
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRevisiting salicylidene-based anion receptors†
Sandeep Kumar Dey *a,
Sonam Kumaria,
Sonal Mandrekara,
Shashank N. Mhaldara,
Sarvesh S. Harmalkara and
Christoph Janiakb
*a,
Sonam Kumaria,
Sonal Mandrekara,
Shashank N. Mhaldara,
Sarvesh S. Harmalkara and
Christoph Janiakb
aSchool of Chemical Sciences, Goa University, Taleigao Plateau, Goa 403206, India. E-mail: sandeepdey@unigoa.ac.in; Tel: +91-7387633550
bInstitute of Inorganic and Structural Chemistry, Heinrich-Heine University, 40204, Düsseldorf, Germany. E-mail: janiak@uni-duesseldorf.de; Tel: +49-2118112286
First published on 17th November 2021
Abstract
Several salicylidene-based colorimetric and fluorimetric anion sensors are known in the literature. However, our 1H-NMR experimental results (in DMSO-d6) showed hydrolysis of imine (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–) bonds in salicylidene-based receptors (SL, CL1 and CL2) in the presence of quaternary ammonium salts (n-Bu4N+) of halides (Cl− and Br−) and oxo-anions (H2PO4−, HSO4− and CH3COO−). The mono-salicylidene compound CL1 showed the most extensive –NCH– bond hydrolysis in the presence of anions. In contrast, the di-salicylidene compound CL2 and the tris-salicylidene compound SL showed comparatively slow hydrolysis of –NCH– bonds in the presence of anions. Anion-induced imine bond cleavage in salicylidene compounds could easily be detected in 1H-NMR due to the appearance of the salicylaldehyde –CHO peak at 10.3 ppm which eventually became more intense over time, and the –NCH– peak at 8.9–9.0 ppm became considerably weaker. Furthermore, the formation of the salicylidene O–H⋯X− (X− = Cl−/Br−) hydrogen-bonded complex, peak broadening due to proton-exchange processes and keto–enol tautomerism have also been clearly observed in the 1H-NMR experiments. Control 1H-NMR experiments revealed that the presence of moisture in the organic solvents could result in gradual hydrolysis of the salicylidene compounds, and the rate of hydrolysis has further been enhanced significantly in the presence of an anion. Based on 1H-NMR results, we have proposed a general mechanism for the anion-induced hydrolysis of imine bonds in salicylidene-based receptors.
CH–) bonds in salicylidene-based receptors (SL, CL1 and CL2) in the presence of quaternary ammonium salts (n-Bu4N+) of halides (Cl− and Br−) and oxo-anions (H2PO4−, HSO4− and CH3COO−). The mono-salicylidene compound CL1 showed the most extensive –NCH– bond hydrolysis in the presence of anions. In contrast, the di-salicylidene compound CL2 and the tris-salicylidene compound SL showed comparatively slow hydrolysis of –NCH– bonds in the presence of anions. Anion-induced imine bond cleavage in salicylidene compounds could easily be detected in 1H-NMR due to the appearance of the salicylaldehyde –CHO peak at 10.3 ppm which eventually became more intense over time, and the –NCH– peak at 8.9–9.0 ppm became considerably weaker. Furthermore, the formation of the salicylidene O–H⋯X− (X− = Cl−/Br−) hydrogen-bonded complex, peak broadening due to proton-exchange processes and keto–enol tautomerism have also been clearly observed in the 1H-NMR experiments. Control 1H-NMR experiments revealed that the presence of moisture in the organic solvents could result in gradual hydrolysis of the salicylidene compounds, and the rate of hydrolysis has further been enhanced significantly in the presence of an anion. Based on 1H-NMR results, we have proposed a general mechanism for the anion-induced hydrolysis of imine bonds in salicylidene-based receptors.
Introduction
Schiff bases have widely been explored as chelating ligands in metal coordination chemistry for several decades,1 and their metal complexes are considered to be promising candidates for a variety of applications related to catalytic activities,2 photoluminescent properties3 and biomedical applications.4 However, with the importance of anion recognition chemistry5 being increasingly recognized as an important discipline of supramolecular chemistry,6 numerous salicylidene Schiff bases have been studied for anion sensing applications in the past two decades.7,8 Interestingly, most salicylidene-based anion sensors are known for colorimetric detection of fluoride (F−) in aprotic solvents such as dimethyl sulfoxide (DMSO) and acetonitrile (CH3CN).7 The mechanism of sensing has been attributed to either –OH⋯F− hydrogen bonding or –OH deprotonation by the fluoride ion. Some of these Schiff bases have also been reported to sense other basic anions like acetate (CH3COO−) and dihydrogenphosphate/hydrogenphosphate (H2PO4− ⇆ HPO42− +H+)9 in addition to F−, depending on the choice of solvent(s).8 However, no crystal structures to validate the formation of receptor-anion complexes or deprotonated receptor are reported in any of the published literature, to the best of our knowledge. Recently, we have reported that salicylidene Schiff bases are not stable in the presence of fluoride ion and undergo imine bond (–NCH–) hydrolysis in aprotic polar media in the presence of hard-to-avoid traces of moisture (2F− + H2O ⇆ OH− + HF2−) to generate deprotonated salicylaldehyde and the respective amine.10 Thus, due to the apparently unavoidable imine bond hydrolysis in the presence of fluoride and water under ambient conditions,10,11 it would not seem easy to obtain crystal structures of a hydrogen bonded receptor–fluoride complex or deprotonated receptor in the presence of a fluoride salt.
Anion-induced hydrolysis of –NCH– bond was first observed by Wang and co-workers in a diketopyrrolopyrrole-derived Schiff base compound in the presence of hydrogensulfate (HSO4−).12 Their results are particularly important in the context of understanding the mechanism of anion sensing by Schiff bases, which may undergo hydrolysis in the presence of certain anionic species. Several chromogenic cyanide (CN−) sensors based on the nucleophilic additions of CN− to –NCH– or OCR– bonds are reported in the literature.13 Further, numerous fluoride chemodosimeter (reactive sensors) are also known in the literature.14 This suggests that the reaction between an anion and a Schiff base cannot be ruled out, and detailed experimental investigations are necessary to come to a definite conclusion.
It is important to note that the anion sensing studies of the reported salicylidene-based receptors by UV-vis spectroscopy were mostly carried out with a large excess of anions (from 5 equiv. to as high as 50 equiv. of tetrabutylammonium salts) which will result in faster hydrolysis of –NCH– bonds in comparison to the 1H-NMR experiments which were often carried out with 1–5 equivalents of tetrabutylammonium (n-Bu4N+) salt added into the Schiff base solution.7,8 The detailed 1H-NMR investigations have revealed the limitations of salicylidene-based receptors as anion sensors in aprotic polar media, which has not been addressed before in the literature.
In continuation of our previous work,10 herein we report the solution-state instability of salicylidene-based receptors SL, CL1 and CL2 (Scheme 1) in the presence of n-Bu4N+X− (X− = Cl−, Br−, CH3COO−, HSO4− and H2PO4−) under non-inert conditions. Similar to the previously observed results for fluoride ion induced hydrolysis of –NCH– bonds,10 addition of other halides and oxo-anions to a solution of salicylidene Schiff base (DMSO-d6, 99.9% D atom) showed hydrogen bond formation between the salicylidene –OH and an anion, followed by keto–enol tautomerism, and eventually hydrolysis of the –NCH– bond(s) in NMR experiments. Control experiments revealed that the addition of n-Bu4N+ salt can significantly enhance the rate of –NCH– bond hydrolysis in moist DMSO-d6. Based on several 1H-NMR experimental results, we propose a general mechanism for the anion-induced hydrolysis of –NCH– bonds in salicylidene compounds.
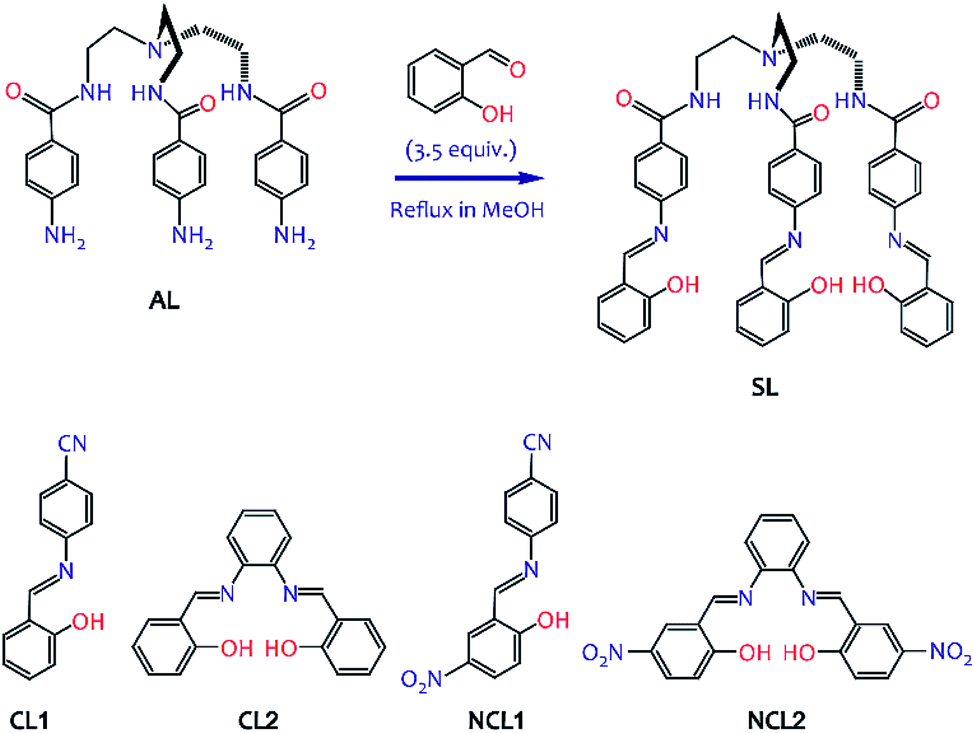 | ||
| Scheme 1 Molecular structures of salicylidene Schiff bases studied here (synthesis details are provided in the ESI†). | ||
Results and discussions
We have recently shown the complete hydrolysis of tris-salicylidene compound SL (Scheme 1) in the presence of excess (n-Bu4N+)F− (10 equivalents) in DMSO-d6, and also proposed a mechanism for fluoride ion induced –NCH– bond hydrolysis under non-inert conditions.10 In order to investigate the effect of other halides and oxo-anions on –NCH– bond hydrolysis, we have carried out 1H-NMR experiments of SL in the presence of 10 equiv. of (n-Bu4N+)Cl−, (n-Bu4N+)Br−, (n-Bu4N+)HSO4−, (n-Bu4N+)H2PO4− and (n-Bu4N+)CH3COO− in DMSO-d6. 1H-NMR spectra of SL mixed with either (n-Bu4N+)Cl−, (n-Bu4N+)H2PO4− or (n-Bu4N+)CH3COO− showed gradual hydrolysis of –NCH– bonds forming salicylaldehyde (C7H6O2) and AL (Scheme 1). Whereas, negligible hydrolysis of SL was observed in the presence of (n-Bu4N+)Br− and (n-Bu4N+)HSO4−. In a control experiment, no hydrolysis was observed to take place for a solution of SL in DMSO-d6, when recorded after 5 days of sample preparation.
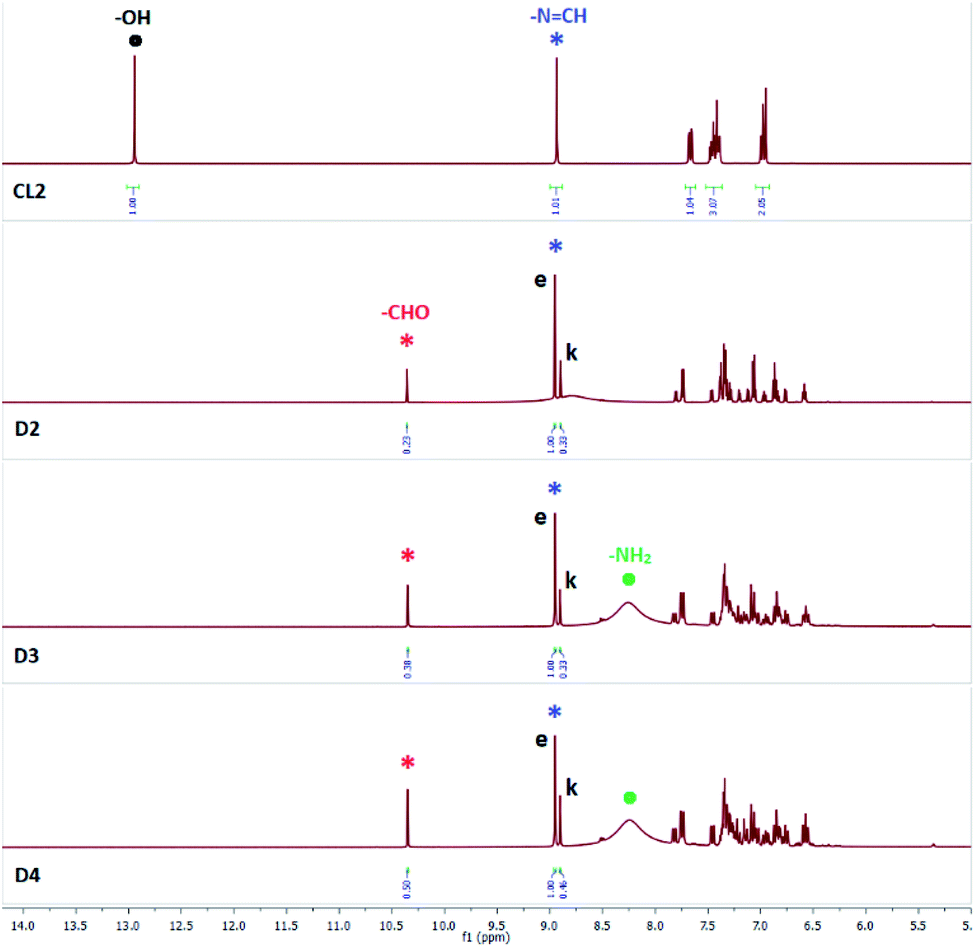
Addition of (n-Bu4N+)Cl− (10 equiv.) to a solution of SL in DMSO-d6 did not show any notable changes in the aromatic region of the 1H-NMR spectrum of SL, recorded after an hour. However, the solution mixture when recorded after 24 hours (D2, Fig. 1) showed the appearance of a salicylaldehyde –CHO signal at 10.3 ppm and salicylaldehyde –OH signal at 11.3 ppm indicating the progress of –NCH– bond hydrolysis in SL. Another new peak at 5.7 ppm for the –NH2 group of AL (Scheme 1) has also been observed, which further suggests the hydrolysis of SL by chloride ion. Due to hydrolysis, intensity of the –NCH– peak at 8.9 ppm was observed to be considerably reduced with concomitant splitting of the singlet peak in the presence of chloride ion (D2, Fig. 1). The occurrence of a triplet-like peak for –NCH– proton may be attributed to the presence of enol and keto tautomers of SL which are hydrogen bonded to chloride ions (Scheme 2) and appear as two closely spaced peaks in addition to the peak of free SL. About 27% hydrolysis was observed to be completed after 24 hours as revealed from the integral values of –CHO and –NCH– peaks of the spectrum (D2, Fig. 1). The solution mixture when recorded on subsequent days showed further hydrolysis of SL, however rather slowly showing 33% hydrolysis after 72 hours (D3 and D4, Fig. 1). With hydrolysis in progress, the –OH peak of SL at 12.8 ppm gets broadened presumably due to the slow proton-exchange processes with moisture adsorbed by the DMSO-d6 solution containing excess (n-Bu4N+)Cl− (Fig. 1), both of which are hygroscopic in nature.
 | ||
| Fig. 1 Aromatic region of 1H-NMR (DMSO-d6) spectra of SL in the presence of 10 equivalents of (n-Bu4N+)Cl−, recorded after 24 hours (D2), 48 hours (D3), and after 72 hours (D4). Signals labelled with red and black dots indicate –OH peaks of salicylaldehyde (red) and SL (black) respectively. Signals labelled with blue and green dots indicate –NH and –NH2 peaks of SL (blue) and AL (green) respectively. Signals labelled with red and blue asterisk indicate –CHO and –NCH– protons of salicylaldehyde (red) and SL (blue) respectively (additional spectra are provided in the ESI†). | ||
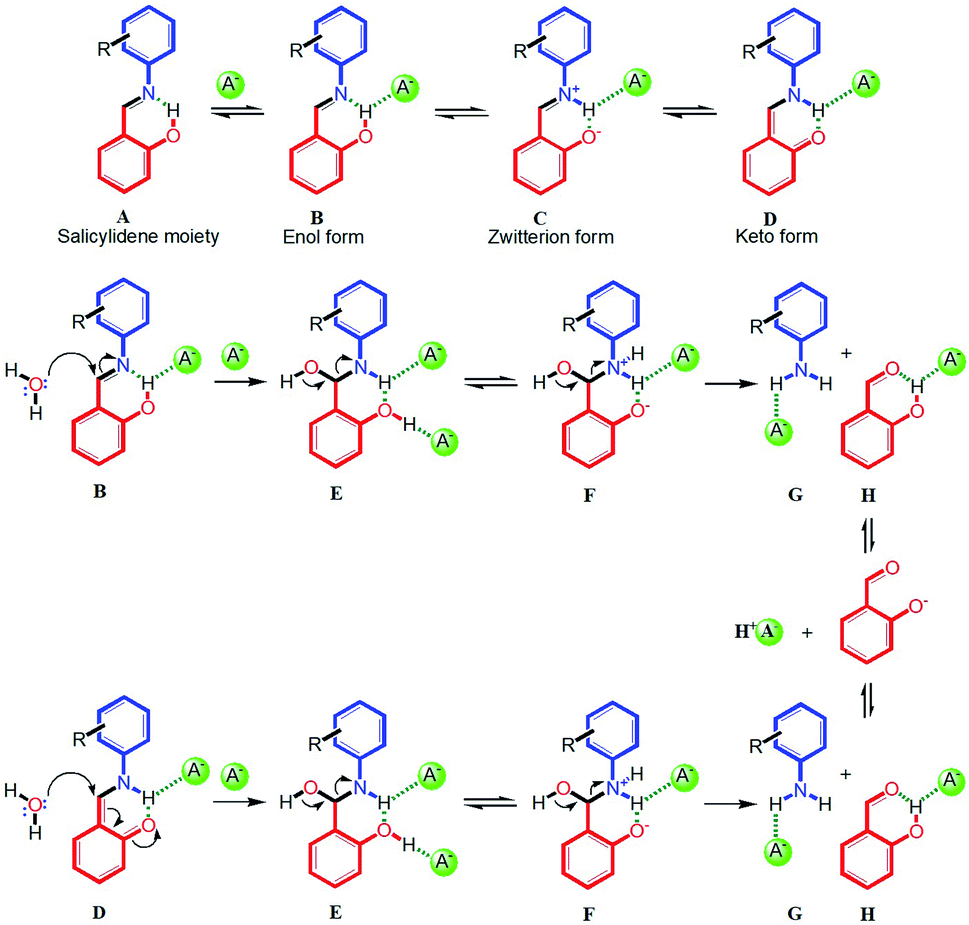 | ||
| Scheme 2 Plausible mechanism for the anion-induced hydrolysis of imine bond in salicylidene Schiff bases widely studied as anion sensors (A− = Cl−, Br−, H2PO4−, HSO4− or CH3COO−). Hydrogen bonds are shown in green dotted lines. | ||
In contrast, 1H-NMR spectrum of SL mixed with (n-Bu4N+)Br− (10 equiv.) in DMSO-d6 did not show any hydrolysis, when recorded after 24 hours (Fig. S15, ESI†). The solution mixture when recorded after 5 days showed 3% hydrolysis of SL (Fig. S15 and S16, ESI†), indicating that the tripodal salicylidene-based receptor is much more stable in the presence of bromide as compared to chloride and fluoride.10
Addition of (n-Bu4N+)H2PO4− (10 equiv.) to a solution of SL in DMSO-d6 showed immediate disappearance of the salicylidene –OH signal at 12.8 ppm indicating fast proton-exchange processes with H2PO4− ions. Further, appearance of a new peak at 10.3 ppm suggested the formation of salicylaldehyde by –NCH– bond hydrolysis (D1, Fig. 2). However, the salicylaldehyde –OH peak at 11.3 ppm (observed in the presence of Cl− in Fig. 1) has not been observed here, further suggests proton-exchange between H2PO4− ions and phenolic –OH groups. The solution mixture when recorded after 24 hours (D2, Fig. 2) showed 44% hydrolysis of SL as revealed from the integral values of –CHO and –NCH– peaks of the spectrum. Negligible hydrolysis was observed to take place in the same sample analysed on the subsequent days (D4, Fig. 2). The –NH2 peak of AL has experienced broadening and appeared downfield shifted at around 8.0 ppm (observed at 5.7 ppm in the presence of Cl−) likely due to strong hydrogen bond interactions with H2PO4− ions.15 In a control experiment, addition of one equivalent of (n-Bu4N+)H2PO4− to a solution of AL in DMSO-d6 resulted in broadening of –NH2 signal (Fig. S40, ESI†). Broadening of –NH2 signal of AL (at 5.7 ppm) has also been observed in the presence of one equivalent of (n-Bu4N+)Cl− (Fig. S41, ESI†).
 | ||
| Fig. 2 Aromatic region of 1H-NMR (DMSO-d6) spectra of SL in the presence of 10 equivalents of (n-Bu4N+)H2PO4−, recorded after 1 hour (D1), 24 hours (D2) and after 72 hours (D4). Signals labelled with red and blue asterisk indicate –CHO and –NCH– protons of salicylaldehyde (red) and SL (blue) respectively. Signal labelled with black dot represent –OH proton of SL (additional spectra are provided in the ESI†). | ||
Addition of (n-Bu4N+)HSO4− (10 equiv.) to a solution of SL in DMSO-d6 also showed immediate disappearance of the –OH signal due to fast proton-exchange processes. However, hydrolysis was observed to be very slow as compared to H2PO4− and the appearance of salicylaldehyde –CHO peak at 10.3 ppm could hardly be observed even after 24 hours. Only 13% hydrolysis of SL was observed to be completed after 5 days, beyond which no further hydrolysis was observed (Fig. S17 and S18, ESI†).
A solution of SL and (n-Bu4N+)CH3COO− (10 equiv.) in DMSO-d6 when recorded after one hour showed appearance of a salicylaldehyde –CHO signal and disappearance of the salicylidene –OH signal (D1, Fig. 3). The solution mixture showed 33% hydrolysis of SL after 48 hours beyond which no further hydrolysis was observed (D3, Fig. 3). The –NCH– singlet (at 8.9 ppm) has split into two closely spaced signals suggesting the presence of both enol and keto forms of SL (Fig. 3). Similar splitting of –NCH– singlet has also been observed for the solution SL mixed with (n-Bu4N+)H2PO4− (Fig. 2).
 | ||
| Fig. 3 Aromatic region of 1H-NMR (DMSO-d6) spectra of SL in the presence of 10 equivalents of (n-Bu4N+)CH3COO−, recorded after 1 hour (D1), 24 hours (D2) and after 48 hours (D3). Signals labelled with red and blue asterisk indicate –CHO and –NCH– protons of salicylaldehyde (red) and SL (blue) respectively. Signals labelled with black and blue dots indicate –OH and –NH peaks of SL (additional spectra are provided in the ESI†). | ||
Thus, the salicylidene-based tripodal receptor SL is largely stable in the presence of excess Br− and HSO4− as compared to Cl−, CH3COO− and H2PO4−. Since, no significant hydrolysis of SL has been observed after 48 hours in the presence of Cl−, CH3COO− and H2PO4− (Table S1, ESI†) the equilibrium constant has been calculated considering the concentrations of SL and hydrolysis products (AL and salicylaldehyde) after 72 hours. The equilibrium constant (K) for the hydrolysis of SL in the presence of different tetrabutylammonium salts is expressed by the equation K[H2O]3 = [AL] × [C7H6O2]3/[SL] (mol3 L−3), since the water peak could not be observed distinctly in several of the 1H-NMR experiments (DMSO-d6) of SL mixed with a tetrabutylammonium salt. We believe that this includes the concentration of water in the calculation of K, so that the K values for a salicylidene compound will reflect a conclusive comparison for different anions under identical experimental conditions. Thus, K[H2O]3 for the solution mixtures of SL with Cl−, CH3COO− and H2PO4− (10 equiv.) were calculated to be 2.411 × 10−5, 3.115 × 10−5 and 1.262 × 10−4 mol3 L−3, respectively (Table S3, ESI†).
Anion-induced hydrolysis of imine bond was also observed in the mono-salicylidene compound CL1 (Scheme 1). In a control experiment, no hydrolysis was observed to take place for a solution of CL1 in DMSO-d6, when recorded after 5 days of sample preparation. Similar mono-salicylidene Schiff bases have previously been reported as colorimetric fluoride sensors in aprotic solvent media under ambient conditions.7a–d,8a–i 1H-NMR experiments revealed that a solution of CL1 mixed with (n-Bu4N+)Cl− (5 equiv.) in DMSO-d6 showed 84% hydrolysis after 24 hours (Fig. 4a and b), as calculated from the integral values of the salicylaldehyde –CHO peak (10.3 ppm) and the imine –NCH– peak (9.0 ppm). The salicylaldehyde –OH peak appeared at 11.3 ppm whereas, the salicylidene –OH peak at 12.5 ppm has disappeared in the presence of excess chloride ions.
 | ||
| Fig. 4 Aromatic region of 1H-NMR (DMSO-d6) spectra of CL1 in the presence of 5 equivalents of (n-Bu4N+)Cl− and (n-Bu4N+)H2PO4− recorded after 1 hour (D1) and after 24 hours (D2). Signals labelled with red and blue asterisk indicate –CHO and –NCH– protons of salicylaldehyde (red) and CL1 (blue) respectively. Signals labelled with red and black dots indicate –OH peaks of salicylaldehyde (red) and CL1 (black) respectively (additional spectra are provided in the ESI†). | ||
Unlike the tris-salicylidene compound SL which showed negligible hydrolysis in the presence of bromide ions, a solution of CL1 mixed with (n-Bu4N+)Br− (5 equiv.) in DMSO-d6 showed 70% hydrolysis after 5 days (Fig. S23, ESI†). Similarly, CL1 in the presence of (n-Bu4N+)HSO4− (5 equiv.) showed 75% hydrolysis in DMSO-d6 after 5 days (Fig. S24, ESI†). Notably, the –NH2 peak of 4-aminobenzonitrile (hydrolysis product) has experienced broadening in the presence of hydrogensulfate, but appeared as a sharp peak at 6.3 ppm in the presence of bromide (Fig. S25, ESI†). However, the –NH2 peak of 4-aminobenzonitrile was not observed in the presence of chloride possibly due to significant peak broadening. Strong hydrogen bonds between –NH protons and anions often lead to peak broadening with shifts in 1H-NMR signals, which is a case of dynamic anion coordination i.e., the exchange of complexed and un-complexed guest (anion) is within the NMR time scale.15
Similar to the spectral changes of SL (Fig. 2), addition of (n-Bu4N+)H2PO4− (5 equiv.) to a solution of CL1 in DMSO-d6 resulted in the disappearance of the –OH signal at 12.5 ppm and appearance of the salicylaldehyde –CHO signal at 10.3 ppm (Fig. 4b and c). The broad peak at 8–8.5 ppm can be assigned to the –NH2 group of 4-aminobenzonitrile (a hydrolysis product) which was observed at 6.3 ppm in the presence of (n-Bu4N+)Br−. The significant downfield shift of the –NH2 peak with concomitant broadening is an indication towards strong hydrogen bonding between –NH2 group and H2PO4− ions. The –NH2 peak has experienced further downfield shift with time and the –NCH– peak at 8.9 ppm has partly merged with the broad –NH2 peak after 24 hours (Fig. 4d). Thus, the percentage of hydrolysis could not be determined accurately from the spectrum recorded after 24 hours, and roughly 75% hydrolysis of CL1 was calculated from the integral values of –CHO (10.3 ppm) and –NCH– (8.9 ppm) peaks (Fig. 4d). In order to accurately estimate the percentage of hydrolysis, the 1H-NMR analysis of a DMSO-d6 solution of CL1 and (n-Bu4N+)H2PO4− (5 equiv.) were carried out every hour. With hydrolysis in progress, the broad –NH2 peak was observed to shift downfield and begin to merge with the –NCH– peak after 2 hours. Peak integral values showed 50% hydrolysis of CL1 after an hour and roughly 75% hydrolysis was completed after 5 hours suggesting that no further hydrolysis has occurred after 5 hours (Fig. S51, ESI†). In a control experiment, addition of less than one equivalent of (n-Bu4N+)H2PO4− to a solution of 4-aminobenzonitrile showed downfield shift of the broad –NH2 peak from 4.0 ppm to 6.2 ppm suggesting hydrogen bonding interactions of –NH2 group with the oxo-anion (Fig. S42, ESI†).
Addition of (n-Bu4N+)CH3COO− (5 equiv.) to a solution of CL1 in DMSO-d6 showed 30% hydrolysis after an hour and complete hydrolysis was observed when the solution mixture was recorded after 48 hours (Fig. S26 and S27, ESI†). The –NH2 peak of 4-aminobenzonitrile appeared as a broad signal at 5.2 ppm and the salicylaldehyde –CHO peak appeared at 10.3 ppm.
Salicylidene-based receptor CL2 has previously been reported as a colorimetric fluoride sensor in DMSO.7e In our experiments, a DMSO-d6 solution of CL2 mixed with (n-Bu4N+)Cl− (5 equiv.) showed progressive hydrolysis of –NCH– bond as evident from the appearance of a salicylaldehyde –CHO peak at 10.3 ppm and a salicylaldehyde –OH peak at 11.3 ppm in the 1H-NMR spectrum (D2, Fig. 5). Further, the –NH2 peak at 5.4 ppm could also be observed for the corresponding amine product confirming hydrolysis of CL2. The –OH signal of CL2 appearing at 13.0 ppm became significantly weaker and broad in the presence of chloride indicative of slow proton-exchange processes with moisture adsorbed by the DMSO-d6 solution containing (n-Bu4N+)Cl−. The –NCH– singlet at 9.0 ppm (CL2 in Fig. 5) has split into two distinct peaks occurring at 8.9 and 9.0 ppm (D2, Fig. 5) due to partial tautomerization of the enol (e) form to the keto (k) form of CL2 (Scheme 2). In addition to the hydrolysis products, the presence of hydrogen bonded adducts formed between the –OH groups of CL2 and Cl− could also be observed in the 1H-NMR spectra (Fig. 5). Two new peaks corresponding to two different types of hydrogen bonded O–H⋯Cl− adducts appeared at 10.35 and 12.35 ppm. Due to the presence of two types of hydrogen bonded adducts in solution, two additional peaks have also appeared for the imine –NCH– proton at 8.4 and 10.0 ppm, each of which splits into two distinct peaks due to keto–enol tautomerism. The –NCH– peaks at 8.9(k)/9.0(e) ppm are associated with free CL2. Identical –NCH– peaks observed at 8.4(k)/8.5(e) and 9.9(k)/10(e) ppm correspond to the O–H⋯Cl− hydrogen bonded signals at 10.35 and 12.35 ppm, respectively (D2, Fig. 5).
 | ||
| Fig. 5 Aromatic region of 1H-NMR spectra (DMSO-d6) of CL2 in the presence of 5 equivalents of (n-Bu4N+)Cl−, recorded after 24 hours (D2) and after 72 hours (D4). Signals labelled with e and k indicate the enol and keto forms of CL2 respectively. Signals labelled with red and blue asterisk indicate –CHO and –NCH– protons of salicylaldehyde (red) and CL2 (blue) respectively. Signals labelled with blue hexagons represent –OH signals for O–H⋯Cl− hydrogen bonded complexes and the corresponding keto–enol peaks for the H-bond complexes. Signals labelled with red and black dots indicates –OH peaks of salicylaldehyde (red) and CL2 (black) respectively (additional spectra are provided in the ESI†). | ||
The solution mixture when recorded after 72 hours (D4, Fig. 5) showed complete disappearance of the salicylidene –OH signal at 13.0 ppm and intensity of the –NCH– peaks at 8.9(k)/9.0(e) ppm was significantly quenched. Peak broadening of the hydrogen bonded O–H⋯Cl− signal at 12.35 ppm was also observed. However, no change in peak intensity and integral values were observed for the –NCH– peaks at 8.4(k)/8.5(e) ppm and the corresponding hydrogen bonded –OH signal at 10.35 ppm that appears partially merged with the –CHO peak. The extent of hydrolysis could not be determined accurately due to the presence of multiple species in the solution containing CL2 and (n-Bu4N+)Cl− (D2 and D4, Fig. 5). Due to the weaker basicity and lower hydration enthalpy of Cl−, free –OH peaks and hydrogen bonded –OH peaks of CL2 could be observed in the presence of Cl−, which were not observed in the presence of more basic anions (F− and CO32−).10
Similar spectral changes have also been observed for a solution of CL2 mixed with (n-Bu4N+)Br− (5 equiv.) in DMSO-d6. The solution mixture when recorded after 5 days showed the presence of a salicylaldehyde –CHO peak, an amine –NH2 peak and three distinct peaks for the –NCH– proton, each of which splits into two distinct signals due to keto–enol tautomerism. The peak at 9.0 ppm corresponds to –NCH– of free CL2, and two additional peaks observed at 8.9 and 8.2 ppm represent –NCH– of two different hydrogen bonded O–H⋯Br− adducts of CL2 (Fig. S36, ESI†). Peak integral values of the –CHO and three –NCH– signals showed roughly 20% hydrolysis after 5 days. However, in a control experiment, no hydrolysis was observed to take place for a solution of CL2 in DMSO-d6, when recorded after 5 days of sample preparation.
A solution of CL2 and (n-Bu4N+)HSO4− (5 equiv.) in DMSO-d6 showed only 12% hydrolysis after 5 days. The salicylidene –OH peak was observed to be broadened due to proton-exchange with HSO4−, while the –CHO and –NH2 peaks of hydrolysis products could distinctly be observed at 10.3 and 5.4 ppm suggesting slow hydrolysis (Fig. S37, ESI†). Three distinct signals for the –NCH– proton (at 9.0, 8.9 and 8.2 ppm) has also been observed in this case.
Addition of (n-Bu4N+)H2PO4− (5 equiv.) to a solution of CL2 in DMSO-d6 showed disappearance of the –OH peak at 13.0 ppm and appearance of the salicylaldehyde –CHO peak at 10.3 ppm (D2, Fig. 6), similar to the observed spectral changes of SL and CL1 in the presence of (n-Bu4N+)H2PO4− (Fig. 2 and 4). Further, the –NCH– peak at 9.0 ppm (CL2 in Fig. 6) has split into two distinct signals occurring at 8.9 and 9.0 ppm (D2, Fig. 6) due to the presence of keto (k) and enol (e) forms of CL2, which was also observed in the presence of (n-Bu4N+)Cl−. The enol form is present in greater proportion as compared to the keto form. The solution mixture when recorded after 48 hours showed further hydrolysis and a broad peak at 8.0–8.5 ppm was observed for the –NH2 group of regenerated amine. The –NH2 signal appeared as a sharp and intense peak at 5.4 ppm for CL2 mixed with either (n-Bu4N+)Cl− or (n-Bu4N+)Br−, but in the presence of (n-Bu4N+)H2PO4−, the –NH2 signal appeared broad and much more downfield shifted likely due to the strong hydrogen bonding interactions between –NH2 group and H2PO4− ions (basicity of H2PO4− > Cl−).15 About 25% hydrolysis was observed to be completed after 72 hours (D4, Fig. 6).
 | ||
| Fig. 6 Aromatic region of 1H-NMR spectra (DMSO-d6) of CL2 in the presence of 5 equivalents of (n-Bu4N+)H2PO4−, recorded after 24 hours (D2), 48 hours (D3) and after 72 hours (D4). Signals labelled with e and k indicate the enol and keto forms of CL2 respectively. Signals labelled with red and blue asterisk indicate –CHO and –NCH– peaks of salicylaldehyde (red) and CL2 (blue) respectively. Signals labelled with black and green dots indicate –OH and –NH2 peaks of CL2 (black) and amine product (green) respectively (additional spectra are provided in the ESI†). | ||
Although keto and enol tautomer peaks could clearly be observed in the spectrum of CL2 mixed with an excess of (n-Bu4N+)X− in DMSO-d6, yet we have carried out variable temperature 1H-NMR analysis of a solution of CL2 and (n-Bu4N+)H2PO4− in CDCl3 with the objective to better understand the anion-induced hydrolysis mechanism of salicylidene Schiff bases. However, no additional information could be extracted from the spectra of the solution mixture recorded at room temperature, 0 °C and at −20 °C. No splitting of the imine –NCH– peak (8.62 ppm) was observed at room temperature and at 0 °C after 1 hour and 6 hours of sample preparation in CDCl3 (Fig. S52 and S53, ESI†). In contrast, the spectra obtained at −20 °C showed minor splitting and broadening of the –NCH– peak, which is less prominent in comparison to the well separated sharp peaks of keto and enol forms observed in the spectra recorded in DMSO-d6.
Addition of (n-Bu4N+)CH3COO− (5 equiv.) to a solution of CL2 in DMSO-d6 showed only 10% hydrolysis after an hour and about 35% hydrolysis was observed to be completed after 48 hours (Fig. S34 and S35, ESI†). Disappearance of the salicylidene –OH peak, appearance of the –CHO peak and splitting of the –NCH– peak into two closely spaced signals has also been observed here.
The equilibrium constant (K) for the hydrolysis of CL2 in the presence of (n-Bu4N+)H2PO4− and (n-Bu4N+)CH3COO− are expressed by the equation K[H2O] = [APIP] × [C7H6O2]/[CL2] mol L−1 (APIP = 2-aminophenyl iminophenol, hydrolysis product), since the water peak could not be observed in several of the 1H-NMR experiments (DMSO-d6) of CL2 and hence, the concentration of water could not be determined from some of the spectra. Thus, K[H2O] for the solution mixtures of CL2 with CH3COO− and H2PO4− (10 equiv.) were calculated to be 1.884 × 10−2 and 8.333 × 10−3 mol L−1, respectively (Table S4, ESI†).
The mono-salicylidene Schiff base NCL1 (Scheme 1) is not a stable compound in the solution-state. Unlike CL1, NCL1 was observed to undergo hydrolysis of –NCH– bond in DMSO-d6 (in the absence of any anion). Structural comparison of NCL1 with CL1 (Scheme 1) suggests that the two-electron withdrawing aromatic rings (benzonitrile and nitrophenol) attached to the imine bond possibly enhance the hydrolytic susceptibility of NCL1 and facilitate –NCH– bond cleavage in DMSO-d6 in the presence of hard-to-avoid traces of moisture adsorbed by the solvent. The presence of moisture (H2O) can be observed in the 1H-NMR spectrum of NCL1 (at 3.36 ppm) which showed about 70% hydrolysis after 15 min. of sample preparation (Fig. S9, ESI†). The presence of moisture has also been detected in the 1H-NMR spectra of SL, CL1 and CL2 in DMSO-d6, which indeed did not induce any hydrolysis of the salicylidene-based compounds under ambient conditions.
The bis-salicylidene Schiff base NCL2 (Scheme 1) has previously been reported as a colorimetric fluoride sensor in DMSO using 10−5 mol L−1 solutions.8j However, NCL2 is not sufficiently soluble in DMSO-d6 and other deuterated solvents (CDCl3, CD3CN and CD3OD) to characterize the compound by NMR spectroscopy. We have observed that the addition of an excess of (n-Bu4N+)F− or (n-Bu4N+)H2PO4− (5 equiv.) resulted in complete solubilization of the compound in DMSO-d6. NCL2 was characterized in the presence of (n-Bu4N+)H2PO4− where no immediate hydrolysis of –NCH– bond was observed, and six peaks for six different sets of –CH protons in the aromatic region of the spectrum confirmed the purity of the compound (Fig. S8, ESI†). However, the salicylidene –OH peak could not be observed due to fast proton-exchange between phenolic –OH and H2PO4− ions, as observed in the previous experiments. A solution of NCL2 mixed with (n-Bu4N+)F− in DMSO-d6 showed about 35% hydrolysis after an hour and 47% hydrolysis was observed to be completed after 24 hours (Fig. S39, ESI†).
We have also carried out some 1H-NMR control experiments in order to validate the role of an anion in the hydrolysis of salicylidene Schiff bases. The following experiments are confirmative of the key role that anions play in the hydrolysis of salicylidene compounds in the presence of moisture. Addition of 15 equiv. of H2O to a DMSO-d6 solution of CL1 did not show any immediate hydrolysis of the compound in an hour, but showed 20% hydrolysis after 6 hours. The solution mixture when recorded after 24 hours showed 80% hydrolysis, and 95% hydrolysis was observed to be completed after 48 hours (Fig. S44, ESI†). Interestingly, addition of one equiv. of (n-Bu4N+)Cl− to another DMSO-d6 solution of CL1 containing 15 equiv. of H2O showed complete hydrolysis of CL1 within an hour (Fig. S43, ESI†). In contrast, 84% hydrolysis of CL1 was observed to be completed after 24 hours in the presence of 5 equiv. of (n-Bu4N+)Cl− in DMSO-d6 (Fig. 4). Similar experiments have also been performed with CL2 and SL. Addition of 15 equiv. of H2O to a DMSO-d6 solution of CL2 showed only 18% and 26% hydrolysis after 24 and 72 hours respectively (Fig. 7). However, addition of one equiv. of (n-Bu4N+)Cl− to another DMSO-d6 solution of CL2 containing 15 equiv. of H2O showed 45% and 71% hydrolysis after 24 and 72 hours respectively (Fig. 8). In a similar experiment, addition of one equiv. of (n-Bu4N+)Cl− to a DMSO-d6 solution of SL containing 30 equiv. of H2O showed 62% hydrolysis after 6 hours (Fig. S46, ESI†). Whereas, only 7% hydrolysis was completed for a solution of SL in DMSO-d6 containing 30 equiv. of H2O after 6 hours (Fig. S45, ESI†). Further, splitting of the –NCH– signals of SL and CL2 have also been observed in the presence of water in DMSO-d6, indicating that the hydrolysis proceeds via keto–enol tautomerism of salicylidene compounds. Thus, our control experiments suggest that the anions can significantly enhance the rate of hydrolysis in salicylidene compounds in moist solvent.
 | ||
| Fig. 7 Aromatic region of 1H-NMR spectra (DMSO-d6) of CL2 in the presence of 15 equivalents of H2O, recorded after 1 hour (D1), 24 hours (D2), 48 hours (D3) and after 72 hours (D4). Signals labelled with e and k indicate the enol and keto forms of CL2 respectively. Signals labelled with red and blue asterisk indicate –CHO and –NCH– peaks of salicylaldehyde (red) and CL2 (blue) respectively. | ||
 | ||
| Fig. 8 Aromatic region of 1H-NMR spectra (DMSO-d6) of CL2 in the presence of 15 equivalents of H2O and 1 equivalent of (n-Bu4N+)Cl−, recorded after 1 hour (D1), 24 hours (D2), 48 hours (D3) and after 72 hours (D4). Signals labelled with e and k indicate the enol and keto forms of CL2 respectively. Signals labelled with red and blue asterisk indicate –CHO and –NCH– peaks of salicylaldehyde (red) and CL2 (blue) respectively. Signals labelled with black and green dots indicate –OH and –NH2 peaks of CL2 (black) and amine product (green) respectively. | ||
Based on the 1H-NMR experimental results, here we propose a plausible and general mechanism for anion-induced hydrolysis of salicylidene Schiff bases in the presence of moisture (Scheme 2). In the first step, the salicylidene –OH group which is involved in intramolecular hydrogen bonding with imine nitrogen (A in Scheme 2), forms an O–H⋯A− hydrogen bonded complex (B in Scheme 2).16 The fact that the peak position of the salicylidene –OH proton in SL did not shift upon addition of halides (Cl− and Br−) suggest that the intramolecular O–H⋯N hydrogen bond persists in the presence of these anions. However, broadening or disappearance of the salicylidene –OH peak in the presence of anions can be attributed to proton-exchange processes in the solution-state. In the second step, the hydrogen bonded enol form (B in Scheme 2) can partly tautomerize to the hydrogen bonded keto form (D in Scheme 2) via a transient zwitterion form (C in Scheme 2).11 Splitting of the –NCH– peak (at 8.9/9.0 ppm) into two closely spaced signals were observed in the 1H-NMR experiments of SL and CL2 in the presence of anions, indicating the presence of both enol and keto forms.10 The proton-exchange processes possibly facilitate the keto–enol tautomerism in the presence of anions, which was also observed in the 1H-NMR spectra of SL and CL2 in the presence of water (see ESI†). In the next step, moisture present in the DMSO-d6 solution mixture can behave as a nucleophile and attack on the imine carbon of the hydrogen bonded enol form (B) or the keto form (D) to generate a hydrogen bonded intermediate of hydroxy(phenylamino)methyl phenol (E in Scheme 2). Due to the acidic nature of phenolic –OH and the presence of a nearby –NH group, intermediate form E may possibly exist in an equilibrium with the zwitterionic form F, similar to B and C. As anions are present in excess in the solution, it is very likely that both the –NH and –OH protons will engage in hydrogen bonding with anions. The participation of the amine –NH proton in hydrogen bonding with an anion has also been observed in the 1H-NMR experiments, where downfield shift (with or without concomitant broadening) of –NH peak was observed as hydrolysis progressed. Finally, formation of the CO double bond by the loss of the O–H proton in E or F results in the cleavage of the HC–NH bond to generate salicylaldehyde and respective aromatic amine (G and H in Scheme 2). Depending upon the basicity of the anion, the regenerated salicylaldehyde can either exist in its neutral form, where the –OH group will be hydrogen bonded to the anion (H in Scheme 2) or in its deprotonated phenolate form.
However, we are also having the opinion that based on a specific anion under investigation, the mechanism of –NCH– bond hydrolysis may somewhat vary.
It is important to mention again that, both DMSO-d6 and some tetrabutylammonium salts used in the 1H-NMR experiments are hygroscopic in nature, and it is always difficult to avoid the presence of moisture in the prepared samples under ambient conditions. All experiments performed in the reported literatures were under ambient conditions, and no mention has been made that the experiments were carried out under inert conditions.7,8
Conclusion
In conclusion, we have proven that the salicylidene Schiff bases (SL, CL1 and CL2) undergo gradual hydrolysis of imine bonds in the presence of tetrabutylammonium salts of halides and oxo-anions in moist aprotic solvent. Bromide and hydrogensulfate ions were observed to show minimal effect on the hydrolysis of the tris-salicylidene compound SL due to the presence of multiple imine bonds and weakly basic nature of these anions having low hydration enthalpy. The extent of hydrolysis in the salicylidene Schiff bases is largely dependent on the structure of the receptor (number of imine bonds), quantity of anions (n-Bu4N+ salts) used in the NMR experiments and moisture adsorbed by the DMSO-d6 solution mixture containing (n-Bu4N+)X−.Formation of the receptor–anion hydrogen bonded complex and subsequent keto–enol tautomerism could clearly be observed in the 1H-NMR experiments of CL2 mixed with (n-Bu4N+)X− salts of halides (chloride/bromide) and oxo-anions. Similar to CL2, splitting of the –NCH– peak has also been observed in the 1H-NMR experiments of SL in the presence of chloride, acetate and dihydrogenphosphate ions (although less prominent), suggesting the presence of both keto and enol tautomers of SL in solution. The mono-salicylidene compound CL1 undergoes much faster anion-induced hydrolysis as compared to CL2 and SL, and no keto–enol tautomerism was observed in the case of CL1.
Thus, due to the apparently unavoidable hydrolysis of the salicylidene Schiff bases in the presence of anions under ambient conditions, it is of utmost concern to validate the solution-state stability of Schiff bases prior to anion sensing studies. Overall, our experimental results demonstrate the limitations of the salicylidene Schiff bases as anion sensors, and suggests a plausible mechanism for the anion-induced hydrolysis of –NCH– bonds via receptor-anion hydrogen bonding and keto–enol tautomerism based on 1H-NMR studies.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
SKD acknowledges the Department of Science and Technology (DST), New Delhi, India for providing financial support through INSPIRE Faculty award (DST/INSPIRE/04/2016/001867). SKD and CJ thank the Alexander von Humboldt Foundation, Bonn, Germany for providing the opportunity for research collaboration. We thank Mrs Birgit Tommes for her great help in obtaining the NMR spectra at HHU Dusseldorf.Notes and references
- (a) X. Liu and J.-R. Hamon, Coord. Chem. Rev., 2019, 389, 94–118 CrossRef CAS; (b) M. Karmakar and S. Chattopadhyay, J. Mol. Struct., 2019, 1186, 155–186 CrossRef CAS; (c) J. Zhang, L. Xu and W.-Y. Wong, Coord. Chem. Rev., 2018, 355, 180–198 CrossRef CAS; (d) C. M. da Silva, D. L. da Silva, L. V. Modolo, R. B. Alves, M. A. de Resende, C. V. B. Martins and A. de Fátima, J. Adv. Res., 2011, 2, 1–8 CrossRef; (e) M. Nath and P. K. Saini, Dalton Trans., 2011, 40, 7077–7121 RSC; (f) W. A. Zoubi, S. G. Mohamed, A. A. S. Al-Hamdani, A. P. Mahendradhany and Y. G. Ko, RSC Adv., 2018, 8, 23294–23318 RSC.
- (a) P. G. Cozzi, Chem. Soc. Rev., 2004, 33, 410–421 RSC , and references therein; ; (b) K. C. Gupta and A. C. Sutar, Coord. Chem. Rev., 2008, 252, 1420–1450 CrossRef CAS; (c) R. Drozdzak, B. Allaert, N. Ledoux, I. Dragutan, V. Dragutan and F. Verpoort, Adv. Synth. Catal., 2005, 347, 1721–1743 CrossRef CAS; (d) P. Adaeo, M. L. Kuznetsov, S. Barroso, A. M. Martins, F. Avecilla and J. C. Pessoa, Inorg. Chem., 2012, 51, 11430–11449 CrossRef PubMed; (e) P. Das and W. Linert, Coord. Chem. Rev., 2016, 311, 1–23 CrossRef CAS; (f) D. Gong, B. Wang, X. Jia and X. Zhang, Dalton Trans., 2014, 43, 4169–4178 RSC.
- (a) C. M. Che, S. C. Chan, H. F. Xiang, M. C. Chan, Y. Liu and Y. Wang, Chem. Commun., 2004, 1484–1485 RSC; (b) T. Sano, Y. Nishio, Y. Hamada, H. Takahashi, T. Usuki and K. Shibata, J. Mater. Chem., 2000, 10, 157–161 RSC; (c) S. H. Li, F. R. Chen, Y. F. Zhou, J. N. Wang, H. Zhang and J. G. Xu, Chem. Commun., 2009, 4179–4181 RSC; (d) S. A. Lee, G. R. You, Y. W. Choi, H. Y. Jo, A. R. Kim, I. Noh, S. J. Kim, Y. Kim and C. Kim, Dalton Trans., 2014, 43, 6650–6659 RSC.
- (a) Y. Sun, Y. Lu, M. Bian, Z. Yang, X. Ma and W. Liu, Eur. J. Med. Chem., 2021, 211, 113098 CrossRef CAS PubMed; (b) S. Parveen, Appl. Organomet. Chem., 2020, 34, e5687 CrossRef CAS; (c) M. S. More, P. G. Joshi, Y. K. Mishra and P. K. Khanna, Mater. Today Chem., 2019, 14, 100195 CrossRef CAS PubMed; (d) M. T. Kaczmarek, M. Zabiszak, M. Nowak and R. Jastrzab, Coord. Chem. Rev., 2018, 370, 42–54 CrossRef CAS; (e) M. Hajrezaie, M. Paydar, C. Y. Looi, S. Z. Moghadamtousi, P. Hassandarvish, M. S. Salga, H. Karimian, K. Shams, M. Zahedifard, N. A. Majid, H. M. Ali and M. A. Abdulla, Sci. Rep., 2015, 5, 9097–9104 CrossRef CAS PubMed.
- K. Bowman-James, A. Bianchi and E. Garcia-Espana, Anion Coordination Chemistry, Wiley-VCH, 1st edn, 2011 Search PubMed.
- J. W. Steed and J. L. Atwood, Supramolecular Chemistry, Wiley, New York, 2nd edn, 2000 Search PubMed.
- (a) R. Sivakumar, V. Reena, N. Ananthi, M. Babu, S. Anandan and S. Velmathi, Spectrochim. Acta, Part A, 2010, 75, 1146–1151 CrossRef PubMed; (b) A. Bhattacharyya, S. C. Makhal, S. Ghosh and N. Guchhait, Spectrochim. Acta, Part A, 2018, 198, 107–114 CrossRef CAS PubMed; (c) J. Li, H. Lin, Z. Cai and H. Lin, Spectrochim. Acta, Part A, 2009, 72, 1062–1065 CrossRef PubMed; (d) K. Liu, X. Zhao, Q. Liu, J. Huo, H. Fu and Y. Wang, J. Photochem. Photobiol., B, 2014, 138, 75–79 CrossRef CAS PubMed; (e) Q. Li, Y. Guo, J. Xu and S. Shao, Sens. Actuators, B, 2011, 158, 427–431 CrossRef CAS; (f) R. Arabahmadi, M. Orojloob and S. Amani, Anal. Methods, 2014, 6, 7384–7393 RSC.
- (a) D. Sharma, R. K. Bera and S. K. Sahoo, Spectrochim. Acta, Part A, 2013, 105, 477–482 CrossRef CAS PubMed; (b) L. Zang and S. Jiang, Spectrochim. Acta, Part A, 2015, 150, 814–820 CrossRef CAS PubMed; (c) S. Dalapati, S. Jana and N. Guchhait, Spectrochim. Acta, Part A, 2014, 129, 499–508 CrossRef CAS PubMed; (d) S. Suganya and S. Velmathi, J. Mol. Recognit., 2013, 26, 259–267 CrossRef CAS PubMed; (e) P. Alreja and N. Kaur, Inorg. Chim. Acta, 2018, 480, 127–131 CrossRef CAS; (f) Y. Hijji and G. Wairia, Proc. SPIE, 2005, 60070B CrossRef; (g) Y. M. Hijji, B. Bararea, A. P. Kennedy and R. Butcher, Sens. Actuators, B, 2009, 136, 297–302 CrossRef CAS; (h) S. Daniel, Bull. Mater. Sci., 2020, 43, 157 CrossRef CAS; (i) N. Yildirim and M. Yildiz, J. Turk. Chem. Soc., Sect. A, 2018, 5, 1271–1278 CrossRef CAS; (j) S. Dalapati, M. A. Alam, S. Jana and N. Guchhait, J. Fluorine Chem., 2011, 132, 536–540 CrossRef CAS; (k) X. Huang, Y. He, Z. Chen and C. Hu, Chin. J. Chem., 2009, 27, 1526–1530 CrossRef CAS; (l) V. Reena, S. Suganya and S. Velmathi, J. Fluorine Chem., 2013, 153, 89–95 CrossRef CAS.
- E. A. Katayev, Y. A. Ustynyuk and J. L. Sessler, Coord. Chem. Rev., 2006, 250, 3004–3037 CrossRef CAS.
- S. K. Dey and C. Janiak, RSC Adv., 2020, 10, 14689–14693 RSC.
- (a) M. Enamullah, A.-C. Chamayou, K. S. Banu, A. C. Kautz and C. Janiak, Inorg. Chim. Acta, 2017, 464, 186–194 CrossRef CAS; (b) B. M. Drašković, G. A. Bogdanović, M. A. Neelakantan, A.-C. Chamayou, S. Thalamuthu, Y. S. Avadhut, J. Schmedt auf der Günne, S. Banerjee and C. Janiak, Cryst. Growth Des., 2010, 10, 1665–1676 CrossRef.
- L. Wang, L. Yang and D. Cao, J. Fluoresc., 2014, 24, 1347–1355 CrossRef CAS PubMed.
- (a) Y. Sun, Y. Liu and W. Guo, Sens. Actuators, B, 2009, 143, 171–176 CrossRef; (b) S. K. Padhana, M. B. Podha, P. K. Sahub and S. N. Sahu, Sens. Actuators, B, 2018, 255, 1376–1390 CrossRef; (c) T. M. Asha, E. Shiju, C. Keloth and M. R. P. Kurup, Appl. Organomet. Chem., 2020, e5520 CAS; (d) F. Wang, L. Wang, X. Chen and J. Yoon, Chem. Soc. Rev., 2014, 43, 4312–4324 RSC; (e) Y. Ding, T. Li, W. Zhu and Y. Xie, Org. Biomol. Chem., 2012, 10, 4201–4207 RSC; (f) P. R. Lakshmi, R. Manivannan, P. Jayasudha and K. P. Elango, Anal. Methods, 2018, 10, 2368–2375 RSC; (g) Z. Xu, X. Chen, H. N. Kim and J. Yoon, Chem. Soc. Rev., 2010, 39, 127–137 RSC.
- (a) J. Cao, C. Zhao, P. Feng, Y. Zhang and W. Zhu, RSC Adv., 2012, 2, 418–420 RSC; (b) S. Yang, Y. Liu and G. Feng, RSC Adv., 2013, 3, 20171–20178 RSC; (c) S. Xu, K. Chen and H. Tian, J. Mater. Chem., 2005, 15, 2676–2680 RSC; (d) B. Zhu, F. Yuan, R. Li, Y. Li, Q. Wei, Z. Ma, B. Du and X. Zhang, Chem. Commun., 2011, 47, 7098–7100 RSC; (e) P. Chen, W. Bai and Y. Bao, J. Mater. Chem. C, 2019, 7, 11731–11746 RSC; (f) L. Fu, F.-L. Jiang, D. Fortin, P. D. Harvey and Y. Liu, Chem. Commun., 2011, 47, 5503–5505 RSC; (g) Shweta, A. Kumar, Neeraj, S. K. Asthana and K. K. Upadhyay, New J. Chem., 2017, 41, 5098–5104 RSC; (h) J. Chen, C. Liu, J. Zhang, W. Ding, M. Zhou and F. Wu, Chem. Commun., 2013, 49, 10814–10816 RSC; (i) B. Li, C. Zhang, C. Liu, J. Chen, X. Wang, Z. Liu and F. Yi, RSC Adv., 2014, 4, 46016–46019 RSC.
- (a) S. K. Dey and G. Das, Dalton Trans., 2011, 40, 12048–12051 RSC; (b) N. Busschaert, M. Wenzel, M. E. Light, P. Iglesias-Hernandez, R. Perez-Tomas and P. A. Gale, J. Am. Chem. Soc., 2011, 133, 14136–14145 CrossRef CAS PubMed; (c) A. Basu, S. K. Dey and G. Das, RSC Adv., 2013, 3, 6596–6605 RSC; (d) M. E. Khansari, M. H. Hasan, C. R. Johnson, N. A. Williams, B. M. Wong, D. R. Powell, R. Tandon and M. A. Hossain, ACS Omega, 2017, 2, 9057–9066 CrossRef PubMed; (e) S. K. Dey, Archana, S. Pereira, S. S. Harmalkar, S. N. Mhaldar, V. V. Gobre and C. Janiak, CrystEngComm, 2020, 22, 6152–6160 RSC.
- S. K. Patil and D. Das, ChemistrySelect, 2017, 2, 6178–6186 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of Schiff bases and characterization, 1H-NMR experiments in the presence of n-Bu4N+ salts. See DOI: 10.1039/d1ra07677a |
| This journal is © The Royal Society of Chemistry 2021 |