
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBicyclo[6.1.0]nonyne carboxylic acid for the production of stable molecular probes†
Tony Rady a,
Michel Mossera,
Marc Nothisena,
Stephane Erbb,
Igor Dovgana,
Sarah Cianféranib,
Alain Wagner*a and
Guilhem Chaubet*a
a,
Michel Mossera,
Marc Nothisena,
Stephane Erbb,
Igor Dovgana,
Sarah Cianféranib,
Alain Wagner*a and
Guilhem Chaubet*a
aBio-Functional Chemistry (UMR 7199), LabEx Medalis, University of Strasbourg, 74 Route du Rhin, 67400 Illkirch-Graffenstaden, France. E-mail: alwag@unistra.fr; chaubet@unistra.fr
bLaboratoire de Spectrométrie de Masse BioOrganique (LSMBO), LabEx Medalis, Université de Strasbourg, CNRS, IPHC UMR 7178, 67000 Strasbourg, France
First published on 17th November 2021
Abstract
Bicyclo[6.1.0]non-4-yn-9-ylmethanol (BCN alcohol) is the most prominent strained-alkyne scaffold in chemical biology. Described herein is the synthesis of an oxidized analogue – BCN acid – whose facile functionalization via amide bond formation yields more stable derivatives than the classically encountered carbamates.
Introduction
The ability to effect precise chemical reactions in biological media has always possessed vast applications, from the labelling of biomolecules to the detection and quantification of reactive metabolites, or the controlled liberation of an active substance. Because of the inherent complex molecular composition of the biological environment, performing synthetic chemistry in such media raised the issue of chemoselectivity: how can a chemical reaction occur selectively between certain reactants without interfering with surrounding biomolecules? Ever since the early 2000s, tremendous efforts have been made to address this question, resulting in the development of bioorthogonal reactions. These take advantage of reactive groups that are usually inert toward chemical functions naturally present in biological systems but highly reactive toward another abiotic reactant. One of the best examples of such bioorthogonal combinations is the azide – cycloalkyne duet, reacting with one another via strain-promoted azide–alkyne cycloaddition (SPAAC). While the kinetics can be considered sluggish when compared to other bioorthogonal reactions, the stability of the resulting triazole products, along with its simplicity of operation and low toxicity participated in making SPAAC a privileged tool for a myriad of applications in chemical biology. As this reaction gained momentum, it was logically accompanied by the development of other families of cycloalkynes, whose design was driven by two major principles: (i) a good stability-to-reactivity balance, with cycloalkynes being sufficiently stable to be used in aqueous environment but reactive enough not to necessitate the adjunction of a catalyst; and (ii) limited lipophilicity, so as to be soluble in biological media and minimise non-specific binding to biomolecules.1 These stringent selection parameters explain why only a handful of strained alkynes have been reported to date for bioorthogonal applications, mainly belonging to the family of cyclooctynes. One of its most prominent members is undoubtedly the endo isomer of bicyclo[6.1.0]nonyne (BCN) alcohol derivative 1 (Fig. 1) developed in 2010.2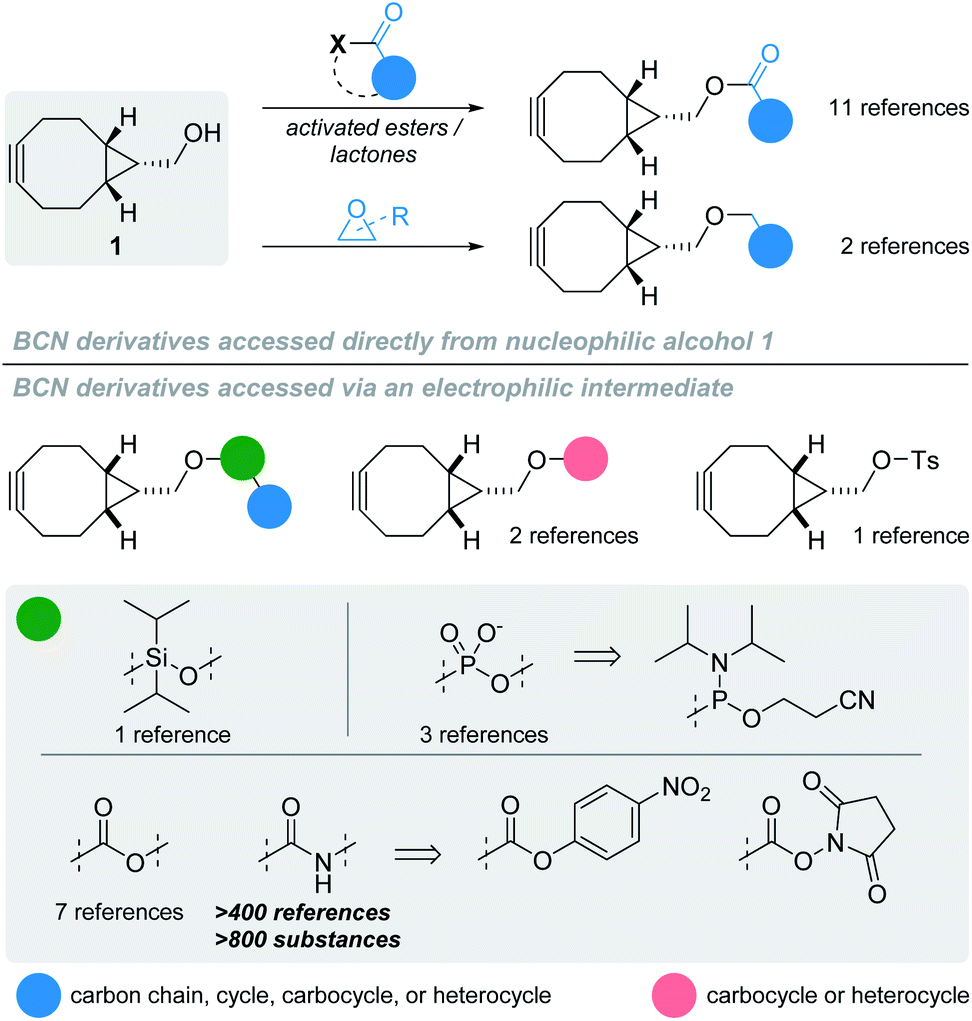 | ||
| Fig. 1 Structure of BCN 1 derivatives and general overview of their synthesis according to the literature. | ||
With a simple primary alcohol as a reactive handle, several chemical strategies were developed to equip this scaffold with relevant payloads. While O-acylation or O-alkylation would seem to be the most appropriate way of accessing valuable BCN derivatives rapidly, only a handful of such procedures have been reported – via direct reaction with activated esters or ring-opening polymerization of lactones and epoxides, respectively (Fig. 1; top part).3 Counterintuitively, the preferential strategy for modifying the primary alcohol of BCN 1 is via its transient conversion to electrophilic species of varied structures (Fig. 1, bottom part), allowing the coupling with a second nucleophile, essentially alcohols and amines. Several options have been investigated over the years to do so, via the incorporation of heteroatom spacers such as silicon or phosphorus,4–6 the use of the Mitsunobu reaction,7,8 or the mechanochemical conversion to BCN tosylate.9 However, applications employing such strategies can be considered scarce in comparison to the use of activated carbonates for the generation of more stable carbonates and carbamates, the latter representing the most commonly encountered type of BCN derivatives. Given that the nucleophilic character of BCN alcohol 1 is hardly used for anything other than the generation of intermediate electrophilic groups, it would seem logical to assume that oxidized and electrophilic analogues of 1 would be a preferable option for the simple preparation of BCN derivatives. However, only two references dealing with BCN aldehyde 2 (Fig. 2) can be found in the scientific literature: one reporting it as an intermediate toward the formation of BCN-containing α-amino acids,10 and the second – more in line with our postulate – as an effective way to yield BCN amine derivatives via reductive amination.11 Even more surprisingly, a SciFinder search for BCN acid 3 led to no result. Intrigued by this observation, we thus decided to synthesize and evaluate the potential of BCN acid 3 as a surrogate for BCN alcohol 1.
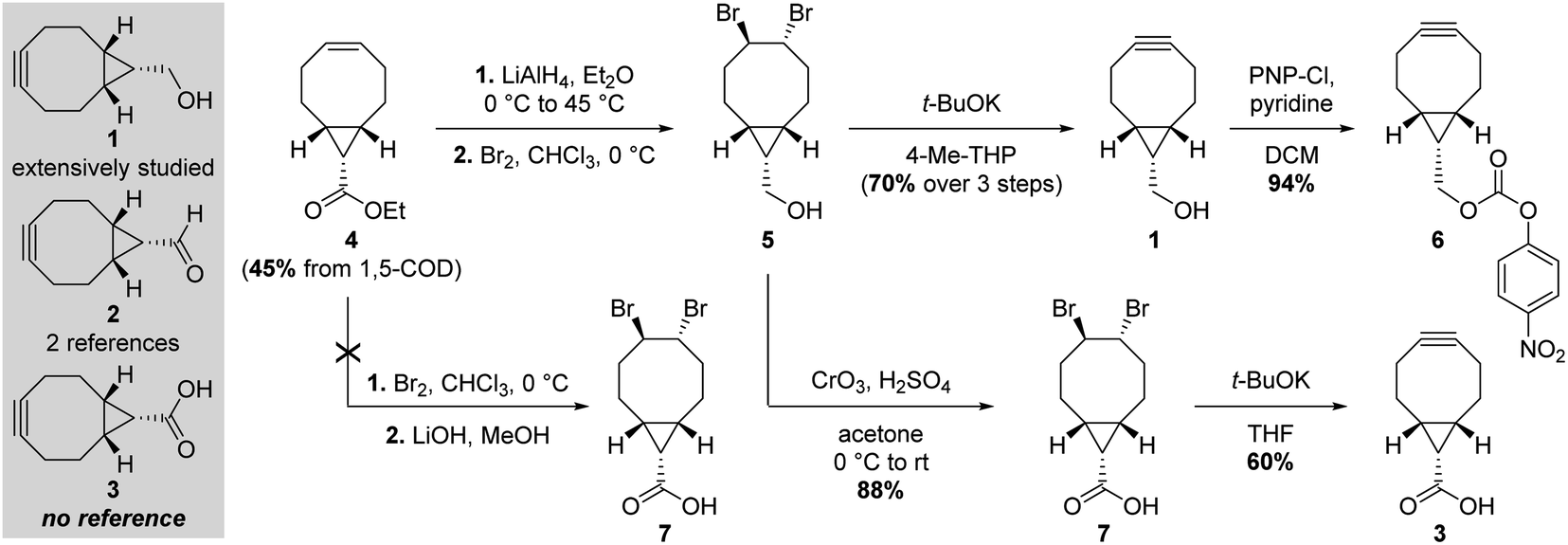 | ||
| Fig. 2 Comparative syntheses of activated BCN 6 and BCN acid 3. | ||
Results and Discussion
We started our synthetic work by slightly tweaking reported procedures (Fig. 2). Starting from 1,5-cyclooctadiene (1,5-cod), our initial goal was to take advantage of the ester group in 4 introduced upon cyclopropyl formation following Fox and coworkers' procedure,12 assuming that a dibromination–saponification–double elimination sequence would deliver the desired BCN acid 3. To our dismay, this approach proved not to be feasible, due to the formation of several side products in the course of the saponification step leading to 7, urging us to redirect our efforts. We thus decided to proceed with the classical route toward BCN 1 until formation of primary alcohol 5, which we envisioned oxidizing to the corresponding carboxylic acid. Jones reagent turned out to be an effective oxidizer, delivering pure 7 in multigram scale and excellent yield after a single recrystallization. Interestingly, no sign of cyclopropyl decomposition was observed under such seemingly harsh conditions, even after prolonged reaction. The final alkyne formation, via the well-documented potassium tert-butoxide-mediated double elimination of the 1,2-dibromide moiety, delivered the expected product 3, which can be stored indefinitely at −20 °C under argon without any apparent decomposition. It is worth stressing that this route shares similar overall yield and number of steps as the one leading to the activated carbonate 6 – the reactive BCN derivative mostly employed in the literature.With BCN 3 in hand, we then set out to evaluate SPAAC kinetics with the model benzyl azide, in accordance with standard procedures (Fig. 3).13 Quantitative conversion to triazole 8 was observed in a mixture of acetonitrile and water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, v/v), with a second-order rate constant of 0.20 M−1 s−1, almost identical to that of BCN alcohol 1 under the same conditions and in the range of values classically observed with other cyclooctynes for this transformation.14,15 Similar to its reduced analogue 1, BCN 3 was also found to undergo rapid inverse electron-demand Diels–Alder (iEDDA) reactions,16 being converted to pyridazine 9 quantitatively in just 30 minutes at room temperature when reacted with 3,6-di-2-pyridyl-1,2,4,5-tetrazine.
:2, v/v), with a second-order rate constant of 0.20 M−1 s−1, almost identical to that of BCN alcohol 1 under the same conditions and in the range of values classically observed with other cyclooctynes for this transformation.14,15 Similar to its reduced analogue 1, BCN 3 was also found to undergo rapid inverse electron-demand Diels–Alder (iEDDA) reactions,16 being converted to pyridazine 9 quantitatively in just 30 minutes at room temperature when reacted with 3,6-di-2-pyridyl-1,2,4,5-tetrazine.
 | ||
| Fig. 3 Reactivity of BCN 3. In the frame: bioorthogonal reactions with benzyl azide (SPAAC) and 3,6-di-2-pyridyl-1,2,4,5-tetrazine (iEDDA) and amide bond formation. Outside the frame: conjugation of native trastuzumab followed by functionalization with a TAMRA fluorophore and native mass spectrum of the resulting conjugate 14. | ||
Having validated that the presence of a carboxylic acid on the BCN scaffold did not alter the alkyne reactivity, we then sought to derivatize 3 in order to demonstrate that it could be an interesting candidate for applications in chemical biology. In a model experiment, 3 was reacted with benzylamine under classical coupling conditions, effectively delivering BCN amide 10, which gave an identical rate constant of 0.20 M−1 s−1 in our SPAAC model, under the same conditions as previously described. To prove that amide bond formation was not limited solely to small molecules, we also decided to evaluate the conjugation of trastuzumab – a therapeutic antibody targeting overexpressed HER2 receptors in breast cancer cells – with para-nitrophenyl ester derivative 12 (Fig. 3). Following a ‘plug-and-play’ strategy,17,18 we next functionalized our trastuzumab-BCN conjugates by SPAAC with the azide-containing TAMRA fluorophore 13. Gratifyingly, excellent yield and average degree of conjugation were obtained for conjugates 14 (62% and 2.7, respectively; Fig. 2), demonstrating the ability of BCN acid 3 to deliver immunoconjugates effectively.
Being now able to derivatize the BCN ring with amide groups rather than carbamates – as it would be with activated BCN alcohol 6 – we next reflected on the benefits it could offer in terms of stability. Indeed, it is well known that carbamates are less stable than amides, as epitomized by a resonance energy 3–4 kcal mol−1 lower due to the stereoelectronic perturbations caused by the extra oxygen,19 a property that has been used to design prodrugs of various active substances.20 Interestingly, besides cyclic and unsubstituted (i.e. R–O(CO)NH2) compounds, all other carbamate classes were found to be unstable in biological media, leading to substantial hydrolysis both in vitro and in vivo.21 While this property could be desirable in certain circumstances, it is hardly the case when BCN 1 is incorporated into probes or payloads via a carbamate group, as a certain stability is expected from this type of linkage.
To compare the stability of BCN amide and carbamate, we thus designed two FRET probes only differing by this functional group, incorporating TAMRA as a fluorophore and BHQ-2 as a quencher. Upon incubation in various media, monitoring the appearance of TAMRA fluorescence at 580 nm thus serves as an indirect evaluation of cleavage between the fluorophore and its quencher, a strategy that we employed in the past for the development of various cleavable linkers.22–25 Amide and carbamate probes 15 and 16, respectively, were thus designed and easily accessed in good yields (see ESI†). Their stability was then evaluated under various conditions and compared to that of simple ether probe 17. While all probes were found to be perfectly stable at different pH and in human plasma (see ESI†), marked differences were observed in vitro. Indeed, upon incubation with three different human cancer cell lines – i.e. breast adenocarcinoma SKBR3 and MDA-MB-231, and uterus adenocarcinoma HeLa – carbamate probe 16 was systematically found to give higher mean fluorescence intensity values than its amide counterpart 15 (Fig. 4). This was attributed to a differential cleavage in cellulo and confirmed by the probes' stability in cell culture medium (see ESI†). Such an apparent cellular lability was unexpected and in stark contrasts with the results observed with amide probe 15, which was found to be as stable as the ether probe 17, indicating a lack of influence of the BCN-triazole group in the observed cleavage of 16. This implies that for biological applications of BCN probes necessitating prolonged incubation, the use of carbamate linkers should be avoided to the benefit of the more robust amide group, especially whenever stability is a crucial parameter.
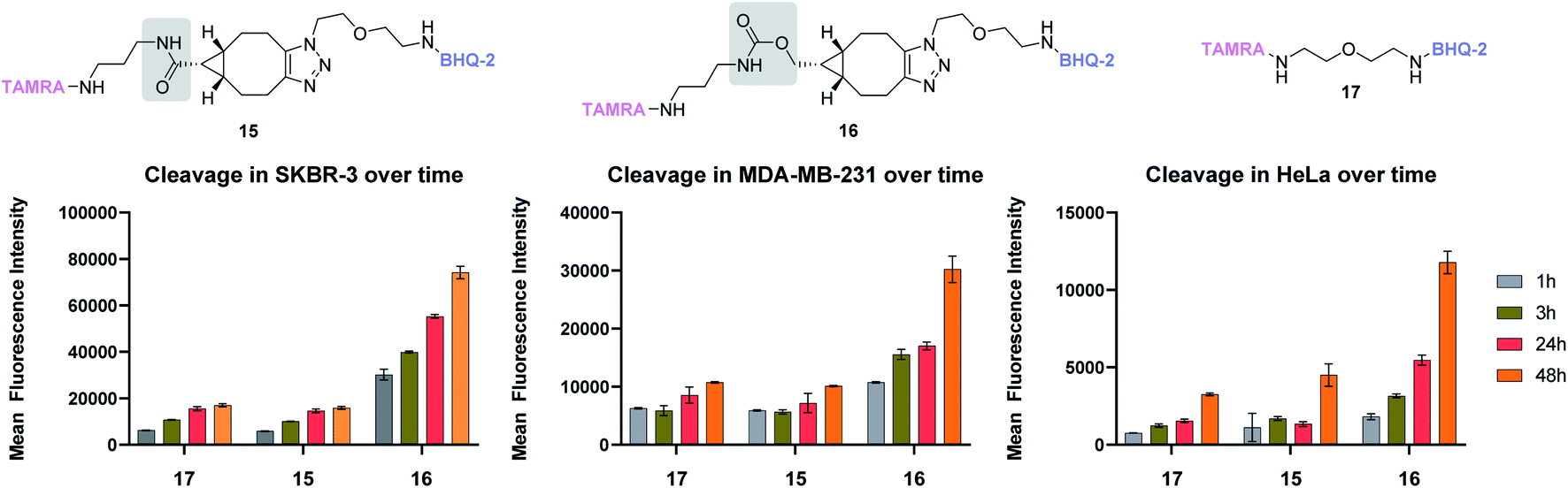 | ||
| Fig. 4 Stability studies of amide and carbamate-based BCN probes 15 and 16, respectively, and ether probe 17 in different cancer cell lines over time. Each measurement was done in triplicate. | ||
Conclusions
In summary, we reported the first synthesis of a BCN equipped with a carboxylic acid. We showed that it shared a similar reactivity with its primary alcohol analogue, the most studied member of the BCN family. The presence of this carboxylate allows a convenient derivatisation via amide bond formation. Most notably, we proved that the resulting compounds were far more stable in cultured cells than classically employed carbamate derivatives. Thus, we believe that BCN acid 3 is an effective surrogate for BCN alcohol 1, and is a more judicious alternative when designing molecular probes for which hydrolytic stability in biological media is key.Author contributions
TR took care of conceptualization, investigation, methodology, validation, visualization and writing of the original draft. MM took care of methodology (upscale synthesis) and validation. MN took care of investigation and supervision (cell culture and stability studies). SE took care of formal analysis (mass spectrometry), data curation and visualization. ID took care of methodology (probe synthesis). SC took care of supervision and validation. AW took care of funding acquisition, resources, supervision and reviewing & editing of the final manuscript. GC took care of conceptualization, data curation, methodology, project administration, resources, supervision, visualization, writing of the original draft and reviewing & editing of the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the CNRS, the University of Strasbourg, the “Agence Nationale de la Recherche”, Sanofi, and the French Proteomic Infrastructure (ProFI; ANR-10-INBS-08-03).Notes and references
- J. C. Jewett, E. M. Sletten and C. R. Bertozzi, J. Am. Chem. Soc., 2010, 132, 3688–3690 CrossRef CAS PubMed.
- J. Dommerholt, S. Schmidt, R. Temming, L. J. A. Hendriks, F. P. J. T. Rutjes, J. C. M. vanHest, D. J. Lefeber, P. Friedl and F. L. van Delft, Angew. Chem., Int. Ed., 2010, 49, 9422–9425 CrossRef CAS PubMed.
- T. Vojkovsky, B. Sullivan and K. N. Sill, Polymer, 2016, 105, 72–78 CrossRef CAS.
- S. Ameta, J. Becker and A. Jäschke, Org. Biomol. Chem., 2014, 12, 4701–4707 RSC.
- A. M. Jawalekar, S. Malik, J. M. M. Verkade, B. Gibson, N. S. Barta, J. C. Hodges, A. Rowan and F. L. Van Delft, Molecules, 2013, 18, 7346–7363 CrossRef CAS PubMed.
- E. M. Schneider, M. Zeltner, V. Zlateski, R. N. Grass and W. J. Stark, Chem. Commun., 2016, 52, 938–941 RSC.
- N. Stuhr-Hansen, C.-D. Vagianou and O. Blixt, Chem. –Eur. J., 2017, 23, 9472–9476 CrossRef CAS PubMed.
- H. Fujita, J. Dou, N. Matsumoto, Z. Wu and J. S. Lindsey, New J. Chem., 2020, 44, 719–743 RSC.
- M. Glassner, S. Maji, V. R. de la Rosa, N. Vanparijs, K. Ryskulova, B. G. D. Geest and R. Hoogenboom, Polym. Chem., 2015, 6, 8354–8359 RSC.
- X. Li, Z. Liu and S. Dong, RSC Adv., 2017, 7, 44470–44473 RSC.
- E. H. P. Leunissen, M. H. L. Meuleners, J. M. M. Verkade, J. Dommerholt, J. G. J. Hoenderop and F. L. van Delft, ChemBioChem, 2014, 15, 1446–1451 CrossRef CAS PubMed.
- J. G. K. O'Brien, S. R. Chintala and J. M. Fox, J. Org. Chem., 2018, 83, 7500–7503 CrossRef PubMed.
- M. F. Debets, S. S. van Berkel, J. Dommerholt, A. J. Dirks, F. P. J. T. Rutjes and F. L. van Delft, Acc. Chem. Res., 2011, 44, 805–815 CrossRef CAS PubMed.
- J. Dommerholt, F. P. J. T. Rutjes and F. L. van Delft, Top. Curr. Chem., 2016, 374, 16 CrossRef PubMed.
- J. Dommerholt, S. Schmidt, R. Temming, L. J. A. Hendriks, F. P. J. T. Rutjes, J. C. M. vanHest, D. J. Lefeber, P. Friedl and F. L. van Delft, Angew. Chem., Int. Ed., 2010, 49, 9422–9425 CrossRef CAS PubMed.
- B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Chem. Soc. Rev., 2017, 46, 4895–4950 RSC.
- I. Dovgan, S. Ursuegui, S. Erb, C. Michel, S. Kolodych, S. Cianférani and A. Wagner, Bioconjugate Chem., 2017, 28, 1452–1457 CrossRef CAS PubMed.
- A. Maruani, M. E. B. Smith, E. Miranda, K. A. Chester, V. Chudasama and S. Caddick, Nat. Commun., 2015, 6, 6645 CrossRef CAS PubMed.
- D. Kaur, P. Sharma and P. V. Bharatam, J. Mol. Struct.: THEOCHEM, 2005, 757, 149–153 CrossRef CAS.
- A. K. Ghosh and M. Brindisi, J. Med. Chem., 2015, 58, 2895–2940 CrossRef CAS PubMed.
- F. Vacondio, C. Silva, M. Mor and B. Testa, Drug Metab. Rev., 2010, 42, 551–589 CrossRef CAS PubMed.
- G. Leriche, M. Nothisen, N. Baumlin, C. D. Muller, D. Bagnard, J.-S. Remy, S. A. Jacques and A. Wagner, Bioconjugate Chem., 2015, 26, 1461–1465 CrossRef CAS PubMed.
- E. Tobaldi, I. Dovgan, M. Mosser, J.-M. Becht and A. Wagner, Org. Biomol. Chem., 2017, 15, 9305–9310 RSC.
- I. Dovgan, S. Kolodych, O. Koniev and A. Wagner, Sci. Rep., 2016, 6, 30835 CrossRef CAS PubMed.
- S. A. Jacques, G. Leriche, M. Mosser, M. Nothisen, C. D. Muller, J.-S. Remy and A. Wagner, Org. Biomol. Chem., 2016, 14, 4794–4803 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectra. See DOI: 10.1039/d1ra07905k |
| This journal is © The Royal Society of Chemistry 2021 |