopropanol, ethanol and methanol (Table 2, entries 7–9). These results suggested that the solvents with high polarity produced high nitrobenzene conversion, which should be due to the high dispersion of the Co–Nx/C-800-AT catalyst in the solvent. The low conversion of nitrobenzene in water was due to the limited solubility of nitrobenzene in the basic solution.
It was noted that aniline was the sole product when the hydrogenation of nitrobenzene was performed in the non-protic solvents (Table 2, entries 2–6), while azobenzene, azoxybenzene and aniline were both produced in alcohol solvents (Table 2, entries 7 and 8). The absence of azobenzene in the non-protic solvents should be due to the fact that these solvents cannot react with NaOH to generate the organic bases to promote the transformation of nitrobenzene into azobenzene, while this was not the case for the reaction in alcohol solvents. Generally speaking, the reactivity of the alcohols with NaOH to generate the corresponding organic base increased in the order methanol, ethanol and isopropanol. Although the reaction of isopropanol with NaOH can generate the largest amount of the base, it produced the lowest conversion of nitrobenzene as well as the lowest selectivity to azobenzene among the reactions in methanol, ethanol and isopropanol. The reason should be that the organic base from the reaction of isopropanol and NaOH had a large steric hindrance, and thus it cannot efficiently promote the transformation of nitrobenzene into azobenzene. To further confirm this, the reaction in ethanol was also performed by the use of sodium isopropoxide, and it also provided a lower conversion than that by the use of NaOH (Table 2, entry 8 vs. entry 10).
The effect of the hydrogen pressure was also studied for the hydrogenation of nitrobenzene into azobenzene, and the results are shown in Fig. 1. It was noted that the hydrogen pressure showed a great effect on the nitrobenzene conversion. A low nitrobenzene conversion of 43.4% was obtained under atmospheric H2 pressure within 2 h at 90 °C. Then, the conversion of nitrobenzene gradually increased to 65.7% and 93.8% within 2 h under a H2 pressure of 2.5 bar and 5 bar, respectively. Full nitrobenzene conversion was achieved after 2 h at 10 bar H2 pressure. The increase of the hydrogen pressure resulted in an increase of the hydrogen concentration in the reaction solvent, and thus the concentration of H2 around the Co active sites increased, leading to a higher reaction rate. The increase of the hydrogen pressure resulted in an increase of the selectivity to aniline, while the selectivity to azoxybenzene decreased with an increase of H2 pressure.
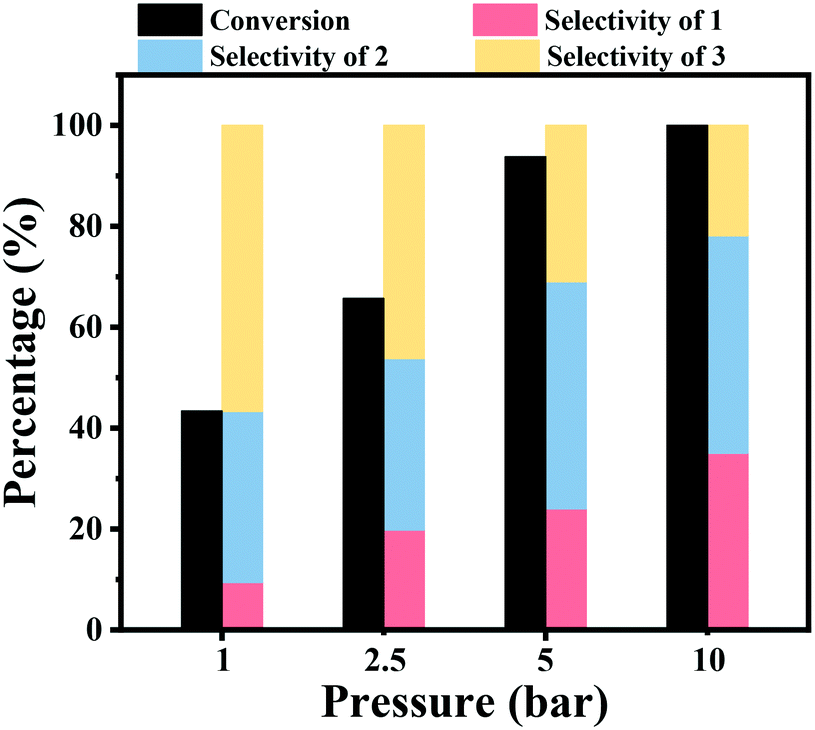 | ||
| Fig. 1 The effect of hydrogen pressure on the hydrogenation of nitrobenzene. Reaction conditions: nitrobenzene (1 mmol), Co–Nx/C-800-AT catalyst (40 mg), solvent (10 mL), 90 °C, H2 (1–10 bar), NaOH (0.5 mmol), 2 h. | ||
The effect of the reaction temperature on the hydrogenation of nitrobenzene over the Co–Nx/C-800-AT catalyst was studied. As shown in Fig. 2, this reaction was sensitive to the reaction temperature. The nitrobenzene conversion was found to greatly increase with an increase of the reaction temperature. A low nitrobenzene conversion of 23.0% was achieved within 2 h at a reaction temperature of 50 °C and 2.5 bar H2 pressure, and then it greatly increased to 45.4% and 65.7% at reaction temperatures of 70 °C and 90 °C, respectively. By further increasing the reaction temperature to 110 °C, nitrobenzene was almost quantitatively converted within 2 h. At higher reaction temperature, the concentration of H2 in the reaction solution should be lower. However, the conversion of nitrobenzene increased with the increase of the reaction temperature, which means that the increase of the reaction temperature accelerated the reaction rate by increasing the contact efficiency of the active molecules. Similarly, the increase of the selectivity to azobenzene should be caused by the increase of the contact efficiency of the active molecules of nitrosoarene (PhNO) and phenylhydroxylamine (PhNHOH) to form the azoxybenzene intermediate. The selectivity to azobenzene increased with an increase of the reaction temperature from 9.0% at 50 °C to 78.9% at 110 °C.
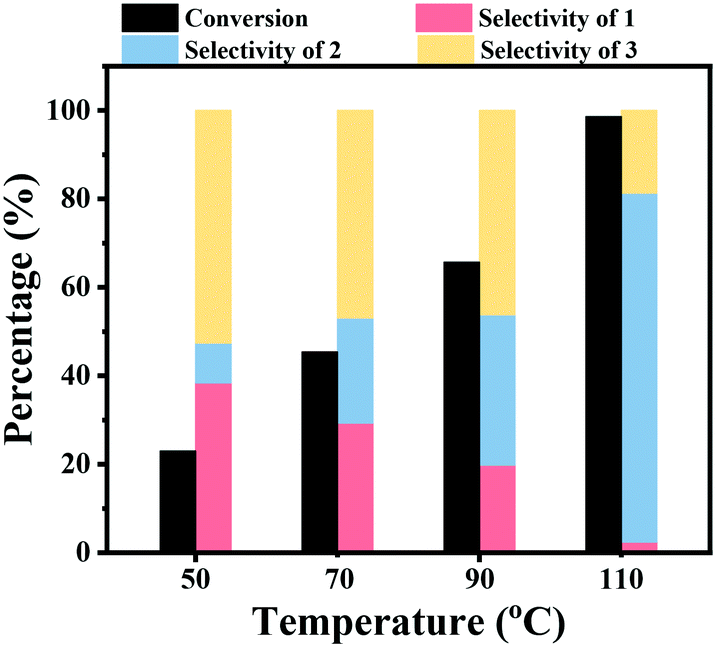 | ||
| Fig. 2 The effect of reaction temperature on the hydrogenation of nitrobenzene. Reaction conditions: nitrobenzene (1 mmol), Co–Nx/C-800-AT catalyst (40 mg), solvent (10 mL), H2 (2.5 bar), NaOH (0.5 mmol), 2 h. | ||
Kinetics of the reaction pathway
To give more insights into the transformation of nitrobenzene into azobenzene, the kinetic curve of the product distribution was studied at 110 °C and 2.5 bar H2 pressure. As shown in Fig. 3, the amount of azoxybenzene reaches its maximum after 1 h, with nitrobenzene being quickly consumed at an early reaction stage. Azoxybenzene was gradually reduced to azobenzene during the reaction process, but the quantity of aniline remains unchanged. The content of azobenzene gradually increased with an increase of the reaction time, and it reached a high yield of 97.5% within 3 h at 110 °C and 2.5 bar H2 pressure.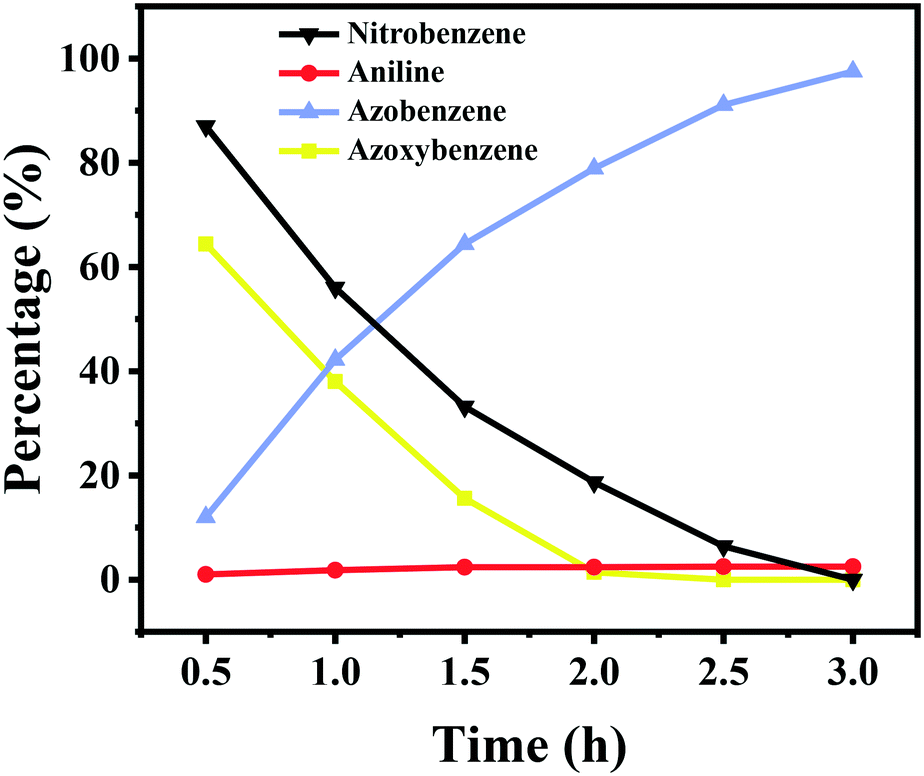 | ||
| Fig. 3 Time course of the product distribution in the hydrogenation of nitrobenzene. Reaction conditions: nitrobenzene (1 mmol), Co–Nx/C-800-AT catalyst (40 mg), ethanol (10 mL), H2 (2.5 bar), NaOH (0.5 mmol), 110 °C. | ||
During the reaction process, no other intermediates except azoxybenzene were observed, and the reason should be due to the fact that the other intermediates are not stable under the reaction conditions. According to our previous work,20 the conversion of nitrobenzene into aniline proceeded via a direct pathway (Scheme 1), namely the hydrogenation of nitrobenzene into nitrosoarene (PhNO), the hydrogenation of nitrosoarene into phenylhydroxylamine (PhNHOH), and the final hydrogenation of phenylhydroxylamine to aniline. In the presence of a base, the nitrobenzene hydrogenation pathway was changed. The reaction energy of the condensation of phenylhydroxylamine and nitrosoarene was lower, the pathway of hydrogenation of phenylhydroxylamine to aniline was inhibited (Scheme 1), and azoxybenzene was more easily produced in the alkaline system.24,25 Then, the hydrogenation of azoxybenzene gave the final product azobenzene. Azobenzene was stable under our reaction conditions, and it cannot be further reduced into aniline.
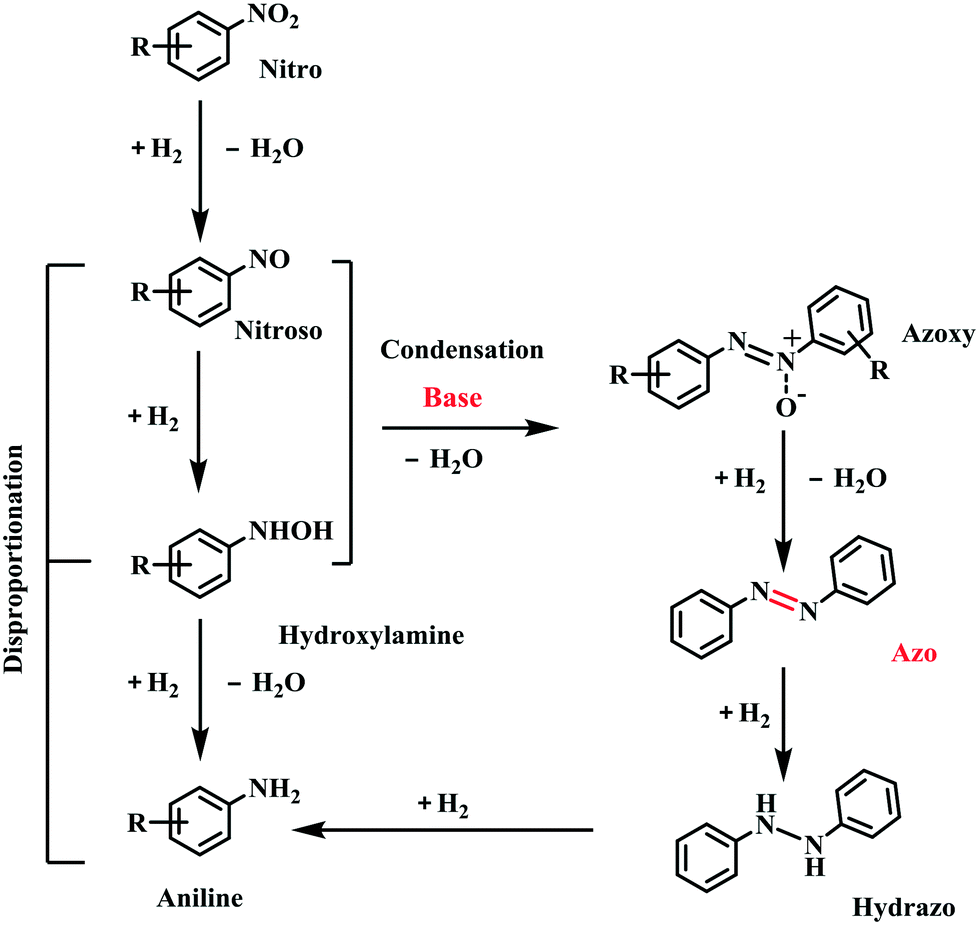 | ||
| Scheme 1 Reaction pathways of the reduction of nitrobenzene. | ||
Substrate scope for this reaction
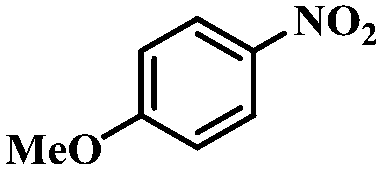
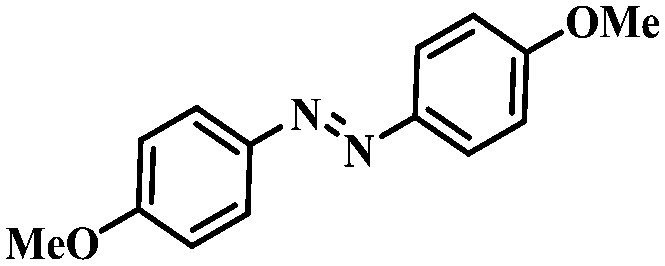
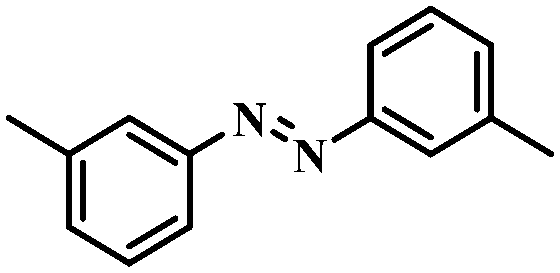
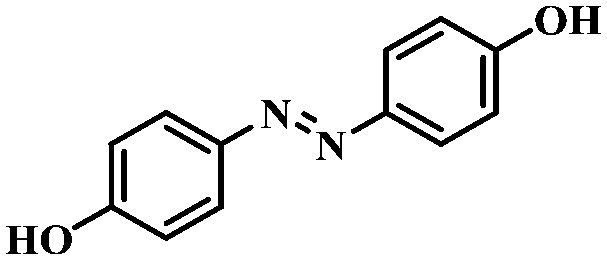
Under the optimized conditions for the 100% transformation of nitrobenzene, the developed method was also applied for other substrates. As shown in Table 3, the reductive coupling of different kinds of nitro compounds afforded the corresponding azo compounds. The steric hindrance of the substituted group affected the activity of the substrates. p-Nitrotoluene and m-nitrotoluene demonstrated higher catalytic activity than o-nitrotoluene. To get a high yield of the corresponding azo compounds, prolonging the reaction time to 6 h was required for o-nitrotoluene. The electron-properties of the substituted groups also showed a great effect on the activity of the substrates. Generally, the substrates with electron-donating groups were more active than the substrates with electron-withdrawing groups. More importantly, other para-substituted substrates containing methoxy, nitrile, hydroxyl, or carbonyl groups also gave high yields without the reduction of these functional groups. Therefore, the Co–Nx/C-800-AT catalyst also demonstrated a good tolerance to other functional groups.| Entry | Nitro compounds | Azo compounds | Time (h) | Con. (%) | Sel. (%) |
|---|---|---|---|---|---|
| a Reaction conditions: nitro compound (1 mmol), Co–Nx/C-800-AT catalyst (40 mg), ethanol (10 mL), H2 (2.5 bar), NaOH (0.5 mmol), 110 °C. | |||||
| 1 |
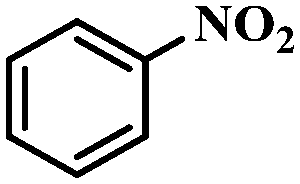
|
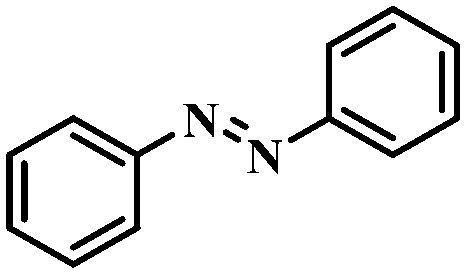
|
3 | 100 | 97.5 |
| 2 |
|
|
2.5 | 100 | 94.6 |
| 3 |
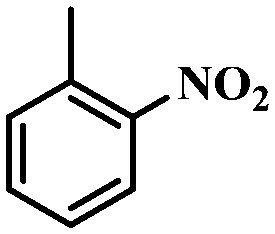
|

|
6 | 100 | 84.6 |
| 4 |
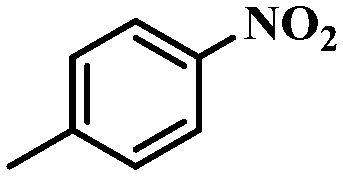
|
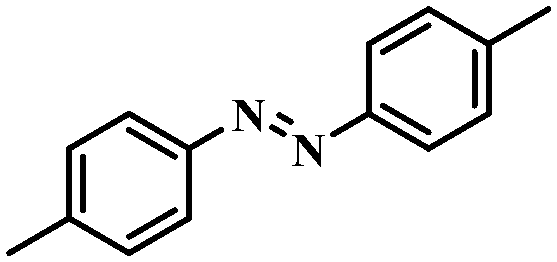
|
3 | 100 | 96.6 |
| 5 |
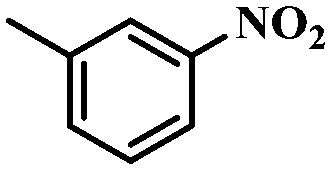
|
|
4 | 100 | 93.0 |
| 6 |
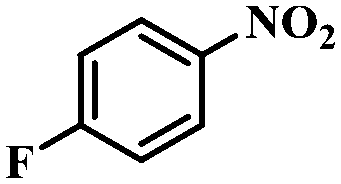
|
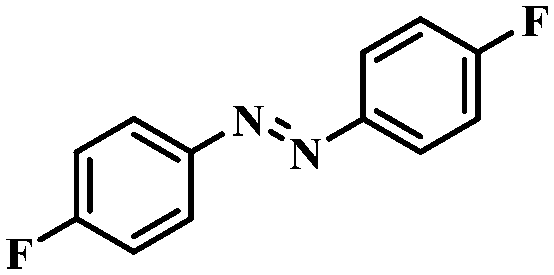
|
4 | 100 | 94.0 |
| 7 |
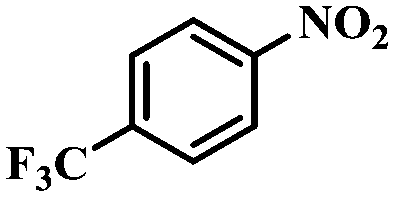
|
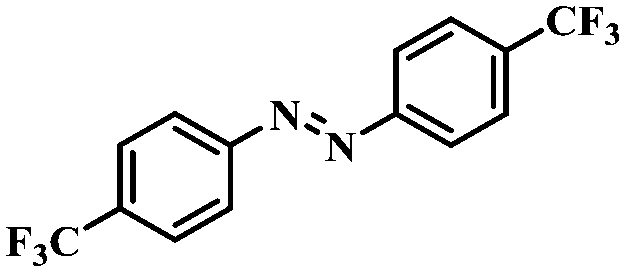
|
4 | 100 | 92.2 |
| 8 |
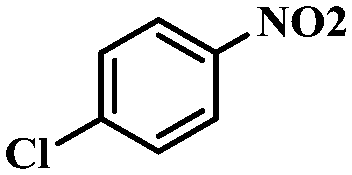
|
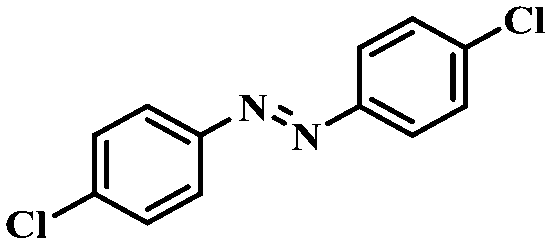
|
4 | 100 | 94.8 |
| 9 |
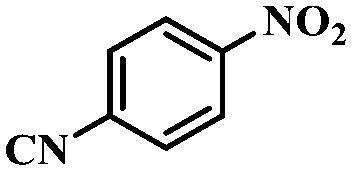
|
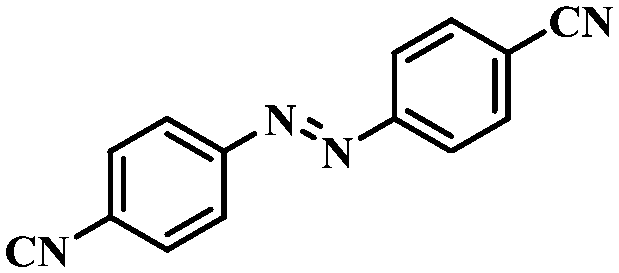
|
3.5 | 100 | 90.3 |
| 10 |
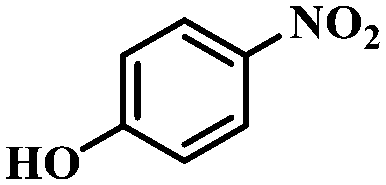
|
|
3 | 100 | 96.2 |
| 11 |
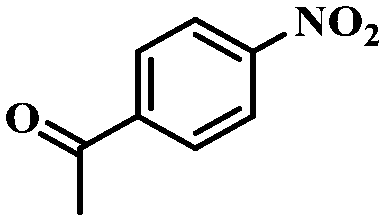
|

|
3.5 | 100 | 98.3 |
| 12 |
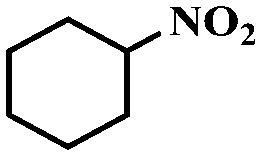
|
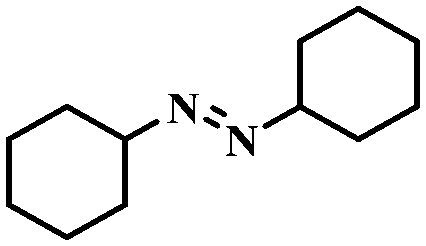
|
4 | 100 | 76.8 |
| 13 |

|
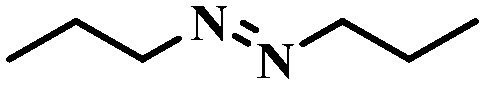
|
4 | 100 | 70.4 |
Stability of the catalyst
Finally, the stability of the Co–Nx/C-800-AT catalyst towards the hydrogenation of nitrobenzene into azoxybenzene was studied (Fig. 4). Recycling experiments were performed at 110 °C and 2.5 bar H2 pressure for 3 h. After each reaction cycle, the catalyst was separated by centrifugation, and washed with water and ethanol, respectively. Then, the spent catalyst was directly used for the next run. As illustrated in Fig. 4, full conversion of nitrobenzene could be achieved in each run, and the selectivity to azoxybenzene remained almost the same at around 97.5%, indicative of the high durability of the Co–Nx/C-800-AT catalyst. The high stability was due to the fact that the Co atoms in the Co–Nx/C-800-AT catalyst were chemically bonded with nitrogen atoms, and thus the catalyst was highly stable, which is in contrast with the traditional supported metal nanoparticles. Besides, ICP-AES tests also showed no detectable metal leaching after the reaction. To further demonstrate the heterogeneous nature of the Co–Nx/C-800-AT catalyst towards nitrobenzene reduction into azoxybenzene, we conducted a hot filtration test for this reaction. After 30 min under the standard reaction conditions, 35.5% nitrobenzene conversion was reached. Then, the Co–Nx/C-800-AT catalyst was removed from the reaction solution by centrifugation. After the removal of the catalyst, the solution was still kept under the same reaction conditions. As shown in Fig. 5, the concentration of nitrobenzene had no obvious change. These results further confirm that the Co–Nx/C-800-AT catalyst was highly stable towards the nitrobenzene reduction into azoxybenzene in ethanol with the use of Cs2CO3 as the additive. | ||
| Fig. 4 The recycling experiments of the Co–Nx/C-800-AT catalyst. Reaction conditions: nitrobenzene (1 mmol), Co–Nx/C-800-AT catalyst (40 mg), ethanol (10 mL), H2 (2.5 bar), NaOH (0.5 mmol), 110 °C, 3 h. | ||
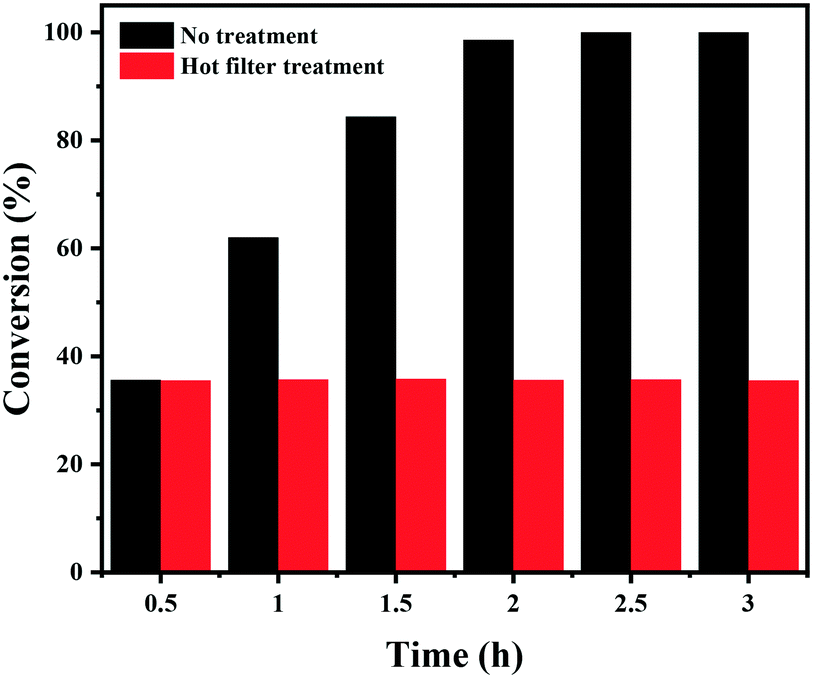 | ||
| Fig. 5 The hot filtration test of the Co–Nx/C-800-AT catalyst. Reaction conditions: nitrobenzene (1 mmol), Co–Nx/C-800-AT catalyst (40 mg), ethanol (10 mL), H2 (2.5 bar), NaOH (0.5 mmol), 110 °C. | ||
Conclusions
In conclusion, we have developed a new method for the reductive transformation of nitro compounds into azo compounds by the use of a heterogeneous non-noble metal catalyst in ethanol under environmentally benign conditions. Different reaction parameters have been investigated, and it was found that the reaction solvents and the base played crucial roles in both the substrate conversion and the product selectivity. Azo compounds were achieved in high yields from 70.4 to 98.3% in ethanol at 110 °C and 2.5 bar H2 pressure with NaOH as an additive. The reaction has a broad scope of reaction substrates, and a variety of functional groups can tolerate the reaction conditions well. Moreover, the Co–Nx/C-800-AT catalyst demonstrated excellent stability and can be recovered and reused through simple phase separation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21872175 & 21922513) and the Basic Scientific Research of Central Colleges, South-Central University for Nationalities (CZY 19005).References
- B. Pandian, R. Arunachalam, S. Easwaramoorthi and J. R. Rao, J. Cleaner Prod., 2020, 256, 120455 CrossRef CAS.
- S. Turlapati, S. B. N, V. K. Vishwakarma, A. S. Achalkumar and G. Hegde, New J. Chem., 2020, 44, 5731–5738 RSC.
- J. Moreira, V. B. Lima, L. A. Goulart and M. R. V. Lanza, Appl. Catal., B, 2019, 248, 95–107 CrossRef CAS.
- K. Haghbeen and E. W. Tan, J. Org. Chem., 1998, 63, 4503–4505 CrossRef CAS.
- Z. L. Zhu and J. H. Espenson, J. Org. Chem., 1995, 60, 1326–1332 CrossRef CAS.
- C. Tie, J. C. Gallucci and J. R. Parquette, J. Am. Chem. Soc., 2006, 128, 1162–1171 CrossRef CAS.
- R. A. Cox, I. Onyido and E. Buncel, J. Am. Chem. Soc., 1992, 114, 1358–1363 CrossRef CAS.
- S. Jang, B. J. Jung, M. J. Kim, W. Lee and D. P. Kim, React. Chem. Eng., 2019, 10, 1752–1756 RSC.
- A. Grirrane, A. Corma and H. Garcia, Science, 2008, 322, 1661–1664 CrossRef CAS.
- J. T. Wang, X. C. Yu, C. Y. Shi, D. J. Lin, J. Li, H. L. Jin, X. Chen and S. Wang, Adv. Synth. Catal., 2019, 361, 3525–3531 CrossRef CAS.
- N. Sakai, S. Asama, S. Anai and T. Konakahara, Tetrahedron, 2014, 70, 2027–2033 CrossRef CAS.
- Q. W. Liu, J. W. Zhang, F. S. Xing, C. C. Cheng, Y. W. Wu and C. J. Huang, Appl. Surf. Sci., 2020, 500, 144214 CrossRef CAS.
- J. Li, S. Y. Song, Y. Long, L. L. Wu, X. Wang, Y. Xing, R. C. Jin, X. G. Liu and H. J. Zhang, Adv. Mater., 2018, 30, 1704416 CrossRef.
- K. Chaiseeda, S. Nishimura and K. Ebitani, ACS Omega, 2017, 2, 7066–7070 CrossRef CAS.
- H. Chen, S. Y. Yao, W. W. Lin, Z. H. Zhang, X. B. Hu, X. Liu, B. H. Yan, K. Q. Chen, Y. Qin, Y. M. Zhu, X. Y. Lu, P. Ouyang, J. Fu and J. G. Chen, Chem. Eng. J., 2020, 390, 124603 CrossRef CAS.
- C. T. Kuo, Y. B. Lu, L. Kovarik, M. Engelhard and A. M. Karim, ACS Catal., 2019, 9, 11030–11041 CrossRef CAS.
- H. Q. Li, X. Liu, Q. Zhang, S. S. Li, Y. M. Liu, H. Y. He and Y. Cao, Chem. Commun., 2015, 51, 11217–11220 RSC.
- Q. W. Liu, Y. Xu, X. Q. Qiu, C. J. Huang and M. Liu, J. Catal., 2019, 370, 55–60 CrossRef CAS.
- B. P. Liu, S. H. Yan, A. T. Zhang, Z. Q. Song, Q. X. Sun, B. B. Huo, W. R. Yang, C. J. Barrow and J. Q. Liu, ChemNanoMat, 2019, 5, 784–791 CrossRef CAS.
- P. Zhou, L. Jiang, F. Wang, K. J. Deng, K. L. Lv and Z. H. Zhang, Sci. Adv., 2017, 3, e1601945 CrossRef.
- Z. P. Zhang, X. Y. Wang, K. Yuan, W. Zhu, T. Zhang, Y. H. Wang, J. Ke, X. Y. Zheng, C. H. Yan and Y. W. Zhang, Nanoscale, 2016, 8, 15744–15752 RSC.
- Z. Q. Yan, X. Y. Xie, Q. Song, F. L. Ma, X. Y. Sui, Z. Y. Huo and M. M. Ma, Green Chem., 2020, 4, 1301–1307 RSC.
- Y. Y. Teng, X. K. Wang, M. Wang, Q. G. Liu, Y. Q. Shao, H. M. Li, C. H. Liang, X. Chen and H. L. Wang, Catalysts, 2019, 9, 339 CrossRef.
- Z. P. Zhang, X. Y. Wang, K. Yuan, W. Zhu, T. Zhang, Y. H. Wang, J. Ke, X. Y. Zheng, C. H. Yan and Y. W. Zhang, Nanoscale, 2016, 8, 15744–15752 RSC.
- L. Hu, X. Q. Cao, L. Y. Shi, F. Q. Qi, Z. Q. Guo, J. M. Lu and H. W. Gu, Org. Lett., 2011, 13, 5640–5643 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |