
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of monovalent salt concentration and peptide secondary structure in peptide-micelle binding†
Suvankar Ghosha,
Gopal Panditb,
Swapna Debnathb,
Sunanda Chatterjee *b and
Priyadarshi Satpati*a
*b and
Priyadarshi Satpati*a
aDepartment of Biosciences and Bioengineering, Indian Institute of Technology Guwahati, Guwahati 781039, Assam, India. E-mail: psatpati@iitg.ac.in; Fax: +91-361-2582249; Tel: +91-361-2583205
bDepartment of Chemistry, Indian Institute of Technology Guwahati, Guwahati 781039, Assam, India. E-mail: sunanda.c@iitg.ac.in; Tel: +91-361-2583310
First published on 17th November 2021
Abstract
Recently, we reported a cationic 14 residue peptide LL-14 (LKWLKKLLKWLKKL) with salt-sensitive broad-spectrum antimicrobial potency. However, the mechanism of its salt (NaCl) sensitivity remained unclear. In this study, we have reported computational (∼14.2 μs of MD) and experimental (CD, fluorescence) investigations to examine the salt-sensitivity and the role of peptide secondary structure on LL-14 binding to simple membrane mimetic (SDS, DPC) systems. LL-14 was shown to adopt a random coil (Pc) conformation in water and α-helical conformation (Ph) in the peptide:SDS micelle complex, accompanied by tryptophan burial, using both simulations and experiments. Simulations successfully deconvoluted the LL-14:micelle binding event in terms of secondary structure (random coil Pc versus helix Ph) and gave atomic insight into the initial and final LL-14:SDS complexes. Electrostatics drove the N-terminus (L1 and K2) of LL-14 (Pc or Ph) to bind the SDS micellar surface, initiating complex formation. LL-14 in amphipathic Ph conformation bound faster and buried deeper into the SDS micelle relative to Pc. Increasing NaCl concentration incrementally delayed LL-14:micelle binding by shielding the overall charges of the interacting partners. LL-14 binding to the SDS micelle was significantly faster relative to that of the zwitterionic DPC micelle due to electrostatic reasons. Cationic α-helical amphipathic peptides (with positively charged N-terminus) with low salt-ion concentration seemed to be ideal for faster SDS binding.
Introduction
Antimicrobial peptides (AMPs) have drawn significant attention as potential antimicrobial and therapeutic agents.1–6 The rapid rise of antibiotic resistance in microbes and the development of multi-drug resistant bacteria have resulted in a therapeutic deadlock.7–11 Additionally, the slow development of new classes of antibiotics, have augmented to the complexity of the problem. This necessitates the immediate development of alternate classes of therapeutic molecules that can combat antimicrobial infections. AMPs are small polypeptides (∼10–50 amino acids long) present in various kingdoms of life starting from microbes, insects, crustaceans, plants, animals and humans which are endowed with the ability to fight antimicrobial infections.12 Their presence in nature for millions of years, as the first line of host-defence against microbial infections caused by bacteria, fungi, yeast, viruses etc., indicate slow development of resistance against them. Additionally, AMPs are biocompatible and upon degradation forms amino acid residues in comparison to the toxic metabolites generated by the other classes of compounds. Thus AMPs are promising as an alternative to conventional antibiotics. AMPs have variable sequences, secondary structures and modes of action (majority of AMPs are membrane-active). However, despite the huge potential of the AMPs as alternate antimicrobial therapeutics, there has only been limited commercial translation of these molecules till date. This is primarily owing to the salt sensitivity of the antimicrobial potency of the AMPs and their short serum stability due to susceptibility to protease degradation. Researchers around the world are trying to develop synthetic AMPs which can circumvent these inherent problems of natural AMPs to develop robust economically viable therapeutics. Additionally, despite computational and experimental advancement, the rational design of selective potent AMPs remains limited13 due to the lack of molecular details of peptide-membrane interactions. Understanding the mechanism of interaction between AMPs and the microbial membranes in atomic terms is the key to designing new efficient membrane-active AMPs.MD simulations partially fill up the gap by offering an atomistic understanding of the same, using simple models.14–32 It has been understood that electrostatics is one of the major driving forces that lead to the microbial membrane affinity of the cationic AMPs.33 MD simulation studies on AMP-membrane mimetic systems offer insights into attributes like antimicrobial potency, cytotoxicity, specificity etc.25,34–36 Experimental investigations including circular dichroism (CD), fluorescence spectroscopy, nuclear magnetic resonance (NMR), isothermal calorimetry (ITC), microscopy (FESEM, CLSM etc), biochemical assays etc. have been used extensively in gaining insights into the mechanism of antimicrobial activity of AMPs.37 Thus, the combination of computational and experimental investigations may accelerate the discovery of therapeutic AMPs.
No systematic investigation has been done to date for understanding the salt sensitivity of the AMPs. Though some salt tolerant AMPs are reported in the literature, they are mostly results of serendipitous discovery rather than of rational design. Most AMPs lose their activity in the presence of physiological salt concentrations which severely limits the commercialization of AMPs. For example, various peptides viz., β-defensins (except for β-defensin III) present in humans, are prone to salt-induced inactivation.38–40 In our present study, we have tried to understand the reason for the salt sensitivity of a synthetic AMP, LL-14 that was earlier reported by our group.41 Cationic fourteen residue long membranolytic LL-14 (Fig. 1), was shown to have high activity (MIC99 ∼1–10 μM) against several strains of ESKAPE pathogens and fungal strains.41 However, the activity of LL-14 was salt-sensitive against several microbes (P. aeruginosa, S. aureus, C. neoformans) leading to a partial loss of activity at physiological NaCl concentrations.
 | ||
| Fig. 1 Structure of (a) Sodium Dodecyl Sulphate (SDS) (b) positively charged LL-14 (–N terminal as –NH3+ and C-terminal is capped as –CONH2). The random-coil (Pc) and α-helical structure of LL-14 are highlighted in the box. Helical wheel representation of the α-helical LL-14 with amino acid sequence highlighting the amphipathicity. | ||
Combining molecular simulations and experimental studies, we have attempted to understand the salt sensitivity and the role of peptide secondary structural features on the antimicrobial potency of LL-14 using simple membrane mimetic micelles. Fluorescence and CD experiments confirmed that the free peptide was unstructured in water and attained α-helical (Ph) conformation only after micelle binding. All-atom classical MD simulations were performed with LL-14 peptide (in two different conformations: (A) random coil or unstructured:Pc, and (B) amphipathic α-helical:Ph) in presence and absence of membrane mimetic system (SDS micelles: mimicking negatively charged prokaryotic membranes) in water with varying monovalent salt (NaCl) concentrations. MD simulations suggested that free LL-14 in water preferred random coil (Pc) conformation in lines with fluorescence and CD experiments. Simulations provided atomic insight into the initial binding complex LL-14(Pc):SDS micelles. However, peptide conformational change in response to micelle binding (LL-14(Pc):SDS → LL-14(Ph):SDS) was not observed within the time-scale of our MD simulations. To gain insight into the LL-14(Ph):SDS complex, simulations were performed to study α-helical (Ph) peptide binding to SDS and the resulting LL-14(Ph):SDS complex agreed with our conclusions from fluorescence experiment. Although, free LL-14 in the α-helical (Ph) peptide was not experimentally characterized and unrealistic, but the study of Ph binding to SDS was important for two reasons: (1) structural information about final LL-14(Ph):SDS complex was obtained, and (2) comparison of the initial binding events in terms of the peptide secondary structure (Pc binding to SDS versus Ph binding to SDS) was possible. The results indicated that salt (NaCl) systematically delayed the initial SDS binding event and secondary structure was crucial for initial binding (Ph binds faster than Pc).
Methods
Molecular dynamics setup
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 atoms, consisting of ∼32665 water molecules and 7 chloride ions (Table S1†).000 atoms were considered for MD simulations of SDS in water. The final snap of the post-equilibrated production dynamics was selected for the peptide-micelle binding study. DPC micelle was modelled in the same way as SDS.000 atoms were considered for MD simulations (Table S1†).000 steps) by the steepest descent algorithm. After minimization, the resulting structure was subjected to 200 ps equilibration (that included molecular dynamics with 100 ps NVT followed by 100 ps NPT ensemble). Initial velocities were assigned from a Maxwell velocity distribution at 300 K. After attaining equilibration, MD was performed using NPT ensemble. During equilibration, temperature was controlled using modified Berendsen thermostat (V-rescale,53 coupling with a time constant of 0.1 ps at 300 K). Simulations were done with 2 fs time-step, 300 K temperature and 1 bar pressure. Modified Berendsen thermostat53,54 and Parrinello–Rahman algorithm55 (for pressure coupling with the compressibility of 4.5 × 10−5 bar−1 and a time constant of 2 ps) were used to maintain the temperature and pressure of the simulation system. Periodic boundary conditions were used in all simulations. Long-range electrostatic interactions were treated using the particle-mesh Ewald algorithm.56 Short-range van der Waals interactions were truncated at 1.2 nm. Structures were collected for analysis at an interval of every 10 ps.
000 atoms, consisting of ∼32665 water molecules and 7 chloride ions (Table S1†).000 atoms were considered for MD simulations of SDS in water. The final snap of the post-equilibrated production dynamics was selected for the peptide-micelle binding study. DPC micelle was modelled in the same way as SDS.000 atoms were considered for MD simulations (Table S1†).000 steps) by the steepest descent algorithm. After minimization, the resulting structure was subjected to 200 ps equilibration (that included molecular dynamics with 100 ps NVT followed by 100 ps NPT ensemble). Initial velocities were assigned from a Maxwell velocity distribution at 300 K. After attaining equilibration, MD was performed using NPT ensemble. During equilibration, temperature was controlled using modified Berendsen thermostat (V-rescale,53 coupling with a time constant of 0.1 ps at 300 K). Simulations were done with 2 fs time-step, 300 K temperature and 1 bar pressure. Modified Berendsen thermostat53,54 and Parrinello–Rahman algorithm55 (for pressure coupling with the compressibility of 4.5 × 10−5 bar−1 and a time constant of 2 ps) were used to maintain the temperature and pressure of the simulation system. Periodic boundary conditions were used in all simulations. Long-range electrostatic interactions were treated using the particle-mesh Ewald algorithm.56 Short-range van der Waals interactions were truncated at 1.2 nm. Structures were collected for analysis at an interval of every 10 ps.MD simulations were performed with the peptides (Pc, Ph) in the aqueous phase, initially placed away from the micelle surface in three different orientations relative to the micelle surface (Fig. S1a†). Each simulation model (Setup A, Setup B, and Setup C, Fig. S1a†) was subjected to multiple independent MD simulations, using randomized initial velocities (rep1, rep2…, Table S2†) at various salt (NaCl) concentrations (Table S2†). 5 independent replicas were considered for the calculation of temporal averages. First direct contact between the LL-14 and SDS micelle was defined as the distance between the positively charged nitrogen of LL-14 and the oxygen head group of the SDS, and fixed to ∼0.35 nm (cut-off distance for hydrogen bonds in proteins).58 Analysing multiple independent MD trajectories, we estimated the distance (dCOM) between the centre of mass of LL-14 (Pc or Ph) and the centre of mass of SDS micelle as ∼3.47 ± 0.01 nm, which represented the first contact (Fig. S1b†) distance between them. It should be noted that the choice of centre-of-mass cut-off distance as 3.47 nm for describing first LL-14:SDS contact was the best estimate with a standard deviation of 0.01 nm. Simulations of free peptides (Pc, Ph) and the free micelles (SDS, DPC) in the water were performed. Each replica was subjected to post-equilibrated MD simulations for a minimum of 10 ns to a maximum of 6 μs (Table S2†). Total ∼14.2 μs simulation was considered for analysis which confirmed convergence of our reported data.
Results
Structural and dynamical properties of LL-14 and SDS micelle
MD simulations of the free peptide (Pc or Ph) and micelle in water provided clues about their size, shape, dynamics. Simulations of free peptide in water starting with two different conformations (i.e., extended:Pc or α-helical:Ph) showed that (1) Pc remained as random coil all along the MD trajectory (up to 1 μs, see Mov1_Pc_in_Water.mpg), (2) Ph (helix) conformation started melting after 800 ns (Mov2_Ph_in_Water.mpg). The results showed that free peptide preferred random coil conformation in water (Fig. 1). Distance of selected atoms to the centre of mass (COM) of SDS micelle (Fig. 2a) indicated that counter-ion (Na+) distribution was severely affected upon peptide binding. The average distance between Na+ and the COM of SDS-micelle increased upon peptide binding and was strikingly large in the case of Pc binding relative to Ph. The results suggested deeper binding of Ph into the SDS micelle core, leading to a closer approach of the counter-ions relative to Pc binding. | ||
| Fig. 2 Average structural and dynamical parameters obtained from simulation. (a) Distance of selected atoms (Fig. 1a) from the centre of mass of the SDS micelle. (b) Oxygen (H2O) to sulphur (SDS) radial distribution function. (c) Representative snapshots of free SDS, Pc bound SDS, and Ph bound SDS. Na+ (Yellow sphere), peptide (dark grey), SDS (aliphatic: light grey, sulphate: red). Waters and hydrogens not shown for clarity. Eccentricity (e), radius of gyration (Rg), micelle radius (Rs), solvent accessible surface area (SASA), diffusion coefficient (D) of Na+ are given. (d) Residue-wise root mean square fluctuation (RMSF) of the heavy atoms of LL-14 in the absence and presence of SDS micelle. Error bars (standard deviation) are shown either as vertical bars (a) or reported after ± (c). | ||
The radial distribution functions (RDF: oxygen atom of water molecules around the sulphur atoms of the SDS micelle) calculated from MD simulation indicated that the first coordination sphere of water was located 0.38 nm away from the sulphur atom of the micelle (Fig. 2b). Beyond the first coordination shell, the water distribution was unstructured and relatively less dense than the bulk. Peptide binding displaced water from the first coordination sphere, resulting in a dip of the RDF peak (Fig. 2b). The effective radius32,59,60 or size (Rs) of the micelle can be defined as the average of two distances, (a) the centre of mass of the micelle to the sulphur atoms of the head group (1.98 nm, Fig. 2a and b) head group sulphur to the first peak of water oxygen radial distribution function (0.38 Å, Fig. 2b) minus the radius of water (0.14 nm). Our estimated effective radius of the micelle was 2.22 nm, which was in excellent agreement with the X-ray scattering experiment60 (2.23 nm). Micelle radius32,59 is also defined as:
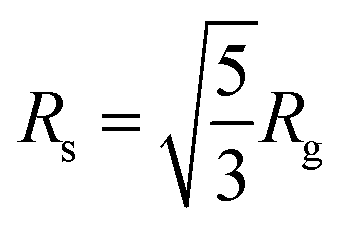 | (1) |
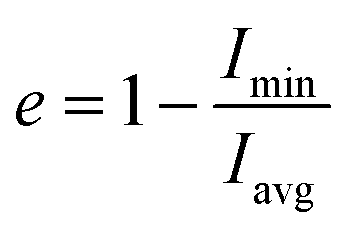 | (2) |
 (of free SDS micelle in water, Fig. 2c) were 0.14 ± 0.05 and 1.31 ± 0.12 respectively (close to previous studies:
(of free SDS micelle in water, Fig. 2c) were 0.14 ± 0.05 and 1.31 ± 0.12 respectively (close to previous studies:  ranging between 1.02–1.13).25,32,63 The eccentricity of the micelle was found to be independent of the salt concentration (Table S3†).
ranging between 1.02–1.13).25,32,63 The eccentricity of the micelle was found to be independent of the salt concentration (Table S3†).
Quantification of solvent accessible surface area (SASA)64 also gave insight into the structural properties of the micelle. From each MD snapshot, the ions, water, peptide were first removed. Then a spherical probe molecule with the radius of 0.14 nm (mimicking water) was rolled over the surface of the micelle. The area formed by the centre of the probe quantified the total accessible surface area (SASA). The average value of the solvent-accessible surface area (SASA) of the peptide-free micelle was 104.5 nm2 (Fig. 2c), which differed significantly relative to a perfect sphere of radius 2.22 nm having a SASA of 61.9 nm2. The surface roughness of the SDS micelle and its non-spherical nature resulted in nearly double the SDS surface area relative to that of a perfect sphere. SASA of the SDS increased significantly (Fig. 2c) in response to interaction with helical peptide (Ph), indicating severe structural distortion and solvent exposure. MD simulations with helical (Ph) and coiled (Pc) peptide in SDS micelle revealed that the former penetrated the SDS micelles, whereas, the latter lied on the SDS surface (Fig. 2c). Peptide binding displaced the counter ions from the micelle surface to the bulk and increased the average distance between the counter-ions (Na+) and COM of SDS micelle (Fig. 2a). Pc acted as a barrier (by lying over the SDS surface, Fig. 2c) between the counter ions (Na+) and SDS surface, increasing the bulk Na+ concentration resulting in the largest average distance between the SDS and Na+ ions (Fig. 2a). Penetration of the peptide (Ph) into SDS micelle resulted in a lower average distance between Na+ and COM of SDS (Fig. 2c and a). The flexibility of the peptide chain was estimated by calculating the root mean square fluctuation (RMSF) of the heavy-atoms of the amino acid residues in the absence and presence of micelle (Fig. 2d). RMSF plots revealed the following features: (1) α-helical peptide Ph had smaller RMSF relative to its random-coil-like Pc analogue. Fluctuations of the amino acid residues were expected to be more pronounced in the random-coil like conformations and less pronounced in the structurally rigid α-helical conformations. (2) A noticeable reduction in the flexibility (particularly in the –N and –C terminal of LL-14) was observed in the case of Ph peptide binding to SDS. Micelle induced loss of LL-14 flexibility was less pronounced for Pc relative to Ph analogue. Thus, the secondary structure of the peptide turned out to play a key role in the structure and dynamics of LL-14:SDS complexes. Secondary structural content of the peptide was estimated (Fig. S2†) using GROMACS analysis tool (gmx do_dssp) that uses DSSP software.65 No significant change in the peptide conformation was observed in response to SDS binding. Peptide (Ph/Pc) more or less retain its conformation (helical/Coil) after SDS micelle binding during the course of our simulations.
Salt effect in peptide-micelle (SDS) binding
The distance between the centre of mass of the peptide and the centre of mass of the micelle (dCOM) was plotted as a function of time (Fig. 3). Distance dCOM ∼3.47 nm indicated first direct contact (see Method) between peptide and micelle. The results revealed several remarkable features (Fig. 3). Firstly, the temporal decay of dCOM was much faster for α-helical (Ph) LL-14 relative to its unstructured random-coil-like (Pc) conformation. This highlighted the importance of secondary structure in peptide-micelle binding kinetics. Secondly, the presence of monovalent salt drastically delayed the peptide-micelle interaction. The salt-induced time delay was independent of the structure of the peptide (helical, Ph or random-coil Pc), indicating a general feature of the salt effect in peptide-micelle binding. An increase in salt concentration (0.0% → 0.5% → 1.0% w/v) systematically delayed the peptide-micelle binding. Thirdly, LL-14 bound to SDS micelle in a rapid (initial contact within 4 ns of MD) and irreversible manner. Fourthly, the distance between the centre of mass of the peptide and micelle was more or less constant after 10 ns, indicating structural convergence of the peptide-micelle complex. The robustness of the above features was confirmed from multiple trajectories (Table S2†), that differed in their initial velocities and relative positions. It should be noted that a single peptide in an explicit cubic water box of dimension ∼14 nm referred to a concentration of ∼605 μM in the MD simulation model. Thus, the concentration of peptide in MD was much larger than the MIC against several ESKAPE pathogens (1–10 M) and in CD experiments (100 μM).41 High concentration of peptide resulted in faster binding of peptide to micelle (within 5 ns for various MD models). Using a larger water-box could be an alternative for lowering the peptide concentration, but would incur huge computational cost, rendering it impractical. The systematic salt-induced delay in the interaction time was a robust feature observed from several MD studies (confirmed from multiple simulations of various MD models, see method). Thus it can be claimed that the general principle of salt-induced delay was captured by our simulation setup with moderate computational cost. | ||
| Fig. 3 Temporal averaged distance between the centre of mass of a LL-14 and the centre of mass of the SDS micelle (dCOM) as a function of time. (a) Initial binding event. Peptide: micelle initial contact distance is represented as green line at dCOM ∼3.47 nm. (b) Plateau of dCOM vs. time plot (after 10 ns) indicate structural convergence of the final peptide: micelle complex. Error bars are not shown for clarity (see Fig. S3† for detail). | ||
Structural insight into the sequential peptide-micelle binding event from MD simulations
Several prominent structural features were observed from MD simulations. Firstly, initial LL-14:SDS contact was formed by involving the positively charged N-terminal of the peptide and the negatively charged micelle surface (Fig. 4a and d). We considered initial contact when the distance between the positively charged nitrogen of the peptide and the oxygen head group of the micelle was 0.35 nm. Secondly, in the finally converged LL-14:SDS complex, the helical peptide (Ph) was engulfed by the micelle (except at the terminals), whereas the random-coil-like peptide (Pc) lied over the surface of the micelle (Fig. 4b and e). We found that the peptide-micelle binding followed a sequential order. First, electrostatic interaction between negatively charged SDS surface with the positive charge of peptide brought them close together. Distance plot between positively charged nitrogen's of the peptide and the centre of mass of SDS (Fig. S4†) during the MD trajectories, including visual examination of MD snaps confirmed that the N-terminal, and the side-chains of K2 of LL-14 played a key role in establishing the initial contact (Fig. 4a and d). Next, additional favourable electrostatic interactions (involving other positively charged lysine's and phosphate head group of SDS) and hydrophobic interactions (involving aliphatic/aromatic side chains of the peptide and aliphatic chain of micelle) led to a stable peptide-SDS complex (Fig. 4b and e).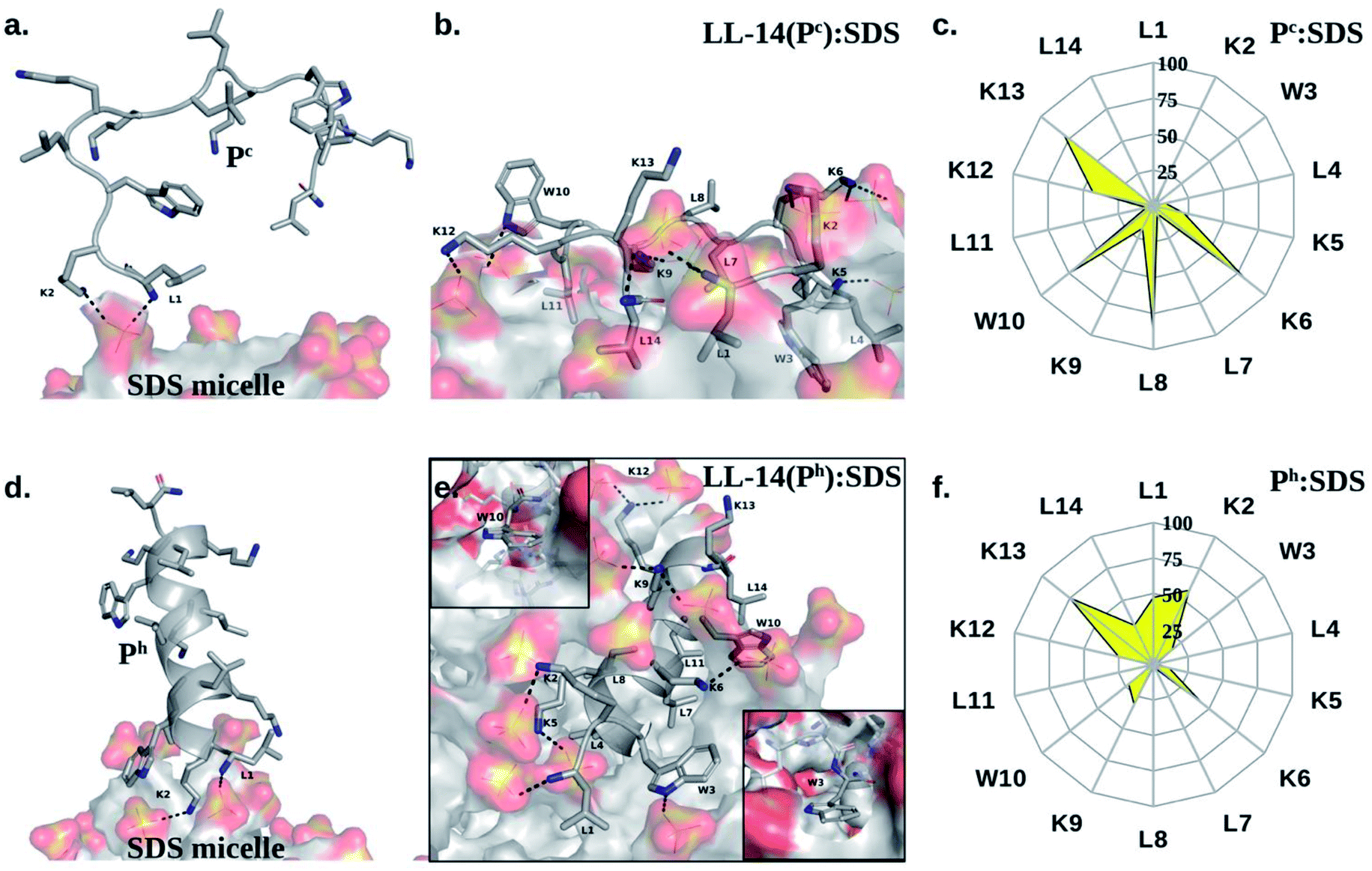 | ||
| Fig. 4 Structures from the peptide: micelle (SDS) binding event, with distinct secondary structures of the peptide (Pc or Ph) as initial condition. (a and d): peptide: SDS-micelle initial contact. Micelle surface forms direct interactions with the NH3+ of the N-terminal (L1) and side-chain of K2 of the LL-14 peptide both in the (a) Pc and (d) Ph conformations. (b and e) Final structure of the peptide: micelle complex. Interaction network is shown as dotted line for (b) Pc:micelle and (e) Ph:micelle complex respectively. For clarity, van der Waals radii of the micelle was reduced by 50% in (e). The local environment around W3 and W10 are explicitly shown without van der Waals reduction in the in-set (e). (c and f) Residue-wise solvent exposure (in percentage) of LL-14 in the final complex was estimated as: (SASA of amino acid residue in SDS:LL-14 complex/SASA of the amino acid residue in LL-14) × 100. Trajectory averaged percent solvent exposure (yellow) is shown in the net-plot with contours of constant percent exposure. Waters and hydrogens not shown for clarity in the structures. | ||
The peptide backbone of Pc satisfied its hydrogen bonding requirement by forming an H-bond with waters or sulphates of the SDS micelle or both (Fig. S5†). Out of the two tryptophan's (W3 and W10), only one tryptophan (W3) of Pc was buried in the hydrophobic aliphatic pocket of SDS (Fig. 4b and c). Side-chain of four residues (K6, L8, W10, and K13) of Pc showed more than 50% solvent exposure (Fig. 4b and c) while being complexed with micelle. In Ph, the backbone was involved in secondary structure formation (i.e., helix), thus, unavailable for interaction with water or SDS. Ph was mostly buried inside the micelle, except for residues K2, K13 (Fig. 4e and f). Note, both the tryptophans (W3, W10) were buried into the hydrophobic core of the SDS micelle in the Ph:SDS complex (Fig. 4e and f). Solvent exposure of Pc in Pc:micelle complex was much larger relative to Ph in Ph:micelle complex (Fig. 4c and f). We estimated the hydration of the hydrophobic part of the SDS micelle in Pc:micelle and Ph:micelle complexes. Hydration of the hydrophobic parts (Pc:micelle = 87.1 ± 8.6, Ph:micelle = 98.6 ± 8.7) was estimated by averaging the number of water molecules (from the last 20 ns of 50 ns of MD trajectory) within the commonly used cut-off of 0.35 nm (ref. 25) of the non-polar moiety (C1–C12 of Fig. 1a) of each SDS molecule. The higher water exposure of the lipid portion of the micelle in the Ph:micelle relative to Pc:micelle complex, indicated higher hydrophobic destabilization of the micelle in the former.
Notably, irrespective of the secondary structural nature of the peptide (Pc or Ph), electrostatic interaction between the positively charged N-terminal of the peptide and the negatively charged anionic head group of the micelle lipid was the driving force for the initial binding (Fig. 4a and d). Structural convergence in terms of the observed atomic interaction network in the final complex (Fig. 4b and e) was confirmed from multiple independent MD simulations (Fig. S1a and Table S2†). Simulations with various peptide (Ph or Pc) orientation relative to the micelle surface were found to be converged to the same final structure of the peptide:micelle complex (Fig. 4b and e) and independent of the salt concentration (Fig. S6†). Reasonable sampling (>14 μs of MD) and converged structure of the final peptide:micelle complex indicated that the reported MD structures of the peptide:micelle complex represented the true minima on the free energy hypersurface.
MD simulation with a neutral DPC micelle (zwitterionic lipid that mimics mammalian membrane) showed that Pc was able to bind DPC micelle surface after 870 ns of dynamics (Mov3_Pc_DPC.mpg), the delay being attributed to the obvious electrostatic reasons. The results highlighted the importance of favourable electrostatic interactions in facilitating peptide:micelle interaction as reported in earlier studies.33 Though LL-14 peptide was found to be cytotoxic,41 MD studies suggested that the interaction of LL-14 with the mammalian membranes was much slower compared to that with the microbial membranes. A detailed structural analysis of the peptide:DPC binding is outside the scope of this paper, and will be reported elsewhere.
The driving force for the initial LL-14:SDS binding event was investigated by plotting the electrostatic (Coulomb) and Lennard-Jones (van der Waals is the attractive term) energy as a function of time (Fig. 5). The results showed that electrostatic energy profile, decayed faster for Ph relative to Pc, whereas there was a little difference in the decay of Lennard-Jones energy for Ph and Pc during the initial binding event (i.e., within 2 ns of MD, see Fig. 2a), highlighting the electrostatic origin for the faster binding of Ph relative to Pc as observed in Fig. 3. Though both the “electrostatic” and “van der Waals” energies were favourable (negative sign, Fig. 5), the former was the primary contributor to the overall interaction potential energy. The favourable electrostatic term was attributed to the interactions between the positively charged side-chains of peptide and negatively charged SDS surface whereas, the favourable Lennard-Jones potential energy indicated favourable van der Waals interactions involving placement of hydrophobic residues of the peptide (Trp, Leu, aliphatic chain of Lys) into the aliphatic core of SDS micelle.
 | ||
| Fig. 5 Electrostatic and Lennard-Jones interaction energy as a function of time during the initial stage of peptide: micelle (SDS) binding. The peptides in distinctly different conformations (Pc or Ph) were subjected to MD simulation. Long-range cut-off of 1.4 nm was used to compute the energies. | ||
Secondary structural insight from CD experiments
CD experiments (Fig. 6a) confirmed that free LL-14 in water adopted a random coil like (Pc) conformation with a negative peak at 200 nm. The result directly corroborated with the helix melting of free LL-14 observed in the MD simulation (Mov2_Ph_in_Water.mpg). However, in the presence of SDS micelles, a positive peak at 195 nm and two negative peaks at 208 nm and 222 nm were observed (characteristic features of significant helical content), which indicated helical conformation of LL-14 in the SDS micelle bound state. Thus, CD experiments suggested SDS micelle induced conformational change of LL-14 peptide (Pc free in water → Ph bound to SDS). Furthermore, the stability of the LL-14:SDS complex was investigated by CD experiments in the presence of chaotropic reagent urea. Experiments were performed either by adding urea into the LL:14-SDS complex, or by adding LL-14 into the solution of urea and SDS micelle. Results confirmed that LL-14 in complex with SDS retained its helical conformation (Ph bound to SDS) even in presence of urea and was independent of the order of addition of urea in the experiments. | ||
| Fig. 6 (a) CD spectra of free SDS micelle (as control, broken line) and LL-14 in various conditions (presence or absence of SDS micelle and urea, solid lines). CD spectra indicate characteristic SDS induced LL-14 conformational change form random coil to helix (Pc:Solid black → Ph:Solid colours) even in the presence of urea. (b) Tryptophan Fluorescence emission spectra of free LL-14 (black), LL-14 in presence of SDS (red), and LL-14 in presence of SDS and salt (red). Blue shift in LL-14 emission observed upon addition of SDS micelle in presence or absence of salt (NaCl). | ||
Fluorescence studies and tryptophan burial in the final peptide:micelle complex
LL-14 binding to SDS micelle was investigated by monitoring the intrinsic fluorescence emission maxima of the tryptophan residues of LL14 in the presence and absence of SDS micelle (Fig. 6b). The free LL-14 peptide in water, upon being excited at λmaxex = 280 nm showed an excitation emission with λmaxem at 358 nm. Upon addition of 30 mM of SDS micellar solution (above CMC = 8.5 mM) to the peptide, λmaxem of LL-14 shifted to 341 nm. This large blue shift of λmaxem of about 17 nm in the SDS miceller system indicated the change in the microenvironment of tryptophan residues. In other words, the blue shift in the presence of SDS micelles suggested the peptide-micelle binding with the insertion of the tryptophan side chains into the hydrophobic core of the micelle. A blue shift (20 nm, λmaxem at 338 nm) was also observed in presence of 150 mM NaCl, thus indicating a salt-independent feature of LL-14 binding to SDS micelle. The more or less similar value of the blue shift (20 nm/17 nm in presence/absence of NaCl) indicated that tryptophan burial into the SDS hydrophobic core was independent of salt. Presence of a single peak at the shifted position (338 nm/341 nm in presence/absence of NaCl) indicated that both the tryptophan residues (W3 and W10) of LL-14 were in the similar environment in the micelle bound state. MD structures of the converged LL-14(Ph):SDS micellar complex, also showed burial of both the tryptophan residues (W3 and W10) in the SDS micelles thus corroborating the experimental observations. Thus, the fluorescence data indirectly proved that the structure of the final peptide:micelle complex was salt-independent and similar to the LL-14(Ph):SDS micellar complex observed from MD simulations.Discussion
The mechanism of membrane-active AMP induced cell-death is a multi-step process that entails AMP binding to membrane, membrane induced secondary structure formation of the peptide (coil → helix is most common), local peptide aggregation at the membrane and subsequent membrane destabilization leading to cell lysis.17 Unfortunately, peptide aggregation and cell lysis are still non-tractable by current computational power due to inherent complexity and large time-scale associated with the event. However, all-atom classical MD simulations are useful for investigating the AMP binding to simple membrane mimetic systems and can shed light on the general aspects (viz., structure, dynamics and kinetics of peptide:micelle binding) of AMP induced cell death. These information would be useful in designing peptide antimicrobials with improved activity and selectivity against pathogenic microbes.Broad-spectrum cationic antimicrobial peptide41 LL-14 (Fig. 1) was selected and its binding to membrane mimetic systems was investigated in atomic details using MD simulations and experiments (CD, fluorescence). Simulations of free LL-14 in water showed that the helix (Ph) conformation melted after ∼800 ns of MD, and LL-14 preferred random-coil (Pc) conformation. CD experiments also revealed that free LL-14 adopted a random coil (Pc) structure in water. Fast LL-14:SDS binding (within 4 ns of MD) (Fig. 3) relative to slow helix melting (∼800 ns), allowed us to study LL-14:SDS binding event considering the secondary structure of the peptide (i.e., Pc versus Ph binding to SDS, despite the fact that Ph was not the preferred conformation of the free peptide in water). The results suggested that the secondary structure of the peptide played a crucial role in SDS micelle binding kinetics (Fig. 3) and insertion (Fig. 4). Helical (Ph) LL-14 established faster initial contact with SDS and was deeply inserted into the micelle in the final stage of binding relative to its coil analogue (Pc). Thus, the secondary structure of peptide, was crucial for the fine-tuning of the speed of the initial peptide:micelle binding event. Electrostatic interaction between the negatively charged head-group of SDS and positively charged nitrogen's of peptide (particularly N-terminal and side-chain of K2) was the key factor for interaction. This was followed by further hydrophobic stabilization (placement of aliphatic/aromatic side-chains of the amino acids into the aliphatic interior of the micelle) and additional electrostatic interactions. The settled peptide (Ph) was found to be under the micelle surface (“hot-dog” like geometry), while Pc was found to be lying on the micelle surface. MD structure of the final converged LL-14:SDS complex showed burial of both the tryptophan residues (W3, W10) of LL-14 into the hydrophobic core of the SDS micelles in the case of the Ph-SDS complex (Fig. 4e), contrary to that of the Pc-micelle complex, where only one of the tryptophan side chains was buried (Fig. 4b).
CD experiments suggested SDS induced helical conformation of the peptide in the final LL-14:SDS complex (Fig. 6). The peptide retained its helical conformation (Ph) in the LL-14:SDS complex even after 24 hours of urea exposure, suggesting the stability of the complex. Furthermore, LL-14 addition to urea containing SDS micellar solution also supported Ph conformation of LL-14. Thus, SDS induced LL-14 conformational change (Pc → Ph) was a consistent and robust feature. We observed a blue shift in the emission maxima of the tryptophan fluorescence (Fig. 6), in the presence of the SDS micellar system, suggesting the burial of the side chains of tryptophan in the hydrophobic inner core of the micelles. The observation of distinct single peak indicated similar environment around both the Tryptophan's (W3 and W10) of LL-14 upon micelle binding. Thus, fluorescence experiments corroborated with the observed final MD structures of LL-14(Ph):SDS complex (Fig. 4e).
It is worth mentioning that, we did not observe Pc→Ph conformational change in response to micelle binding during our finite simulation time-scale (even extending the simulation up to 6 μs, see Mov4_Pc_SDS.mpg). Thus, the micelle induced LL-14 folding was not computationally tractable and certainly limited by the time scale and simplified description of the system. Nonetheless, MD simulation of Pc binding to SDS provided atomic insight into the thermodynamically hidden (experimentally unresolved) high energy state, i.e., LL-14(Pc):SDS complex. Moreover, MD simulation of Ph binding to SDS was successful in providing atomic model (i.e., LL-14(Ph):SDS complex, Fig. 4e) for the experimentally characterized state (Fig. 6). A large activation barrier between the high energy (LL-14(Pc):SDS complex) and low energy (i.e., LL-14(Ph):SDS complex) state might hinder the observation of micelle induced LL-14 conformational change (Pc → Ph) during the course of MD simulations. Interestingly, MD simulations not only provided atomic insight into the various stages (viz., initial (Fig. 4b), final (Fig. 4e) complex) of peptide binding and/insertion process but was also useful in understanding the kinetic aspects (Fig. 3). An increase in salt (NaCl) concentration shielded the charges of both the SDS micelle and peptide resulting in a delayed binding process (Fig. 3). The final structure of the peptide-micelle complex was well converged and independent of the salt concentration. Thus, although the salt delayed the forward-rate (kon) of the LL-14:SDS initial binding event, the atomic interaction network observed in the final peptide-micelle complex was independent of the salt concentration. Fluorescence experiments showed a similar amount of blue shift in the λmax of the Trp fluorescence emission in the presence of SDS both in the absence and presence of salt, suggesting similar amount of Trp side chain burial in the LL14:SDS complex. This was completely in alignment with the findings from the simulation (Fig. 4e). Though, the difference in the interaction times of the peptide with the SDS micelles was small (∼1 ns), it should be remembered that the simulations were performed at a much higher concentration (∼6× MIC) due to the constraint in the computational expenditure. Inspite of the absolute value of the delay time, our results definitely showed the trend of the effect of salt ions. It may be hypothesized that slowing down of the initial peptide-micelle binding process might facilitate microbial progeny growth, leading to the impaired activity of LL-14 in the presence of salt. It should be noted, that our argument did not underestimate the crucial effect of hydrophobic stabilization in peptide-micelle/membrane interactions. It was an important component of antimicrobial effect, but came into play only after the initial peptide-micelle binding event. The present results also emphasized the importance of terminals of the peptide in rendering the activity of the AMPs, consistent with the previous experimental study.33 We had demonstrated earlier that the conversion of the free C-terminal to the amidated analogue in a small synthetic AMP P4, improved the antimicrobial potency many folds. Thus, α-helical conformation of any cationic peptide and lower salt (NaCl) concentration would be ideal conditions for fast peptide-micelle (SDS) binding.
MD studies based on simple micelle models certainly have limitations in addressing issues like the effect of various other salts (monovalent, divalent etc.), importance of peptide-lipid ratios, role of proteins embedded in the bacterial membrane, inadequate representation of microbial lipids etc. Nevertheless, systematic MD simulations emphasized the importance of (1) the secondary structure and the N-terminal of LL-14 for establishing fast initial contact with SDS, and (2) salt in delaying the peptide-micelle association.
Thus the above discussions indicated that: (i) peptide-SDS micelle binding was sequential and electrostatics drove the initial binding followed by van der Waals stabilization. (ii) Presence of salt (NaCl) delayed the initial peptide:micelle contact by shielding the overall charge of the interacting partners. (iii) Peptide in α-helical conformation (Ph) bound faster to the SDS micelle than its random-coil analogue (Pc). Thus, peptide:micelle binding kinetics depended on both the salt concentration and the nature of the peptide (Pc or Ph). (iv) The converged MD structure of the final LL-14(Ph):SDS complex (Fig. 4e) corroborated with the fluorescence experiments. The computational analysis complemented our experimental studies by providing structural and dynamical insight into the experimentally unresolved/unrealized high-energy states, like, free Ph in water and final Pc:SDS complex (Fig. 4b). LL-14 binding to DPC micelle (zwitterionic eukaryotic membrane mimic) was observed to be much slower relative to SDS micelle binding.
The CD and fluorescence experiments were performed in the SDS micellar environment (above CMC concentration) at pH = 7. SDS is highly acidic with a pKa = −1.5.66 Thus, it is unlikely to alter the protonation state of the SDS molecule (negatively charged) around pH = 7. Thus, MD simulations were performed for charged SDS micelle at the same pH. However, the critical micelle concentration (CMC) of the SDS depends on the pH (ref. 67) and the salt concentration.68 Self-assembly behaviour (micelle-bilayer) of lauric acid (fatty acid) was investigated extensively by Morrow et al., employing continuous constant pH molecular dynamics (CpHMD) simulations.69,70 It was shown that lauric acid self-assembly was pH dependent (low pH favoured bilayer and high pH favoured micelle). Although, lauric acid is different from SDS as the former contain –COOH group whereas the later contain –SO4 group, but the results were of general importance to the surfactant assembly. Vila Viçosa et al. investigated71 the titration curve of oleic acid (carboxylic acid with 18 carbons) bilayer using extended CpHMD method (CpHMD-L). The computed titration curves were in good agreement with the experimental data. The adopted methodology (CpHMD-L) is very promising for studying more realistic pH sensitive lipid bilayers. Similar to SDS, the 14-mer AMP (LKWLKKLLKWLKKL) considered in this study contained titratable lysine side-chains and amino N-terminal. The pKa of the side-chain of lysine (amino acid) and N-terminal of Leucine (amino acid) are 10.53 and 9.6 respectively.72 At neutral pH = 7 the titratable residues (of the free peptide) were expected to be positively charged, thus, simulations were performed with +7 charged 14-mer peptide. However, large shifts in pKa values (∼5 units) of lysine residues buried inside a protein interior was reported by Isom et al.73 Titration events were shown to alter the AMP:micelle binding event.74–79 In the present study however, salt-bridge interaction was reported to be formed involving negatively charged SDS surface and NH3+ tips of peptide. The ion-pair (SO4−:NH3+) might alter the charge state to form (SO4H:NH2) in the anhydrous hydrophobic core, investigating which is beyond the scope of the present study. Thus, continuous constant pH molecular dynamics simulations might be useful for understanding the role of pH in the mechanism of peptide:micelle binding.
Conclusion
In this paper we have successfully established the link between the salt concentration (as well as secondary-structure of peptide) and the speed of peptide:micelle binding, and achieved structural convergence of the final peptide:micelle complex that was complemented by experimental observations. Salt (NaCl) induced slowing down of the kinetics of the peptide micelle interaction might be one of the possible reasons for the salt sensitivity of the AMPs in general. A detailed thermodynamic analysis (binding affinities at various salt concentrations) of peptide:micelle interaction will be reported in a separate study. We expect that the observations reported here will be relevant for other cationic peptides and micelle mimetic systems in general.Abbreviations
| AMP | Anti-microbial peptide |
| CD | Circular dichroism |
| CMC | Critical micelle concentration |
| DPC | Dodecylphosphocholine |
| MD | Molecular dynamics |
| NMR | Nuclear magnetic resonance |
| PDB | Protein data bank |
| PME | Particle mesh Ewald |
| RMSD | Root mean square deviation |
| RMSF | Root mean square fluctuation |
| SDS | Sodium dodecyl sulphate |
| Trp | Tryptophan |
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
SG, GP, SD thanks MHRD, Govt. of India for funding. PARAM-ISHAN, Bimolecular Simulation Lab (BSL) of Department of Biosciences and Bioengineering and BIF facility (Supported by DBT, Project Code: BT/BI/12/064/2012 (NER-BIF)) of IIT Guwahati is gratefully acknowledged for providing the computing facility. SC acknowledges Department of Biotechnology (BT/PR21251/NNT/28/1067/2016) for funding.References
- J. Lakshmaiah Narayana and J. Y. Chen, Antimicrobial peptides: Possible anti-infective agents, Peptides, 2015, 72, 88–94 CrossRef CAS PubMed.
- A. A. Bahar and D. Ren, Antimicrobial Peptides, Pharmaceuticals, 2013, 6, 1543–1575 CrossRef PubMed.
- N. Strempel, J. Strehmel and J. Overhage, Potential Application of Antimicrobial Peptides in the Treatment of Bacterial Biofilm Infections, Curr. Pharm. Des., 2014, 21, 67–84 CrossRef PubMed.
- H. K. Kang, C. Kim, C. H. Seo and Y. Park, The therapeutic applications of antimicrobial peptides (AMPs): a patent review, J. Microbiol., 2017, 55, 1–12 CrossRef CAS PubMed.
- M. D. Seo, H. S. Won, J. H. Kim, T. Mishig-Ochir and B. J. Lee, Antimicrobial Peptides for Therapeutic Applications: A Review, Molecules, 2012, 17, 12276–12286 CrossRef CAS PubMed.
- R. Sinha and P. Shukla, Antimicrobial Peptides: Recent Insights on Biotechnological Interventions and Future Perspectives, Protein Pept. Lett., 2018, 26, 79–87 CrossRef PubMed.
- V. M. Dcosta, C. E. King, L. Kalan, M. Morar, W. W. L. Sung, C. Schwarz, D. Froese, G. Zazula, F. Calmels, R. Debruyne, G. B. Golding, H. N. Poinar and G. D. Wright, Antibiotic resistance is ancient, Nature, 2011, 477, 457–461 CrossRef CAS PubMed.
- K. Bush, P. Courvalin, G. Dantas, J. Davies, B. Eisenstein, P. Huovinen, G. A. Jacoby, R. Kishony, B. N. Kreiswirth, E. Kutter, S. A. Lerner, S. Levy, K. Lewis, O. Lomovskaya, J. H. Miller, S. Mobashery, L. J. V. Piddock, S. Projan, C. M. Thomas, A. Tomasz, P. M. Tulkens, T. R. Walsh, J. D. Watson, J. Witkowski, W. Witte, G. Wright, P. Yeh and H. I. Zgurskaya, Tackling antibiotic resistance, Nat. Rev. Microbiol., 2011, 9, 894–896 CrossRef CAS PubMed.
- G. Taubes, The bacteria fight back, Science, 2008, 356–361 CrossRef CAS PubMed.
- C. Willyard, The drug-resistant bacteria that pose the greatest health threats, Nature, 2017, 543, 15 CrossRef CAS PubMed.
- E. D. Brown and G. D. Wright, Antibacterial drug discovery in the resistance era, Nature, 2016, 529, 336–343 CrossRef CAS PubMed.
- J. D. Steckbeck, B. Deslouches and R. C. Montelaro, Antimicrobial peptides: new drugs for bad bugs?, Expert Opin. Biol. Ther., 2014, 14, 11–14 CrossRef CAS PubMed.
- M. Zasloff, Antimicrobial peptides of multicellular organisms, Nature, 2002, 415, 389–395 CrossRef CAS PubMed.
- P. La Rocca, P. C. Biggin, D. P. Tieleman and M. S. P. Sansom, Simulation studies of the interaction of antimicrobial peptides and lipid bilayers, Biochim. Biophys. Acta, Biomembr., 1999, 1462, 185–200 CrossRef CAS.
- D. P. Tieleman and M. S. P. Sansom, Molecular dynamics simulations of antimicrobial peptides: from membrane binding to trans-membrane channels, International Journal of Quantum Chemistry, John Wiley and Sons Inc., 2001, vol. 83, pp. 166–179 Search PubMed.
- H. Khandelia, A. A. Langham and Y. N. Kaznessis, Driving engineering of novel antimicrobial peptides from simulations of peptide-micelle interactions, Biochim. Biophys. Acta, Biomembr., 2006, 1758, 1224–1234 CrossRef CAS PubMed.
- Y. Shai, Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by α-helical antimicrobial and cell non-selective membrane-lytic peptides, Biochim. Biophys. Acta, Biomembr., 1999, 1462, 55–70 CrossRef CAS.
- H. Khandelia and Y. N. Kaznessis, Molecular dynamics investigation of the influence of anionic and zwitterionic interfaces on antimicrobial peptides' structure: Implications for peptide toxicity and activity, Peptides, 2006, 27, 1192–1200 CrossRef CAS PubMed.
- H. Khandelia and Y. N. Kaznessis, Molecular dynamics simulations of the helical antimicrobial peptide ovispirin-1 in a zwitterionic dodecylphosphocholine micelle: Insights into host-cell toxicity, J. Phys. Chem. B, 2005, 109, 12990–12996 CrossRef CAS PubMed.
- H. Khandelia and Y. N. Kaznessis, Molecular dynamics simulations of helical antimicrobial peptides in SDS micelles: What do point mutations achieve?, Peptides, 2005, 26, 2037–2049 CrossRef CAS PubMed.
- S. K. Kandasamy and R. G. Larson, Molecular dynamics study of the lung surfactant peptide SP-B1-25 with DPPC monolayers: Insights into interactions and peptide position and orientation, Biophys. J., 2005, 88, 1577–1592 CrossRef CAS PubMed.
- S. K. Kandasamy and R. G. Larson, Chemistry and Physics of Lipids, Binding and insertion of α-helical anti-microbial peptides in POPC bilayers studied by molecular dynamics simulations, Chem. Phys. Lipids, 2004, 132, 113–132 CrossRef CAS PubMed.
- C. Appelt, F. Eisenmenger, R. Kühne, P. Schmieder and J. A. Söderhäll, Interaction of the antimicrobial peptide cyclo(RRWWRF) with membranes by molecular dynamics simulations, Biophys. J., 2005, 89, 2296–2306 CrossRef CAS PubMed.
- C. M. Shepherd, H. J. Vogel and D. P. Tieleman, Interactions of the designed antimicrobial peptide MB21 and truncated dermaseptin S3 with lipid bilayers: Molecular-dynamics simulations, Biochem. J., 2003, 370, 233–243 CrossRef CAS PubMed.
- A. D. MacKerell, Molecular dynamics simulation analysis of a sodium dodecyl sulfate micelle in aqueous solution: Decreased fluidity of the micelle hydrocarbon interior, J. Phys. Chem., 1995, 99, 1846–1855 CrossRef CAS.
- C. M. Shepherd, K. A. Schaus, H. J. Vogel and A. H. Juffer, Molecular dynamics study of peptide-bilayer adsorption, Biophys. J., 2001, 80, 579–596 CrossRef CAS PubMed.
- T. Wymore and T. C. Wong, The structure and dynamics of acth (1–10) on the surface of a sodium dodecylsulfate (sds) micelle: A molecular dynamics simulation study, J. Biomol. Struct. Dyn., 2000, 18, 461–476 CrossRef CAS PubMed.
- T. Wymore and T. C. Wong, Molecular dynamics study of substance P peptides partitioned in a sodium dodecylsulfate micelle, Biophys. J., 1999, 76, 1213–1227 CrossRef CAS PubMed.
- M. F. Lensink, B. Christiaens, J. Vandekerckhove, A. Prochiantz and M. Rosseneu, Penetratin-membrane association: W48/R52/W56 shield the peptide from the aqueous phase, Biophys. J., 2005, 88, 939–952 CrossRef CAS PubMed.
- J. P. Ulmschneider, Charged Antimicrobial Peptides Can Translocate across Membranes without Forming Channel-like Pores, Biophys. J., 2017, 113, 73–81 CrossRef CAS PubMed.
- Q. Wang, G. Hong, G. R. Johnson, R. Pachter and M. S. Cheung, Biophysical properties of membrane-active peptides based on micelle modeling: A case study of cell-penetrating and antimicrobial peptides, J. Phys. Chem. B, 2010, 114, 13726–13735 CrossRef CAS PubMed.
- C. D. Bruce, M. L. Berkowitz, L. Perera and M. D. E. Forbes, Molecular dynamics simulation of sodium dodecyl sulfate micelle in water: Micellar structural characteristics and counterion distribution, J. Phys. Chem. B, 2002, 106, 3788–3793 CrossRef CAS.
- G. Pandit, H. Ilyas, S. Ghosh, A. P. Bidkar, S. Abdul Mohid, A. Bhunia, P. Satpati and S. Chatterjee, Insights into the Mechanism of Antimicrobial Activity of Seven-Residue Peptides, J. Med. Chem., 2018, 61, 7614–7629 CrossRef CAS PubMed.
- N. A. Berglund, T. J. Piggot, D. Jefferies, R. B. Sessions, P. J. Bond and S. Khalid, Interaction of the Antimicrobial Peptide Polymyxin B1 with Both Membranes of E. coli: A Molecular Dynamics Study, PLoS Comput. Biol., 2015, 11, e1004180 CrossRef PubMed.
- J. Li, S. Liu, R. Lakshminarayanan, Y. Bai, K. Pervushin, C. Verma and R. W. Beuerman, Molecular simulations suggest how a branched antimicrobial peptide perturbs a bacterial membrane and enhances permeability, Biochim. Biophys. Acta, Biomembr., 2013, 1828, 1112–1121 CrossRef CAS PubMed.
- P. Das, T. Sercu, K. Wadhawan, I. Padhi, S. Gehrmann, F. Cipcigan, V. Chenthamarakshan, H. Strobelt, C. dos Santos, P. Y. Chen, Y. Y. Yang, J. P. K. Tan, J. Hedrick, J. Crain and A. Mojsilovic, Accelerated antimicrobial discovery via deep generative models and molecular dynamics simulations, Nat. Biomed. Eng., 2021, 1–11 Search PubMed.
- G. Pandit, K. Biswas, S. Ghosh, S. Debnath, A. P. Bidkar, P. Satpati, A. Bhunia and S. Chatterjee, Rationally designed antimicrobial peptides: Insight into the mechanism of eleven residue peptides against microbial infections, Biochim. Biophys. Acta, Biomembr., 2020, 1862, 183177 CrossRef CAS PubMed.
- M. J. Goldman, G. M. Anderson, E. D. Stolzenberg, U. P. Kari, M. Zasloff and J. M. Wilson, Human β-defensin-1 is a salt-sensitive antibiotic in lung that is inactivated in cystic fibrosis, Cell, 1997, 88, 553–560 CrossRef CAS PubMed.
- J.-R. C. García, A. Krause, S. Schulz, F.-J. Rodríguez-Jiménez, E. Klüver, K. Adermann, U. Forssmann, A. Frimpong-Boateng, R. Bals and W.-G. Forssmann, Human β-defensin 4: a novel inducible peptide with a specific salt-sensitive spectrum of antimicrobial activity, FASEB J., 2001, 15, 1819–1821 CrossRef.
- R. Bals, X. Wang, Z. Wu, T. Freeman, V. Bafna, M. Zasloff and J. M. Wilson, Human β-defensin 2 is a salt-sensitive peptide antibiotic expressed in human lung, J. Clin. Invest., 1998, 102, 874–880 CrossRef CAS PubMed.
- G. Pandit, N. Chowdhury, S. Abdul Mohid, A. P. Bidkar, A. Bhunia and S. Chatterjee, Effect of Secondary Structure and Side Chain Length of Hydrophobic Amino Acid Residues on the Antimicrobial Activity and Toxicity of 14-Residue-Long de novo AMPs, ChemMedChem, 2021, 16, 355–367 CrossRef CAS PubMed.
- L. Schrodinger, The PyMOL Molecular Graphics System, Version 2.4.0, 2010 Search PubMed.
- J. Lee, X. Cheng, J. M. Swails, M. S. Yeom, P. K. Eastman, J. A. Lemkul, S. Wei, J. Buckner, J. C. Jeong, Y. Qi, S. Jo, V. S. Pande, D. A. Case, C. L. Brooks, A. D. MacKerell, J. B. Klauda and W. Im, CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field, J. Chem. Theory Comput., 2016, 12, 405–413 CrossRef CAS PubMed.
- Y. Croonen, E. Geladé, M. Van Der Zegel, M. Van Der Auweraer, H. Vandendriessche, F. C. De Schryver and M. A. Kuleuven, Influence of Salt, Detergent Concentration, and Temperature on the Fluorescence Quenching of 1-Methylpyrene In Sodium Dodecyl Sulfate with m-Dlcyanobenzene, J. Phys. Chem., 1983, 87, 1426–1431 CrossRef CAS.
- A. R. Rakitin and G. R. Pack, Molecular Dynamics Simulations of Ionic Interactions with Dodecyl Sulfate Micelles, J. Phys. Chem. B, 2004, 108, 2712–2716 CrossRef CAS.
- S. Lebecque, J. M. Crowet, M. N. Nasir, M. Deleu and L. Lins, Molecular dynamics study of micelles properties according to their size, J. Mol. Graphics Modell., 2017, 72, 6–15 CrossRef CAS PubMed.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, GROMACS: Fast, flexible and free, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- J. B. Klauda, R. M. Venable, J. A. Freites, J. W. O'Connor, D. J. Tobias, C. Mondragon-Ramirez, I. Vorobyov, J. A. D. MacKerell and R. W. Pastor, Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types, J. Phys. Chem. B, 2010, 114, 7830–7843 CrossRef CAS PubMed.
- R. B. Best, X. Zhu, J. Shim, P. E. M. Lopes, J. Mittal, M. Feig and J. A. D. MacKerell, Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral angles, J. Chem. Theory Comput., 2012, 8, 3257–3273 CrossRef CAS PubMed.
- S. R. Durell, B. R. Brooks and A. Ben-Naim, Solvent-induced forces between two hydrophilic groups, J. Phys. Chem., 1994, 98, 2198–2202 CrossRef CAS.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, LINCS: A linear constraint solver for molecular simulations, J. Comput. Chem., 1997, 1463–1472 CrossRef CAS.
- B. Hess, P.-LINCS: A parallel linear constraint solver for molecular simulation, J. Chem. Theory Comput., 2008, 4, 116–122 CrossRef CAS PubMed.
- G. Bussi, D. Donadio and M. Parrinello, Canonical sampling through velocity rescaling, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. Van Gunsteren, A. Dinola and J. R. Haak, Molecular dynamics with coupling to an external bath, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- M. Parrinello and A. Rahman, Strain fluctuations and elastic constants, J. Chem. Phys., 1982, 76, 2662–2666 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- A. A. Langham, H. Khandelia and Y. N. Kaznessis, How can a β-sheet peptide be both a potent antimicrobial and harmfully toxic? Molecular dynamics simulations of protegrin-1 in micelles, Biopolymers, 2006, 84, 219–231 CrossRef CAS PubMed.
- T. Kajander, P. C. Kahn, S. H. Passila, D. C. Cohen, L. Lehtiö, W. Adolfsen, J. Warwicker, U. Schell and A. Goldman, Buried charged surface in proteins, Structure, 2000, 8, 1203–1214 CrossRef CAS.
- S. Bogusz, R. M. Venable and R. W. Pastor, Molecular dynamics simulations of octyl glucoside micelles: Structural properties, J. Phys. Chem. B, 2000, 104, 5462–5470 CrossRef CAS.
- R. Itri and L. Q. Amaral, Distance distribution function of sodium dodecyl sulfate micelles by X-ray scattering, J. Phys. Chem., 1991, 95, 423–427 CrossRef CAS.
- D. Bendedouch, S. H. Chen and W. C. Koehler, Structure of ionic micelles from small angle neutron scattering, J. Phys. Chem., 1983, 87, 153–159 CrossRef CAS.
- S. Salaniwal, S. T. Cui, H. D. Cochran and P. T. Cummings, Molecular simulation of a dichain surfactant/water/carbon dioxide system. 1. Structural properties of aggregates, Langmuir, 2001, 17, 1773–1783 CrossRef CAS.
- J. Shelley, K. Watanabe and M. L. Klein, Simulation of a sodium dodecylsulfate micelle in aqueous solution, Int. J. Quantum Chem., 1990, 38, 103–117 CrossRef.
- B. Lee and F. M. Richards, The interpretation of protein structures: Estimation of static accessibility, J. Mol. Biol., 1971, 55, 379 CrossRef CAS.
- W. Kabsch and C. Sander, Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features, Biopolymers, 1983, 22, 2577–2637 CrossRef CAS PubMed.
- K. D. Ristroph and R. K. Prud’homme, Hydrophobic ion pairing: encapsulating small molecules, peptides, and proteins into nanocarriers, Nanoscale Adv., 2019, 1, 4207–4237 RSC.
- A. Rahman and C. W. Brown, Effect of pH on the critical micelle concentration of sodium dodecyl sulphate, J. Appl. Polym. Sci., 1983, 28, 1331–1334 CrossRef CAS.
- M. Abe, K. Kato and K. Ogino, Effects of inorganic electrolytes and of pH on micelle formation of amphoteric-anionic mixed surfactant systems, J. Colloid Interface Sci., 1989, 127, 328–335 CrossRef CAS.
- B. H. Morrow, P. H. Koenig and J. K. Shen, Self-Assembly and Bilayer–Micelle Transition of Fatty Acids Studied by Replica-Exchange Constant pH Molecular Dynamics, Langmuir, 2013, 29, 14823–14830 CrossRef CAS PubMed.
- B. H. Morrow, P. H. Koenig and J. K. Shen, Atomistic simulations of pH-dependent self-assembly of micelle and bilayer from fatty acids, J. Chem. Phys., 2012, 137, 194902 CrossRef PubMed.
- D. Vila-Viçosa, V. H. Teixeira, A. M. Baptista and M. Machuqueiro, Constant-pH MD Simulations of an Oleic Acid Bilayer, J. Chem. Theory Comput., 2015, 11, 2367–2376 CrossRef PubMed.
- D. R. Lide, CRC handbook of chemistry and physics: a ready-reference book of chemical and physical data Search PubMed.
- D. G. Isom, C. A. Castañeda and B. R. Cannon, Large shifts in pKa values of lysine residues buried inside a protein, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 5260–5265 CrossRef CAS PubMed.
- D. dos S. Alvares, I. B. S. Martins, T. G. Viegas, M. S. Palma, A. S. de Araujo, S. J. de Carvalho and J. R. Neto, Modulatory Effects of Acidic pH and Membrane Potential on the Adsorption of pH-Sensitive Peptides to Anionic Lipid Membrane, Membr, 2021, 11, 307 CrossRef PubMed.
- D. Vila-Viçosa, T. F. D. Silva, G. Slaybaugh, Y. K. Reshetnyak, O. A. Andreev and M. Machuqueiro, Membrane-Induced pKa Shifts in wt-pHLIP and Its L16H Variant, J. Chem. Theory Comput., 2018, 14, 3289–3297 CrossRef PubMed.
- V. H. Teixeira, D. Vila-Viçosa, P. B. P. S. Reis and M. Machuqueiro, pKa Values of Titrable Amino Acids at the Water/Membrane Interface, J. Chem. Theory Comput., 2016, 12, 930–934 CrossRef CAS PubMed.
- P. R. Magalhães, M. Machuqueiro and A. M. Baptista, Constant-pH Molecular Dynamics Study of Kyotorphin in an Explicit Bilayer, Biophys. J., 2015, 108, 2282–2290 CrossRef PubMed.
- C. A. Carvalheda, S. R. R. Campos and A. M. Baptista, The Effect of Membrane Environment on Surfactant Protein C Stability Studied by Constant-pH Molecular Dynamics, J. Chem. Inf. Model., 2015, 55, 2206–2217 CrossRef CAS PubMed.
- C. A. Carvalheda, S. R. R. Campos, M. Machuqueiro and A. M. Baptista, Structural Effects of pH and Deacylation on Surfactant Protein C in an Organic Solvent Mixture: A Constant-pH MD Study, J. Chem. Inf. Model., 2013, 53, 2979–2989 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra06772a |
| This journal is © The Royal Society of Chemistry 2021 |