
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effects of kaolinite layer expansion and impurities on the solid-state reaction of kaolinite†
Shingo Machida *,
Ken-ichi Katsumata and
Atsuo Yasumori
*,
Ken-ichi Katsumata and
Atsuo Yasumori
Department of Material Science and Technology, Faculty of Advanced Engineering, Tokyo University of Science, 6-3-1 Niijuku, Katsushika-ku, Tokyo 125-8585, Japan. E-mail: shingo.machida@rs.tus.ac.jp
First published on 30th November 2021
Abstract
Expanded kaolinite without impurities was found to serve as a suitable raw material for the rapid formation of metastable CaAl2Si2O8 with the suppression of byproduct formation. This was accomplished based on the solid-state reaction of the kaolinite with calcium carbonate promoted by mechanical grinding.
Introduction
Various inorganic solids can be formed via solid-state reactions induced by grinding two or more raw materials together to activate and increase their surface area.1–3 These grinding effects are especially advantageous in the case of layered inorganic solids4 since these materials comprise stacked layers whose stacking order and structure can be disrupted by grinding.5–9 The degree of disruption can be increased as these inorganic layers expand upon intercalation, a process in which guest species are inserted between stacked layers to form intercalation compounds comprising alternately stacked inorganic and guest species layers.4 Therefore, intercalation compounds may have applications in solid-state reactions in which the absence of impurities is desirable.Herein, we report the effects of the expansion of kaolinite layers and of the presence of impurities on the solid-state reaction of this material. Kaolinite, a layered aluminosilicate having the formula Al2Si2O5(OH)4, is well-known as a raw material for the synthesis of various inorganic solids.10–13 Each neutral and asymmetrical layer within kaolinite comprises an AlO2(OH)4 sheet and a SiO4 sheet. These sheets form hydrogen bonds at kaolinite interlayers where neither ions nor molecules are originally present. Additionally, kaolinite undergoes intercalation of salts and neutral molecules bearing polar groups. These groups break/weaken hydrogen bonds between AlO2(OH)4 and SiO4 sheets at pristine kaolinite interlayers, to expand kaolinite layers and to form intercalation compounds.14–32 These compounds easily undergo the disruption of the stacking order by grinding compared to pristine kaolinite.7,9 Among compounds prepared using kaolinite intercalation chemistry, methoxy-modified kaolinite (MeO-Kaol) is organically-modified kaolinite with the formula Al2Si2O5(OH)4−x(OCH3)x (in which x never exceeds 1)16,17 and can accommodate water molecules under ambient conditions in a stable manner. In this scenario, the basal spacing of the material is expanded from 0.72 nm of pristine kaolinite to 0.86 nm.16,17 MeO-Kaol is also able to intercalate methanol molecules when immersed in this solvent, to increase the basal spacing to 1.12 nm.17 Interestingly, white and brown regions of the kaolinite are distinguishable following the methoxylation process,17 with the latter color likely due to the presence of impurities. The study reported herein assessed the solid-state reaction of MeO-Kaol with calcium carbonate (CaCO3), a low-cost material, to form a metastable CaAl2Si2O8 phase. Metastable CaAl2Si2O8 containing anorthite, the stable phase of CaAl2Si2O8, can be produced by the calcination of a mixture of ground CaCO3 and Georgia kaolinite (GK), a brown kaolinite, for 12 h.10 Metastable CaAl2Si2O8, a series of layered inorganic solids having the formula RAl2Si2O8 (where R is an alkali metal cation)33, exhibits phosphorescence when doped with rare earth ions.34 Additionally, the CaO–Al2O3–SiO2 glass–ceramic precipitated metastable CaAl2Si2O8 crystals shows improved mechanical properties and unique fracture behavior35 that is currently under investigaiton. Amorphous metastable CaAl2Si2O8 has also been shown to efficiently remove heavy metal ions from solutions in conjunction with a minimal increase in pH.11 Based on these applications, it would be helpful to develop a means of efficiently synthesizing this material. The present research initially examined the effects of kaolinite layer expansion using GK, a well-crystallized kaolinite that displays significant intercalation capabilities.19 The effects of impurities were subsequently assessed in trials with Kanpaku kaolinite (KP), a white kaolinite, during which this material was treated with acid and impurities were removed during methoxylation.17 These different kaolinites were ground with CaCO3 then subjected to calcination to form metastable CaAl2Si2O8. The calcination mechanism was investigated by trials using halloysite, a tubular aluminosilicate with the same formula as kaolinite, as a raw material. It should be noted that, in the halloysite structure, the AlO2(OH)4 and SiO4 surfaces appear at interiors and exteriors of the tubes respectively.36
Experimental
In these trials, 5 g portions of GK (KGa-1b, obtained from the Source Clays Repository of the Clay Material Society of the U.S.A.) or KP (JSCC-1101c, obtained from the Clay Science Society of Japan) were soaked in 50 mL of 1 M hydrochloric acid for three days to produce the specimens referred to as A-KP and A-GK, respectively. The methoxy modifications of GK, KP and A-KP were conducted according to a previously reported method17,18 to give the products designed herein as Me-GK, Me-KP and Me-A-KP. During these preparations, the upper and lower regions of centrifuged products, which exhibited different colorations, were separated in a manner similar to that in a previous study.18 These upper and lower regions are denoted herein as U-X and L-X, where X represents the type of kaolinite and the products. Following these procedures, KP and Me-KP were dispersed in methanol and allowed to stand for a day. A sample of methanol-dispersed KP also underwent immediate centrifugation.A quantity of the kaolinites or halloysite (obtained from Sigma-Aldrich Co. Ltd.) was mixed with CaCO3 (obtained from Hayashi Pure Chemical Int., Ltd.) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio using an agate mortar and pestle to generate the specimens referred to herein as the ungrounded raw materials. A 720 mg quantity of each mixture were subsequently dispersed in 8 mL of methanol and wet milled in a planetary ball mill at 250 rpm for 12 h using a resin vessel (12.5 mL) and 120 silicon carbide balls (3 mm in diameter). After milling, the solids were centrifuged at 4000 rpm for 1 min and then dried at 80 °C for 1 h to provide the specimens referred to herein as the ground raw materials. After drying, portions of these products were calcined at 900 °C for 1.5, 3, or 4.5 h to prepare the products denoted as Y + C-1.5, 3, or 4.5 h, where Y and C represent raw materials and CaCO3, respectively.
:1 molar ratio using an agate mortar and pestle to generate the specimens referred to herein as the ungrounded raw materials. A 720 mg quantity of each mixture were subsequently dispersed in 8 mL of methanol and wet milled in a planetary ball mill at 250 rpm for 12 h using a resin vessel (12.5 mL) and 120 silicon carbide balls (3 mm in diameter). After milling, the solids were centrifuged at 4000 rpm for 1 min and then dried at 80 °C for 1 h to provide the specimens referred to herein as the ground raw materials. After drying, portions of these products were calcined at 900 °C for 1.5, 3, or 4.5 h to prepare the products denoted as Y + C-1.5, 3, or 4.5 h, where Y and C represent raw materials and CaCO3, respectively.
X-ray diffraction (XRD) patterns (XRD-6100, Shimadzu) were acquired from kaolinite and the products to assess the stacking order and crystalline phases. Fourier-transform infrared (IR) spectra (FT/IR-4100, JASCO) were obtained by KBr method to investigate the degree of structural breakdown of the products. Field-emission scanning electron microscopy (FE-SEM; Supra40 microscope, Zeiss) was used to characterize particle sizes and morphologies.
Results and discussion
The brown color of GK changed only minimally following the acid treatment to produce A-GK, while the original KP was white (Fig. 1a); thus, the methoxylation of A-GK was not conducted (see Experimental). Brown and relatively white regions were observed in the products after methoxy modification of GK and KP (Fig. 1b and c). Both U-Me-GK and L-Me-GK were brown, whereas U-Me-KP was white (Fig. 1d). Additionally, the proportion of the brown region decreased after methoxy modification of A-KP (Fig. 1e). A brown region did not appear when methanol-dispersed KP and Me-KP were allowed to stand for a day, and after which the former was immediately centrifuged (Fig. 1f–h). U-Me-A-KP + C-4.5 h was white, while Me-GK + C-4.5 h and KP + C-4.5 h appeared light yellow and pink, respectively (Fig. 1i). | ||
| Fig. 1 Photographs of (a) GK (left), A-GK (middle) and KP (right), (b) Me-GK after centrifugation during preparation process, (c) Me-KP after centrifugation, (d) U-Me-GK (left), L-Me-GK (middle) and U-Me-KP (right), (e) Me-A-KP after centrifugation, (f) methanol-dispersed Me-KP after standing for one day, (g) methanol-dispersed KP after standing, (h) centrifuged KP after being dispersed in methanol, and (i) U-Me-A-KP + C-4.5 h (left), Me-GK + C-4.5 h (middle) and KP + C-4.5 h (right). | ||
It is well known that kaolinite can undergo incomplete intercalation,9 which is demonstrated by the continued presence of an XRD peak corresponding to a d value of 0.72 nm (due to the original kaolinite layers) following intercalation. The ratio of the intensity of the diffraction line due to expanded kaolinite layers to that of the line due to the original kaolinite layers can therefore be used as an indicator of the extent of intercalation.20–23 In general, GK generally exhibits greater intercalation capability than KP.19
Fig. 2 presents XRD patterns for ground and unground GK mixed with CaCO3. After grinding, the ratio of the intensity of the diffraction line (d = 0.86 nm) due to the basal spacing of Me-GK to that of the (020) diffraction line due to the lateral atomic arrangement of the kaolinite layers decreased relative to the ratio of the intensity of the diffraction line (d = 0.72 nm) due to the basal spacing of pristine kaolinite to this same (020) line.24 In order to easily understand the degree of stacking order in each raw material, we tentatively show the relative intensities of reflection due to (001), basal spacings, with respect to those due to (020) (I(001)/I(020)); 4.8 for GK mixed with CaCO3 before grinding, 3.8 for that after grinding, 11 for Me-GK mixed with CaCO3, and 4.2 for that after grinding. Thus, the stacking order in MeO-GK was greatly perturbed compared with that in pristine kaolinite. The relative intensities of the reflections ascribed to CaCO3 also decrease after grinding.
 | ||
| Fig. 2 XRD patterns for GK mixed with CaCO3 (a) before and (b) after grinding, and for Me-GK mixed with CaCO3 (c) before and (d) after grinding. Reflections due to CaCO3 are indicated by filled circles in (a). | ||
Fig. S1† shows IR spectra in the OH stretching region of GK, Me-GK, and GK-based ground raw materials. The kaolinite spectrum (Fig. S1a†) generally exhibits three OH stretching bands at 3696, 3670, and 3653 cm−1 due to inter-layer hydroxyl groups and one OH stretching band due to inner-layer hydroxyl group. The former three bands are perturbed by intercalation of guest species, while the latter cannot undergo such perturbation. When the intercalation reaction proceeds, the intensities of the three former bands relative to the later band decrease compared to those obtained from the kaolinite spectrum.14,37,38 Additional OH stretching bands/shoulders due to interlayer hydroxyl groups hydrogen-bonded to the guest species can appear at lower wavenumbers.17,37,38 In this study, the MeO-Kaol spectrum (Fig. S1b†) fits well with that reported previously.17 Meanwhile, four OH stretching bands are broaden when kaolinite undergoes mechanical grinding due to the structural breakdown.7 In the present study, the profiles of ground raw materials are slightly broadened compared to GK and Me-GK (Fig. S1c and d†). The degrees are lesser extent than those reported in previous studies.7,39 Compared to GK, Me-GK exhibits the more spectrum broadening after grinding (Fig. S1c and d†).
Fig. 3 shows XRD patterns obtained from the original KP, the KP-based products, and the raw materials after grinding with CaCO3. There are no significant differences in the patterns generated by KP and A-KP (Fig. 3a and b), although the latter includes a weak reflection resulting from quartz25 (Fig. 3b). This quartz reflection also appears in the XRD pattern for L-Me-A-AP (Fig. 3d) but not in that for U-Me-A-AP (Fig. 3c). The reflections due to impurities40 acquired from U-Me-KP and L-Me-KP (Fig. S2†) were less intense in the pattern obtained from U-Me-A-KP (Fig. 3c). After grinding, the profile changes for KP or U-Me-A-KP mixed with CaCO3 by grinding (Fig. 3e and f) are similar to those for GK or Me-GK mixed with CaCO3 by grinding as shown in Fig. 2c and d.
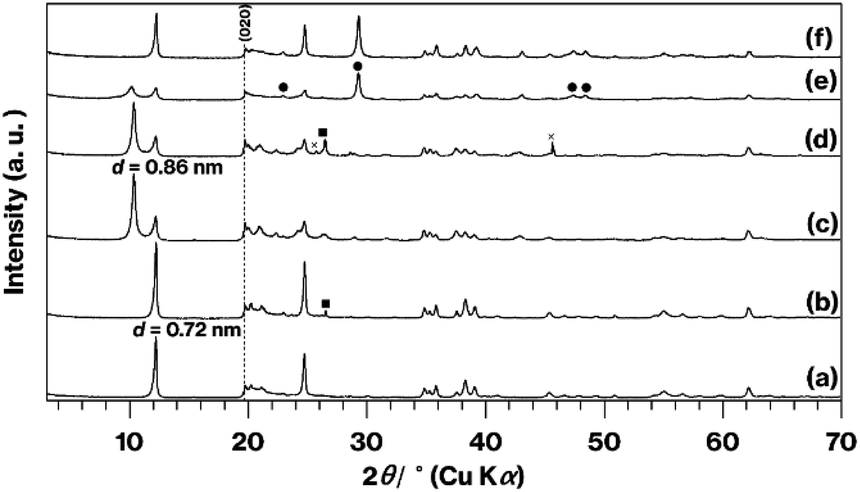 | ||
| Fig. 3 XRD patterns for (a) KP, (b) A-KP, (c) U-Me-A-KP, (d) L-Me-A-KP, (e) ground U-Me-A-KP mixed with CaCO3, and (f) ground KP mixed with CaCO3. Reflections due to CaCO3 are indicated by filled circles and squares, while the cross mark represents an unknown phase. | ||
Fig. 4 provides XRD patterns for the calcined products. Compared with GK + C-3 h, which essentially generated a halo XRD pattern (Fig. 4a), Me-GK + C-3 h (Fig. 4b) provided a pattern that agrees with that reported for metastable CaAl2Si2O8.10 Additionally, there are some weak diffraction lines due to anorthite10 in this pattern (Fig. 4b). Compared with the Me-GK + C-3 h pattern (Fig. 4b), the intensities of the reflections resulting from metastable CaAl2Si2O8 and anorthite are greater in the Me-GK + C-4.5 h pattern (Fig. 4c). In addition, relative to the Me-GK + C-4.5 h pattern (Fig. 4c), the intensities of the reflections due to metastable CaAl2Si2O8 are slightly increased while those of the anorthite reflections are decreased for U-Me-A-KP + C-4.5 h (Fig. 4d).
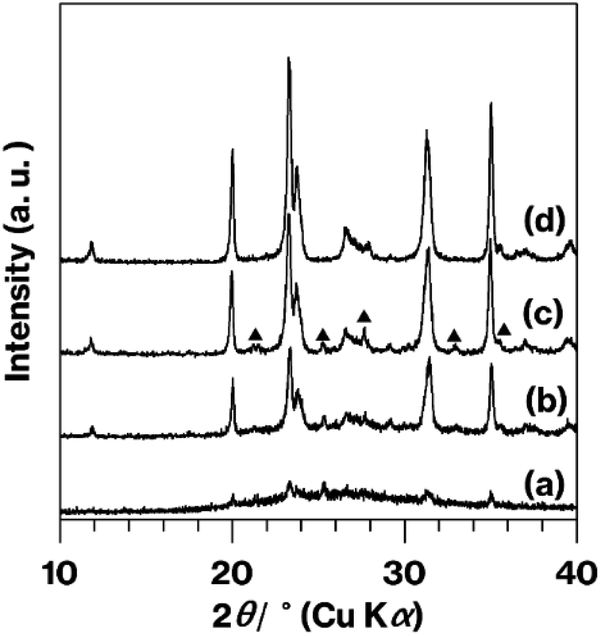 | ||
| Fig. 4 XRD patterns for (a) GK + C-3 h, (b) Me-GK + C-3 h, (c) Me-GK + C-4.5 h, and (d) U-Me-A-KP + C-4.5 h. Reflections due to anorthite are indicated by filled triangles in (c). | ||
The patterns obtained from Me-GK + C-3 h, Me-GK + C-4.5 h and U-Me-A-KP + C-4.5 h (Fig. 4b–d) are similar to those reported previously, which indicates the formation of metastable CaAl2Si2O8 and a small amount of anorthite after a 12 h calcination at 900 °C following the planetary milling of a mixture of GK and CaCO3 for 12 h.10 The U-Me-A-KP pattern in particular displays weaker anorthite reflections. Note that the ground GK and CaCO3 sample that was calcined at 800 °C for 12 h generated a halo XRD pattern as a consequence of the decomposition of the CaCO3 and dehydroxylation of the kaolinite to produce metakaolinite, an amorphous layered aluminosilicate.26,41
Prior study determined that the grinding induced the breakdown of both Al–O–Si and Si–O–Si bonds to form new hydroxyl groups with adsorbed water, based on the mass loss due to H2O release estimated using thermogravimetry (TG) analysis.10 It is also known that a portion of the hydroxyl groups in MeO-Kaol is replaced with methoxy groups.16,17 The TG curve obtained from the MeO-Kaol shows overlapping mass losses due to kaolinite dehydroxylation and degradation of methoxy groups.16,17,25 CHN analysis is therefore required to determine the carbon content due to methyl groups.16,17,25 However, the amount of methoxy groups in the ground raw materials cannot be detected since they contain CaCO3 which releases carbons due to the decarbonation by heating. Additionally, the amounts of protons in methoxy groups cannot be also detected since those in water due to the dehydroxylation of kaolinite are overlapped. Therefore, the present study cannot clarify the mass loss related to the removal of H2O from the material. Although kaolinite hydrates can be prepared without methoxylation27 and could be used as a raw material for wet grinding in water, kaolinite hydrates are often unstable under ambient conditions as a result of the release of interlayer water, which can destroy the stacking order.28 That is, deintercalation of water molecules can lead to the appearance of disordered stacking. The release of guest species can shift the dehydroxylation of kaolinite to lower temperatures,9 and so it is difficult to assess the true mass loss associated with the release of H2O generating from the breaking of kaolinite bonds such as Al–O–Si bonds. Concerning the breakdown of kaolinite structure by grinding, the effect is lesser extent compared to those reported in previous studies7,39 based on IR spectra (Fig. S1†). This permits us to focus the difference in the staking order disrupted by grinding between expanded and pristine kaolinites.
The solid-state reaction of kaolinite involves metakaolinite.3 Although metakaolinite generally does not produce XRD diffraction lines because it has an amorphous layered structure, we recently succeeded in the formation of orderly stacked metakaolinite and revealed the destruction of the ordered structure upon manual grinding.26 Thus, before grinding and calcination of such raw materials, it would be beneficial to assess the effects of pre-heating kaolinite to form metakaolinite with various stacking orders. Additionally, the reaction of CaCO3 with metakaolinite is worth further investigation. Our group therefore intends to continue our study of such materials. In this scenario, the use of organic solvents as media for wet grinding could be helpful to suppress the rehydration of metakaolinite.42
Based on the appearance of the various products (Fig. 1) and the XRD patterns (Fig. 3), the presence of impurities in the kaolinite was eliminated by combining the acid treatment, methoxy modification and centrifugation when KP was used. The separation of the white and brown regions by centrifugation was not possible without methoxylation and natural sedimentation, although the details of the associated mechanisms are not yet clear. The XRD patterns for the calcined products (Fig. 4) showed a lack of impurities when U-Me-A-KP was employed as raw materials for the rapid formation of metastable CaAl2Si2O8, with suppression of the anorthite phase and a product that was truly white (Fig. 1i). In general, the brown color of aluminosilicate clay minerals is due to impurities and small amounts of Fe substituted for Al or Mg. This substituted Fe likely remains in the calcined products, based on the results for Me-GK and KP (Fig. 1i). In the case of the GK, however, the more stronger acid treatment may decompose the kaolinite structure. The data also indicate that quartz in the KP could be removed by the present procedures (Fig. 3 and S2†). Additionally, based on the reduced structural and morphological decompositions (Fig. S3 and S4†) of halloysite, in which the tubular exteriors have SiO4 surfaces, the reaction of CaCO3 with a silica-like surface appears to promote the formation of an anorthite phase. The impurities in synthetic kaolinite comprise small amounts of boehmite and gibbsite,43,44 although the intercalation capability of this material has been examined solely using dimethylsulfoxide21 as an effective molecule for direct intercalation between kaolinite layers.15,16,20–22 Thus, when expanded synthetic kaolinites are used as raw materials in solid-state reactions, the intercalation capacity of each material should be investigated in detail using various polar molecules, as conducted in previous studies using GK and KP.15–21,23–28 Additionally, a centrifugation to separate impurities as conducted in the present study should be examined. Therefore, the use of other kaolinites could require further basic studies.
The present XRD patterns (Fig. 2 and 4) and previous results10 (see two paragraphs before) reveal the rapid formation of metastable CaAl2Si2O8 when using expanded kaolinite as a raw material. It is well known that methanol molecules cannot intercalated between kaolinite layers.27 Thus, the expansion of the kaolinite layers upon methanol accommodation facilitated the destruction of the stacking order of the kaolinite layers when MeO-Kaol underwent wet grinding in methanol. The degree of stacking disorder observed in this study was also greater than that reported in a previous study that assessed the wet grinding of GK and CaCO3 in water using a planetary ball mill.10
When the calcined product was prepared using halloysite as the raw material, the XRD pattern for halloysite + C-4.5 h (Fig. S3a†) included reflections due to metastable CaAl2Si2O8, along with anorthite reflections that were more intense compared with the XRD patterns shown in Fig. 4. There were no significant differences in the patterns (Fig. S3b and c†) and the tube morphology (Fig. S4†) of the halloysite before and after grinding.
Since kaolinite occurs naturally, it will unavoidably have variable levels of crystallinity. Effect of crystallinity on the present results as well as kaolinite intercalation capability are thus one of interests, while particle size distributions and impurities are also different for each kaolinite produced in nature. Note that the kaolinite intercalation capability depends on the lateral sizes of kaolinite platy particles.14 Additionally, to the best of our knowledge, even in the case of synthetic kaolinite, the size and distributions are not well controlled. Meanwhile, methoxy-modified kaolinite has been used as a versatile intermediate to intercalate various organic molecules and polymers15–18,24–26 and expansion of the kaolinite layers up to a spacing of approximately 4 nm has been reported.24 This level of expansion could also be sufficient to destroy the stacking order of the kaolinite layers during grinding. Methoxy-modified kaolinite can also be prepared by intercalation of kaolinite with urea,29 which is as inexpensive as methanol. Therefore, the present results demonstrate the potential industrial uses of kaolinite.
Conclusions
In summary, we have demonstrated the effects of kaolinite layer expansion and of the presence of impurities on the rapid formation of metastable CaAl2Si2O8 with suppressed byproduct formation via a solid-state reaction of kaolinite, especially MeO-Kaol, with CaCO3. The present method can be applied to kaolinite, other layered inorganic solids and/or their intercalation compounds ground with various raw materials such as carbonates.45–48Author contributions
Shingo Machida: conceptualization, data curation, investigation, project administration, writing-original draft, supervision. Ken-ichi Katsumata: project administration. Atsuo Yasumori: project administration.Conflicts of interest
There are no conflicts to declare.Notes and references
- G. Cohn, Chem. Rev., 1948, 42, 527 CrossRef CAS PubMed.
- D. Tan and F. Garcia, Chem. Soc. Rev., 2019, 48, 2274 RSC.
- D. N. Rainer and R. E. Morris, Dalton Trans., 2021, 50, 8995 RSC.
- M. Ogawa, K. Saito and M. Sohmiya, Dalton Trans., 2014, 43, 10340 RSC.
- H. Takahashi, Bull. Chem. Soc. Jpn., 1959, 32, 235 CrossRef CAS.
- H. Takahashi, Bull. Chem. Soc. Jpn., 1959, 32, 252 CrossRef CAS.
- R. L. Frost, J. Kristof, E. Mako and W. Martens, Langmuir, 2002, 18, 6491 CrossRef CAS.
- K. Okada, N. Watanabe, K. V. Jha, Y. Kameshima, A. Yasumori and K. J. D. MacKenzie, Appl. Clay Sci., 2003, 23, 329 CrossRef CAS.
- A. Weiss, Angew. Chem., Int. Ed., 1963, 12, 697 CrossRef.
- K. Okada, N. Watanabe, K. V. Jha, Y. Kameshima, A. Yasumori and K. J. D. MacKenzie, J. Mater. Chem., 2003, 550 RSC.
- B. Zsirka, A. Táborosi, P. Szabó, R. K. Szilagyu, E. Horváth, T. Juzsakova, D. Fertig and J. Kristóf, Langmuir, 2017, 33, 3354 CrossRef PubMed.
- T. Abdullahi, Z. Harun and M. H. D. A. Othman, Adv. Powder Technol., 2017, 28, 1827 CrossRef CAS.
- Hartati, D. Prasetyoko, M. Santoso, I. Qoniah, W. L. Leaw, P. B. D. Firda and H. Nur, J. Chin. Chem. Soc., 2020, 67, 911 CrossRef CAS.
- G. Lagaly, M. Ogawa and I. Dékány, Developments in Clay Science Volume 5A Handbook of Clay Science, ed. F. Bergaya and G. Lagaly, Elsevier, Oxford UK, 2nd edn, 2013, pp. 435–445 Search PubMed.
- C. Detellier, Chem. Rec., 2018, 18, 868 CrossRef CAS PubMed.
- J. J. Tunney and C. Detellier, J. Mater. Chem., 1996, 6, 1679 RSC.
- Y. Komori, H. Endo, R. Takenawa, S. Hayashi, Y. Sugahara and K. Kuorda, Langmuir, 2000, 16, 5506 CrossRef CAS.
- S. Machida, M. Sohmiya, Y. Ide and Y. Sugahara, Langmuir, 2018, 34, 12694 CrossRef CAS PubMed.
- C. Morimoto, K. Tomita and M. Kawano, Rep. Fac. Sci., Kagoshima Univ., Earth Sci. Biol., 1996, 29, 21 CAS.
- S. Olejnik, L. A. G. Aylmore, A. M. Posner and J. P. Quirk, J. Phys. Chem., 1968, 72, 241 CrossRef CAS.
- S. Olejnik, A. M. Posner and J. P. Quirk, Clay Miner., 1970, 8, 421 CrossRef CAS.
- S. Satokawa, R. Miyawaki, S. Tomura and Y. Shibasaki, Clay Sci., 1997, 10, 231 CAS.
- S. Machida, G. Régis and Y. Sugahara, Langmuir, 2019, 35, 13553 CrossRef CAS PubMed.
- Y. Kuroda, K. Ito, K. Itabashi and K. Kuroda, Langmuir, 2011, 27, 2028 CrossRef CAS PubMed.
- S. Machida, N. Idota and Y. Sugahara, Dalton Trans., 2019, 48, 11663 RSC.
- S. Machida, K. Katsumata and A. Yasumori, RSC Adv., 2021, 11, 23090 RSC.
- P. M. Costanzo and R. F. Giese Jr, Clays Clay Miner., 1990, 38, 160 CrossRef CAS.
- P. M. Costanzo, R. F. Giese Jr and M. Lipisicas, Clays Clay Miner., 1984, 32, 419–428 CrossRef CAS.
- H. Cheng, X. Hou, X. Li and R. L. Frost, Appl. Clay Sci., 2015, 109–110, 55 CrossRef CAS.
- R. L. Frost, J. T. Kloprogge, T. H. T. Tran and J. Kristof, Am. Mineral., 1998, 83, 1182 CrossRef CAS.
- H. Hwang, D. Seoung, Y. Lee, Z. Liu, H.-P. Liermann, H. Cynn, T. Vogt, C.-C. Kao and H.-K. Mao, Nat. Geosci., 2017, 10, 947 CrossRef CAS.
- A. Basu and M. Mookherjee, ACS Earth Space Chem., 2021, 5, 834 CrossRef CAS.
- J. Töpel-Schadt, W. F. Müller and H. Pentinghaus, J. Mater. Sci., 1978, 13, 1809 CrossRef.
- K. Shinha, B. Pearson, S. R. Casolco, J. E. Garay and O. A. Graeve, J. Am. Ceram. Soc., 2009, 92, 2504 CrossRef.
- K. Maeda, K. Akatsuka, G. Okuma and A. Yasumori, Crystals, 2021, 206(11), 393 CrossRef.
- W. O. Yah, A. Takahara and Y. M. Lvov, J. Am. Chem. Soc., 2012, 134, 1853 CrossRef CAS PubMed.
- J. Kristóf, R. L. Frost, A. Felinger and J. Mink, J. Mol. Struct., 1997, 410–411, 119 Search PubMed.
- E. Horváth, J. Kristóf and R. L. Frost, Appl. Spectrosc. Rev., 2010, 45, 130 CrossRef.
- R. L. Frost, Clay Miner., 1997, 32, 65 CrossRef CAS.
- A. Guatame-García, M. Buxton, F. Deon, C. Lievens and C. Hecker, Miner. Process. Extr. Metall. Rev., 2018, 39, 420 CrossRef.
- A. K. Chakraborty, Phase Transformation of Kaolinite Clay, ed. A. K. Chakraborty, Springer, India, 2014, pp. 195–202 Search PubMed.
- J. Rocha, J. Adams and J. Klinowski, J. Solid State Chem., 1990, 89, 260 CrossRef CAS.
- S. Satokawa, R. Miyawaki, Y. Osaki, S. Tomura and Y. Shibasaki, Clays Clay Miner., 1996, 44, 417 CrossRef CAS.
- S. Satokawa, Y. Osaki, S. Samejima, R. Miyawaki, S. Tomura, Y. Shibasaki and Y. Sugahara, Clays Clay Miner., 1994, 42, 288 CrossRef CAS.
- M. Mahloujifar and M. Mansournia, Mater. Res. Express, 2019, 6, 025040 CrossRef.
- K. Okada, H. Arai, Y. Kameshima, A. Yasumori and K. J. D. Mackenzie, Mater. Lett., 2003, 57, 3354 Search PubMed.
- K. Okada, M. Nemoto, Y. Kameshima and A. Yasumori, J. Ceram. Soc. Jpn., 2000, 108, 898 CrossRef CAS.
- K. Sumi, Y. Kobayashi and E. Kato, J. Ceram. Soc. Jpn., 1998, 106, 89 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra07762g |
| This journal is © The Royal Society of Chemistry 2021 |