
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImmobilization of glucose oxidase on bioinspired polyphenol coatings as a high-throughput glucose assay platform
Suhair Sunoqrot *a,
Amani Al-Hadida,
Ahmad Manasrahb,
Ruba Khnoufc and
Lina Hasan Ibrahima
*a,
Amani Al-Hadida,
Ahmad Manasrahb,
Ruba Khnoufc and
Lina Hasan Ibrahima
aDepartment of Pharmacy, Faculty of Pharmacy, Al-Zaytoonah University of Jordan, Amman 11733, Jordan. E-mail: suhair.sunoqrot@zuj.edu.jo; Fax: +962 64291423; Tel: +962 64291511 ext. 197
bDepartment of Mechanical Engineering, Faculty of Engineering and Technology, Al-Zaytoonah University of Jordan, Amman 11733, Jordan
cDepartment of Biomedical Engineering, Faculty of Engineering, Jordan University of Science and Technology, Irbid 22110, Jordan
First published on 13th December 2021
Abstract
Glucose oxidase (GOx) is an enzyme with important industrial and biochemical applications, particularly in glucose detection. Here we leveraged the oxidative self-polymerization phenomenon of simple polyphenols (pyrogallol or catechol) in the presence of polyethylenimine (PEI) to form adhesive coatings that enabled GOx immobilization on conventional multi-well plates. Immobilization was verified and optimized by directly measuring GOx activity inside the coated wells. Our results showed that incorporating PEI in polyphenol coatings enhanced their enzyme immobilization efficiency, with pyrogallol (PG)-based coatings displaying the greatest enzyme activity. The immobilized enzyme maintained similar affinity to glucose compared to the free enzyme. GOx-immobilized PG/PEI-coated wells exhibited intermediate recycling ability but excellent resistance to urea as a denaturing agent compared to the free enzyme. GOx-immobilized 96-well plates allowed the construction of a linear glucose calibration curve upon adding glucose standards, with a detection limit of 0.4–112.6 mg dL−1, which was comparable to commercially available enzymatic glucose assay kits. The assay platform was also capable of effectively detecting glucose in rat plasma samples. Our findings present a simple enzyme immobilization technique that can be used to construct a glucose assay platform in a convenient multi-well format for high-throughput glucose quantification.
Introduction
Glucose oxidase (GOx; β-D-glucose:oxygen 1-oxidoreductase; EC 1.1.3.4) is a dimeric flavoprotein composed of two identical subunits of 80 kDa each.1 GOx is commercially produced from the fungi Aspergillus niger and Penicillium glaucum. It utilizes molecular oxygen to catalyze the oxidation of β-D-glucose to gluconic acid, coupled to the reduction of the flavin adenine dinucleotide (FAD) cofactor into FADH2. To regenerate the enzyme, FADH2 is oxidized by oxygen (O2), producing FAD and hydrogen peroxide (H2O2).2 Owing to its reaction mechanism, GOx has been used in a wide variety of applications, most importantly glucose sensing. As the reaction metabolizes glucose, GOx has been extensively used in glucose-sensing devices where it oxidizes blood glucose and the produced electrons from the oxidation reaction are harvested to produce an electrical current, which then is transformed into a readable result.3Enzyme immobilization can be defined as the confinement of an enzyme to a phase different from the one for substrates and products.4 Immobilization has emerged as a powerful tool to improve the stability, reusability, and cost-effectiveness of catalytic enzymes.5 Examples of matrices for enzyme immobilization include organic and inorganic supports, as well as hybrid and composite materials.6 GOx is one of the most widely investigated immobilized enzymes. Beside its use in glucose detection, GOx has been investigated for applications in the chemical, pharmaceutical, food, biotechnology, textile, and other industries.7 For these applications, GOx has been immobilized on various types of surfaces using a variety of techniques such as adsorption, physical entrapment, and covalent attachment.8 Recent examples include GOx immobilization on tannin-coated NiFe2O4 magnetic nanoparticles for dye removal,9 entrapment within poly(vinyl alcohol)/cyclodextrin hydrogels for continuous glucose monitoring,10 and immobilization on metal–organic frameworks for glucose detection.11
In addition to its use in glucose sensors, GOx forms an essential component of commercially available enzymatic glucose assay kits. In addition to GOx, these kits contain a peroxidase such as horseradish peroxidase (HRP), a chromogenic peroxidase substrate, and glucose standards. The reaction of GOx with glucose yields H2O2, which in turn is used by the peroxidase to oxidize its substrate leading to a color change. The intensity of the color can then be used to determine unknown glucose concentrations based on a glucose standard calibration curve. These kits typically contain amounts sufficient for 100 assays (e.g., enough for one 96-well plate) and often cost more than the price of the individual kit components. Additionally, if the kit is not used all at once it becomes prone to cross-contamination.
Upon the emergence of mussel-inspired polydopamine coatings,12,13 attention has also turned to plant polyphenols. Several of these chemically diverse compounds have been investigated as precursors for substrate-independent functional coatings in various applications.14–16 These coatings are typically formed upon oxidation-mediated self-polymerization of the polyphenols, which can conveniently be triggered in mild alkaline solutions.17 The reactivity of the oxidized polyphenols has been leveraged to graft various surface ligands and biomolecules without requiring conventional chemical activation or toxic crosslinkers.18 The robustness of polyphenol coatings may be further enhanced by incorporating polyamines, producing composite coatings with physicochemical properties comparable to polydopamine.19,20 In this work, we hypothesized that we could devise a simplified way to immobilize GOx directly inside multi-well plates using bioinspired adhesive coatings composed of simple polyphenols such as pyrogallol (PG) and catechol (CT). These coatings were reinforced by incorporating a polyamine such as branched polyethylenimine (PEI) in the coating solution, allowing the primary amines of PEI to react with the oxidized polyphenols, mimicking mussel-inspired adhesion.21,22 Similarly, GOx could adhere to the polyphenol coating due the abundance of nucleophiles (free amines and thiols) within its amino acid side chains, in addition to noncovalent interactions. Through a series of characterization experiments, we show for the first time that bioinspired coatings formed from simple polyphenols reinforced with PEI can efficiently immobilize GOx without loss of affinity or stability, enabling glucose quantification in a high-throughput manner.
Experimental
Materials
Glucose oxidase (GOx) from Aspergillus niger (Type X-S, 149![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 units per g solid), polyethyleneimine (PEI, branched, average Mw 25000 by light scattering), pyrogallol (PG), bicine, gallic acid, and the Folin–Ciocalteu reagent (10 N) were obtained from Sigma-Aldrich (St. Louis, MO, USA). Catechol (CT) and phosphate-buffered saline (PBS, 10× solution) were purchased from Fisher (Waltham, MA, USA). Glucose, horseradish peroxidase (HRP, RZ 3.0), and 3,3′,5,5′-tetramethylbenzidine (TMB) were procured from Santa Cruz Biotechnology (Dallas, TX, USA). Urea was obtained from Panreac Quimica (Barcelona, Spain). Ultrapure water was prepared using a 5-UV water purification system (EMD Millipore).
500 units per g solid), polyethyleneimine (PEI, branched, average Mw 25000 by light scattering), pyrogallol (PG), bicine, gallic acid, and the Folin–Ciocalteu reagent (10 N) were obtained from Sigma-Aldrich (St. Louis, MO, USA). Catechol (CT) and phosphate-buffered saline (PBS, 10× solution) were purchased from Fisher (Waltham, MA, USA). Glucose, horseradish peroxidase (HRP, RZ 3.0), and 3,3′,5,5′-tetramethylbenzidine (TMB) were procured from Santa Cruz Biotechnology (Dallas, TX, USA). Urea was obtained from Panreac Quimica (Barcelona, Spain). Ultrapure water was prepared using a 5-UV water purification system (EMD Millipore).
Immobilization of GOx on polyphenol-coated multi-well plates
Polyphenol-mediated GOx immobilization on the surfaces of standard tissue culture-treated 24-well plates (SPL Life Sciences, Korea) was carried out as previously described with some modifications.18 First, an adhesive coating of polyphenols was deposited on the bottoms of the well plates by relying on the auto-oxidation of the polyphenol precursors in mild alkaline buffer, leading to the formation of surface-adherent films. Co-deposition of polyphenols with PEI was also employed to obtain reinforced films. Coating solutions were freshly prepared by dissolving PG and CT in 0.1 M bicine buffer (with 0.6 M NaCl, pH 7.8) at a concentration of 2 mg mL−1. A PEI stock solution was also prepared at 2 mg mL−1 in 0.1 M bicine buffer (with 0.6 M NaCl, pH 7.8). Two hundred and fifty microliters of the same bicine buffer were added to the wells in duplicate, followed by adding 250 μL of the PG or CT coating solutions to the same wells. For co-deposition with PEI, 250 μL of the PEI stock solution were added to another set of wells in duplicate, followed by adding 250 μL of the PG or CT coating solutions to the same wells. A third set of wells was treated with 500 μL of 1 mg mL−1 PEI in bicine buffer as a control. Thus, all treated wells contained 500 μL of coating solution and the final concentration of each polyphenol and PEI was 1 mg mL−1. The plates were then wrapped with parafilm and placed in an orbital shaking incubator (Biosan ES-20, Riga, Latvia) operating at 50 rpm for 4 h at 25 °C. After 4 h, all the solutions were removed, and the wells were washed with ultrapure water three times. Immediately after, 500 μL of GOx (1 mg mL−1 in PBS) were added to each of the coated wells and to a set of uncoated wells as a control. The plates were then wrapped with parafilm and placed back in the orbital shaking incubator for another 20 h of incubation at 50 rpm and 25 °C. After 20 h, each well was washed with ultrapure water three times, the plates were wrapped with parafilm, and stored at 4 °C till further characterization. To determine the effect of GOx concentration on the amount immobilized, the coating steps were performed in 24-well plates as described above, except that before adding GOx, serial dilutions of the GOx stock solution in PBS (0.125, 0.250, 0.500, and 1.000 mg mL−1) were prepared directly in the coated wells and the immobilization was carried out as described above.Total phenol content of polyphenol-coated plates
The amount of polyphenol (PG or CT) adhered to the surface of the plates was measured immediately after coating before GOx immobilization. The method was adapted from the Folin–Ciocalteu total phenol assay.23,24 Briefly, the Folin–Ciocalteu reagent was diluted 1:10 in ultrapure water. Gallic acid standards were prepared in the range 12.5–200.0 μg mL−1 in ultrapure water, and 125 μL of each dilution were placed in a separate well of an untreated 24-well plate. Then, 125 μL of ultrapure water were added to each polyphenol-coated well. Five hundred microliters of the Folin–Ciocalteu reagent were added to each well containing gallic acid standards and the blank (125 μL of ultrapure water), and polyphenol-coated wells. After 5 min, 500 μL of 10% sodium bicarbonate were added to the same wells, and the plates were incubated in the dark for 30 min. The absorbance of the plates was read at 765 nm using a Synergy HTX multi-mode microplate reader (Biotek, Winooski, VT, USA). After subtracting the blank reading, the absorbance of the wells containing gallic acid dilutions was plotted against gallic acid concentration (μg mL−1) to obtain a calibration curve. The polyphenol content in the coated wells was then determined based on the gallic acid calibration curve and expressed as gallic acid equivalents (GAE). The experiment was performed at least three times and duplicate wells were tested each time.
Activity assay of immobilized GOx
GOx activity was determined directly in the well plates by measuring the rate of H2O2 formation upon adding a fixed amount of glucose to each well. GOx catalyzes the oxidation of glucose, resulting in the formation of gluconic acid and H2O2. The rate of H2O2 formation can in turn be monitored by adding HRP and TMB. HRP catalyzes H2O2-mediated oxidation of TMB to produce a blue colored product with a distinct absorbance peak at 650 nm (Scheme 1).25 All activity assays were conducted using a Synergy HTX multi-mode microplate reader (Biotek) equipped with Gen5 software version 3.08. The measurement temperature was set to 25 °C, and the protocol was set to the kinetic mode. Readings were taken at 1 min intervals for a total of 10 min, where each interval included shaking for 10 s before taking the measurement at 650 nm. For the activity assay in 24-well plates, the reaction mixture was prepared to a final volume of 450 μL per well and the total volume of the reaction mixture was adjusted according to the total number of wells to be tested. Each 450 μL of reaction mixture were composed of 25 μL HRP (from a 1 mg mL−1 stock solution in PBS), 5 μL TMB (from a 10 mg mL−1 stock solution in DMSO), and 420 μL PBS. The glucose substrate solution was separately prepared at 2.25 mg mL−1 (12.5 mM) in PBS. The concentration of the glucose solution was chosen to obtain a linear reaction rate that could be measured spectrophotometrically within 5–10 min. After setting up the microplate reader and adding the reaction mixture to the wells, the plate was placed in the microplate reader, 50 μL of the glucose substrate solution were pipetted into each well, and the kinetic measurements were immediately started. After the last kinetic interval, the absorbance readings were plotted versus time. The reaction rate of each well was then obtained by linear regression analysis of the absorbance versus time plots. Each experiment was performed at least three times and duplicate wells were tested each time. To quantify the actual amount of GOx immobilized in each well, a calibration curve was constructed from the reaction rate versus free GOx concentration. Briefly, serial dilutions of free GOx were prepared in PBS, and then the activity assay was performed in untreated 24-well plates by mixing 50 μL of GOx standards with 25 μL HRP (from a 1 mg mL−1 stock solution in PBS), 5 μL TMB (from a 10 mg mL−1 stock solution in DMSO), and 370 μL PBS (total volume = 450 μL per well). The activity assay was initiated by adding 50 μL of 2.25 mg mL−1 glucose to each well, and the reaction rate for each GOx concentration was obtained as described above.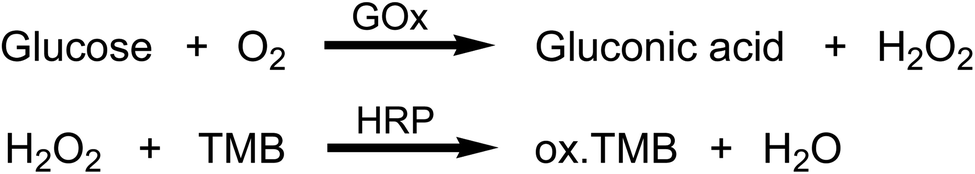 | ||
| Scheme 1 Enzymatic cascade reactions used for the detection of GOx activity. | ||
Determination of the Michaelis–Menten constant (Km)
Km for immobilized GOx was determined from the Michaelis–Menten plot. First, serial dilutions of glucose (0.28–9.00 mg mL−1 in PBS; equivalent to 1.56–50.00 mM) were prepared. Then, 450 μL of reaction mixture (25 μL HRP, 5 μL TMB, and 420 μL PBS) were added to each well of GOx-immobilized 24-well plates in duplicate. After setting up the microplate reader in the kinetic mode, 50 μL of each glucose dilution were added to the wells and the measurements were started. The reaction rates were then plotted versus glucose concentration and the plot was fitted to the Michaelis–Menten equation using Graphpad Prism 7. The same experiment was performed on free GOx in untreated 24-well plates by mixing 50 μL free GOx (31.25 μg mL−1 in PBS), 25 μL HRP, 5 μL TMB, and 370 μL PBS in each well, followed by adding different concentrations of glucose and measuring the reaction rate. All experiments were performed at least three times.Stability of immobilized GOx
The stability of immobilized GOx was determined after repeated cycles of glucose oxidation and after incubation with urea as a denaturing agent. The recycling ability of immobilized GOx was tested by performing the activity assay as described above which was designated as cycle 1. Immediately after, the plates were washed with ultrapure water three times and the activity assay was repeated for a total of five cycles, washing the wells after each cycle before adding a fresh reaction mixture. The results were expressed as the relative reaction rate % compared to the control wells (cycle 1). Stability in the presence of urea was determined by incubating freshly prepared GOx-immobilized plates with 500 μL of urea (1, 2, 4, and 5 M in PBS) for 24 h at 25 °C. After washing the wells with ultrapure water three times, the activity assay was performed as described above and the results were expressed as the relative reaction rate % compared to the control wells (without urea treatment). Stability of free GOx in the presence of urea was conducted by mixing the enzyme at an amount equivalent to immobilized GOx with 500 μL of urea (1, 2, 4, and 5 M in PBS). The samples were incubated for 24 h at 25 °C. Afterwards, the activity assay was performed by mixing 50 μL of each sample with the reaction mixture (25 μL HRP, 5 μL TMB, and 370 μL PBS) in 24-well plates, followed by adding 50 μL of 2.25 mg mL−1 glucose to each well and initiating the activity assay as described above. All the stability assays were performed at least three times and duplicate wells were tested each time.Glucose assay
To assess the utility of GOx-immobilized well plates as a high-throughput glucose assay platform, GOx was immobilized on polyphenol-coated 96-well plates. Polyphenol coating and GOx immobilization were carried out as described above for 24-well plates, except that the volumes of the reagents were adjusted to achieve a final volume of 100 μL instead of 500 μL per well. Serial dilutions of glucose were prepared in PBS in the range 4.4–1125.0 μg mL−1 (equivalent to 24.4–6250.0 μM). To test the assay platform's selectivity to glucose, plasma samples from three healthy adult male Wistar rats were diluted 1:1 in PBS and used alongside the glucose dilutions. The plasma samples were obtained from control animals treated with saline as part of a previously approved study (Al-Zaytoonah University of Jordan's Institutional Review Board decision no. 1/2/2020-2021). All experiments were conducted in accordance with the Helsinki guidelines for animal research. The glucose assay was carried out similar to the GOx activity assay, except serial dilutions of glucose were used in the reaction mixture instead of using a fixed glucose concentration. The reaction mixture was prepared by mixing 5 μL HRP, 1 μL TMB, and 84 μL PBS (for a total of 90 μL per well). The total volume of the reaction mixture was adjusted according to the total number of wells to be assayed, including blanks, glucose dilutions, and plasma samples. Ninety microliters of the reaction mixture were then added to each well, followed by adding 10 μL of each glucose dilution or plasma sample in duplicate. The plate was immediately transferred to an orbital shaking incubator (50 rpm, 25 °C) and incubated for 30 min. After 30 min, 20 μL of a stop solution (0.16 M sulfuric acid) were added to each well to convert the TMB blue product to a more stable, yellow-colored chromogen. The plate was transferred to a microplate reader and the absorbance was read at 450 nm. A glucose calibration curve was constructed by linear regression analysis of the absorbance at 450 nm versus glucose concentration. The same plasma samples were analyzed using a commercial glucose assay kit for comparison (ab272532, Abcam, Cambridge, UK) following the manufacturer's instructions.
Statistical analysis
All results were expressed as the mean ± SD from at least three independent experiments. Statistical differences were analyzed by 1- or 2-way analysis of variance (ANOVA), followed by Tukey's or Sidak's multiple comparisons tests, respectively, where p < 0.05 was considered statistically significant.Results and discussion
GOx immobilization in polyphenol-coated multi-well plates
GOx was immobilized in multi-well plates via polyphenol adhesive coatings as illustrated in Fig. 1. Polyphenol-mediated surface modification has emerged as a simple technique to alter the surface properties of various substrates under ambient conditions, without the need for crosslinkers or harsh reaction conditions.14 In addition to the now popular polydopamine surface coating strategy, plant polyphenols have also been investigated as coating precursors. Examples include tannic acid (TA), epigallocatechin gallate (EGCG), and catechin.17 Simple polyphenols such as gallic acid, PG, and CT have also been shown to form surface coatings through oxidative self-polymerization.26 Polyphenol coatings have been used to impart a variety of functionalities to the substrate surface, including antifouling, targeting, sensing, and catalysis.16 Several enzymes have previously been immobilized on similar surfaces, enabled by multiple types of interactions between the enzyme and the oxidized polyphenol coatings, such as nucleophilic addition reactions, H-bonding, and van der Waals interactions.18 GOx has been immobilized on TA/PEI coated microcapsules and nanoparticles, for glucose sensing25 and for anticancer drug delivery,27 respectively. To best of our knowledge, no attempt has been made to immobilize GOx on surfaces coated with simple polyphenols, nor has the immobilization been performed in conventional multi-well plates.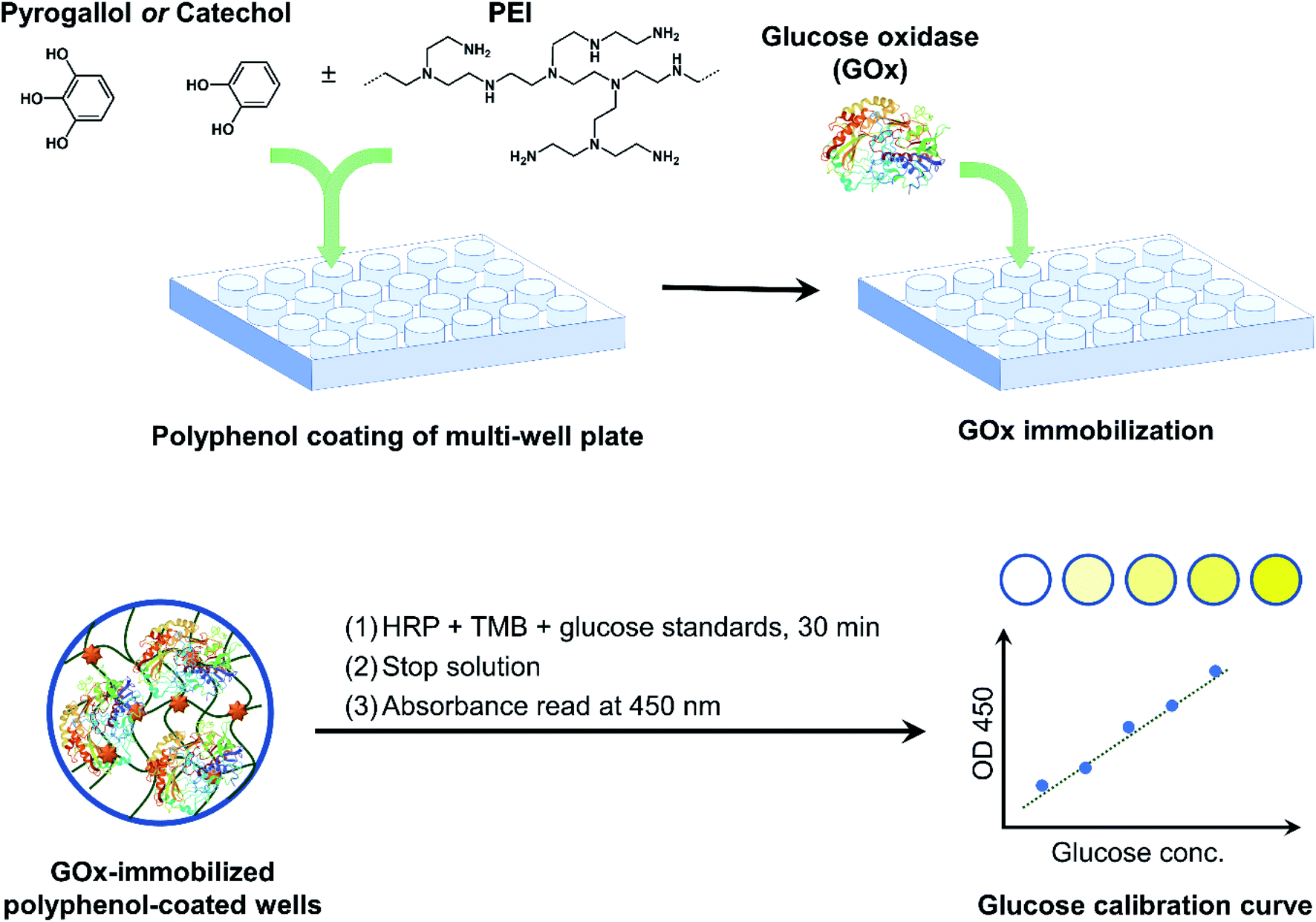 | ||
| Fig. 1 Overview of GOx immobilization in multi-well plates via bioinspired polyphenol chemistry. GOx-immobilized plates were then used as a colorimetric glucose assay platform. | ||
PG and CT with and without PEI were used to coat the wells of conventional microplates through oxidative self-polymerization of the polyphenols in a mildly alkaline buffer. The plates were composed of tissue culture-treated polystyrene, rendering the wells hydrophilic. The coating conditions including the buffer composition and coating time were chosen based on previous work.18,28 The geometry of the substrate (wells of a multi-well plate) made it difficult to directly verify successful surface modification after polyphenol coating through typical surface chemical analysis techniques (e.g., infrared and/or X-ray photoelectron spectroscopy). Instead, we relied on measuring the polyphenol content inside each well using the Folin–Ciocalteu method. As shown in Fig. 2, CT-coated wells were associated with the lowest polyphenol content (0.6 ± 1.2 μg mL−1 GAE), indicating their weak adhesive properties, unlike PG which demonstrated a higher polyphenol content of 4.7 ± 1.2 μg mL−1 GAE. This finding may be attributed to the presence of three phenolic hydroxyl groups in PG while CT has only two. When PEI was added to the CT coating solution, more CT could be incorporated in the coating as the GAE per well was significantly increased to 7.9 ± 2.2 μg mL−1 (p < 0.01). Likewise, the addition of PEI to the PG precursor solution resulted in a significant increase in GAE from 4.7 ± 1.2 to 33.1 ± 8.1 μg mL−1 per well (p < 0.0001). Moreover, PG/PEI-coated wells were associated with the greatest GAE per well across all groups.
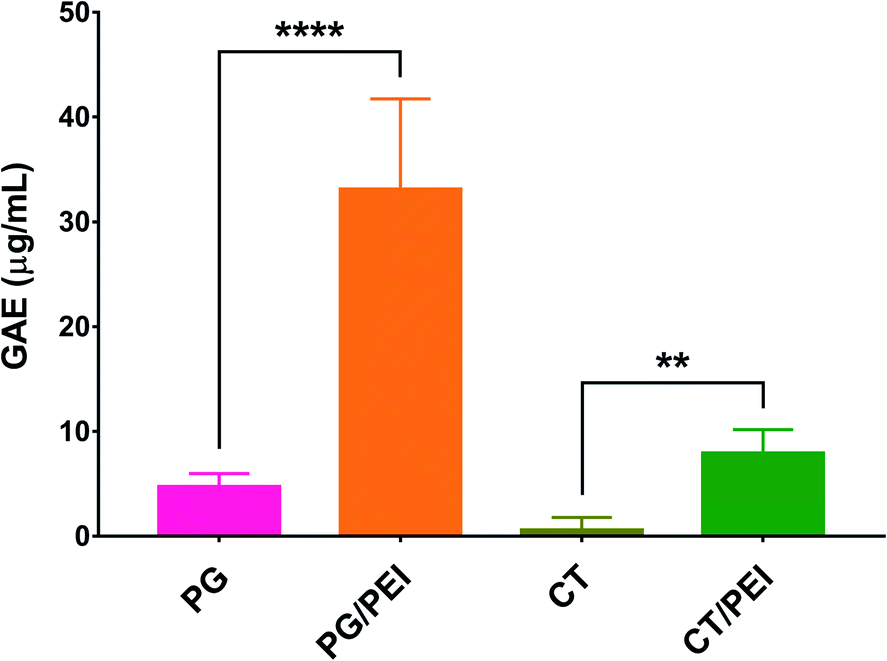 | ||
| Fig. 2 Total phenol content of 24-well plates coated with polyphenols (PG or CT) with or without co-deposition with PEI. Results are expressed as gallic acid equivalents (GAE) in μg mL−1. **p < 0.01 and ****p < 0.0001 based on 1-way ANOVA followed by Tukey's multiple comparisons test. | ||
Oxidation of polyphenols such as CT and PG leads to the formation of reactive ortho-quinones and α-hydroxy-ortho-quinones, respectively. These intermediates can behave as electrophiles, allowing nucleophilic addition reactions to occur on the aromatic ring. They can also act as nucleophiles, capable of reacting with various electrophiles including other molecules of the same polyphenol, as well as (hetero)dienes and/or dienophiles in Diels–Alder-type cycloaddition reactions.29 As demonstrated in Fig. 2, the addition of a polyamine such as PEI mediated the incorporation of more polyphenol molecules in the coating. This may be attributed to the abundance of primary amines in PEI which can undergo Michael addition and/or Schiff's base formation reactions with the oxidized polyphenols, further strengthening the coating layer through crosslinking (Scheme 2).19,25 PEI, a polycation, may also adhere to the polyphenol coating via cation–π interactions with the partially negative aromatic rings of PG and CT.30,31 Our findings are consistent with previous reports. For example, Zhang et al. described the formation of hollow microcapsules by generating a polyphenol coating on sacrificial CaCO3 microspheres through the oxidative coupling of TA in an alkaline buffer.25 When TA was used alone, the microcapsules were fragile and disassembled upon removing the CaCO3 template. Therefore, TA was co-deposited with PEI to obtain a more mechanically stable coating. Furthermore, Kim et al. reported that using separated catechols and polyamines (as opposed to dopamine where the catechol and amine moieties are on the same molecule) can result in stronger surface adhesion. This was due to the suppression of cohesive interactions between the polyphenol molecules, which promoted their adhesion to the substrate surface, allowing the formation of highly ordered nanocoatings even on surfaces with high curvature (e.g. nanoparticles).32
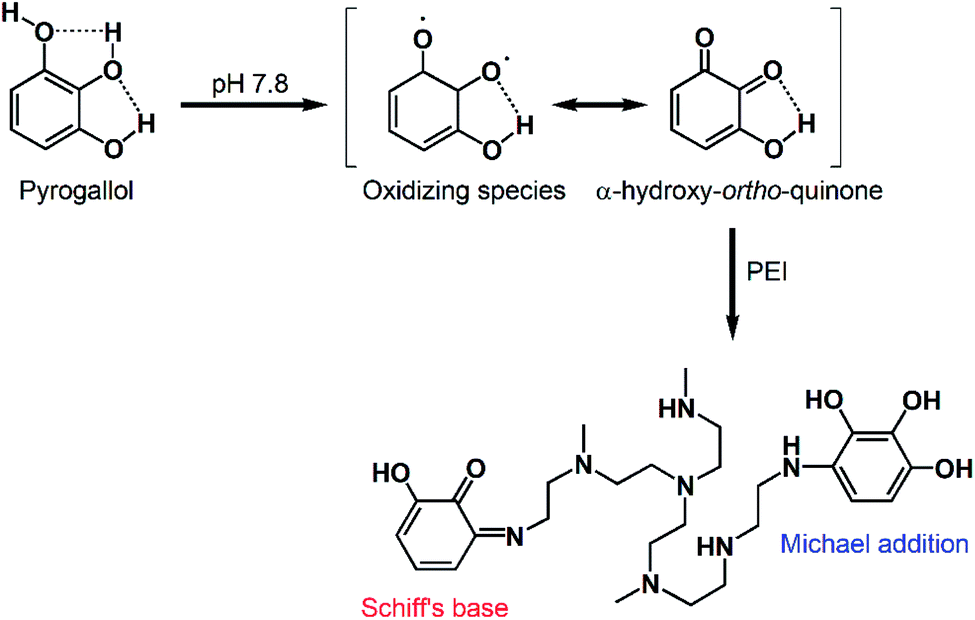 | ||
| Scheme 2 Proposed reaction mechanism between oxidized PG and PEI, leading to the formation of crosslinked coatings via Schiff's base and/or Michael addition reactions. | ||
Next, GOx was immobilized on polyphenol-coated wells by a simple incubation process under ambient conditions. To ensure that GOx immobilization was indeed mediated by adhesion to the polyphenol coating, control groups included physically adsorbed GOx on untreated wells and wells pretreated with PEI alone. Immobilization was verified by measuring GOx activity inside the wells by relying on the rate of formation of the HRP chromogenic substrate (TMB) after adding a fixed amount of glucose to each well. Note that all activity assays were carried out at pH 7.4. Excessive gluconic acid formation and its potential effect on the pH of the system could be easily detected by the change in color of the oxidized TMB product from blue to yellow/brown. The pH effects were minimized by optimizing the glucose concentration and limiting the duration of the kinetic experiment, so that only readings where the oxidized TMB product exhibited a blue color were used in determining enzyme activity. Representative kinetic plots are shown in Fig. 3A, and the corresponding reaction rates are presented in Fig. 3B. The actual amount of immobilized GOx (Fig. 3C) was determined from a calibration curve of reaction rate versus free GOx concentration, where the activity assay was conducted under the same conditions as for the immobilized enzyme. Physically adsorbed GOx on untreated wells exhibited negligible activity with a reaction rate of 0.006 ± 0.002 ΔOD650 per min, which was below the detection limit of the GOx calibration curve (i.e., the amount of GOx immobilized was almost zero) (Fig. 3C). On the other hand, GOx adsorbed on wells pretreated with PEI showed a slightly faster reaction rate of 0.030 ± 0.013 ΔOD650 per min corresponding to 0.7 ± 0.5 μg GOx per well. Nonetheless, the amount was not significantly different from free GOx adsorbed on untreated wells. PEI adhered to untreated wells most likely by electrostatic interactions, which also mediated the adsorption of GOx even after repeated washing. However, the binding was not reproducible as evidenced by the large standard deviation (SD) values.
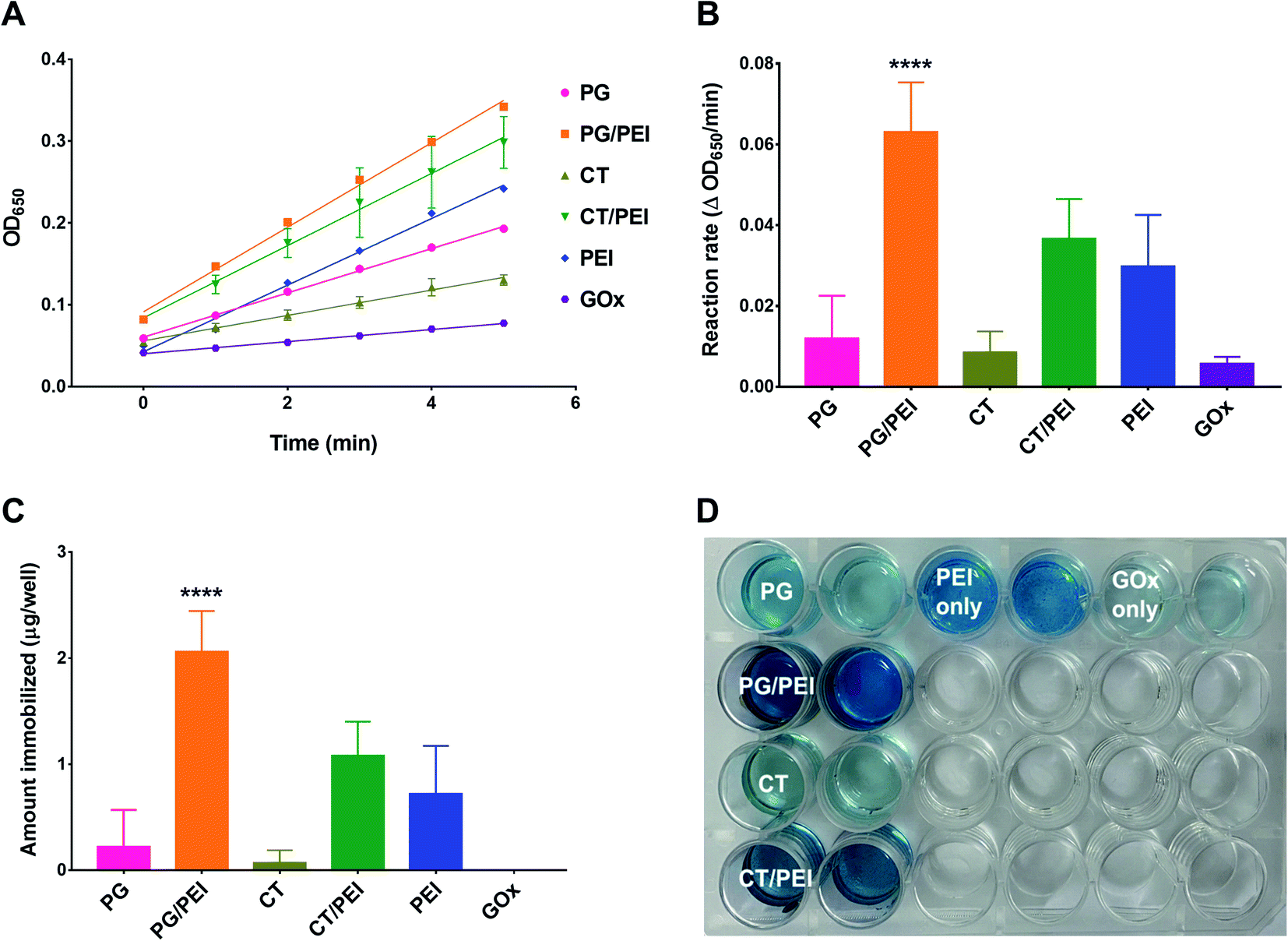 | ||
| Fig. 3 Verification of GOx immobilization on polyphenol-coated surfaces by direct measurement of enzyme activity. (A) Representative GOx activity assay results after immobilizing GOx (1 mg mL−1) on polyphenol-coated surfaces of 24-well plates with or without co-deposition with PEI. GOx immobilized on PEI-only and uncoated surfaces was also tested for comparison. Results are presented as the absorbance of the blue TMB product (OD650) versus time; (B) GOx activity represented by the reaction rates obtained from the slopes of the regression lines in panel (A); (C) amount of GOx (μg per well) immobilized on polyphenol-coated surfaces corresponding to panel (B); (D) representative GOx-immobilized 24-well plate immediately after conducting the GOx activity assay. Results are expressed as mean ± SD from three trials, ****p < 0.0001 based on 1-way ANOVA followed by Tukey's multiple comparisons test. | ||
PEI is used in enzyme immobilization by carbodiimide-mediated amide bond formation with enzymes or their supports.33 This approach is more favored over nonspecific electrostatic interactions to achieve a more stable bond. In this work, we followed an alternative approach by relying on the bioinspired interaction between amines and catechols to form a reinforced adhesive coating for GOx immobilization. As a result, PEI-incorporated CT and PG coatings were associated with significantly greater activities compared to non-PEI-containing counterparts. GOx immobilized on CT-coated wells was associated with a reaction rate of 0.009 ± 0.005 ΔOD650 per min, corresponding to 0.07 ± 0.12 μg GOx per well. The activity was similar to GOx immobilized on PG-coated wells, where the measured enzyme activity was 0.012 ± 0.011 ΔOD650 per min, which was equivalent to 0.22 ± 0.34 μg GOx per well. The activities and amounts of GOx immobilized on these two surfaces did not significantly differ from physically adsorbed GOx on untreated and PEI-pretreated wells, indicating the weak binding between GOx and polyphenol-only surfaces. On the other hand, GOx immobilized on CT/PEI-coated wells exhibited a reaction rate of 0.037 ± 0.010 ΔOD650 per min, corresponding to 1.08 ± 0.32 μg GOx per well. GOx immobilized on PG/PEI-coated wells resulted in the highest enzymatic activity and the greatest amount of GOx immobilized, producing a reaction rate of 0.063 ± 0.012 ΔOD650 per min and 2.06 ± 0.38 μg GOx per well. The relative activities of the different surfaces were also evident upon visual inspection of the wells at the end of the kinetic experiment, where the intensity of the blue-colored TMB product was proportional to GOx activity (Fig. 3D).
Overall, incorporating PEI in polyphenol coatings enhanced their enzyme immobilization efficiency compared to polyphenol-only wells, which likely resulted from a combination of factors. First, PEI could mediate electrostatic interactions with the enzyme as evidenced by observing a certain degree of GOx activity in PEI-only wells. Second, PEI enhanced the surface adhesion of polyphenol molecules through crosslinking and/or electrostatic interactions as discussed earlier. This in turn contributed to a greater degree of enzyme immobilization, particularly in PG/PEI-coated wells, by affording additional interactions with GOx such as hydrogen bonding, π–π stacking, and van der Waals interactions. The free thiols and primary amines in the amino acid side chains of GOx may have also acted as nucleophiles, reacting with the oxidized aromatic rings of PG and CT. Having shown the highest activity, PG/PEI-coated surfaces were used for further experiments.
To optimize the concentration of GOx in the immobilization solution, PG/PEI-coated wells were incubated with different GOx concentrations before conducting the activity assay. As depicted in Fig. 4, the concentration of GOx which produced the highest activity was 1 mg mL−1, resulting in a reaction rate of 0.067 ± 0.011 ΔOD650 per min, which corresponded to 2.06 ± 0.38 μg GOx per well. Diluting the GOx solution by half resulted in a proportional reduction in activity as well as the amount immobilized. However, the values obtained did not significantly differ upon further dilution of the GOx initial solution. The discrepancy between the final amount of immobilized GOx compared to the initial amount added may be attributed to the bulkiness of the enzyme, causing steric hindrance against binding to the flat nonporous well surface. At the same time, a high concentration was needed for optimum binding, most probably to increase the number of collisions during immobilization. Consequently, the concentration of GOx in the immobilization solution was fixed at 1 mg mL−1.
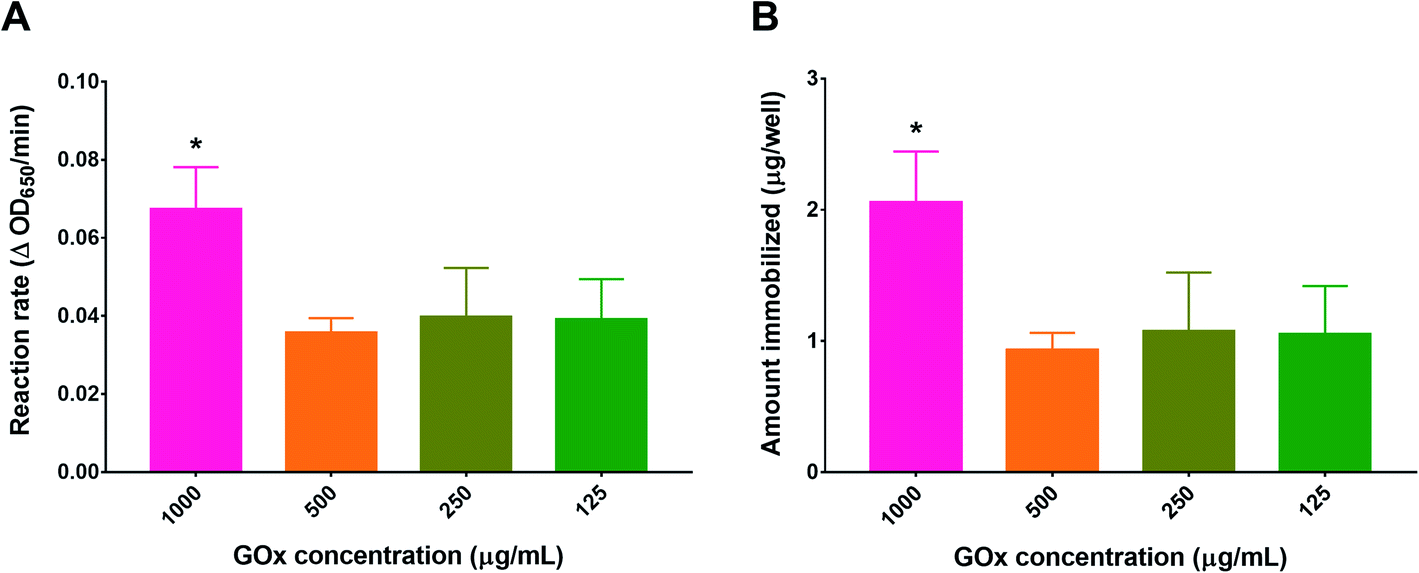 | ||
| Fig. 4 Optimization of the GOx concentration used during immobilization on PG/PEI-coated surfaces of 24-well plates. (A) GOx reaction rates expressed as the change in the absorbance of the blue TMB product at 650 nm with time as a function of GOx concentration, and (B) the corresponding amount of immobilized GOx (μg per well). Results are expressed as mean ± SD from three trials, *p < 0.05 based on 1-way ANOVA followed by Tukey's multiple comparisons test. | ||
Determination of the Michaelis–Menten constant (Km)
We examined the effect of GOx immobilization on its affinity to its substrate, glucose. Immobilized enzymes can exhibit a reduction in their substrate affinity depending on the immobilization method used as well as the morphology and topology of the supports.34 In this work, GOx was immobilized on a nonporous surface through a combination of interactions that may have included covalent bonding. Enzyme affinity was determined by incubating PG/PEI-coated GOx-immobilized wells with different glucose concentrations and performing the activity assay. The reaction rates at each glucose concentration were obtained from linear regression analysis of OD650 versus time. The rates were then plotted against glucose concentration and fitted to the Michaelis–Menten model. An amount of free GOx approximating the amount immobilized in the wells was similarly incubated with glucose for comparison. As illustrated in Fig. 5, the Michaelis–Menten plots for the free and immobilized enzyme were superimposable, with almost identical maximum velocities (Vm). Importantly, the Km value for the immobilized enzyme was found to be 24.64 ± 6.26 mM, which was not significantly different compared to the free enzyme (21.13 ± 5.44 mM). This strongly indicates that the immobilization process did not compromise the enzyme kinetics nor the accessibility of the glucose substrate to the enzyme's active sites.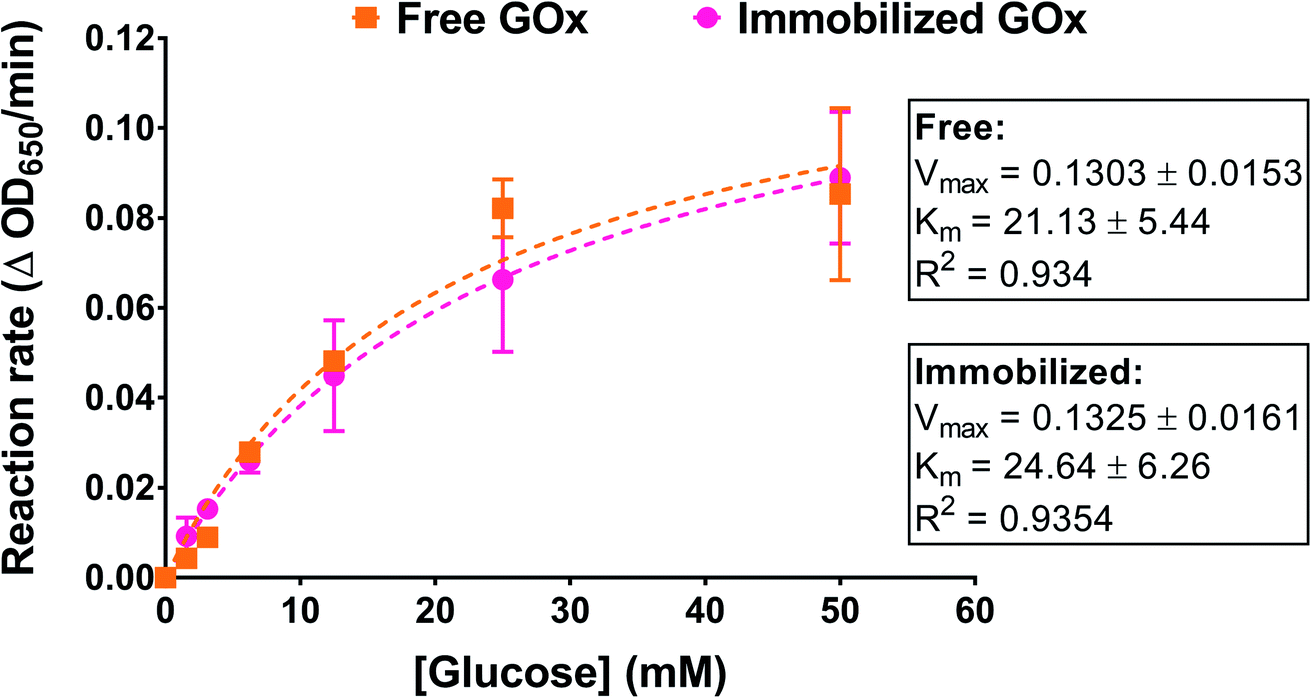 | ||
| Fig. 5 Michaelis–Menten plots of free and immobilized GOx obtained by conducting the activity assay using different glucose concentrations. The concentration of free GOx used was chosen to approximate the actual concentration of immobilized GOx. Results are presented as the mean ± SD of three trials. Curve fitting to the Michaelis–Menten model was performed using GraphPad Prism 7. | ||
Stability of immobilized GOx
One of the rationales for immobilizing enzymes is to be able to regenerate them after each catalytic reaction. Since GOx is a cofactor-dependent enzyme, its regeneration ability is highly dependent on the recycling of the FAD cofactor.35 In this study, we tested the recycling ability of immobilized GOx by reusing the treated wells after washing off the residual substrate and repeating the activity assay. The process was performed for a total of five cycles, and the enzyme activity was calculated as % of the control (cycle 1). As depicted in Fig. 6, the measured activity in the 2nd cycle was significantly reduced to 60.4 ± 16.6% of the control (p < 0.05). After the 3rd, 4th, and 5th cycles, the residual enzyme activities were 39.7 ± 5.5%, 34.4 ± 6.3%, and 30.1 ± 9.4% of the control, respectively. Even though cycles 3 through 5 resulted in a significant reduction in GOx activity relative to cycle 1 (p < 0.01), the associated activities did not significantly differ from each other, indicating that a plateau was reached. The observed reduction in enzyme activity may most likely be attributed to incomplete regeneration of FAD from FADH2. Nonetheless, the activity was not completely diminished and seemed to reach a constant value from the 3rd cycle onward.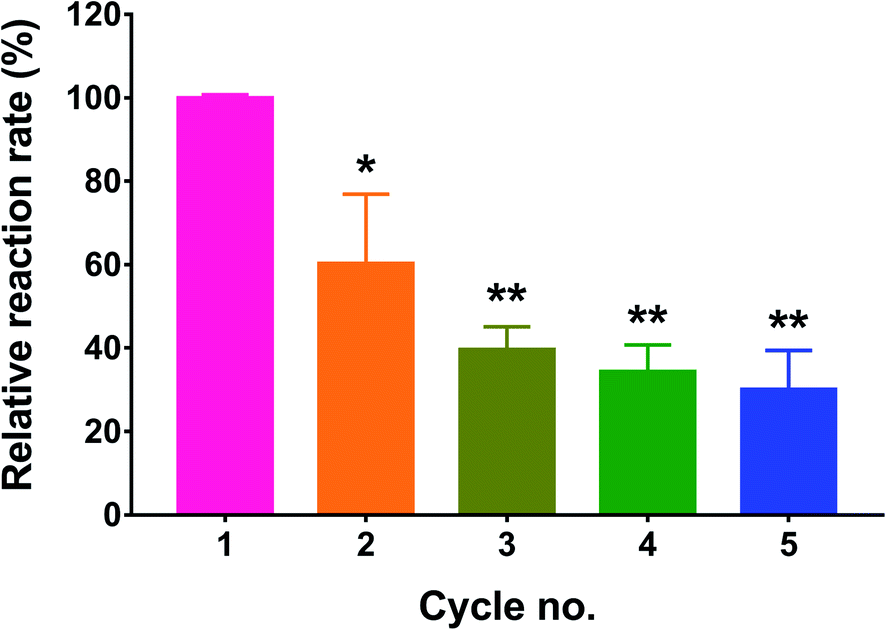 | ||
| Fig. 6 Stability of immobilized GOx after repeated cycles of enzyme activity assays. Results are expressed as the % reaction rate relative to the control (cycle 1) and are presented as the mean ± SD of three trials. *p < 0.05, **p < 0.01 based on 1-way ANOVA followed by Tukey's multiple comparisons test. | ||
Another test for enzyme stability is its ability to resist denaturation in the presence of denaturing agents such as urea. Urea causes denaturation by inducing the dissociation and unfolding of the GOx dimer, resulting in loss of activity.36 In this study, free and immobilized GOx were each incubated with increasing urea concentrations for 24 h, followed by measuring the residual activity compared to the control (no urea). As shown in Fig. 7, the activity of free GOx was markedly affected by even the lowest urea concentration of 1 M, displaying 72.3 ± 1.4% of its original activity (p < 0.01). The relative activity further decreased to 57.4 ± 2.5%, 50.5 ± 6.1%, and 41.1 ± 3.2% of the control as the urea concentration was increased to 2, 4, and 5 M, respectively. Conversely, immobilized GOx was unaffected by urea up to 2 M. However, as the urea concentration was increased to 4 and 5 M, a significant reduction in enzyme activity was observed, reaching 63.2 ± 9.2% (p < 0.01) and 58.6 ± 5.5% (p < 0.001) of the control, respectively. Although urea solutions are basic in nature, the pH had a negligible effect on the activity assay. In the case of the free enzyme incubated with urea, after the incubation, only 50 μL of the enzyme-urea mixture was taken out and mixed with 450 μL of the reaction mixture which was prepared in PBS (pH 7.4). In the case of the immobilized enzyme, the wells were washed three times with water after incubation with urea and before adding the reaction mixture prepared in PBS pH 7.4.
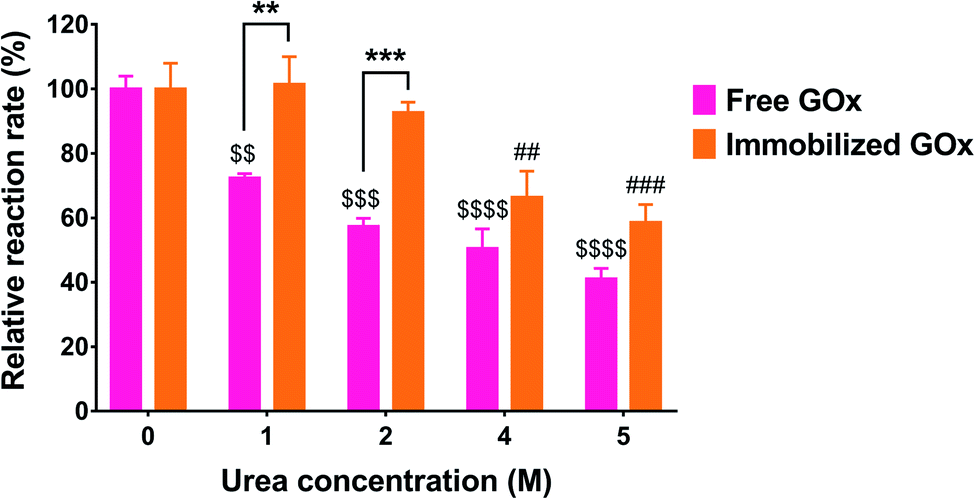 | ||
| Fig. 7 Stability of immobilized GOx compared to the free enzyme after incubation with different concentrations of urea. Results are expressed as the % reaction rate relative to the control (no urea) and are presented as the mean ± SD of three trials. ** and *** denote statistical significance between free and immobilized GOx at p < 0.01 and p < 0.001, respectively. $$, $$$, and $$$$ denote statistical significance between urea-treated and untreated free GOx at p < 0.01, p < 0.001, and p < 0.0001, respectively. ## and ### denote statistical significance between urea-treated and untreated immobilized GOx at p < 0.01 and p < 0.001, respectively. Results were analyzed by 2-way ANOVA followed by Sidak's multiple comparisons test. | ||
According to Akhtar et al., free GOx was not affected by urea up to 2 M, whereas the activity decreased sharply from 100% to about 5% between 2 and 5 M urea, with a complete loss of enzymatic activity observed at 6 M urea and above.36 Rauf et al. reported that GOx immobilized on a cellulose acetate-polymethylmethacrylate membrane retained 50% of its original activity in 2 M urea and was completely denatured in 6 M urea.37 On the other hand, Cao et al. co-immobilized GOx and HRP in microporous silica foam and observed no significant change in enzymatic activity up to 2 M urea. However, the activity was decreased to 50% in 5 M urea, while the activity of the free enzymes dropped to 14% at the same urea concentration.38 The differences observed between our findings and those reported in the literature with regard to the susceptibility of the free enzyme to urea may most likely be attributed to the different enzyme source. Nonetheless, in our study, we compared the same enzyme in its free and immobilized forms. As depicted in Fig. 7, GOx immobilized on PG/PEI-coated wells demonstrated superior stability against denaturation, particularly at 1 M (p < 0.01) and 2 M (p < 0.001) urea, maintaining >90% of its original activity, unlike the free enzyme. Moreover, the immobilized enzyme kept more than 50% of its original activity even at 5 M urea, which strongly supports the effectiveness of the PG/PEI coating in stabilizing GOx against denaturing agents.
GOx-immobilized PG/PEI-coated multi-well plates as a glucose assay platform
To test the potential applicability of our immobilization method in glucose quantification, we adapted the polyphenol coating and GOx immobilization procedures to 96-well plates simply by scaling down the volumes used during coating and immobilization. We chose a 96-well plate format as it is typically employed in commercial glucose assay kits. After immobilizing GOx, the glucose assay was conducted by incubating the wells with a reaction mixture containing HRP and TMB, in addition to a range of glucose concentrations. To ensure the reaction completion and to stabilize the TMB product, the plates were incubated for 30 min followed by adding an acidic stop solution,39 respectively. After reading the absorbance of the plates at 450 nm (λmax for the acidic TMB product), a calibration curve was constructed by plotting each absorbance value versus the corresponding glucose concentration. As illustrated in Fig. 8A, a linear relationship (R2 > 0.99) was obtained within the range 24.4–6250 μM glucose (equivalent to 0.4–112.6 mg dL−1), which was comparable to commercial kits. For example, the GOx-based glucose detection kit offered by Invitrogen (cat. no. EIAGLUC) has a detection range of 0.5–32 mg dL−1, a similar kit from Abcam (cat. no. ab272532) has an assay range of 0.7–300 mg dL−1, and another one from Cayman Chemical (cat. no. 10009528) provides a detection range of 2.5–25 mg dL−1. As a proof-of-concept experiment, we used the developed glucose assay platform to quantify glucose in plasma samples withdrawn from healthy rats and compared the results to a commercial kit. According to the GOx-immobilized platform, the average plasma glucose concentration was found to be 7477 ± 886 μM, corresponding to 135 ± 16 mg dL−1 (Fig. 8B). The result was similar to that obtained using the commercial kit (6937 ± 461 μM, corresponding to 125 ± 8 mg dL−1). This experiment demonstrates the promising potential of the developed platform to detect glucose in biological samples, which may be also expanded to other matrices such as foods and beverages.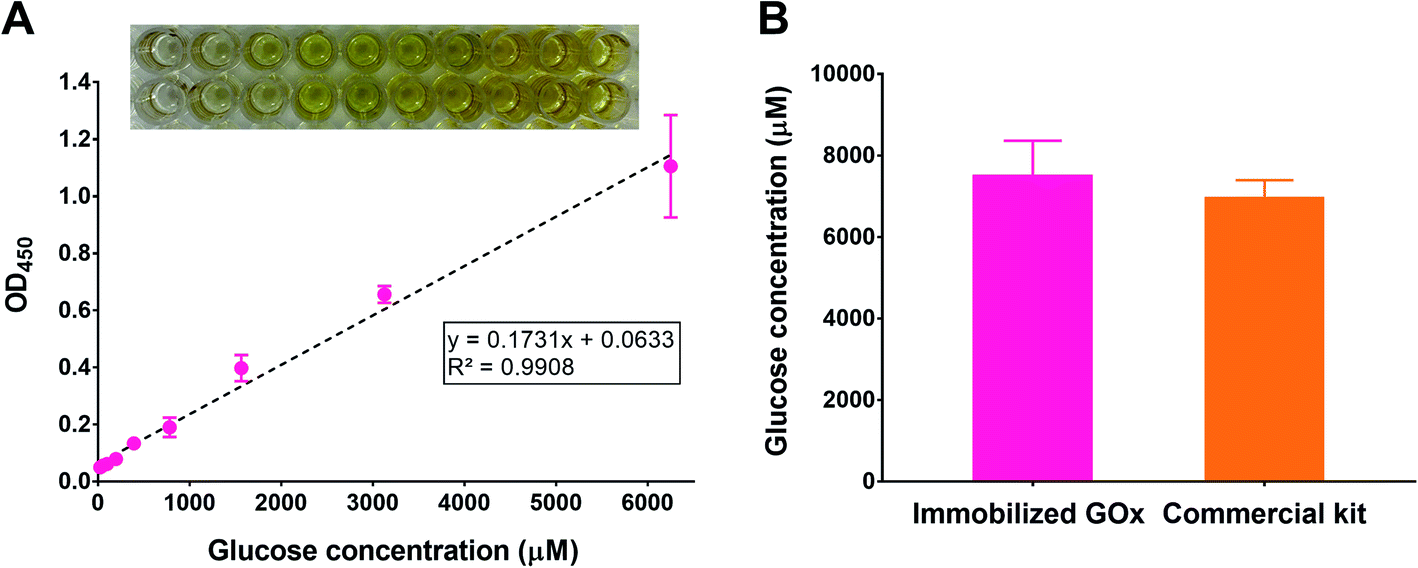 | ||
| Fig. 8 (A) Glucose assay conducted in GOx-immobilized 96-well plates. A calibration curve was obtained by plotting the absorbance of the yellow TMB product at 450 nm versus glucose concentration. Each concentration was assayed in duplicate. Linear regression analysis was performed using GraphPad Prism 7. Top: representative image of a GOx-immobilized 96-well plate after conducting the glucose assay; (B) Average plasma glucose concentrations from three different healthy rats assayed using the developed glucose assay platform and compared to a commercial glucose assay kit. | ||
Advances in materials science continue to inspire research and development efforts in the context of glucose detection platforms. Several of these platforms are based on the selective interaction between GOx and glucose and rely on the generation of optical or electrochemical signals.40 Optical detection methods take advantage of the change in color of an indicator that reflects the concentration of glucose. In addition to colorimetric readouts, recent developments in optical detection involve the generation of fluorescent or luminescent signals in response to glucose to improve the assay sensitivity, selectivity, and/or detection range. For example, a sensor based on upconversion nanoparticles was developed for H2O2-generating analytes such as glucose. The sensor was designed to give out dual colorimetric and fluorescence readouts through a cyclic signal amplification process.41 Electrochemistry-based glucose sensors are based on amperometric detection of the electrons generated during glucose oxidation by the action of GOx, which forms the basis for most glucose biosensors on the market.42 To address the oxygen dependence problem of first-generation amperometric biosensors, Xu et al. reported a nanostructured composite of TiO2 and ceria-cerium phosphate as an electrochemical enzymatic glucose biosensor operating in oxygen-restrictive environments. Electrodes decorated with the nanocomposite exhibited a linear glucose detection range from 0.1–1.7 mM, with a detection limit of 17.1 μM.43 The electrochemical signals may be further enhanced by combining them with luminescent signals. An electrogenerated chemiluminescent (ECL) biosensor was developed by growing TiO2 nanowires on Ti3C2 MXenes and depositing the nanocomposites along with GOx on a glassy carbon electrode. GOx-immobilized electrodes exhibited a detection range of 20 nM–12 mM and a lower detection limit of 1.2 nM.44 Non-enzymatic platforms have also been reported, where glucose detection is achieved either by catalyzing its oxidation using transition metals45,46 or by employing enzyme mimics.47
The key advantage of our developed platform is the simple fabrication procedure compared to the reported glucose detection systems. Moreover, commercial enzymatic glucose assay kits rely on the use of pre-aliquoted stock solutions of GOx, a peroxidase such as HRP, a colorimetric or fluorometric HRP substrate, and glucose standards. While convenient, these kits have drawbacks such as the inherent instability of the enzymes and the risk of cross-contamination between the different stock solutions. In addition, they are typically designed for assays in 96-well plates. Our assay platform can be freshly prepared when needed and scaled up or down depending on the number of samples required and the multi-well plate desired, which allows greater flexibility in designing the assay. Moreover, the enzymes can be stored in powder form and only prepared when needed, which provides a longer shelf-life and minimal risk for cross-contamination between reaction mixture components.
Conclusions
We describe the facile immobilization of GOx on polyphenol-coated multi-well plates. A bioinspired adhesive coating of simple polyphenols (PG or CT) was deposited on tissue-culture grade microplates by relying on the oxidative self-polymerization of polyphenols in a mild alkaline buffer. The coatings were reinforced by adding PEI to the coating solution, which translated to an increase in their enzyme immobilization ability, particularly for PG/PEI coated surfaces. Enzyme activity assays could be directly conducted in the immobilized well plates, highlighting the convenience of the immobilization method. The immobilized enzyme maintained similar affinity to glucose compared to its free form as indicated by Km values. The immobilization process significantly enhanced the enzyme's resistance to denaturation in the presence of urea. A linear glucose calibration curve could be successfully constructed by conducting the glucose assay in GOx-immobilized 96-well plates, where the detection range was comparable to commercially available colorimetric assay kits. The potential application of the platform in bioanalytical assays was further demonstrated by analyzing glucose levels in rat plasma samples, which were found to be comparable to the results obtained using a commercial kit. However, thorough investigation of assay selectivity, accuracy, precision, and other validation parameters is warranted. Although the immobilized GOx gradually lost its activity after repeated cycles of activity assays, the relatively high affinity of GOx to its substrate may support recycling of the immobilized plates albeit with a lower substrate sensitivity. However, the detection sensitivity may be enhanced by employing a fluorescence-emitting substrate. The assay platform may be further optimized by co-immobilization of GOx and HRP and/or using a chromogen that is unaffected by solution pH to avoid the additional the step of adding a stop solution.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Al-Zaytoonah University of Jordan grants no. 11/18/2018-2019 and 15/28/2017-2018. The authors thank Dr Ali Ibrahim (Al-Zaytoonah University of Jordan) for access to the Synergy HTX multi-mode microplate reader and valuable discussions.References
- S. B. Bankar, M. V. Bule, R. S. Singhal and L. Ananthanarayan, Biotechnol. Adv., 2009, 27, 489–501 CrossRef CAS PubMed.
- A. L. Galant, R. C. Kaufman and J. D. Wilson, Food Chem., 2015, 188, 149–160 CrossRef CAS PubMed.
- N. Mano, Bioelectrochemistry, 2019, 128, 218–240 CrossRef CAS PubMed.
- S. Datta, L. R. Christena and Y. R. S. Rajaram, 3 Biotech, 2013, 3, 1–9 CrossRef PubMed.
- M. Bilal, Y. Zhao, S. Noreen, S. Z. H. Shah, R. N. Bharagava and H. M. N. Iqbal, Biocatal. Biotransform., 2019, 37, 159–182 CrossRef CAS.
- J. Zdarta, A. S. Meyer, T. Jesionowski and M. Pinelo, Catalysts, 2018, 8, 92 CrossRef.
- M. K. Dubey, A. Zehra, M. Aamir, M. Meena, L. Ahirwal, S. Singh, S. Shukla, R. S. Upadhyay, R. Bueno-Mari and V. K. Bajpai, Front. Microbiol., 2017, 8, 1032 CrossRef PubMed.
- E. W. Nery and L. T. Kubota, J. Pharm. Biomed. Anal., 2016, 117, 551–559 CrossRef CAS PubMed.
- K. Atacan, N. Güy, S. Çakar and M. Özacar, J. Photochem. Photobiol., A, 2019, 382, 111935 CrossRef CAS.
- G. J. Kim, K. J. Yoon and K. O. Kim, J. Mater. Sci., 2019, 54, 12806–12817 CrossRef CAS.
- Z. Zhao, Y. Huang, W. Liu, F. Ye and S. Zhao, ACS Sustainable Chem. Eng., 2020, 8, 4481–4488 CrossRef CAS.
- J. H. Ryu, P. B. Messersmith and H. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 7523–7540 CrossRef CAS PubMed.
- N. R. Barros, Y. Chen, V. Hosseini, W. Wang, R. Nasiri, M. Mahmoodi, E. P. Yalcintas, R. Haghniaz, M. M. Mecwan, S. Karamikamkar, W. Dai, S. A. Sarabi, N. Falcone, P. Young, Y. Zhu, W. Sun, S. Zhang, J. Lee, K. Lee, S. Ahadian, M. R. Dokmeci, A. Khademhosseini and H.-J. Kim, Biomater. Sci., 2021, 9(20), 6653–6672 RSC.
- D. G. Barrett, T. S. Sileika and P. B. Messersmith, Chem. Commun., 2014, 50, 7265–7268 RSC.
- L. Q. Xu, K.-G. Neoh and E.-T. Kang, Prog. Polym. Sci., 2018, 87, 165–196 CrossRef CAS.
- M. Shin, E. Park and H. Lee, Adv. Funct. Mater., 2019, 29, 1903022 CrossRef CAS.
- T. S. Sileika, D. G. Barrett, R. Zhang, K. H. A. Lau and P. B. Messersmith, Angew. Chem., Int. Ed., 2013, 125, 10966–10970 CrossRef.
- A. M. Sousa, T.-D. Li, S. Varghese, P. J. Halling and K. H. Aaron Lau, ACS Appl. Mater. Interfaces, 2018, 10, 39353–39362 CrossRef CAS PubMed.
- W.-Z. Qiu, G.-P. Wu and Z.-K. Xu, ACS Appl. Mater. Interfaces, 2018, 10, 5902–5908 CrossRef CAS PubMed.
- G. Liu, Z. Jiang, C. Chen, L. Hou, B. Gao, H. Yang, H. Wu, F. Pan, P. Zhang and X. Cao, J. Membr. Sci., 2017, 537, 229–238 CrossRef CAS.
- B. D. B. Tiu, P. Delparastan, M. R. Ney, M. Gerst and P. B. Messersmith, Angew. Chem., Int. Ed., 2020, 59, 16616–16624 CrossRef CAS PubMed.
- S. Suárez-García, J. Sedó, J. Saiz-Poseu and D. Ruiz-Molina, Biomimetics, 2017, 2, 22 CrossRef PubMed.
- S. Sunoqrot, E. Al-Shalabi, L. Hasan Ibrahim and H. Zalloum, Molecules, 2019, 24, 3815 CrossRef CAS PubMed.
- A. R. Althaher, S. A. Oran and Y. K. Bustanji, Trop. J. Nat. Prod. Res., 2021, 5, 1333–1339 CrossRef CAS.
- S. Zhang, Z. Jiang, X. Wang, C. Yang and J. Shi, ACS Appl. Mater. Interfaces, 2015, 7, 19570–19578 CrossRef CAS PubMed.
- F. Behboodi-Sadabad, S. Li, W. Lei, Y. Liu, T. Sommer, P. Friederich, C. Sobek, P. B. Messersmith and P. A. Levkin, Mater. Today Bio, 2021, 10, 100108 CrossRef CAS PubMed.
- K. Du, Q. Liu, M. Liu, R. Lv, N. He and Z. Wang, Nanotechnology, 2019, 31, 015101 CrossRef PubMed.
- S. Geißler, A. Barrantes, P. Tengvall, P. B. Messersmith and H. Tiainen, Langmuir, 2016, 32, 8050–8060 CrossRef PubMed.
- S. Quideau, D. Deffieux, C. Douat-Casassus and L. Pouységu, Angew. Chem., Int. Ed., 2011, 50, 586–621 CrossRef CAS PubMed.
- J. Saiz-Poseu, J. Mancebo-Aracil, F. Nador, F. Busqué and D. Ruiz-Molina, Angew. Chem., Int. Ed., 2019, 58, 696–714 CrossRef CAS PubMed.
- Q. Lyu, N. Hsueh and C. L. Chai, ACS Biomater. Sci. Eng., 2019, 5, 2708–2724 CrossRef CAS PubMed.
- H. Kim, H. Kim, S. H. Kim, J. M. Park, Y. J. Jung, S. K. Kwak and J. Park, Chem. Mater., 2021, 33, 952–965 CrossRef CAS.
- J. J. Virgen-Ortíz, J. C. S. dos Santos, Á. Berenguer-Murcia, O. Barbosa, R. C. Rodrigues and R. Fernandez-Lafuente, J. Mater. Chem. B, 2017, 5, 7461–7490 RSC.
- H. D. Neira and A. E. Herr, Anal. Chem., 2017, 89, 10311–10320 CrossRef CAS PubMed.
- C. J. Hartley, C. C. Williams, J. A. Scoble, Q. I. Churches, A. North, N. G. French, T. Nebl, G. Coia, A. C. Warden, G. Simpson, A. R. Frazer, C. N. Jensen, N. J. Turner and C. Scott, Nat. Catal., 2019, 2, 1006–1015 CrossRef CAS.
- M. S. Akhtar, A. Ahmad and V. Bhakuni, Biochemistry, 2002, 41, 3819–3827 CrossRef CAS PubMed.
- S. Rauf, A. Ihsan, K. Akhtar, M. Ghauri, M. Rahman, M. Anwar and A. Khalid, J. Biotechnol., 2006, 121, 351–360 CrossRef CAS PubMed.
- X. Cao, Y. Li, Z. Zhang, J. Yu, J. Qian and S. Liu, Analyst, 2012, 137, 5785–5791 RSC.
- P. D. Josephy, T. Eling and R. P. Mason, J. Biol. Chem., 1982, 257, 3669–3675 CrossRef CAS.
- H. Lee, Y. J. Hong, S. Baik, T. Hyeon and D.-H. Kim, Adv. Healthcare Mater., 2018, 7, 1701150 CrossRef PubMed.
- H. Chen, Q. Lu, K. He, M. Liu, Y. Zhang and S. Yao, Sens. Actuators, B, 2018, 260, 908–917 CrossRef CAS.
- K. Tian, H. Liu, Y. Dong, X. Chu and S. Wang, Colloids Surf., A, 2019, 581, 123808 CrossRef CAS.
- J. Xu, K. Yang, X. Zhang, Y. Lei, X. Meng and Z. Wang, J. Solid State Electrochem., 2021, 25, 1937–1947 CrossRef CAS.
- Y. Sun, P. Li, Y. Zhu, X. Zhu, Y. Zhang, M. Liu and Y. Liu, Biosens. Bioelectron., 2021, 194, 113600 CrossRef CAS PubMed.
- T. Chen, D. Liu, W. Lu, K. Wang, G. Du, A. M. Asiri and X. Sun, Anal. Chem., 2016, 88, 7885–7889 CrossRef CAS PubMed.
- F. Xie, X. Cao, F. Qu, A. M. Asiri and X. Sun, Sens. Actuators, B, 2018, 255, 1254–1261 CrossRef CAS.
- Q. Chen, M. Liu, J. Zhao, X. Peng, X. Chen, N. Mi, B. Yin, H. Li, Y. Zhang and S. Yao, Chem. Commun., 2014, 50, 6771–6774 RSC.
| This journal is © The Royal Society of Chemistry 2021 |