
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSystematic characterization of metabolic profiles of ingenol in rats by UPLC-Q/TOF-MS and NMR in combination with microbial biotransformation†
Si-Jia Xiao‡
 a,
Shan-Shan Li‡b,
Bin Xiea,
Wei Chena,
Xi-Ke Xua,
Xian-Peng Zu*a and
Yun-Heng Shen*a
a,
Shan-Shan Li‡b,
Bin Xiea,
Wei Chena,
Xi-Ke Xua,
Xian-Peng Zu*a and
Yun-Heng Shen*a
aDepartment of Natural Medicinal Chemistry, School of Pharmacy, Naval Medical University, Shanghai 200433, China. E-mail: zuxianpeng@163.com; shenyunheng@hotmail.com
bSchool of Pharmaceutical Sciences, Yunnan Key Laboratory of Pharmacology for Natural Products, Kunming Medical University, Kunming 650500, Yunnan, China
First published on 23rd November 2021
Abstract
Ingenol, as the precursor of the marketed drug ingenol mebutate, has been proven to have a variety of bioactivities. The purpose of this study was to identify the metabolites of ingenol using ultra-performance liquid chromatography-quadrupole time-of-flight-mass spectrometry (UPLC-Q/TOF-MS) combined with UNIFI software. Plasma, urine and fecal samples of rats were obtained and analyzed. A total of 18 metabolites were detected and identified in rat, including five phase II metabolites (M14–M18). Moreover, as microbial biotransformation is helpful to obtain sufficient reference standards of metabolites, the co-culture of ingenol with the fungus Cunninghamella elegans bio-110930 was also studied and yielded 4 phase I metabolites, in which reference standards of three metabolites were further obtained by preparative scale biotransformation. By matching their retention times, accurate masses, and fragment ions with metabolites in rat, the structures of three metabolites (M2, M3 and M4) were unambiguously confirmed by NMR technology. The results revealed that C. elegans bio-110930 functioned as an appropriate model to mimic and prepare phase I metabolism of ingenol in vivo to a certain extent. It also revealed that hydroxylation, oxygenation, sulfonation, and glucuronidation were the major metabolic pathways of ingenol. Furthermore, the first systematic metabolic study of ingenol is of great significance to elucidate the metabolites and metabolic pathways in vivo, which is helpful to predict metabolites of ingenol in humans, understand the elimination mechanism of ingenol, and clarify its effectiveness and toxicity.
Introduction
Ingenol (Fig. 1), a natural ingenane type of diterpenoid, is one of the active ingredients from the extract of Euphorbia peplus. Ingenol has been shown to bind and activate protein kinase C, and exhibits hepatic- and gastrointestinal-protective,1 anti-tumor2 and anti-HIV activities.3 In particular, ingenol mebutate, a derivative of ingenol, has been successfully marketed as a licensed medicine for the treatment of actinic keratosis.4–6 Considering that ingenol and its ingenane analogues have a variety of bioactivities, it is very significant to study the metabolic profiles of ingenol in vivo for clarifying and understanding efficacy and toxicity of ingenane diterpenoids.7 | ||
| Fig. 1 The chemical structure of ingenol. | ||
According to the guidance of the U.S. Food and Drug Administration (https://www.fda.gov/drugs/guidance-compliance-regulatory-information/guidances-drugs), drug metabolism is a very important part of safety evaluation and a major factor in drug development.8–10 In recent years, the combination of a powerful analytical tool ultra-performance liquid chromatography-quadrupole time-of-flight-mass spectrometry (UPLC-Q/TOF-MS) and a computer-aided identification platform UNIFI (Waters® UNIFI® Scientific Information System), with the advantages of accuracy in mass measurement, high efficacy in separation technique and rapid identification in metabolites, has been widely applied for metabolic research.11 However, there are still two major difficulties in the metabolite investigations. One is to obtain the accurate structures of metabolites, and the other is to obtain sufficient metabolites for further pharmacological and toxicological studies.12 In order to solve these problems, it is essential to obtain the sufficient reference standards of metabolites. Generally, the preparation of metabolites mainly includes chemical methods,12 small experimental animal models,13 microsomal preparations,9,14 enzyme-catalyzed reactions,15,16 and microbial biotransformation,12,17 etc. Compared with other methods, microbial biotransformation is more convenient and cost-effective, especially with the advantage of large-scale preparation in vitro.17–21 Particularly, the fungus Cunninghamella elegans has been shown to possess the cytochrome P450 monooxygenase system similar to mammals, which could promote the reactions catalyzed by human CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A4, thus producing similar metabolic profiles to mammals.22,23 However, the presence and abundance of the metabolites determined by microbial transformation may not be an out-and-out representation of in vivo metabolites. Thus, a combination of these methods was applied in our study.
In this investigation, we aim to utilize UPLC-Q/TOF-MS and UNIFI platform to identify the metabolites of ingenol in rat, and to propose the metabolic pathways of ingenol. A total of 18 metabolites, including five phase II metabolites, were detected and identified. Hydroxylation, oxygenation, sulfonation, and glucuronidation were the predominant metabolic pathways of ingenol in rat. Additionally, microbial biotransformation based on the fungal strain C. elegans bio-110930 was applied to mimic and prepare metabolites of ingenol in vivo, so as to get more detailed metabolism information for predicting metabolites and interpreting structures of metabolites (Fig. 2).
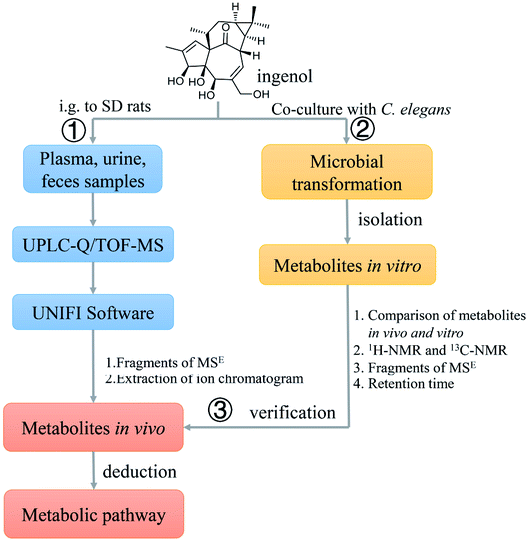 | ||
| Fig. 2 A comprehensive strategy to systematically analyze the metabolites in rat of ingenol. | ||
Experimental section
Chemicals and reagents
Ingenol was purchased from Chengdu Herbpurify Co., Ltd. (Chengdu, China). Sabouraud dextrose broth was procured from Qingdao Hope Bio-Technology Co., Ltd. (cat no. HB0233, China). Sabouraud dextrose agar was obtained from Solarbio (cat no. P9240, China). The fungal strain, C. elegans (bio-110930), was purchased from the Beijing Baioubowei Biotechnology Co., Ltd. (Beijing, China). Column chromatography (CC) was performed using Sephadex LH-20 gel (GE Medical Systems Ltd, Buckinghamshire, U.K). Ethyl acetate and acetone were analytical grade from Shenyang Chemical Reagent Co., Ltd. (Shenyang, China). LC-MS-grade acetonitrile, methanol and formic acid were purchased from Fisher-Scientific (Fair Lawn, NJ, USA) and used in mobile phase and sample preparation. LC-MS-grade leucine enkephalin was obtained from Sigma-Aldrich (MO, USA). Ultra-pure water was purified by a Milli-Q system (Millipore, Bedford, MA, USA). All other reagents were of analytical reagent grade.Instrumentation and analysis condition
For metabolite identification, chromatographic analyses were performed using a Waters Acquity UPLC I-class system (Waters, Milford, MA, USA), equipped with an auto-sampler, a binary solvent delivery system, an online degasser, and a photodiode array detector. An ACQUITY UPLC® HSS T3 column (2.1 mm × 150 mm, 1.8 μm, Waters) with a HSS T3 VanGuard™ Pre-Column 3/Pk (2.1 × 5.0 mm, 1.8 μm) was used. The optimized parameters were set as follows: the mobile phase consisted of eluent A (0.1% formic acid in water, v/v) and eluent B (acetonitrile). The flow rate was 0.3 mL min−1. The column and auto-sampler temperatures were maintained at 40 °C and 4 °C, respectively. The gradient elution program was optimized as follows: 0–15 min, 5–90% B; 15–17 min, 90–100% B; 17–20 min, 100–100% B.The mass spectrometry detection was performed on SYNAPT G2-Si HDMS system, equipped with an electrospray ionization (ESI) source (Waters Corp., Manchester, UK). Negative ion mode was conducted in this analysis. Mass spectrometry conditions were finally set as follows: capillary voltage 2.5 kV, cone voltage 40 V, source temperature 120 °C, and desolvation temperature 400 °C. Nitrogen was used as desolvation and cone gas with a flow rate of 800 and 50 L h−1, respectively, and full-scan mass range was set as m/z 50–1500 Da. In auto mass spectrometry mode, the collision-induced dissociation energies were set at 5 eV for the precursor ion at low-energy mode, and the collision-induced dissociation energies were set from 20 to 40 eV for high-energy mode. Real-time data were calibrated using an external reference (LockSpray™) at a concentration of 0.2 ng mL−1 with an infusion flow rate of 10 μL min−1, generating a reference ion for the negative ion mode (m/z 554.2615) during the UPLC-MS analysis. Data were acquired and processed using MassLynx™ NT 4.1 software (Waters, Milford, MA, USA).
Accurate molecular weights of some metabolites were acquired using an Agilent 6520 Accurate Mass quadrupole time-of-flight mass spectrometer (Q-TOF MS; Agilent Technologies, USA). The capillary voltage of the ion source was set at 3.5 kV in negative ion mode. Nitrogen was used as desolvation and nebulizing gas at a constant temperature of 350 °C. The scan range was set at m/z 100–1500 Da.
The isolation and purification of metabolites were performed using an Agilent 1200 series semi-preparative high performance liquid chromatography (HPLC) system (Palo Alto, CA, USA) consisting of a G1311A quatpump solvent delivery system, G1379A degasser unit, a G1313A auto-sampler and a G1315B DAD detector. The preparation was performed with a Zorbax SB-C18 (5 μm, 9.4 mm × 25 cm) column (Agilent Technologies, US). The wavelength was set at 280 nm.
Nuclear magnetic resonance (NMR) spectra of ingenol and metabolites were measured on Bruker AV-500 spectrometers (Faellanden, Switzerland) using tetramethylsilane as internal standard.
Animals and drug administration
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Naval Medical University and approved by the Animal Ethics Committee of Naval Medical University. Male Sprague–Dawley rats (200–250 g, Shanghai Sippr-BK Laboratory Animal Co., Ltd. (Shanghai, China, license no. SCXK-Shanghai-2018-0006)) were housed in humidity- and temperature-controlled room (50 ± 10% and 22–24 °C) with a 12 h light/dark cycle before the experiment. They were allowed free access to standard laboratory food and water for one week of acclimation, then fasted overnight but with free access to water before experiments. Ingenol was dissolved in 0.5% sodium carboxymethyl cellulose solution and then ultrasonically mixed for 30 min (10 mg mL−1), and was administered orally to rats at the dose of 100 mg kg−1 body weight at a single dose.Sample collection and preparation
Blood samples (0.5 mL) were collected from six rats through the orbital sinus before administration (blank sample) and 0.25, 0.5, 1, 2, 4, 6 and 12 h after administration. Plasma samples were prepared by centrifugation at 4000 rpm for 10 min. A 200 μL aliquot of plasma was mixed with four volumes of acetonitrile for protein precipitation and desalination. After centrifugation, the supernatant was transferred and evaporated to dryness under a nitrogen stream at 30 °C. The residue was re-dissolved in methanol (100 μL) and then was centrifuged at 4 °C and 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min. A 2 μL aliquot of the supernatant was injected into the UPLC-Q/TOF-MS system.
000 rpm for 10 min. A 2 μL aliquot of the supernatant was injected into the UPLC-Q/TOF-MS system.
For urine and feces sampling, 12 rats were divided into administration group and blank group, and placed separately in stainless steel metabolic cages. Urine and feces samples were collected in containers surrounded by ice over 0–6, 6–12 and 12–24 hours before and after drug administration. The mix urine samples were centrifuged at 4000 rpm for 10 min at 4 °C, a 1 mL aliquot of supernatant was mixed with four volumes of acetonitrile. Feces samples (1.0 g) were crushed and then ultrasonically extracted by acetonitrile (10 mL) for 30 min. All the above-mentioned mixtures were centrifuged at 12000 rpm at 4 °C for 10 min individually. The supernatants were transferred to clean tubes and evaporated to dryness at 30 °C under a gentle stream of nitrogen. The residues were dissolved in 100 μL of methanol and then centrifuged at 12000 rpm at 4 °C for 10 min. A 1 μL aliquot of supernatants injected into the UPLC-Q/TOF-MS system for analysis.
Microbial transformation and isolation of ingenol
The biotransformation process was conducted using the fungus C. elegans bio-110930 at two scales: preliminary screening and preparative. Preliminary screening scale biotransformation of ingenol was carried out in 250 mL Erlenmeyer flasks containing 100 mL of liquid medium. The flasks were placed on a rotary shaker (160 rpm, 30 °C). A standard two-stage fermentation protocol was employed in all experiments.24 After 2 days of pre-culture, the substrates 5 mg (dissolved in 0.5 mL acetone) were added into each flask. Taking 1 mL samples on days 0, 2, 4, 7, 10 and 14, samples were centrifuged and degree of transformation was compared to controls on TLC and UPLC-Q/TOF-MS. Culture controls consisted of sterile medium, in which microorganisms were grown under identical conditions without substrate. Substrate controls were composed of sterile medium and the same amount of substrate incubated under the same conditions without microorganisms.The preparative scale biotransformation of ingenol by C. elegans bio-110930 was carried out in 50 Erlenmeyer flasks of 1000 mL (each flask containing 300 mL of medium), and autoclaved at 121 °C for 30 min. The flasks were inoculated with the fungal culture and placed on a shaker (160 rpm) at 28 °C for incubation. After 48 h of pre-culture, ingenol (1.0 g), dissolved in 50 mL acetone, was distributed among the 50 flasks evenly, and then left on a rotary shaker for another 7 days at 28 °C. Finally, the contents of all flasks were combined and filtered, and the aqueous layer was extracted three times with equal volumes of EtOAc. Then the organic layers were combined and evaporated under vacuum.
The crude extract (5.4 g) was eluted with gradient MeOH/H2O (0–30% MeOH, 25 mL min−1, 3 h; 30–50% MeOH, 25 mL min−1, 3 h; 50–70% MeOH, 25 mL min−1, 3 h; 70–100% MeOH, 25 mL min−1, 3 h) on a Buchi reversed phase medium pressure liquid chromatography (RP-MPLC) instrument to yield 6 fractions (Fr. A–Fr. F). On the basis of TLC and HPLC analysis, the metabolites of ingenol were detected in Fr. B–Fr. E. Fr. B (131.4 mg) was separated on Sephadex LH-20 column chromatography (CC) (3 cm × 150 cm) with MeOH/H2O (30%) to give Fr. B1–Fr. B5. Fr. B3 (22.3 mg) was purified by HPLC on a semi-preparative Zorbax SB-C18 column (CH3CN/H2O, 8%, 2 mL min−1, 45 min) to give metabolites M2 (4.5 mg, Rt = 37 min) and M3 (5.8 mg, Rt = 40 min), Fr. D (2.13 g) was applied to Sephadex LH-20 CC (3 cm × 150 cm) with MeOH/H2O (50%) to yield Fr. D1–Fr. D5, Fr. D2 (558.0 mg) was further subjected to Sephadex LH-20 CC (3 cm × 150 cm) to divide into five fractions (Fr. D2.1–Fr. D2.4). The metabolite M4 (Rt = 36.0 min, 2.0 mg) were obtained by semi-preparative HPLC (CH3CN/H2O, 20%, 2 mL min−1) from Fr. D2.2 (17.5 mg).
Results and discussion
Mass fragmentation behavior analyses of ingenol
For the purpose of obtaining a comprehensive fragmentation behavior of ingenol, the standard solution of ingenol was firstly analyzed via UPLC-Q/TOF-MS, which was helpful for the identification of ingenol metabolites.The parent drug ingenol had a deprotonated molecular ion [M − H]− at m/z 347.1857 in negative ion mode. In the MS/MS spectrum, it had characteristic and the most abundant fragment ion at m/z 329.1766, derived from the loss of water, which further lost a water to form the ion at m/z 311.1646. The fragment ions at m/z 317.1752 and m/z 299.1648 were generated by loss of CH2O from the fragments at m/z 347.1857 and m/z 329.1766, respectively. Moreover, the fragment at m/z 283.1698 resulted from the ion at m/z 299.1653 by loss of O. The loss of H2O (m/z 329.1769) is the characteristic product ion of ingenol. Mass spectrum and the fragmentation scheme for ingenol are shown in Fig. 3. The 1H-NMR and 13C-NMR spectra data of ingenol are listed in Tables 1 and 2, with the carbon position labeled as shown in Fig. 1.
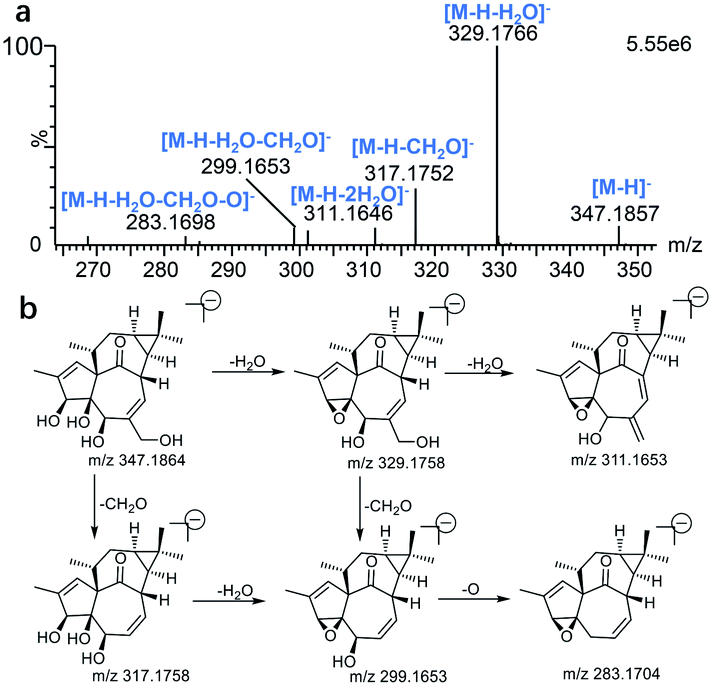 | ||
| Fig. 3 The Mass spectrum (a) and proposed fragmentation pathways (b) of ingenol. | ||
| No. | Ingenola | M2a | M3a | M4a |
|---|---|---|---|---|
| a Measured at 500 MHz in methanol-d4. | ||||
| 1 | 5.80 (q, 1.6) | 5.79 (brd., 1.10) | 5.81 (brd, 1.30) | 6.08 (brt., 1.42) |
| 3 | 4.34 (s) | 4.35 (s) | 4.34 (s) | 4.54 (s) |
| 5 | 3.63 (s) | 3.65 (s) | 3.63 (s) | 3.68 (s) |
| 7 | 5.96 (m) | 6.00 (m) | 5.98 (m) | 5.97 (m) |
| 8 | 4.28 (m) | 4.32 (brd, 4.38) | 4.42 (m) | 4.30 (m) |
| 11 | 2.42 (m) | 2.48 (m) | 2.44 (m) | 2.48 (m) |
| 12 | 2.38 (m) | 2.40 (m) | 2.47 (m) | 2.41 (m) |
| 1.73 (dt, 6.30, 15.6) | 1.74 (dt, 5.70, 15.30) | 1.85 (m) | 1.76 (m) | |
| 13 | 0.68 (td, 6.3, 8.7) | 0.82 (td, 6.34, 8.56) | 0.85 (m) | 0.69 (m) |
| 14 | 0.84 (dd, 8.4, 11.9) | 1.00 (dd, 4.38, 8.56) | 0.98 (dd, 6.79, 7.87) | 0.85 (dd, 8.35,11.85) |
| 16 | 1.06 (s) | 3.26 (d, 11.18) | 1.13 (s) | 1.06 (s) |
| 3.24 (d, 11.18) | ||||
| 17 | 1.12 (s) | 1.18 (s) | 3.74 (d, 11.64) | 1.12 (s) |
| 3.70 (d, 11.64) | ||||
| 18 | 0.94 (d, 7.10) | 0.95 (d, 7.07) | 0.95 (d, 7.09) | 0.97 (d, 7.05) |
| 19 | 1.82 (brd, 1.40) | 1.82 (brd., 1.10) | 1.82 (brd., 1.30) | 4.23 (dd, 1.42, 14.65); 4.18 (dd, 1.65, 14.65) |
| 20 | 4.10 (d, 13.6) | 4.11 (d, 13.75) | 4.10 (d, 13.35) | 4.10 (d,13.70) |
| 4.04 (d, 13.20) | 4.04 (d, 13.75) | 4.03 (d, 13.35) | 4.05 (d, 13.70) | |
| Position | Ingenola | M2a | M3a | M4a |
|---|---|---|---|---|
| a Measured at 125 MHz in methanol-d4. | ||||
| 1 | 129.54 (d) | 129.30 (d) | 129.50 (d) | 129.50 (d) |
| 2 | 141.15 (s) | 141.3 (s) | 141.2 (s) | 145.3 (s) |
| 3 | 80.76 (d) | 80.70 (d) | 80.7 (d) | 77.2 (d) |
| 4 | 86.02 (s) | 86.0 (s) | 86.1 (s) | 86.1 (s) |
| 5 | 75.05 (d) | 75.1 (d) | 74.9 (d) | 74.8 (d) |
| 6 | 143.99 (s) | 144.0 (s) | 144.0 (s) | 144.0 (s) |
| 7 | 124.41 (d) | 124.2 (d) | 124.2 (d) | 124.3 (d) |
| 8 | 44.96 (d) | 44.4 (d) | 44.4 (d) | 45.0 (d) |
| 9 | 210.67 (s) | 210.7 (s) | 210.2 (s) | 210.5 (s) |
| 10 | 73.98 (s) | 74.1 (s) | 73.9 (s) | 73.5 (s) |
| 11 | 40.57 (d) | 40.5 (d) | 40.7 (d) | 40.6 (d) |
| 12 | 31.84 (t) | 31.5 (t) | 31.6 (t) | 31.9 (t) |
| 13 | 24.47 (d) | 21.3 (d) | 25.0 (d) | 24.4 (d) |
| 14 | 24.31 (d) | 21.2 (d) | 24.5 (d) | 24.3 (d) |
| 15 | 24.98 (s) | 31.3 (s) | 31.1 (s) | 25.1 (s) |
| 16 | 28.94 (q) | 72.5 (t) | 24.5 (q) | 28.9 (q) |
| 17 | 15.80 (q) | 11.6 (q) | 63.2 (t) | 15.8 (q) |
| 18 | 17.55 (q) | 17.7 (q) | 17.2 (q) | 17.5 (q) |
| 19 | 15.54 (q) | 15.5 (q) | 15.6 (q) | 60.6 (t) |
| 20 | 65.53 (t) | 65.5 (t) | 65.5 (t) | 65.5 (t) |
Structural elucidation of ingenol metabolites in rat
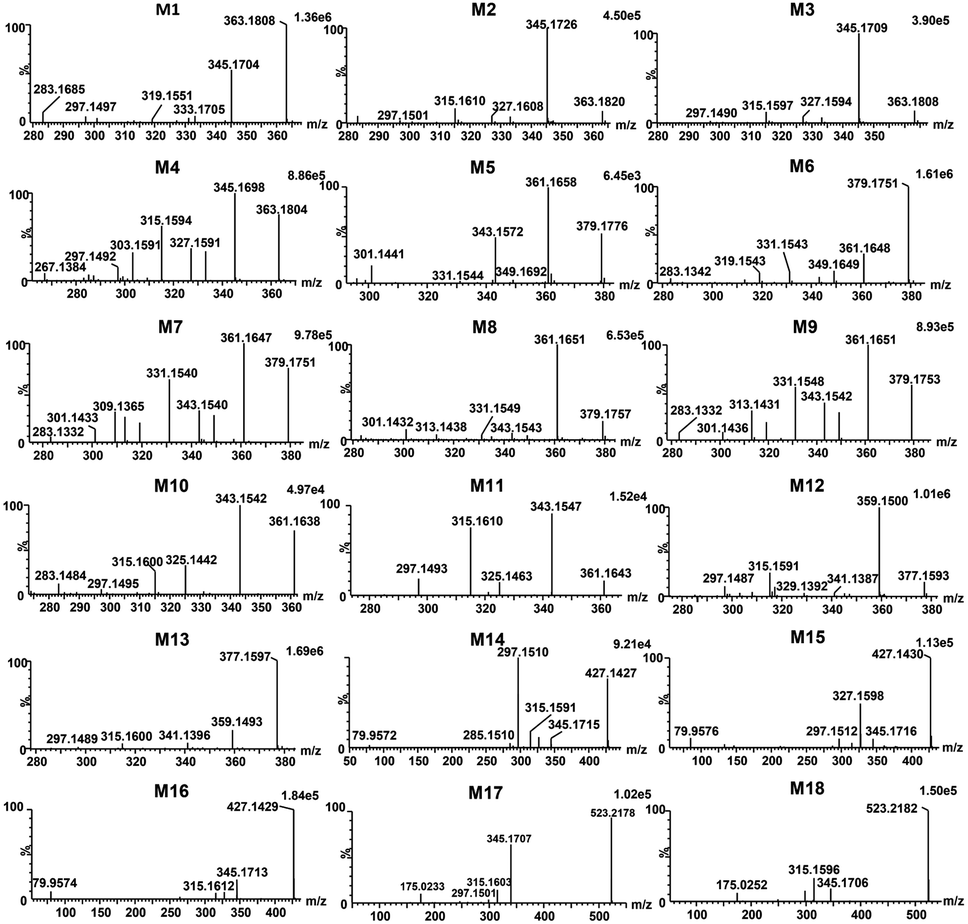
Total ion chromatograms of urine, feces and plasma samples, which were collected from the experimental rats after oral administration of ingenol, were analyzed by UNIFI 4.1 software. The analytes in each sample were compared according to the characteristic mass spectrum behavior (including parent ions, internal cleavage in the ion source, characteristic fragment ions of each metabolite) and retention time. Compared with the peaks in the corresponding blank sample, a total of 18 metabolites identified in rat are listed in Table 3. Extracted ion chromatograms and product ion spectra of metabolites are presented in Fig. 4 and 5. The proposed metabolic pathway of ingenol is shown in Fig. 6. The retention times, precursor molecular ion, key fragments, and distribution of ingenol and its metabolites are listed in Table 3.| No. | Component Name | RT (min) | Formula [M − H]− | Observed m/z | Error (ppm) | MSn | Distribution | Fungi | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Rat | ||||||||||
| Blood | Urine | Faeces | ||||||||
| M1 | M + O | 4.34 | C20H27O6 | 363.1808 | −1.38 | 363.1808, 345.1704, 333.1705, 327.1596, 319.1551, 315.1592, 309.1397, 297.1497 | √ | √ | √ | √ |
| M2 | M + O | 5.00 | C20H27O6 | 363.1820 | 1.93 | 363.1820, 345.1726, 333.1720, 327.1608, 315.1610, 297.1501 | √ | √ | √ | √ |
| M3 | M + O | 5.48 | C20H27O6 | 363.1808 | −1.38 | 363.1808, 345.1709, 333.1704, 327.1594, 315.1597, 301.1803, 297.1490 | √ | √ | √ | √ |
| M4 | M + O | 6.93 | C20H27O6 | 363.1804 | −2.48 | 363.1804, 345.1698, 333.1700, 327.1591, 315.1594, 303.1591, 297.1492, 285.1487, 267.1384 | √ | √ | √ | √ |
| M5 | M + 2O | 3.02 | C20H27O7 | 379.1776 | 3.69 | 379.1776, 361.1658, 349.1692, 343.1572, 331.1544, 301.1441 | √ | √ | √ | — |
| M6 | M + 2O | 3.63 | C20H27O7 | 379.1751 | −2.90 | 379.1751, 361.1648, 349.1649, 343.1539, 331.1543, 313.1433, 319.1543, 301.1440, 283.1342 | √ | √ | √ | — |
| M7 | M + 2O | 3.82 | C20H27O7 | 379.1760 | −0.53 | 379.1760, 361.1656, 349.1655, 343.1548, 331.1553, 319.1550, 313.1442, 301.1443, 283.1332 | √ | √ | √ | — |
| M8 | M + 2O | 3.94 | C20H27O7 | 379.1757 | −1.32 | 379.1757, 361.1651, 349.1648, 343.1543, 331.1549, 313.1438, 301.1433, 283.1326 | √ | √ | √ | — |
| M9 | M + 2O | 4.30 | C20H27O7 | 379.1753 | −2.37 | 379.1753, 361.1651, 349.1651, 343.1542, 331.1548, 319.1544, 313.1431, 301.1436, 283.1332 | √ | √ | √ | — |
| M10 | M + O–2H | 5.94 | C20H25O6 | 361.1643 | −3.88 | 361.1643, 343.1542, 325.1442, 315.1600, 297.1495, 283.1484 | √ | √ | √ | — |
| M11 | M + O–2H | 6.34 | C20H25O6 | 361.1643 | −3.88 | 361.1643, 343.1547, 325.1463, 315.1610, 297.1497 | √ | √ | √ | — |
| M12 | M + 2O–2H | 5.12 | C20H25O7 | 377.1593 | −3.45 | 377.1593, 359.1500, 341.1387, 315.1591, 297.1487 | √ | √ | √ | — |
| M13 | M + 2O–2H | 5.82 | C20H25O7 | 377.1597 | −2.39 | 377.1597, 359.1493, 341.1396, 315.1600, 297.1489 | √ | √ | √ | — |
| M14 | M + SO3 | 4.46 | C20H27O8S | 427.1426 | −1.40 | 427.1426, 345.1715, 327.1598, 315.1591, 297.1510, 285.1510, 79.9572 | — | — | √ | — |
| M15 | M + SO3 | 4.68 | C20H27O8S | 427.1430 | −0.47 | 427.1430, 345.1716, 327.1598, 315.1599,79.9576 | — | — | √ | — |
| M16 | M + SO3 | 5.27 | C20H27O8S | 427.1429 | −0.70 | 427.1429, 345.1713, 327.1614, 315.1612, 79.9574 | — | — | √ | — |
| M17 | M + C6H8O6 | 6.88 | C26H35O11 | 523.2174 | −2.10 | 523.2174, 345.1707, 315.1603, 297.1501, 175.0233 | — | √ | — | — |
| M18 | M + C6H8O6 | 7.08 | C26H35O11 | 523.2182 | −0.57 | 523.2182, 345.1706, 315.1596, 175.0252 | — | √ | — | — |
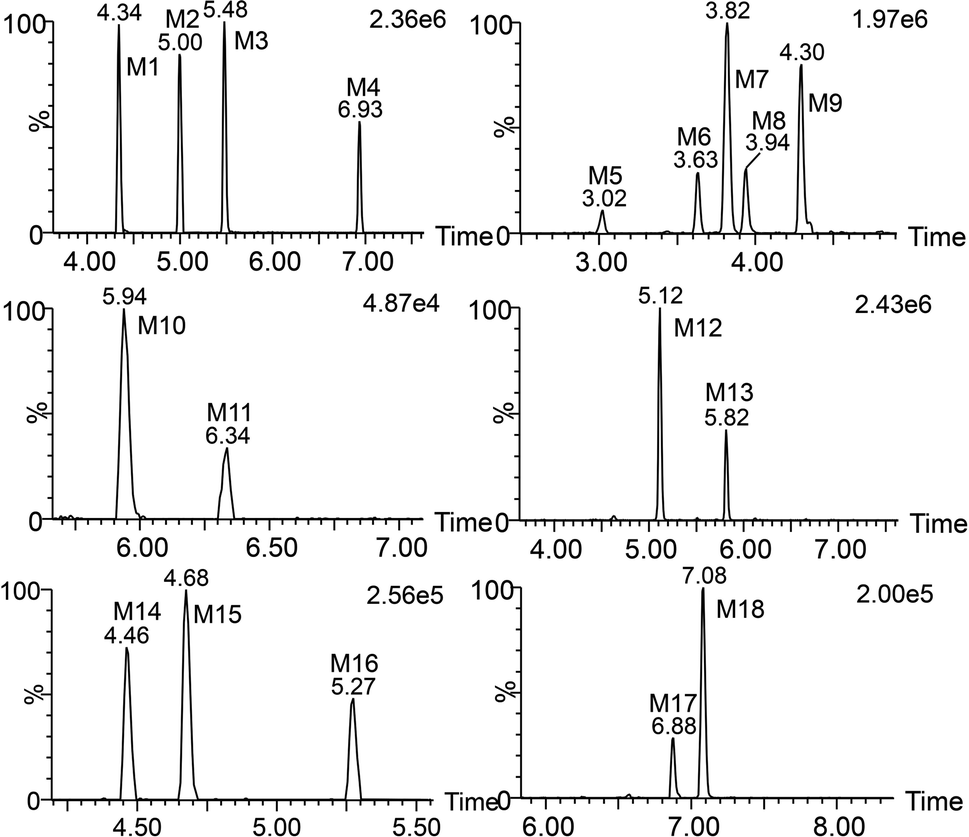 | ||
| Fig. 4 Extracted ion chromatograms of ingenol metabolites in rat. | ||
 | ||
| Fig. 5 Product ion spectra of ingenol metabolites in rat. | ||
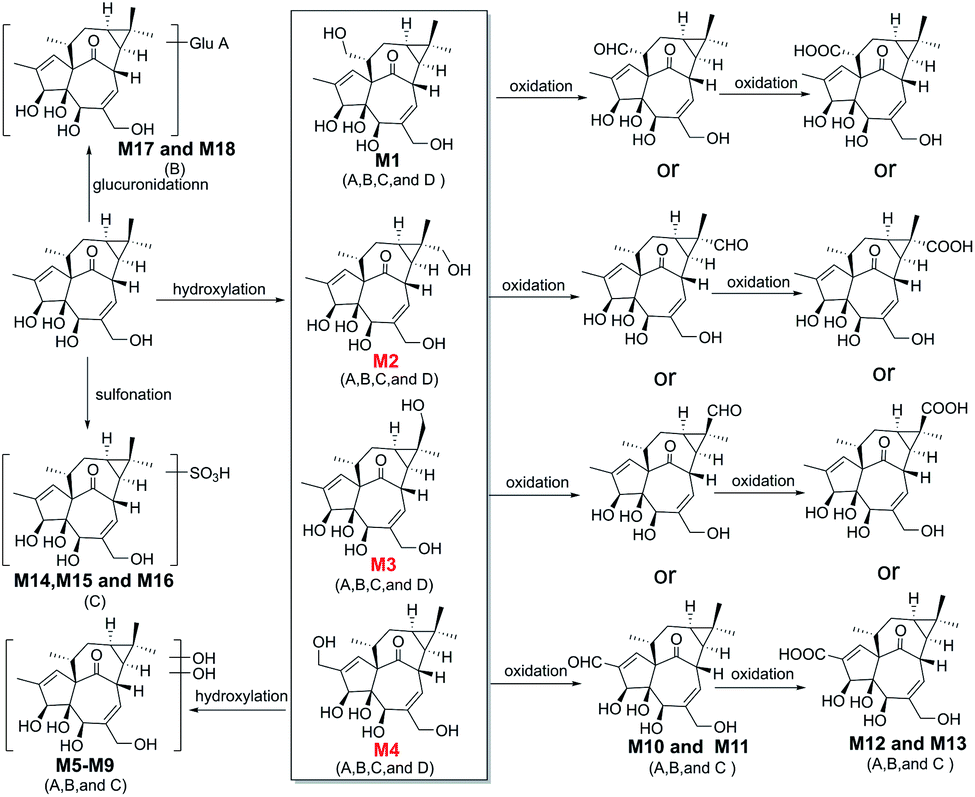 | ||
| Fig. 6 The proposed metabolic pathway of ingenol in rat plasma (A), rat urine (B), rat feces (C), and micro-organism (D). | ||
Metabolites (M1–M4) were detected with the HPLC retention times between 4.34 and 6.93 min. The molecular ion at m/z 363.1808 ([M − H]−) was observed, 16 Da mass of an oxygen atom more than that of ingenol. The fragment ions at m/z 345.1704 and m/z 327.1596 resulted from successive water loss from the ion at m/z 363.1808, and the fragment ions at m/z 333.1705, m/z 315.1592 and m/z 297.1497 were generated by CH2O loss from m/z 363.1808, m/z 345.1704 and m/z 327.1596, respectively. The momohydroxylated products were more likely to lose a CH2O group compared with ingenol. This clearly suggested that the hydroxylated site should be located on the methyl moiety of the molecule, but the exact substituted position remained to be determined.
The structure of M2 was confirmed by comparison of its HPLC retention time and MS/MS fragments with the corresponding reference standard isolated and purified from microbial transformation samples (Fig. 7). The structure of M2 as the reference standard was determined based on NMR analyses. Tables 1 and 2 showed the comparison of the 1H-NMR and 13C-NMR data of metabolite M2 and ingenol. The 1H and 13C-NMR spectroscopic data of M2 displayed the resonances for structural fragments similar to those of ingenol, except that the appearance of an extra hydroxymethyl unit [δH 3.26 (d, 11.18) and 3.24 (d, 11.18); δC 72.5 (t)], instead of the methyl signals for C-16 in ingenol [δH 1.06 (s); δC 28.9 (q)], revealing that an additional hydroxyl group might be substituted at C-16 in M2. Furthermore, in the 13C-NMR spectrum, the resonances of C-15 and C-16 were shifted significantly downfield from δC 24.98 to 31.3, and from δC 28.94 to 72.5, respectively, and the resonance of C-17 was shifted upfield from δC 15.8 to 11.6. As a result, the hydroxyl group was located at C-16. After browsing related research, M2 was characterized as 16-hydroxyl ingenol by comparison of the NMR and HR-MS data with those of the literature.25
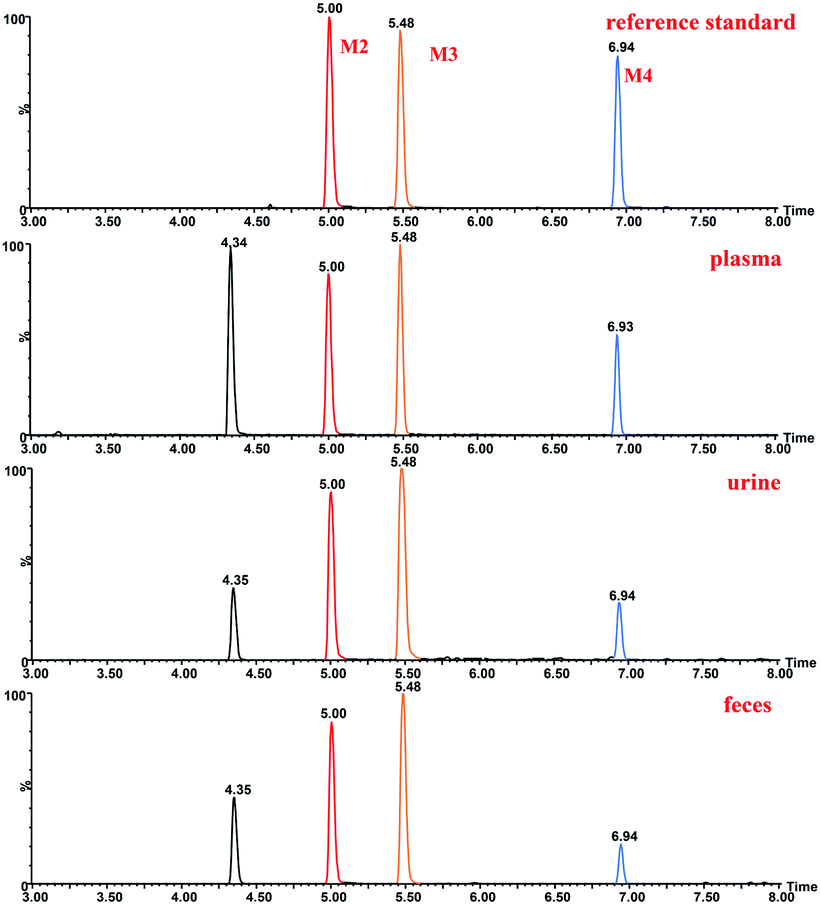 | ||
| Fig. 7 Extracted ion chromatograms of ingenol metabolites in rat and reference standard isolated and purified from microbial transformation samples. | ||
The structure of M3 was confirmed by comparison of its HPLC retention time and MS/MS fragments with the corresponding reference standard isolated and purified from microbial transformation products (Fig. 7). Analysis of the 1H and 13C-NMR spectroscopic data of M3 revealed structural similarity to ingenol (Tables 1 and 2), the appearance of an extra oxygenated methylene unit [δH 3.74 (d, J = 11.64 Hz, H-17a) and 3.70 (d, J = 11.64 Hz, H-17b); δC 63.2], in place of the methyl signals for C-17 (δH 1.12; δC 15.8) in ingenol, suggesting the introduction of one additional hydroxyl group in M3. In addition, the resonance of C-15 appeared at lower field at δC 31.1 and C-16 was shifted upfield to δC 24.5. Consequently, the extra hydroxyl group was attributed to C-17, which was further evidenced by key HMBC correlations from the hydroxymethylene protons at δH 3.74 and 3.70 to C-13, C-14, and C-15, and from H-16 at δH 1.13 to C-13, C-14, C-15 and C-17, together with key NOESY correlations of the hydroxylmethyl protons at δH 3.74 and 3.70 with H-8 (δH 4.42) and H-12 at δH 2.47 (Fig. S1†). Therefore, M3 was assigned as 17-hydroxyl ingenol.
The structure of M4 was confirmed by comparing the HPLC retention time and MS/MS fragments of M4 with the corresponding reference standard isolated and purified from microbial transformation products (Fig. 7). The 1H-NMR spectrum of M4 (Table 1) displayed characteristic signals of only three methyls H-16 (δH 1.06), H-17 (δH 1.12) and H-18 (δH 0.97), with two extra oxygenated protons at δH 4.23 (dd, J = 1.42, 14.65 Hz) and δH 4.18 (dd, J = 1.65, 14.65 Hz), rather than four methyl groups of ingenol, indicating that a methyl was hydroxylated in M4. Compared to the 1H-NMR and 13C-NMR spectra of ingenol (Tables 1 and 2), the signals for a hydroxymethylene occurred at δC 60.6 in 13C-NMR spectrum and at δH 4.23 (dd, J = 1.42, 14.65 Hz) and δH 4.18 (dd, J = 1.65, 14.65 Hz) in 1H-NMR spectrum in M4, instead of the methyl signals for C-19 at δH 1.82 (brd, 1.40) and δC 15.5 in ingenol, suggesting the additional hydroxyl group to be attached to C-19. This coincided with the 13C-NMR resonances observed for downfield shift of C-2 (Δδ + 4.15) and upfield shift of C-3 (Δδ − 3.56) in comparison with those of ingenol. Furthermore, the HMBC correlations of H-19 (δH 4.23 and 4.18) with C-1, C-2 and C-3, revealing the hydroxylation of C-19. Thus, M4 was established as 19- hydroxyl ingenol.
Due to the fragment pathway, the hydroxyl groups of metabolites M1–M4 were substituted on the methyl groups of ingenol. The hydroxyl groups of metabolites M2–M4 occupied at C-16, C-17 and C-19, respectively. Only a methyl group at C-18 was not replaced, therefore, metabolite M1 was most likely to be hydroxylated at the remaining methyl group of ingenol.
Metabolites M5–M9 were predicted to be dihydroxylated derivatives with HPLC retention times between 3.02 and 4.30 min. Their parent ions were detected at m/z 379.1751 ([M − H]−) in negative ion mode, 16 mass unit more than those of M1–M4, suggested the presence of an additional hydroxyl group. The fragment ions at m/z 361.1648 and m/z 343.1539 resulted from successive water loss of the fragment ion at m/z 379.1751, and the fragment ions at m/z 349.1649, m/z 331.1543 and m/z 313.1433 were generated by CH2O loss from the fragment ions at m/z 379.1751, m/z 361.1648 and m/z 343.1539, respectively. The fragment ions at m/z 331.1553 and m/z 313.1442 also resulted from successive H2O loss of the fragment ion at m/z 349.1649. In particular, the fragment ions at m/z 349.1649 and 319.1543 were generated by successive CH2O loss of the ion at m/z 379.1751, in which the ions at m/z 283.1342 and 313.1433 resulted from the ion at m/z 343.1539 in similar mechanism. Consistent with monohydroxylation products of ingenol, they showed a series of product ions resulting from loss of H2O and CH2O. More fragments resulted from loss of CH2O, demonstrating that dihydroxylated ingenol metabolites had more hydroxyl groups at methyl moiety than monohydroxylated metabolites.
Metabolites (M10–M11) were detected 5.94 and 6.34 min. The parent ion was detected at m/z 361.1643 ([M − H]−) in negative ion mode. They were 14 Da more than ingenol and 2 Da less than M1–M4, which were hydroxylation products of ingenol. The fragment ions at m/z 325.1442 and 343.1542 were generated by successive water loss from the ion at m/z 361.1643. The fragment ions at m/z 315.1600 and 297.1495 were generated by CO loss from the ions at m/z 343.1542 and m/z 325.1442, respectively, indicating the existence of an aldehyde group. Furthermore, according to the proposed metabolic pathway of tanshinone IIA by Sun et al.,26 the metabolic modification could take place in the methyl of ingenol, from methyl to primary alcohol, then to aldehyde group. Thus, it is provisionally interpreted that M10 and M11 was produced by transforming methyl of ingenol to aldehyde.
Metabolites (M12–M13), which showed deprotonated ion at m/z 377.1593 ([M − H]−) in negative ion mode with the retention times of 5.12 and 5.82 min, were 30 Da more than ingenol and 2 Da less than dihydroxylation products of ingenol like M5–M9. The fragment ions at m/z 359.1500 and 341.1387 were proposed to result from successive loss of water from the molecular ion at m/z 377.1593. In addition, the fragments at m/z 315.1591 and m/z 297.1487 were generated from the ions at m/z 359.1500 and m/z 341.1387 following loss of CO2, respectively. The fragment spectrum showed a prominent loss of CO2, indicating the existence of carboxyl group. This compound might be produced by the oxidation of M10 or M11, which was further in accordance with our initial speculation. Therefore, it is provisionally interpreted that M12 and M13 was produced by transforming aldehyde of M10 or M11 to carboxyl.
Metabolites (M14–M16) were detected at HPLC retention times between 4.46 and 5.27 min. They showed a molecular ion at m/z 427.1426 ([M − H]−) in negative ion mode, with 80 Da more than ingenol. The fragment ion at m/z 345.1715 provided reliable evidence for identifying metabolites, and molecular ion of SO3 at m/z 79.9576 ([M − H]−) was also observed, demonstrating that a hydroxyl group in ingenol was replaced by SO3. Therefore, they were tentatively identified as sulfonated products of ingenol.
Metabolites (M17 and M18) eluted at about 6.88 and 7.08 min, respectively. They gave the deprotonated molecule ion [M − H]− at m/z 523.2174, 176 Da more than ingenol. The fragment ion at m/z 345.1707 was proposed to result from loss of C6H10O6 from the molecular ion, and the deprotonated ion [M − H]− of C6H8O6 at m/z 175.0233 was also observed, indicating a hydroxyl group of ingenol was replaced by a glucruonide.
Elucidation of the metabolic pathway in rat
In vivo metabolism of ingenol and the metabolic pathways were accordingly proposed using UPLC-Q/TOF-MS, as shown in Fig. 6. In general, the in vivo metabolism of ingenol can be concluded to undergo four pathways. The first pathway is hydroxylation of methyl moiety to form monohydroxylated ingenol (M1–M4), which undergo further metabolism to form dihydroxylated ingenol (M5–M9). The second pathway involves the oxidation of methyl in ingenol, from methyl to primary alcohol, then to aldehyde group (M10–M11), and finally to carboxyl group (M12–M13). The third metabolic pathway is the direct sulfonation of the hydroxyl part in ingenol to form sulfonated adducts (M14 and M16). The fourth metabolic pathway is to directly combine with glucuronide to form glucuronide adducts (M17 and M18). Therefore, hydroxylation, oxidation, sulfonation and glucuronidation are the main metabolic pathways of ingenol in rat.In addition, in this study, phase I metabolites were detected in rat plasma, urine and feces, while phase II metabolites were detected only in urine or feces. Therefore, phase I metabolites of ingenol might be potential components with pharmacological or toxicological effects,27 and phase II metabolites usually increase polarity and reduce liposolubility, so as to accelerate drug excretion from the body and usually play a detoxification role in drug metabolism.28
Conclusions
In the present study, a rapid and reliable analytical method based on UPLC Q/TOF-MS was developed and used for the identification of ingenol metabolites in rat (plasma, urine, and feces) and C. elegans bio-110930 model. A total of 18 metabolites were identified in vivo based on the molecular ions and the MS/MS fragments, including monohydroxylated ingenol (M1–M4), dihydroxylated ingenol (M5–M9), methyl hydroformlated ingenol (M10 and M11), methyl carboxylated ingenol (M12 and M13), sulfate ingenol (M14–M16) and glucuronide ingenol (M17 and M18). Among them, reference standards of three metabolites were further prepared by preparative scale microbial transformation. The structures of these metabolites were confirmed by comparison of their HPLC retention times and MS/MS fragments with the prepared reference standards, whose structures were determined based on 1D and 2D NMR analyses. The three identified metabolites were 16α-hydroxy ingenol (M2), 17β-hydroxy ingenol (M3), and 19-hydroxy ingenol (M4). Furthermore, ingenol was proved to be metabolized mainly via hydroxylation, oxygenation, sulfonation, and glucuronidation. Moreover, C. elegans bio-110930 could be a suitable model to simulate and prepare ingenol phase I metabolites. Thus, an overall description of metabolites of ingenol from rat and C. elegans bio-110930 has been provided. Furthermore, our study provides valuable information in predicting in vivo human metabolites of ingenol.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by The Key Research and Development Program of China (2017YFC1702002, 2017YFC1700200, 2019YFC1711000), the National Natural Science Foundation of China (31870327, 82173704, 82004215, 8200141985, 81573318, 81373301, 81230090, 81520108030), Shanghai Engineering Research Center for the Preparation of Bioactive Natural Products (10DZ2251300), National Science and Technology Major Project of China (2018ZX09731016-005, 2019ZX09201004-003-010) and Shanghai Municipal Health Commission Project (20204Y0326).Notes and references
- Z. Qiao, H. J. Yong, Z. Yi, W. Kan, Z. Min, D. C. Pei, F. Y. Wei, P. T. Yu, H. W. Jian and Z. Li, J. Ethnopharmacol., 2020, 267, 113507 Search PubMed.
- C. M. Hasler, G. Acs and P. M. J. C. R. Blumberg, Cancer Res., 1992, 52, 202–208 CAS.
- K.-J. Hong, H. S. Lee, Y.-s. Kim and S. S. Kim, Public Health Res. Perspect., 2011, 2, 109–114 CrossRef PubMed.
- A. K. Gupta and M. Paquet, J. Cutaneous Med. Surg., 2013, 17, 173–179 CrossRef CAS PubMed.
- D. R. Alchin, Dermatol. Ther., 2014, 4, 157–164 CrossRef PubMed.
- M. Bourcier, L. Stein Gold, L. Guenther, C. M. Andreassen, J. Selmer and G. Goldenberg, J. Dermatol. Treat., 2017, 28, 652–658 CrossRef CAS PubMed.
- I. P. de Sousa, M. V. Sousa Teixeira and N. A. Jacometti Cardoso Furtado, Molecules, 2018, 23, 1387–1419 CrossRef PubMed.
- F. P. Guengerich, Chem. Res. Toxicol., 2009, 22, 237–238 Search PubMed.
- F. Gaunitz, P. Dahm, L. Mogler, A. Thomas, M. Thevis and K. Mercer-Chalmers-Bender, Anal. Bioanal. Chem., 2019, 411, 3561–3579 CrossRef CAS PubMed.
- K. E. Grafinger, A. Wilke, S. Knig and W. Weinmann, Drug Test. Anal., 2019, 11, 721–729 CrossRef CAS PubMed.
- L. Xu, Y. Liu, H. Wu and A. Zhou, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2019, 1137, 121934 CrossRef PubMed.
- K. Li, F. Qin, L. Jing, F. Li and X. Guo, Anal. Bioanal. Chem., 2013, 405, 2619–2634 CrossRef CAS PubMed.
- M. Duan, Q. Li, D. Zhong and Z. Peng, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2018, 1095, 138–141 CrossRef CAS PubMed.
- R. He, J. Fan, R. Chen, D. Guo, M. Zhao, Z. Zhang, C. Liang, M. Chen, H. Song and W. Zhang, Chemosphere, 2021, 264, 128495 CrossRef CAS PubMed.
- T. Nguyen, S.-J. Yeom and C.-H. Yun, Appl. Sci., 2021, 11, 603–626 CrossRef CAS.
- K. Lapham, E. Callegari, J. Cianfrogna, J. Lin, M. Niosi, C. C. Orozco, R. Sharma and T. C. Goosen, Drug Metab. Dispos., 2020, 48, 1350–1363 CrossRef CAS PubMed.
- Y. Ma, P. Sun, Y. Zhao, K. Wang, X. Chang, Y. Bai, D. Zhang and L. Yang, Molecules, 2019, 24, 315–328 CrossRef PubMed.
- K. Yildirim, A. Kuru and E. Küçükbaşol, Biocatal. Biotransform., 2019, 38, 7–14 CrossRef.
- A. Aziz, S. Bano, W. Atia Tul and M. I. Choudhary, Steroids, 2020, 154, 108467 CrossRef CAS PubMed.
- R. Sponchiado, J. M. Sorrentino, N. Olegario, S. S. Oliveira, L. M. Cordenonsi, G. P. Silveira, A. M. Fuentefria, A. S. L. Mendez, M. Steppe and C. V. Garcia, Biomed. Chromatogr., 2019, 33, e4496 CrossRef PubMed.
- C. Chen, K. Song, Y. Zhang, C. Chu, B. Fan, Y. Song, H. Huang and G. Chen, Phytochemistry, 2021, 182, 112608 CrossRef CAS PubMed.
- W. Palmer-Brown, R. Miranda-CasoLuengo, K. H. Wolfe, K. P. Byrne and C. D. Murphy, Sci. Rep., 2019, 9, 9240–9248 CrossRef PubMed.
- S. Asha and M. Vidyavathi, Biotechnol. Adv., 2009, 27, 16–29 CrossRef CAS PubMed.
- Y. Wang, L. Xiang, Z. Wang, J. Li, J. Xu and X. He, Bioorg. Chem., 2020, 101, 103870 CrossRef CAS PubMed.
- J. B. He, H. J. Zhu, G. F. Luo, G. M. Liu, Y. Li, H. Chen, S. P. Chen, X. Lu, G. C. Zhou and Y. X. Cheng, Bull. Korean Chem. Soc., 2010, 31, 2211–2214 CrossRef CAS.
- J. H. Sun, M. Yang, X. M. Wang, M. Xu, A. H. Liu and D. A. Guo, J. Pharm. Biomed. Anal., 2007, 44, 564–574 CrossRef CAS PubMed.
- U. M. Zanger and M. Schwab, Pharmacol. Ther., 2013, 138, 103–141 CrossRef CAS PubMed.
- Z. Zhang and T. Wei, Acta Pharm. Sin. B, 2018, 8, 721–732 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: The spectra including 1D, 2D-NMR, HRESIMS of compounds 2–4. See DOI: 10.1039/d1ra07915h |
| ‡ Si-Jia Xiao and Shan-Shan Li contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |