
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolid-state dye-sensitized solar cells using polymeric hole conductors†
Nick Vlachopoulos *a,
Michael Grätzela and
Anders Hagfeldt*b
*a,
Michael Grätzela and
Anders Hagfeldt*b
aLaboratory of Photonics and Interfaces, Institute of Chemical Sciences & Engineering, École Polytechnique Fédérale de Lausanne, Lausanne, Switzerland. E-mail: nikolaos.vlachopoulos@epfl.ch
bDepartment of Chemistry–Ångström Laboratory, Uppsala University, Box 523, 75120, Uppsala, Sweden. E-mail: anders.hagfeldt@uu.se
First published on 13th December 2021
Abstract
The present review presents the application of electronically conducting polymers (conducting polymers) as hole conductors in solid-state dye solar cells (S-DSSCs). At first, the basic principles of dye solar cell operation are presented. The next section deals with the principles of electrochemical polymerisation and its photoelectrochemical variety, the latter being an important, frequently-used technique for generating conducting polymers and hole conductors in DSSCs. Finally, two varieties of S-DSSC configurations, those of dry S-DSSC and of S-DSSCs incorporating a liquid electrolyte, are discussed.
Introduction
Dye-sensitised solar cells (DSSCs) have been recognised in the last quarter century as a keynote third-generation photovoltaic technology, undergoing systematic investigations both at the academic and industrial level. They are composed of a dye-coated mesoporous oxide photoelectrode (PE), a conductive dark, in the sense of not needing light fort its operation, counter electrode (CE), either metallic or non-metallic with sufficient conductivity, and, in the between, a charge-transport medium (Fig. 1). The latter can be a liquid electrolyte, containing a redox mediator, e.g., iodide/triiodide or a transition metal (CuI/CuII, CoII/CoIII) inorganic coordination complex. Alternatively, in the case of solid-state DSSCs (S-DSSCs) the charge-transport medium (CTM) can be, in the case of an n-type photoelectrode, a solid-state hole conductor (HC) or, alternatively, in the case of a p-type photoelectrode, a solid-state electron conductor.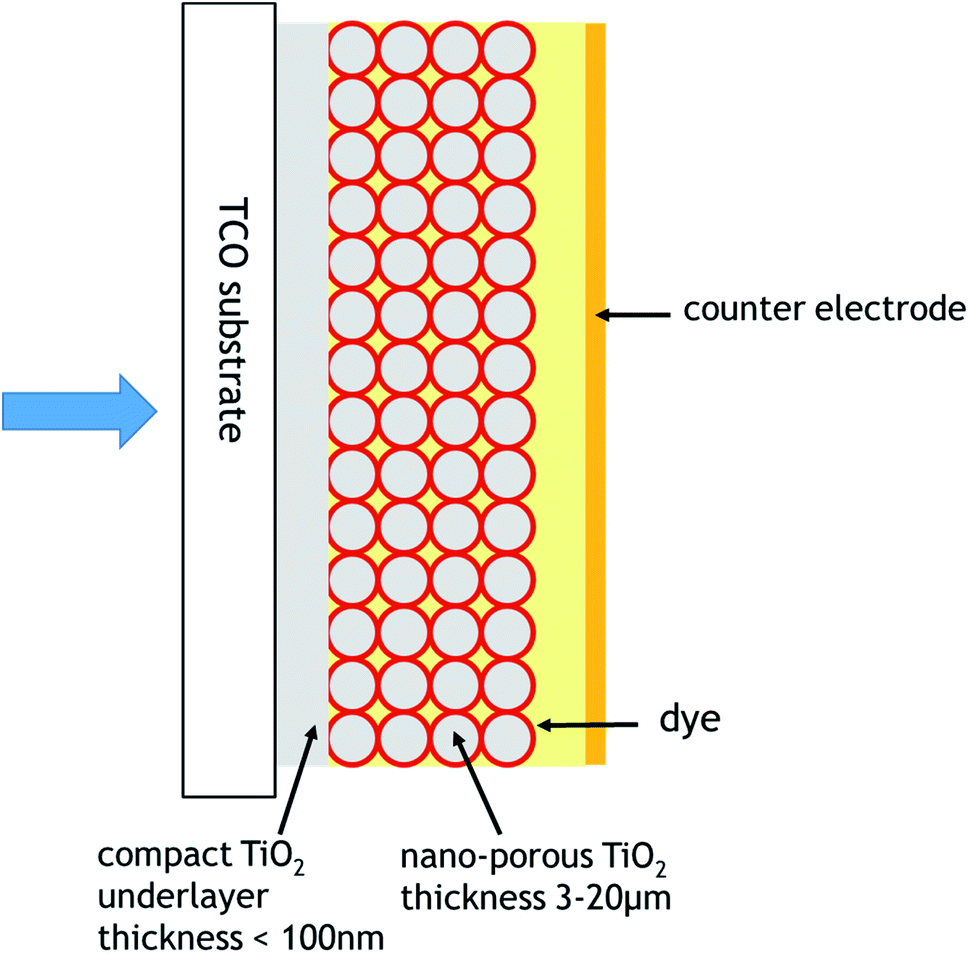 | ||
| Fig. 1 General dye sensitised solar cell configuration. | ||
One of the possibilities of hole conductor is that of an electronically conducting polymer. This publication is devoted to the use of conducting polymers as hole conductors in DSSCs, in recognition of the substantial contributions of Professor Seth Marder, Georgia Institute of Technology, to the science of conducting polymers, to whom the present publication is dedicated. A frequently method used for generating in situ the conducting polymer layer on the photoelectrodes is electrochemical polymerisation under illumination (photoelectrochemical polymerisation).
In the first two sections, the basic features of DSSC operation and the general principles of electrochemical generation of conducting polymers, for either metal electrodes (or non-metal electrodes of or approaching metallic conductivity), without the need of illumination, or illuminated semiconductor electrodes, are presented. Subsequently, particular cases of conducting polymer-based DSSCs are described.
Dye solar cell principles
The dye sensitisation of semiconductor photoelectrodes has been systematically investigated since the 1960s by a number of research groups, mainly in Germany, the UK, the USA, and Japan. This early work has been summarised by Memming.1 However, before the middle 1980s, the emphasis was in using electrodes, single crystal or polycrystalline, with a low roughness factor (quotient of real-to-geometric surface area). As a result, a low fraction of the incident dye was absorbed, and, accordingly, the resulting photocurrent was low. An important breakthrough was the introduction of porous TiO2 dye-sensitized electrodes, with a high roughness factor, resulting to substantially higher dye coverage, by the research group of Michael Grätzel, with the first publication in 1985.2–5 A substantial enhancement of the solar-to-electrical energy conversion DSSC efficiency was achieved by the introduction of dye sensitized colloidal mesoporous TiO2 electrodes by O'Regan and Grätzel,6 confirmed shortly thereafter.7,8 In the followed three decades, a large number of publications in dye sensitisation have been published. For details on the variety of DSSCs studied, a number of reviews and monographs can be consulted.9–25 In addition to TiO2, other semiconductor substrates, e.g., ZnO, SnO2, Nb2O5, NiO, has been considered in DSSC studies. However, with respect to SS-DSSCs studies, TiO2 has been the substrate of preference, and will be considered as the dye support in this article.The advantage of porous electrodes, as compared to smooth electrodes, in which a dye is chemisorbed lies to the fact that, at first, a larger fraction of the incident light is absorbed, due to the larger amount of adsorbed dye adsorbed per unit of geometrical surface area. Moreover, the impinging light beam undergoes multiple reflections within the porous structure, resulting to an even larger fraction of absorbed light. The properties of oxide electrodes used in DSSCs are discussed by Barbé et al.26 and Gerfin et al.27
The DSSCs discussed in this publication are based on a n-type photoelectrode juxtaposed to a dark counter electrode. DSSCs consisting of a p-type photoelectrode and a dark counter electrode, or tandem cells consisting of simultaneously irradiated n-type and p-type photoelectrodes, are not considered here. Usually, the mesoporous TiO2 layer is deposited on transparent conductive oxide-coated glass, quite often F-doped tin oxide, with the light impinging from the glass side of the electrode (back-irradiation). For some applications, with the mesoporous TiO2 deposited on a metal substrate, light has to impinge on a sufficiently transparent counter electrode.
The various processes in a DSSC can be classified as useful, contributing to electricity generation, and deleterious, contributing to a loss of useful energy and limiting the DSSC performance. Useful processes are described by eqn (1)–(11) and deleterious ones by eqn (12)–(18) below.
Upon light absorption, a dye undergoes photoexcitation (Fig. 2):
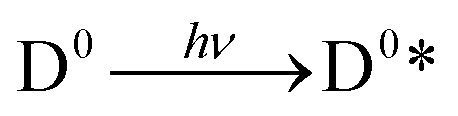 | (1) |
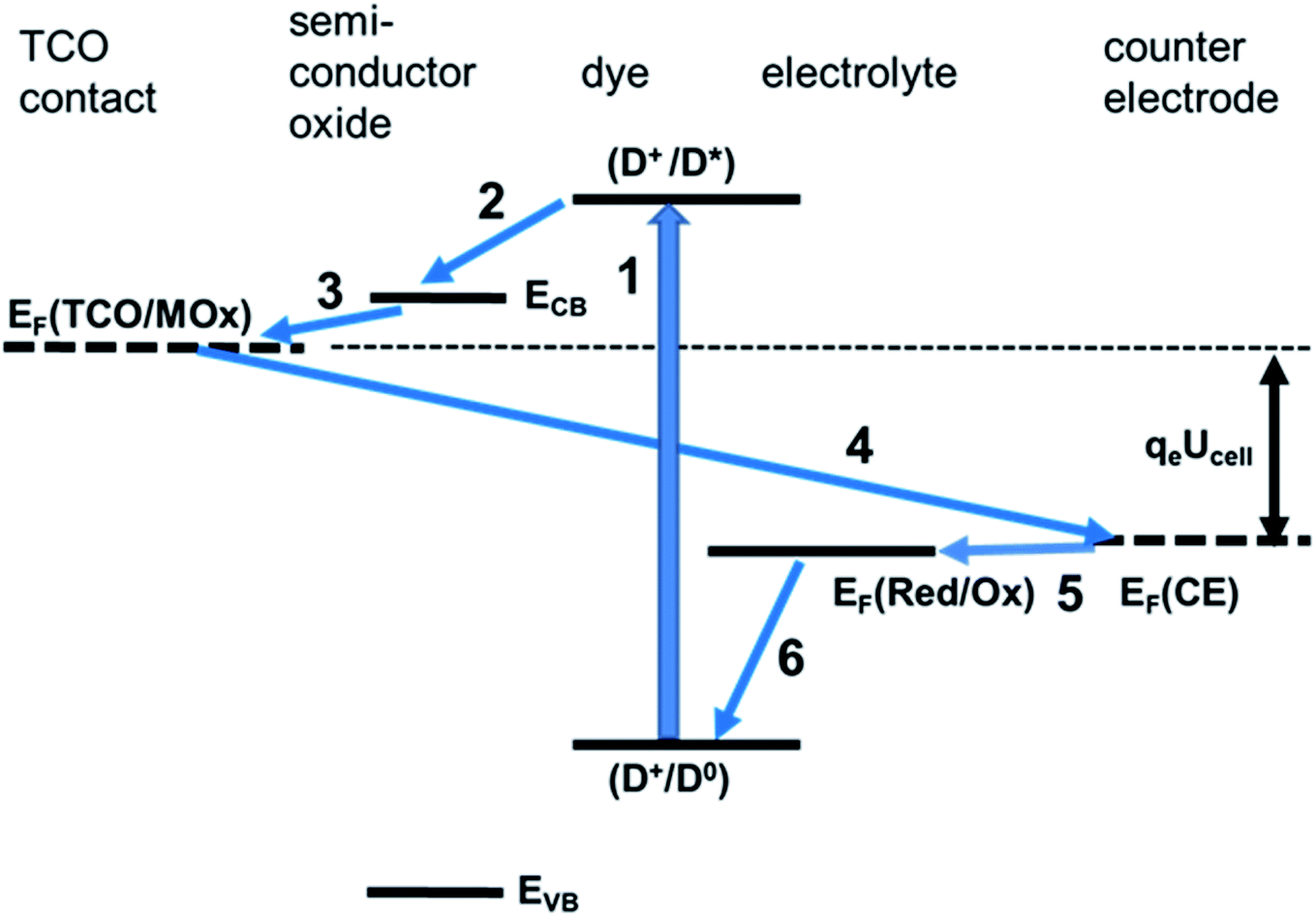 | ||
| Fig. 2 Useful processes in dye solar cell operation. (1) Photoexcitation. (2) Injection. (3) Collection. (4) Flow from photoelectrode to counter electrode. (5) Reaction at the counter electrode. (6) Dye regeneration. | ||
Subsequently, the excited dye undergoes oxidation by injecting an electron into the semiconductor conduction band of the mesoporous TiO2 (Fig. 2):
| D0* → D+ + e−(CB) | (2) |
The superscripts 0 and + in D0 and D+ indicate relative charge and not the charge of the species itself.
The injected electrons are subsequently collected to the photoelectrode contact phase (CT@PE), usually, as mentioned before, is a transparent conducting oxide layer on glass:
| e−(CB) → e−(CT@PE) | (3) |
Electrons flow through the external circuit toward the CE, with the possibility of electricity generation:
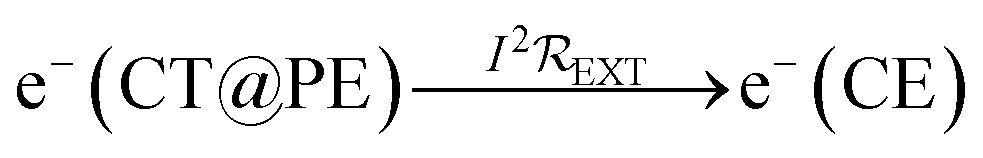 | (4) |
The oxidised dye is regenerated by reacting with oxidising the reduced form of the mediator Med0 or by injecting holes (h+) into the hole conductor:
| D+ + Med0 → D0 + Med+ | (5) |
| D+ + HC → D0 + h+(HC@PE) | (6) |
Med+(PE) or h+(HC) is transported from the photoelectrode to the counter electrode:
| Med+(PE) → Med+(CE) | (7) |
| h+(HC@PE) → h+(HC@CE) | (8) |
At the counter electrode, M+(CE) is back converted to M(CE) or, alternatively, the hole of the hole conductor is filled:
| Med+(CE) + e−(CE) → Med0(CE) | (9) |
| h+(HC@CE) + e−(CE) → HC | (10) |
In the case of a redox mediator in an electrolyte, M0 back diffuses from the counter electrode toward the photoelectrode:
| Med+(CE) → Med+(PE) | (11) |
The net result of processes (1)–(11) is the conversion of sunlight to electricity without any net chemical change in the charge-transport medium.
In addition to the above useful processes, a number of deleterious processes, posing limitations to the performance of a S-DSSC, has to be considered (Fig. 3).
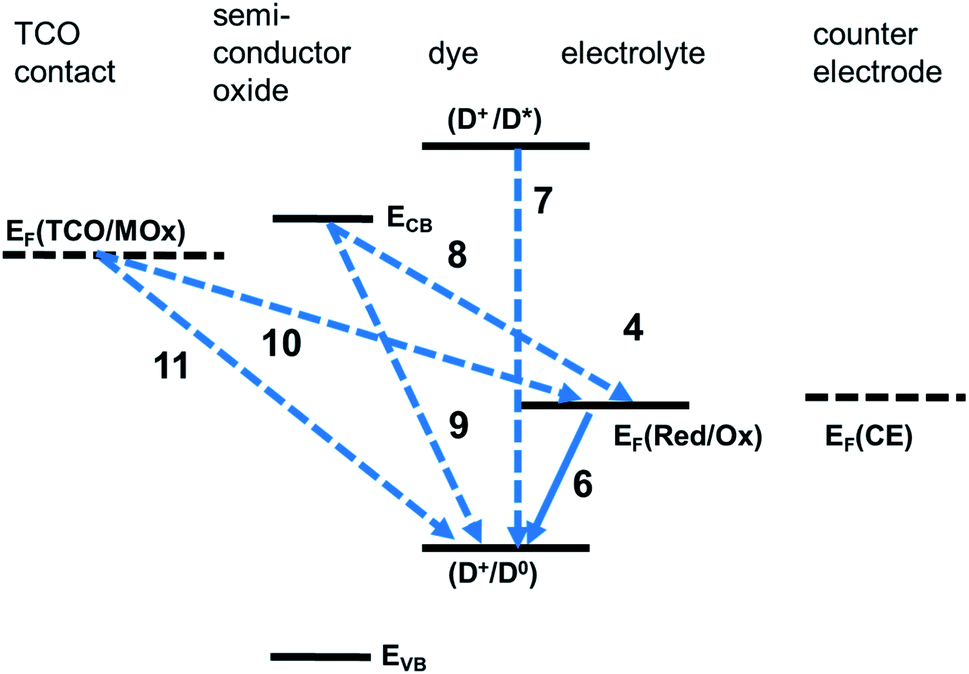 | ||
| Fig. 3 Deleterious processes in dye solar cell operation. (7) Excited dye deactivation. (8) and (9) Back electron transfer from conduction band to oxidised dye (8) or oxidised mediator. (9)–(11) Back electron transfer from substrate to oxidised dye (10) or oxidised mediator (11). | ||
The deexcitation of D0*, with the evolution of heat or luminescent radiation
| D0* → D0 | (12) |
Electrons injected into the semiconductor can undergo back electron transfer reactions with the photo-oxidised dye:
| e−(CB) + D+ → D0 | (13) |
Alternatively, they can be annihilated by reacting with Med+(PE) or with a hole in the case of S-DSSC:
| e−(CB) + Med+(PE) → Med@PE | (14) |
| e−(CB) + h+(HC@PE) → HC | (15) |
Reactions eqn (13)–(15) – can compete with electron collection to the contact to the photoelectrode. The first of them is termed recombination. These processes can be partially suppressed when hydrophobic pendant groups are introduced on the periphery of the dye molecule.
Similarly, electrons at the contact to the photoelectrode can react either with D+ or, alternatively, with Med+(PE) or the hole of the charge-transport medium at the proximity of the photoelectrode [h+(CTM@PE)]:
| e−(CT@PE) + D+ → D0 | (16) |
| e−(CT@PE) + Med+(PE) → Med0(PE) | (17) |
| e−(CT@PE) + h+(CTM@PE) → CTM | (18) |
The occurrence of the latter two processes is due to the fact that the electrolyte or the hole conductor can permeate the pores and come into direct contact with the contact to the photoelectrode. In order to prevent it, a compact oxide blocking layer, or underlayer, of thickness in the order of 10–100 nm, is interposed between the transparent conducting oxide substrate and the mesoporous oxide. In particular, for polymer hole conductors, coupled to dye/TiO2 electrodes, the use of a compact TiO2 underlayer is unavoidable.
In addition to the DSSC operation, the above processes are of relevance to the process of photoelectrochemical polymerisation related to the generation of a polymeric hole conductor at close proximity to the dye, as discussed in the following section.
The generation of current from the incident light beam of dye-sensitised electrodes is characterised by the incident-photon-to-current efficiency (IPCE, φIPCE), defined as the ratio of the electron flux to the external circuit (Je) divided by the flux of incident photons reaching the dye layer (JPH(DYE)):
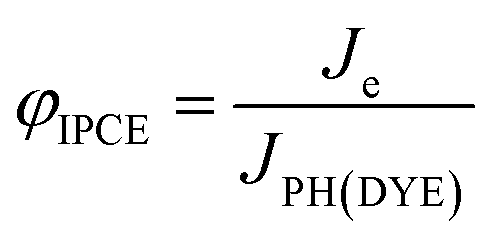 | (19) |
The IPCE, often it is expressed as percentage [φIPCE(%)], is mostly measured for incident monochromatic light. If j is the current density and PLIGHT(DYE) is the irradiance (incident energy flux) reaching the dye layer for monochromatic light of wavelength λ, then, for incident radiation of wavelength it is
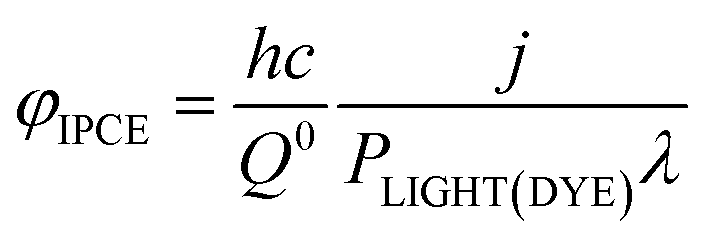 | (20) |
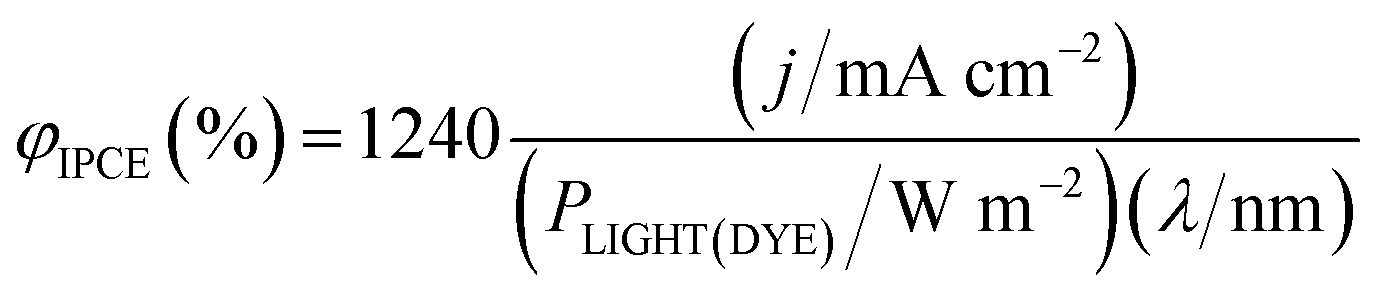 | (21) |
The IPCE is usually measured with the DSSC voltage (Ucell) imposed at a value within a domain where j is independent of Ucell, often at Ucell = 0, where j assumes its short-circuit value (jSC). For efficient DSSC operation, IPCE should be as close to 100% as possible. In the base of negligible back electron transfer reactions from the contact to the photoelectrode, IPCE is expressed in terms of the light harvesting efficiency (φIPCE), the electron injection efficiency φINJ, and the electron collection efficiency (φCOLL) as
| φIPCE = φLHEφINJφCOLL | (22) |
The power conversion efficiency (PCE, φPCE) of the solar cell is expressed as
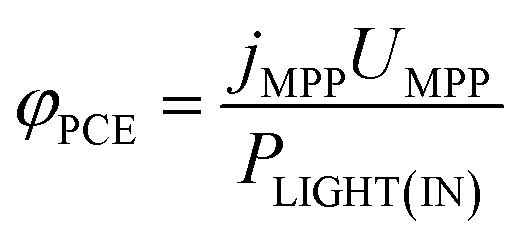 | (23) |
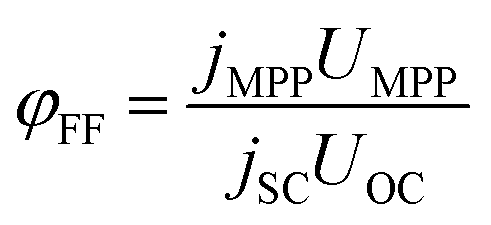 | (24) |
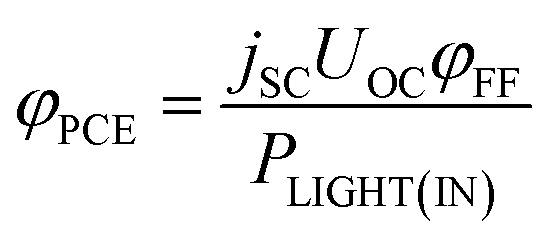 | (25) |
Consider the case of a polychromatic light source, which, for laboratory investigations, usually is a simulated Air Mass 1.5 (AM1.5) sunlight source with spectral irradiance PLIGHT(IN),SP(λ). For sunlight, the latter is tabulated for special conditions, or can be measured. The spectral irradiance for light reaching the dye layer PLIGHT(DYE),SP(λ) is related to PLIGHT(IN),SP(λ) by the wavelength-dependent coefficient rλ equal to the fraction of photons of wavelength λ lost due to reflection at the various interfaces and absorption by the various material phases encountered by photons before they reach the dye layer:
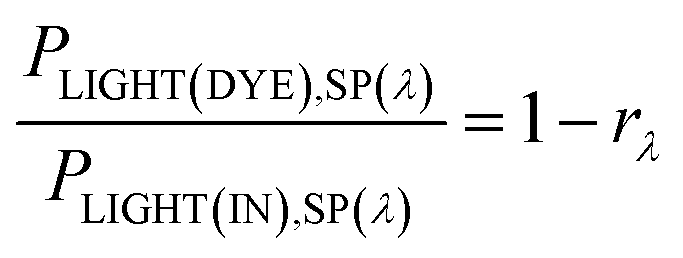 | (26) |
PPH(IN) and PPH(DYE),SP(λ) are related as
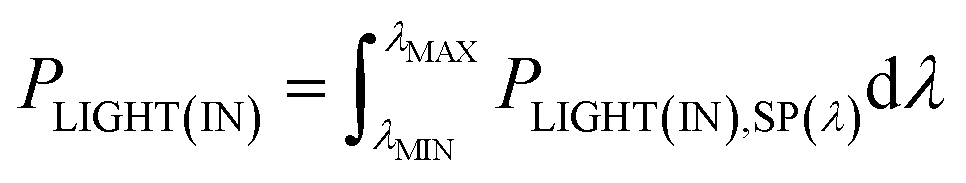 | (27) |
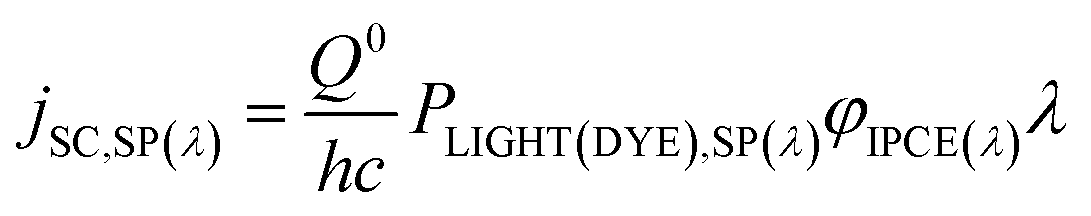 | (28) |
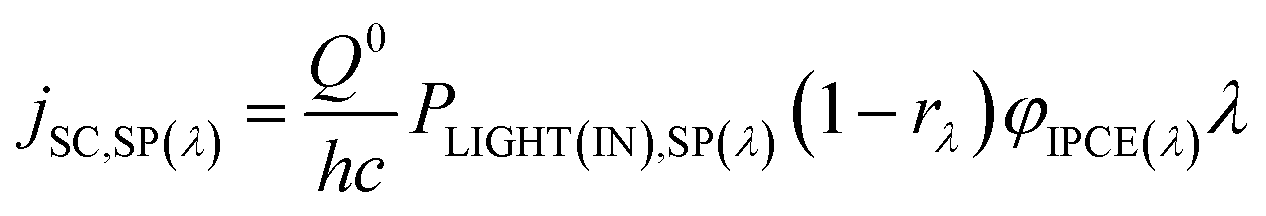 | (29) |
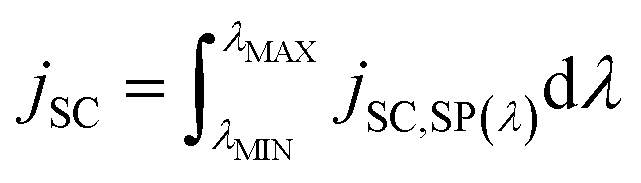 | (30) |
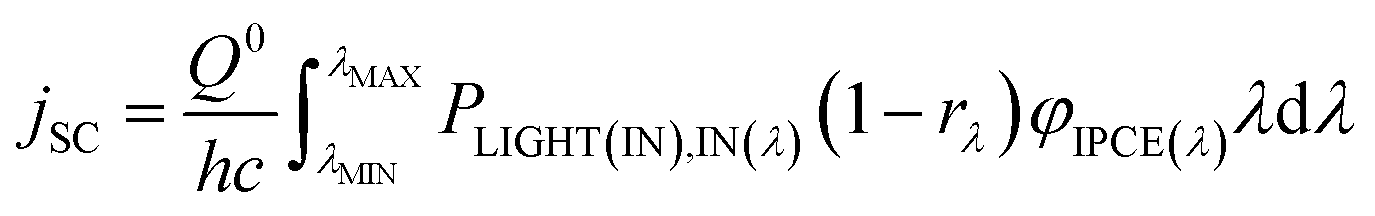 | (31) |
If the φIPCE(λ) vs. spectrum is known, the above equation allows to estimate the jSC on the basis of tabulated AM1.5 (or other) solar energy spectra. A good agreement between the calculated and measured in the laboratory jSC presupposes the linearity of the dependence of jSC on the value of incident irradiance. A sublinear dependence of jSC vs. PLIGHT(λ) could be attributed to transport limitations in the hole conductor layer.
Relatively few of the PCE values reported in the literature have been certified by an accredited solar cell laboratory. In the present article, all reported PCEs are considered as uncertified, unless otherwise specified. For DSSCs with a liquid electrolyte containing a redox mediator the maximal PCE for simulated AM1.5 sunlight is 14.3%;28 the highest certified PCE is 13.0%.29 For SS-DSSC the maximal PCE is 11.0%, obtained with a solidified Cu(I/II) coordination complex as hole conductor.30 For all-solid S-DSSCs based on electronically conducting polymers as hole conductors the maximal PCE is 7.1%, as discussed in the section devoted to the performance of polymer-based S-DSSCs.31
S-DSSC with an electronically conducting polymer hole conductor should be distinguished from DSSC in which a conducting polymer is used as an immobilisation matrix for a liquid electrolyte.32–34 Such cells, in which the CTM is the immobilised liquid electrolyte containing a redox mediator are often termed quasi solid state DSSCs. Alternatively, quite often the conductivity of a solid hole conductor can be enhanced by the addition of a liquid inert, non-electroactive electrolyte; several examples should be cited later in the article. Such solar cells are not solid-state cells in the strict sense. Therefore, a distinction has to be made between all-solid S-DSSC and liquid electrolyte impregnated S-DSSC, with the latter to be henceforth to be denoted as solid–liquid, where the term solid denotes that a solid-state hole conductor is responsible for charge transport from the photoelectrode to the counter electrode and that a non-volatile liquid phase impregnates the solid hole conductor; otherwise, they will be termed dry S-DSSCs.
In addition, electronically conducting polymers, prepared electrochemically or by other methods, have been extensively used as counter electrodes in dye solar cells.22,35–37
Electrochemical and photoelectrochemical polymerisation
Typical electronically conducting polymers of interest which have been studied as hole conductors to DSSC applications include, but are not limited to, polypyrrole, poly(3-hexylthiophene) (P3HT), poly(3-octylthiophene) (P3OT), poly(3,4-ethylenedioxythiophene) (PEDOT) and poly(3,4-ethylenedioxypyrrole) (PEDOP), with the monomers corresponding to the last two polymers denoted as EDOT and EDOP, respectively. Various polymerisation approaches can be applied in order to generate the above polymers, including the chemical, photochemical, and electrochemical polymerisation, or its photoelectrochemical variety, photoelectrochemical polymerisation. In the latter case, the photooxidized dye is regenerated by reacting, according to eqn (5), with the polymerisation precursor, monomer or short oligomer, or with the growing polymer chain, in which case these species replace the mediator (Med). Chemical and photochemical polymerisation require the addition of sacrificial electron donors; photoelectrochemical polymerisation, instead, is considered a “clean” method since it does not have this requirement, with the holes for the oxidative polymerisation originating either from a metal electrode or from the irradiated dye molecules according to eqn (5). For details on the properties of electronically conducting polymers, electrochemical polymerisation mechanisms, and conducting polymer applications, a number of monographs and reviews can be consulted.38–46 Several electrochemical textbooks contain brief but informative introductive sections on conducting polymers.47–49The first step in electrochemical polymerisation is the oxidation of a precursor (P), which can be either a monomer or a short oligomer, often a dimer. The advantage of an oligomer as precursor is the easier polymerisation, requiring a less negative electrode potential in electrochemical polymerisation, or a dye with a less positive standard electrode potential for the D+/D0 redox system.
The first step in electrochemical polymerisation or photoelectrochemical polymerisation in a solution containing only the precursor, in the absence of oligomers, is the generation of a cation radical
| HPH → HPH+˙ + e− | (32) |
| 2(HPH)+˙ → [HP(H)-(H)PH]2+ → HP2H + 2H+ | (33) |
The precursor dimer undergoes further oxidation to a cation radical
| HP2H → (HP2H)+˙ + e− | (34) |
| (HP2H)+˙ + (HPH)+˙ → [HP2(H)-(H)PH]2+ → HP3H + 2H+ | (35) |
| (HP2H)+˙ + (HP2H)+˙ → [HP2(H)-(H)P2H]2+ → HP4H + 2H+ | (36) |
The polymerisation further continues the same way with the formation of longer chains. A polymer chain with m precursor units is oxidised to a radical (HPmH)+˙ which can be coupled either to (HPH)+˙ or to another chain (HPnH)+˙, with n not necessarily equal to m, as follows:
| HPmH → (HPmH)+˙ + e− | (37) |
| (HPmH)+˙ + (HPH)+˙ → [HPm(H)-(H)PH]2+ → HPm+1H + 2H+ | (38) |
| (HPmH)+˙ + (HPnH)+˙ → [HPm(H)-(H)PnH]2+ → HPm+nH + 2H+ | (39) |
It should be pointed out that a fraction of the generated polymer does not attach to the electrode but is either dissolved or precipitates from solution, with the solubility decreasing with polymer length.
At the electrochemical polymerisation at a metal electrode, the end of electrochemical polymerisation or photoelectrochemical polymerisation, the polymer is in the oxidized state, since the electrode potential needed for the oxidation of the deposited polymer is less positive than that needed for electrochemical polymerisation. In such doping, only a fraction of the monomer units M undergoes oxidation. In order to keep the balance, anions A− from solution are incorporated into the polymer. The doping process can be written as
| H–Mn–H + nyA− → H–(M)nny+–HnyA− + nye− | (40) |
Upon transfer of the polymer-coated metal electrode in an inert electrolyte, a successive doping–dedoping cycle can be performed by modulating the electrode potential between a more and less positive value, respectively. An electronically conducting polymer is in fact a mixed conductor, with both electronic and ionic conductivity due to A−.
The same doping reaction takes place at a dye/oxide electrode during photoelectrochemical polymerisation, with the driving force needed for doping being lower than that needed for polymer generation.
Electrochemical polymerisation at a metal electrode can be performed by three methods: (a) by cyclic voltammetry, with the electrode potential shifting between a value where no electrochemical polymerisation takes place and another where polymer generation tales place; (b) at constant electrode potential; and (c) at constant current. In the case of photoelectrochemical polymerisation, only methods (b) and (c) are usually applied. In practice, method (c) may be preferred because it allows for a uniform formation of a polymer layer. In the case of constant-current polymerisation the electrode potential should be monitored because, if assumes a positive value beyond a certain threshold, irreversible polymer oxidation may occur: if the electrode potential surpasses a threshold value, the electrochemical polymerisation or photoelectrochemical polymerisation should be interrupted.
The vacant energy levels effective in photoelectrochemical polymerisation, originating from the photooxidized dye, have an energy above that and, usually, close to that of the redox Fermi level E⊖F(D+/D0), defined as the electrochemical potential per electron of solvated electrons, or, equivalently of electrons of a conductive electrode equilibrating with D+/D0. In a photoelectrochemical polymerisation process, E⊖F(D+/D0) lies substantially below that of the Fermi level of electrons at the metal electrode EF(metal) imposed at photoelectrochemical polymerisation. In comparison, for electrochemical polymerisation at a conductive electrode, electron levels involved in photoelectrochemical polymerisation originate from energy levels close to and below EF(metal). Therefore, a more positive EF(metal), corresponding to a less positive electrode potential, is needed for the photoelectrochemical polymerisation than for electrochemical polymerisation of the same monomer at a metal electrode, justified by the fact that light provides additional energy for polymerisation. The principle of photoelectrochemical polymerisation is presented in Fig. 4.
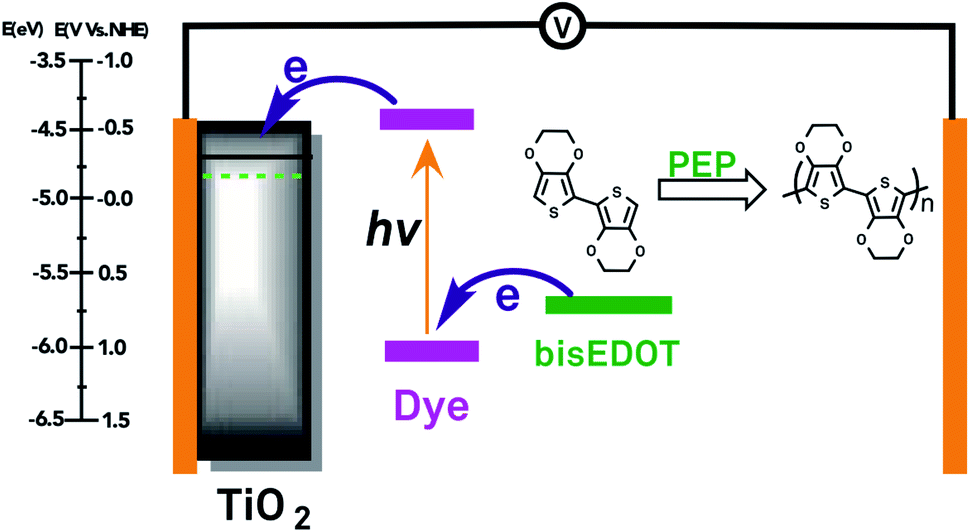 | ||
| Fig. 4 Photoelectrochemical polymerisation. | ||
Dye solar cells based on conducting polymers
In a DSSC based on an electronically conducting polymer as hole conductor, the latter can be either deposited as a polymer material previously generated ex situ or be generated from a precursor by photoelectrochemical or other type of polymerisation. Table 1 presents the chronological evolution of dry-DSSCs and Table 2 of solid–liquid-DSSCs. A list of abbreviations for dyes and for polymers or other compounds is presented in Tables 3 and 4, respectively.| No | Year | Hole cond. | Dye code | Irradiance % of AM1.5 | PCE % | UOC V | ISC mA cm−2 | FF | CE Au | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1998 | PP | N3 | 22 | <0.1 | 0.3 | 0.02 | <0.5 | Au | 50 |
| 2 | 2002 | P3OT | Ru-N719 | 80 | 0.2 | 0.65 | 0.45 | 0.44 | Au | 53 |
| 3 | 2012 | P3HT | D35 | 100 | 3.2 | 0.88 | 6.8 | 0.53 | Ag | 59 |
| 4 | 2012 | P3HT | CYC-B11 | 100 | 3.7 | 0.76 | 6.7 | 0.71 | Au | 58 |
| 5 | 2014 | PEDOT | LEG4 | 100 | 5.6 | 0.91 | 10.8 | 0.57 | Ag | 78 |
| 6 | 2016 | PEDOT | LEG4 | 100 | 7.1 | 0.83 | 13.4 | 0.64 | Ag | 31 |
| No | Year | Hole cond. | Dye code | Irradiance % of AM1.5 | PCE % | UOC V | ISC mA cm−2 | FF | CE | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1998 | PP | N3 | 22 | <0.1 | 0.67 | 0.082 | <0.5 | Au | 50 |
| 2 | 2004 | PEDOT | Ru-N719 | 100 | 0.5 | 0.47 | 2.3 | 0.50 | C | 61 |
| 3 | 2004 | PEDOT | Ru-Z907 | 100 | 0.9 | 0.68 | 2.6 | 0.51 | Au | 62 |
| 4 | 2006 | PEDOT | Ru-HRS-1 | 10 | 2.6 | 0.78 | 4.5 | 0.74 | Au | 63 |
| 5 | 2008 | PEDOT | Ru-Z719 | 100 | 2.9 | 0.75 | 5.3 | 0.73 | Au | 71 |
| 6 | 2011 | PEDOT | Ru-HRS-1 | 100 | 3.3 | 0.78 | 5.7 | 0.72 | Ag | 64 |
| 7 | 2011 | PEDOT* | D149 | 100 | 6.1 | 0.86 | 9.3 | 0.75 | Pt | 74 |
| 8 | 2012 | PEDOT* | D205 | 100 | 7.1 | 0.93 | 10.1 | 0.76 | Au | 73 |
| ADCBZ | Bis(2-(9H-carbazol-9-yl)ethyl)4,4′-(4-(5-(2-cyano-2-carboxyethenyl)thiophen-2-yl)phenylazanediyl)dibenzoate |
| B11 | TBA(Ru[(4-carboxylic acid-4![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) -carboxylate-2,2-bipyridine)(4,4′-bis(5-(hexylthio)-2,2′-bithien-5′-yl)-2,2′-bipyridine)(NCS)2]) -carboxylate-2,2-bipyridine)(4,4′-bis(5-(hexylthio)-2,2′-bithien-5′-yl)-2,2′-bipyridine)(NCS)2]) |
| C106 | RuLL′(NCS)2 (L = 2,2′-bipyridyl-4,4′-dicarboxylic acid; L′ = 4,4′-bis(5-(hexylthio)thiophen-2-yl)-2,2′-bipyridine) |
| C218 | (E)-3-(6-(4-(bis(4-(hexyloxy)phenyl)amino)phenyl)-4,4-dihexyl-4H-cyclopenta[2,1-b:3,4-b′]dithiophen-2-yl)-2-cyanoacrylic acid |
| C220 | 2-Cyano-3-{6-{4-[N,N-bis(4-hexyloxyphenyl)amino]phenyl}-4,4-didodecyl-4Hcyclopenta[2,1-b:3,4-b′]dithiophene-2-yl}acrylic acid |
| D21L6 | 3-(5-(5-(4-(Bis(4-(hexyloxy)phenyl)amino)phenyl)thiophene-2-yl)thiophene-2-yl)-2-cyanoacrylic acid |
| D35 | (E)-3-(5-(4-(bis(20,40-dibutoxybiphenyl-4-yl)amino)phenyl)thiophen-2-yl)-2-cyanoacrylic acid |
| D102 | (5-{4-[4-(2,2-Diphenylvinyl)phenyl]-1,2,3,3a,4,8b-hexahydro-cyclopenta[b]indol-7-ylmethylene}-4-oxo-2-thioxo-thiazolidin-3-yl)acetic acid (indoline dye) |
| D149 | 5-[[4-[4-(2,2-Diphenylethenyl)phenyl]-1,2,3-3a,4,8b-hexahydrocyclopent[b]indol-7-yl]methylene]-2-(3-ethyl-4-oxo-2-thioxo-5-thiazolidinylidene)-4-oxo-3-thiazolidineacetic acid |
| D205 | 5-[[4-[4-(2,2-Diphenylethenyl)phenyl]-1,2,3,3a,4,8b-hexahydrocyclopent[b]indol-7-yl]methylene]-2-(3-octyl-4-oxo-2-thioxo-5-thiazolidinylidene)-4-oxo-3-thiazolidineacetic acid |
| DPP07 | 3-(4-Bromophenyl)-2,5-bis(2-ethylhexyl)-6-(thiophen-2-yl)pyrrolo[3,4-c]pyrrole-1,4(2H,5H)-dione |
| HRS-1 | Cis-Ru(4,4′-di(hexylthienylvinyl))(4,4′-dicarboxy-2,29-bipyridyl)(NCS)2 [dhtbpy = 4,4′-di(hexylthienylvinyl)-2,29-bipyridyl; dcbpy = 4,4′-dicarboxy-2,29-bipyridyl] |
| LEG4 | (E)-3-(6-(4-(bis(2′,4′-dibutoxy-[1,1′-biphenyl]-4-yl)amino)phenyl)-4,4-dihexyl-4H-cyclopenta[2,1-b:3,4-b′]dithiophen-2-yl)-2-cyanoacrylic acid |
| Ru-N3 | Cis-bis(isothiocyanato)bis(2,2′-bipyridyl-4,4′-dicarboxylato)-ruthenium(II) |
| Ru-N719 | Cis-diisothiocyanato-bis(2,2′-bipyridyl-4,4′-dicarboxylato)ruthenium(II)bis(tetrabutylammonium) |
| Ru-Z907 | Cis-diisothiocyanato-(2,2′-bipyridyl-4,4′-dicarboxylic acid)-(2,2′-bipyridyl-4,4′-dinonyl)ruthenium(II) |
| Bis-EDOT | 2,3-Dihydro-5-(2,3-dihydrothieno[3,4-b][1,4]dioxin-5-yl)thieno[3,4-b][1,4]dioxine |
| BMImTFSI | 1-Butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide |
| EDOP | 3,4-Ethylenedioxypyrrole |
| EDOT | 3,4-Ethylenedioxythiophene |
| EMImTFSI | 1-Ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide |
| P3HT | Poly-(3-hexyl)thiophene |
| LiTFSI | Lithium bis(trifluoromethylsulfonyl)imide |
| P3OT | Poly-(3-octyl)thiophene |
| PDTI | Thiophene-isothianaphthene copolymer |
| PEDOT | Poly(3,4-ethylenedioxythiophene) |
| PEDOP | Poly(3,4-ethylenedioxypyrrole) |
| TBP | 4-Tert-butylpyridine |
Unless otherwise indicated, all PCE values refer to simulated full sun irradiation (100 mW cm−2). A general diagram of a S-DSSC based on a conducting polymer hole conductor is presented in Fig. 5.
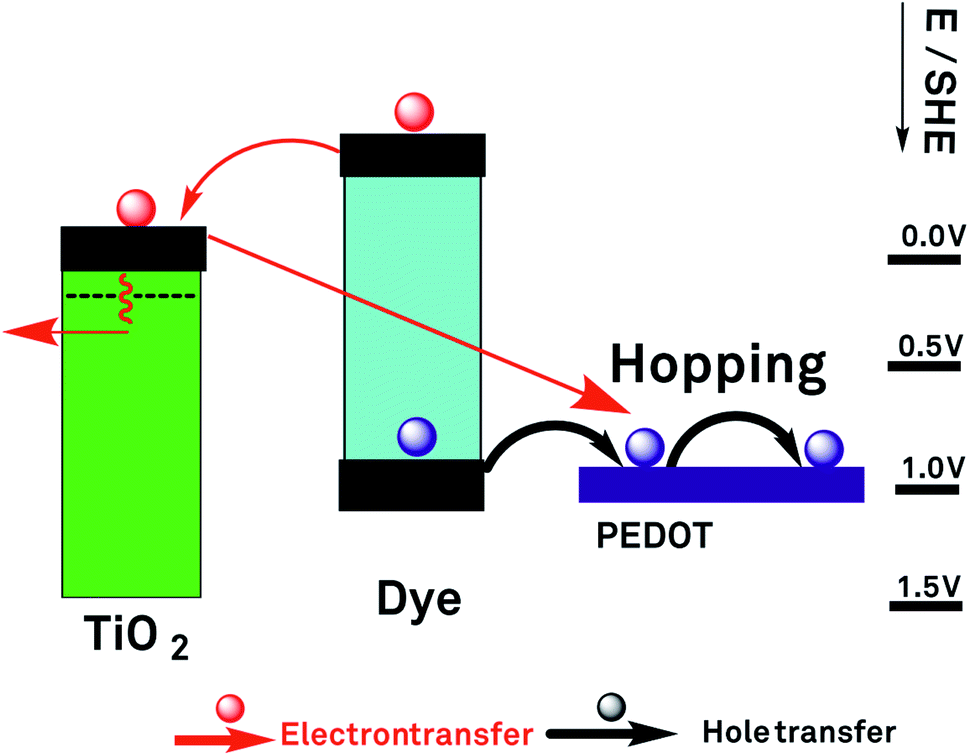 | ||
| Fig. 5 Operation of a solid-state dye sensitised solar cell with PEDOT as hole conductor. | ||
The earliest example of a dry S-DSSC with a conducting polymer as hole conductor has been communicated by the Yanagida group in Japan (year 1998)50,51 with a polypyrrole hole conductor deposited by photoelectrochemical polymerisation in a 3-electrode cell; the dye was the Ru coordination compound Ru-N3 (see Table 3 for all abbreviations for dyes) on the mesoporous TiO2. The counter electrode was a vacuum-evaporated gold (Au) layer. For the best performing cell, the PCE was below 0.1%, with UOC = 0.7 V and jSC = 8 × 10−2 mA cm−2 under simulated full sun irradiation. Henceforth, all PCE values will refer to this condition of irradiation, unless otherwise specified. No blocking underlayer was used between the transparent conducting oxide (transparent conducting oxide) substrate and the mesoporous TiO2 layer.
Gebeyehu et al. (2001–2002)52–54 developed several dry S-DSSCs with dye Ru–N719/mesoporous TiO2 and poly(3-octylthiophene) (P3OT) or a thiophene-isothianaphthene copolymer (PDTI) as hole conductor. The pre-formed polymer, dissolved in an organic solvent, was deposited by spin coating; in the case of PDTI, the solution also contains poly(methyl-methacrylate) (PMMA) in order to improve film formation. The counter electrode was a vacuum-evaporated Au layer. No underlayer was interposed between transparent conducting oxide and dye/mesoporous TiO2. However, in one DSSC configuration, a compact TiO2 layer, of the same quality as the underlayer used in experiments by other research groups, was used as dye support and compared to mesoporous TiO2.54 As expected, in the case of dye/compact TiO2, jSC was lower than for dye/mesoporous TiO2, while the UOC was not higher for dye/compact TiO2 (PLIGHT(IN) = 60 mW cm−2; dye/compact TiO2: φPCE = 0.0011%, UOC = 0.57 V, jSC = 0.027 mA cm−2, and φFF = 0.43; dye/mesoporous TiO2: φPCE = 0.15%, UOC = 0.65 V, jSC = 0.325 mA cm−2, and φFF = 0.4). For the best performing cell, with P3OT, under irradiation of 60 mW cm−2 it was φPCE = 0.16%, with UOC = 0.65 V, jSC = 0.325 mA cm−2, and φFF = 0.44.53
Wang et al. (2006)55 used poly(carboxylated diacetylene) (PDA) as hole conductor for a dry S-DSSC, generated by photochemical polymerisation of a monomer solution impregnating a TiO2/Ru-N3 electrode. A thin TiO2 underlayer was interposed between the transparent oxide layer (transparent conducting oxide) support and the mesoporous TiO2 layer in order to prevent recombination. In the photoelectrochemical polymerisation solution an amount of base tert-butylpyridine (TBP) was added in order to suppress back electron transfer from the conducting polymer; the addition of a base, TBP or other, into either the dye deposition bath or the electrolyte is a common practice for liquid electrolyte-based DSSCs. Since the PDA layer was thin, with the possibility of pinholes, it was coated by a second hole-conducting polymer layer, P3HT, which prevents the contact of the gold counter electrode layer, deposited by vacuum evaporation, with TiO2. The best performance device exhibited a φPCE = 0.8%, with UOC = 0.46 V, jSC = 4.25 mA cm−2, φFF = 0.44.
Johansson et al. (2005)56 investigated the dry solid-state junction between a Ru dye-coated compact TiO2 electrode and a pre-formed PEDOT/PSS polymer, with a Cu foil serving as counter electrode; a graphite layer ensured good contact between Cu and PEDOT–PSS. Several Ru-dyes were considered. The maximum jSC and UOC and under full sun was below 0.1 mA cm−2 and 0.6 V, respectively.
Caramori et al. (2008)57 prepared a solid–liquid-DSSCs with PEDOP as hole conductor generated by photoelectrochemical polymerisation. They used a specially synthesised Ru dye with pyrrole pendant groups so that co-polymerisation occurs between dye and EDOP, resulting to the covalent attachment between dye and hole conductor. A compact TiO2 underlayer was interposed between the transparent conducting oxide substrate and the mesoporous TiO2 dye supporting layer. The counter electrode, Au or C, was pressed against the PEDOP-coated photoelectrode, with a layer of inert non-volatile solvent, LiClO4 in γ-butyrolactone, finally introduced. The PCE of the DSSCs was below 0.1%, with UOC and jSC below 0.5 V and 0.2 mA cm−2, respectively.
SS-DSSCs developed after 2010 usually incorporated a compact TiO2 underlayer. Chen et al. (2012)58 developed a dry DSSCs with the ruthenium dyes Ru-Z907 and Ru-B11 and pre-formed P3HT as hole conductor. The counter electrode was an evaporated Au layer. The highest PCE was 3.0%, obtained with the Ru-B11 sensitiser (UOC = 0.76 V, jSC = 6.71 mA cm−2, φFF = 0.72). The dry S-DSSC cell of Yang et al. (2012)59 used the metal-free organic dye, D35 in conjunction with pre-formed P3HT as hole conductor and an evaporated ag layer as counter electrode, with a resulting PCE of 3.2% (UOC = 0.88 V, jSC = 6.8 mA cm−2, φFF = 0.53). This type of a cell was applied in a hybrid SS-DSSC-electrochemical capacitor system (photocapacitor) for solar energy storage in a collaborative effort of the teams of Anders Hagfeldt (Uppsala, Sweden, physical chemistry) and Pawel Kulesza (Warsaw, Poland, electrochemistry).60
In a series of publications, Shozo Yanagida and collaborators in Osaka, Japan,61–70 describe the development of solid–liquid-DSSCs, with PEDOT as HL, impregnated with a non-volatile electrolyte. The keynote contribution of the Yanagida group was the establishment of photoelectrochemical polymerisation deposition of conducting polymers as a viable method in DSSC research. In all cases, PEDOT was deposited by photoelectrochemical polymerisation, with one exception,67 where chemical polymerisation was applied. Similar solid–liquid DSSCs were subsequent developed by other research groups in Australia71 and Singapore.72–74 The non-volatile electrolyte was either an ionic liquid, EMITFSI61–71 or BMITFSI,70 with LiTFSI and base TBP as additives, or propylene carbonate with LiTFSI.72–74 This treatment resulted to an increase of both UOC and FF due to the creation of a double layer suppressing back electron transfer.
The dyes were either inorganic coordination complexes of ruthenium, Ru-N719,62,66,67 Ru-Z907,61,62,65,68–71 and Ru-HRS-1,63,64 frequently used in DSSC with liquid electrolytes, or organic metal-free dyes, D149 (ref. 72 and 74) and D205.73
In general, bis-EDOT was the precursor, due to the fact that the oxidative power of the dyes used (corresponding to E⊖F(D+/D0) is not sufficient to oxidise EDOT, apart from one case,70 where an EDOT trimer (tri-EDOT) was used. The counter electrode was Au,61,62,66,68,69,72–74 Au/PEDOT,71 Ag,64 Pt,63 and PEDOT.65,70 Due to the presence of the liquid phase, the metal counter electrode layers cannot be deposited by vacuum evaporation, the usual method of counter electrode deposition in S-DSSC, but have to be deposited on a solid substrate, usually transparent conducting oxide glass or polymer, and pressed against the photoelectrode. The highest PCE for solid–liquid-DSSCs is 7.1%,73 with dye D205 (UOC = 0.93 V, jSC = 10.1 mA cm−2, and φFF = 0.76).
The application of photoelectrochemical polymerisation in preparing dry S-DSSCs was pursued by the group of Anders Hagfeldt in Uppsala, Sweden in the middle 2010s,31,75–83 in collaboration with the group of Mohamed Jouini in Paris, France, with the latter contributing to the development of the aqueous photoelectrochemical polymerisation (AQ-photoelectrochemical polymerisation) process84–87 as alternative to the usual organic photoelectrochemical polymerisation process (ORG-photoelectrochemical polymerisation). The hole conductor was mainly PEDOT generated by the photoelectrochemical polymerisation of bis-EDOT dissolved in either an organic or aqueous micellar solution, with the dispersed micelles effectively solubilising the precursor. Bis-EDOT is insoluble in water but can be dispersed in an aqueous micellar solution. In the experiments discussed in this section, the aqueous photoelectrochemical polymerisation solutions contained Triton-X surfactant and LiTFSI. After either nonaqueous or aqueous photoelectrochemical polymerisation, at constant current, the electrode was exposed to a volatile electrolyte containing LiTFSI and base TBP in acetonitrile, followed by drying in air. Subsequent, in all cases apart from one,82 the counter electrode, a Ag layer, was generated by vacuum evaporation. Aitola et al. (2015)82 used a single-wall carbon nanotube (SWCN) counter electrode in place of the evaporate Ag counter electrode, prepared separately, and deposited on the photoelectrode after photoelectrochemical polymerisation, followed by exposure of the whole photoelectrode/counter electrode assembly to a LiTFSI-TBP-acetonitrile solution and air drying.
Comparative data for the performance S-DSSCs prepared by AQ-photoelectrochemical polymerisation and ORG-photoelectrochemical polymerisation are presented in Table 5. The quality of the conducting polymer layers is different in both cases. Porous layers, with shorter chains, is generated by AQ-photoelectrochemical polymerisation of bis-EDOT due to the lower concentration of precursor in the aqueous solution; as polymerisation progresses, it is easier for a precursor cation radical to initiate a new chain rather than add to an existing polymer layer. For the same amount of charge, more, shorter chains are generated by AQ-photoelectrochemical polymerisation than by ORG-photoelectrochemical polymerisation. This fact was confirmed by electrospray mass spectroscopy measurements (Zhang et al., 2015, Anal. Chem.80). Therefore, the contact area between hole conductor and dye layer is larger in the former case. As a result, rate regeneration is faster, resulting to somewhat larger photocurrents for hole conductors generated by AQ-photoelectrochemical polymerisation. On the other hand, due to the larger contact areas the back electron transfer reactions are also faster, resulting to a somewhat lower open-circuit photovoltage. In all, the PCE values in both cases are comparable, under full sun light at around 5–6% for dye LEG4 and 4–5% for dye D35. For Ru-Z907 the PCE is much lower (below 0.2%) for AQ-photoelectrochemical polymerisation compared to ORG-photoelectrochemical polymerisation probably due to the fact that several Ru dyes tend to desorb in presence of water, which preferentially adsorbs on TiO2. The attachment of several organic dyes in TiO2 is more stable.
| No | Dye | Monomer and medium | PCE % | UOC V | ISC mA cm−2 | FF |
|---|---|---|---|---|---|---|
| 1 | D35 | EDOT-ORG | 0.04 | 0.62 | 0.27 | 0.25 |
| 2 | D35 | EDOT-AQ | 3.0 | 0.81 | 6.2 | 0.60 |
| 3 | D35 | Bis-EDOT-ORG | 5.6 | 0.91 | 10.8 | 0.57 |
| 4 | D35 | Bis-EDOT-AQ | 5.2 | 0.84 | 10.9 | 0.56 |
In addition to bis-EDOT, it was shown for the first time that EDOT monomer can be also used for producing a hole conductor layer in DSSCs, with photoelectrochemical polymerisation in aqueous colloidal solution generating sufficiently thick layers. Contrarily, the ORG-photoelectrochemical polymerisation from a nonaqueous EDOT solution generated much thinner layers, resulting to S-DSSCs with a PCE below 0.1% (Table 3). Apart from PEDOT, it was shown for the first time that an efficient DSSC can be constructed based on PEDOP as hole conductor,76 a polymer structurally similar to PEDOT, with –NH replacing –S in the five-member monomer ring: the PCE was two orders of magnitude higher than that of a previous-DSSC with the same polymer as hole conductor.57
The dyes were mainly metal-free organic days, D21L6, D35, LEG4, C218, MK253, DPP07 and, to a lesser extent, ruthenium dyes Ru-B11 and Ru-Z907. The highest PCE, at 7.1% (Zhang et al., 2016, Nano Energy31), was obtained for dry S-DSSC with conducting polymer hole conductors deposited by organic photoelectrochemical polymerisation. Was obtained with dye LEG4 and PEDOT generated by organic photoelectrochemical polymerisation (UOC = 0.830 V, jSC = 13.4 mA cm−2, and φFF = 0.64). For PEDOP as hole conductor generated by ORG-photoelectrochemical polymerisation, the highest PCE was 4.3% (Zhang et al., 2014, ChemPhysChem) for dye D35, under full sun (UOC = 0.825 V, jSC = 8.0 mA cm−2, and φFF = 0.66) and 4.6% under 46% sun (UOC = 0.785 V, jSC = 4.0 mA cm−2, and φFF = 0.53).
Apart from DSSC device studies, a number of fundamental physicochemical characterisation studies was performed, including the aforementioned mass spectroscopy study of Zhang et al., 2015, Anal. Chem.80 as well as investigations involving the charge transport properties of the PEDOT hole conductor by Park et al. (2012)81 and the effect of TiO2 pore size by Zhang et al. (2016, Electrochim. Acta).79
An alternative S-DSSC preparation method for PEDOT-based DSSC, proposed by Delices et al.,88 is the in situ copolymerisation of a dye and a conducting polymer precursor. A metal-free organic dye, functionalised with a carbazole group, was co-polymerised with bis-EDOT by photoelectrochemical polymerisation. The PCE of the resulting dry S-DSSC, at 1%, is rather low; however, this S-DSSC preparation method, guaranteeing intimate contact between dye and hole conductor by covalent attachment, should be further pursued. A similar approach was used before for S-DSSC cells based on the co-polymerisation of a Ru dye and EDOT.57
Finally, the possibility of hybrid solar cells with both a mediator in a liquid electrolyte and a solid hole conductor, proposed by Kim et al. (2011, 2012)89,90 should be mentioned. PEDOT serving as hole conductor for charge transport between photoelectrode and counter electrode, is permeated by an acetonitrile-cased electrolyte containing iodide (I−) as redox mediator. I− is responsible for dye regeneration, being oxidised by photogenerated D+ to triiodide according to the reaction
| 2D+ + 3I−(EL@PE) → 2D0 + I3−(EL@PE) | (41) |
| I3−(EL@PE) + HC → 3I−(EL@PE) + 2h+(HC@PE) | (42) |
Holes diffuse from the vicinity of the photoelectrode to the counter electrode (eqn (8)) and are annihilated by counter electrode electrons (eqn (10)) as in the case of usual S-DSSC operation.
The advantage of this system is that the possible problems due to added I3− are avoided; these include light absorption by coloured I3−, volatility of I2 generated by the disproportionation of I3− to I− and I2, and corrosivity of I3− toward metals which can be used as structural components of DSSCs. This hybrid approach could be extended to other redox mediators.
Conclusion
Solid-state dye sensitised solar cells (S-DSSCs) present several advantages, especially with respect to manufacturing, as compare to these based on a liquid electrolyte, since printing electronics technologies may be readily adapted for their production. Among the various S-DSSCs, these based on electronically conducting polymers are of particular interest. A particular advantage of conducting polymers is the versatility of adapting their molecular structure according to the needs of particular device configurations. In addition to the polymers discussed in this review, other types, for example polymers with pendant redox-active groups, may find applicability in DSSC applications in the near future. In this respect the synergy of scientists in various disciplines, chemical synthesis, materials chemistry, photochemistry–photophysics, and electrochemistry would be essential.Conflicts of interest
There are no conflicts to declare.References
- R. Memming, Prog. Surf. Sci., 1984, 17, 7 CrossRef CAS.
- J. Desilvestro, M. Grätzel, L. Kavan, J. Moser and J. Augustynski, J. Am. Chem. Soc., 1985, 107, 2988 CrossRef CAS.
- N. Vlachopoulos, P. Liska, J. Augustynski and M. Grätzel, J. Am. Chem. Soc., 1988, 110, 1216 CrossRef CAS.
- M. Grätzel and P. Liska, Photo-Electrochemical Cell, US Pat. No 4.927.721, 1990.
- M. K. Nazeeruddin, P. Liska, J. Moser, N. Vlachopoulos and M. Grätzel, Helv. Chim. Acta, 1990, 73, 1788 CrossRef CAS.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- M. K. Nazeeruddin, A. Kay, E. Muller, P. Liska, N. Vlachopoulos and M. Grätzel, J. Am. Chem. Soc., 1993, 115, 6382 CrossRef CAS.
- A. Hagfeldt, B. Didriksson, T. Palmqvist, H. Lindström, S. Södergren, H. Rensmo and S. E. Lindquist, Sol. Energy Mater. Sol. Cells, 1994, 31, 481 CrossRef CAS.
- I. Benesperi, H. Michaels and M. Freitag, J. Mater. Chem. C, 2018, 6, 11903 RSC.
- M. Freitag and G. Boschloo, Curr. Opin. Electrochem., 2017, 2, 111 CrossRef CAS.
- M. Grätzel, Nature, 2001, 414, 338–344 CrossRef PubMed.
- M. Grätzel and J. R. Durrant, in Nanostructured and Photoelectrochemical Systems for Solar Photon Conversion, ed. M. D. Archer and A. J. Nozik, Imperial College Press, London, 2008, vol. 8, pp. 503–536 Search PubMed.
- A. Hagfeldt and M. Grätzel, Chem. Rev., 1995, 95, 49 CrossRef CAS.
- A. Hagfeldt and M. Grätzel, Acc. Chem. Res., 2000, 33, 269 CrossRef CAS PubMed.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- W. Hou, Y. Xiao, G. Han and J.-Y. Lin, Polymers, 2019, 11, 143 CrossRef PubMed.
- K. Kalyanasundaram, Dye-sensitized solar cells, Electrochemical Polymerisation FL Press, Lausanne, Switzerland, 1990 Search PubMed.
- A. Mahmood, J. Energy Chem., 2015, 24, 686–692 CrossRef.
- M. Stojanović, N. Flores-Diaz, Y. Ren, N. Vlachopoulos, L. Pfeifer, Z. Shen, Y. Liu, S. M. Zakeeruddin, J. V. Milić and A. Hagfeldt, Helv. Chim. Acta, 2021, 104, e2000230 CrossRef.
- N. Vlachopoulos, A. Hagfeldt, I. Benesperi, M. Freitag, G. Hashmi, G. Jia, R. A. Wahyuono, J. Plentz and B. Dietzek, Sustainable Energy Fuels, 2021, 5, 367–383 RSC.
- N. Vlachopoulos, J. Zhang and A. Hagfeldt, Chimia, 2015, 69, 41–51 CrossRef CAS PubMed.
- S. Yun and A. Hagfeldt, Counter Electrodes for Dye-sensitized and Perovskite Solar Cells, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2018 Search PubMed.
- S. Yun, J. N. Freitas, A. F. Nogueira, Y. Wang, S. Ahmad and Z. S. Wang, Prog. Polym. Sci., 2016, 59, 1–40 CrossRef CAS.
- J. Zhang, M. Freitag, A. Hagfeldt and G. Boschloo, in Molecular Devices for Solar Energy Conversion and Storage, ed. H. Tian, G. Boschloo and A. Hagfeldt, Springer Nature, Singapore, 1st edn, 2018, ch. 4, pp. 151–185 Search PubMed.
- A. J. McEvoy and M. Grätzel, Sol. Energy Mater. Sol. Cells, 1994, 32, 221–227 CrossRef CAS.
- C. J. Barbé, F. Arendse, P. Comte, M. Jirousek, F. Lenzmann, V. Shklover and M. Grätzel, J. Am. Ceram. Soc., 1997, 80, 3157–3171 CrossRef.
- T. Gerfin, M. Grätzel and L. Walder, Prog. Inorg. Chem., 1996, 44, 345–399 Search PubMed.
- K. Kakiage, Y. Aoyama, T. Yano, K. Oya, J. I. Fujisawa and M. Hanaya, Chem. Commun., 2015, 51, 15894–15897 RSC.
- Best Research-Cell Efficiency Chart-NREL, https://www.nrel.gov/pv/cell-efficiency.html, accessed July 2021.
- Y. Cao, Y. Saygili, A. Ummadisingu, J. Teuscher, J. Luo, N. Pellet, F. Giordano, S. M. Zakeeruddin, J. E. Moser, M. Freitag, A. Hagfeldt and M. Grätzel, Nat. Commun., 2017, 8, 15390 CrossRef PubMed.
- J. Zhang, N. Vlachopoulos, M. Jouini, M. B. Johansson, X. Zhang, M. K. Nazeeruddin, G. Boschloo, E. M. J. Johansson and A. Hagfeldt, Nano Energy, 2016, 19, 455–470 CrossRef CAS.
- Q. Li, Q. Tang, H. Chen, H. Xu, Y. Qin, B. He, Z. Liu, S. Jin and L. Chu, Mater. Chem. Phys., 2014, 144, 287–292 CrossRef CAS.
- Q. Li, X. Chen, Q. Tang, H. Cai, Y. Qin, B. He, M. Li, S. Jin and Z. Liu, J. Power Sources, 2014, 248, 923–930 CrossRef CAS.
- Q. Li, H. Li, X. Jin and Z. Chen, Electrochim. Acta, 2018, 260, 413–419 CrossRef CAS.
- J. Wu, Z. Lan, J. Lin, M. Huang, Y. Huang, L. Fan, G. Luo, Y. Lin, Y. Xie and Y. Wei, Chem. Soc. Rev., 2017, 46, 5975–6023 RSC.
- H. Ellis, N. Vlachopoulos, L. Häggman, C. Perruchot, M. Jouini, G. Boschloo and A. Hagfeldt, Electrochim. Acta, 2013, 107, 45–51 CrossRef CAS.
- K. Saranya, M. Rameez and A. Subramania, Eur. Polym. J., 2015, 66, 207–227 CrossRef CAS.
- A. Elschner, S. Kirchmeyer, W. Lövenich, U. Merker and K. Reuter, PEDOT: Principles and applications of an intrinsically conductive polymer, CRC Press, Boca Raton, Florida, USA, 2010 Search PubMed.
- G. Inzelt, Conducting Polymers, Springer, Berlin, Germany, 2012 Search PubMed.
- J. R. Reynolds, B. C. Thompson and T. A. Skotheim, Conjugated Polymers, CRC Press, Boca Raton, Florida, USA, 2019 Search PubMed.
- G. G. Wallace, P. R. Teasdale, G. M. Spinks and L. A. P. Kane-Maguire, Conductive Electroactive Polymers, CRC Press, Boca Raton, Florida, USA, 2008 Search PubMed.
- J. J. Heinze, B. A. Frontana-Uribe and S. Ludwigs, Chem. Rev., 2010, 110, 4724–4771 CrossRef CAS PubMed.
- J. Heinze, in Organic Electrochemistry, ed. O. Hammerich and B. Speiser, CRC Press, Boca Raton, Florida, USA, 5th edn, 2016, ch. 41, pp. 1571–1604 Search PubMed.
- J. Heinze, Top. Curr. Chem., 1990, 152, 1–47 CrossRef CAS.
- J. Heinze, Synth. Met., 1991, 43, 2805–2823 CrossRef CAS.
- K. Gurunathan, A. V. Murugan, R. Marimuthu, U. P. Mulik and D. P. Amalnerkar, Mater. Chem. Phys., 1999, 61, 173–191 CrossRef CAS.
- V. S. Bagotsky, Fundamentals of Electrochemistry, Wiley, Hobokn, New Jersey, USA, 2005 Search PubMed.
- P. A. Christensen and A. Hamnett, Techniques and Mechanisms in Electrochemistry, Chapman & Hall, London, U.K., 1994 Search PubMed.
- J. Koryta, J. Dvořák and L. Kavan, Principles of Electrochemistry, Wiley, Chichester, UK, 1993 Search PubMed.
- K. Murakoshi, R. Kogure, Y. Wada and S. Yanagida, Sol. Energy Mater. Sol. Cells, 1998, 55, 113–125 CrossRef CAS.
- K. Murakoshi, R. Kogure, Y. Wada and S. Yanagida, Chem. Lett., 1997, 471–472 CrossRef CAS.
- D. Gebeyehu, C. J. Brabec and N. S. Sariciftci, Thin Solid Films, 2002, 403–404, 271–274 CrossRef CAS.
- D. Gebeyehu, C. J. Brabec, N. S. Sariciftci, D. Vangeneugden and R. Kiebooms, Synth. Met., 2002, 125, 279–287 CrossRef CAS.
- D. Gebeyehu, C. J. Brabec, F. Padinger, T. Fromherz, S. Spiekermann, N. Vlachopoulos, F. Kienberger, H. Schindler and N. S. Sariciftci, Synth. Met., 2001, 121, 1549–1550 CrossRef CAS.
- Y. Wang, K. Yang, S. Kim, R. Nagarajan and L. A. Samuelson, Chem. Mater., 2006, 18, 4215–4217 CrossRef CAS.
- E. M. J. Johansson, L. Yang, E. Gabrielsson, P. W. Lohse, G. Boschloo, L. Sun and A. Hagfeldt, J. Phys. Chem. C, 2012, 116, 18070–18078 CrossRef CAS.
- S. Caramori, S. Cazzanti, L. Marchini, R. Argazzi, C. A. Bignozzi, D. Martineau, P. C. Gros and M. Beley, Inorg. Chim. Acta, 2008, 361, 627–634 CrossRef CAS.
- W.-C. Chen, C.-Y. Chen, C.-G. Wu, K.-C. Ho and L. Wang, J. Power Sources, 2012, 214, 113–118 CrossRef CAS.
- L. Yang, U. B. Cappel, E. L. Unger, M. Karlsson, K. M. Karlsson, E. Gabrielsson, L. Sun, G. Boschloo, A. Hagfeldt and E. M. J. Johansson, Phys. Chem. Chem. Phys., 2012, 14, 779–789 RSC.
- M. Skunik-Nuckowska, K. Grzejszczyk, P. J. P. J. Kulesza, L. Yang, N. Vlachopoulos, L. Häggman, E. Johansson and A. Hagfeldt, J. Power Sources, 2013, 234, 91–99 CrossRef CAS.
- N. Fukuri, N. Masaki, T. Kitamura, Y. Wada and S. Yanagida, J. Phys. Chem. B, 2006, 110, 25251–25258 CrossRef CAS PubMed.
- N. Fukuri, Y. Saito, W. Kubo, G. K. R. Senadeera, T. Kitamura, Y. Wada and S. Yanagida, J. Electrochem. Soc., 2004, 151, A1745 CrossRef CAS.
- A. J. Mozer, Y. Wada, K.-J. Jiang, N. Masaki, S. Yanagida and S. N. Mori, Appl. Phys. Lett., 2006, 89, 043509 CrossRef.
- K. Manseki, W. Jarernboon, Y. Youhai, K.-J. Jiang, K. Suzuki, N. Masaki, Y. Kim, J. Xia and S. Yanagida, Chem. Commun., 2011, 47, 3120–3122 RSC.
- M. Lira-Cantu, F. C. Krebs, P. Gomez-Romero and S. Yanagida, Mater. Res. Soc. Symp. Proc., 2008, 1007, 249–257 Search PubMed.
- Y. Saito, N. Fukuri, R. Senadeera, T. Kitamura, Y. Wada, S. Yanagida, Y. F. Saito, N. Senadeera, R. Kitamura, T. Wada and Y. Yanagida, Electrochem. Commun., 2004, 6, 71–74 CrossRef CAS.
- Y. Saito, T. Kitamura, Y. Wada and S. Yanagida, Synth. Met., 2002, 131, 185–187 CrossRef CAS.
- J. Xia, N. Masaki, M. Lira-Cantu, Y. Kim, K. Jiang and S. Yanagida, J. Am. Chem. Soc., 2008, 130, 1258–1263 CrossRef CAS PubMed.
- J. Xia, N. Masaki, M. Lira-Cantu, Y. Kim, K. Jiang and S. Yanagida, J. Phys. Chem. C, 2008, 112, 11569–11574 CrossRef CAS.
- Y. Kim, Y. E. Sung, J. Bin Xia, M. Lira-Cantu, N. Masaki and S. Yanagida, J. Photochem. Photobiol., A, 2008, 193, 77–80 CrossRef CAS.
- A. J. Mozer, D. K. Panda, S. Gambhir, T. C. Romeo, B. Winther-Jensen and G. G. Wallace, Langmuir, 2010, 26, 1452–1455 CrossRef CAS PubMed.
- L. Cai, X. Liu, L. Wang and B. Liu, Polym. Bull., 2012, 68, 1857–1865 CrossRef CAS.
- X. Liu, Y. Cheng, L. Wang, L. Cai and B. Liu, Phys. Chem. Chem. Phys., 2012, 14, 7098–7103 RSC.
- X. Liu, W. Zhang, S. Uchida, L. Cai, B. Liu and S. Ramakrishna, Adv. Mater., 2010, 22, E150–E155 CrossRef CAS PubMed.
- L. Yang, J. Zhang, Y. Shen, B. W. Park, D. Bi, L. Häggman, E. M. J. Johansson, G. Boschloo, A. Hagfeldt, N. Vlachopoulos, A. Snedden, L. Kloo, A. Jarboui, A. Chams, C. Perruchot and M. Jouini, J. Phys. Chem. Lett., 2013, 4, 4026–4031 CrossRef CAS.
- J. Zhang, L. Häggman, M. Jouini, A. Jarboui, G. Boschloo, N. Vlachopoulos and A. Hagfeldt, ChemPhysChem, 2014, 15, 1043–1047 CrossRef CAS PubMed.
- J. Zhang, A. Jarboui, N. Vlachopoulos, M. Jouini, G. Boschloo and A. Hagfeldt, Electrochim. Acta, 2015, 179, 220–227 CrossRef CAS.
- J. Zhang, L. Yang, Y. Shen, B. W. Park, Y. Hao, E. M. J. Johansson, G. Boschloo, L. Kloo, E. Gabrielsson, L. Sun, A. Jarboui, C. Perruchot, M. Jouini, N. Vlachopoulos and A. Hagfeldt, J. Phys. Chem. C, 2014, 118, 16591–16601 CrossRef CAS.
- J. Zhang, M. Pazoki, J. Simiyu, M. B. M. B. Johansson, O. Cheung, L. Häggman, E. M. J. Johansson, N. Vlachopoulos, A. Hagfeldt and G. Boschloo, Electrochim. Acta, 2016, 210, 23–31 CrossRef CAS.
- J. Zhang, H. Ellis, L. Yang, E. M. J. Johansson, G. Boschloo, N. Vlachopoulos, A. Hagfeldt, J. Bergquist and D. Shevchenko, Anal. Chem., 2015, 87, 3942–3948 CrossRef CAS PubMed.
- B.-W. Park, L. Yang, E. M. J. Johansson, N. Vlachopoulos, A. Chams, C. Perruchot, M. Jouini, G. Boschloo and A. Hagfeldt, J. Phys. Chem. C, 2013, 117, 22484–22491 CrossRef CAS.
- K. Aitola, J. Zhang, N. Vlachopoulos, J. Halme, A. Kaskela, A. G. Nasibulin, E. I. Kauppinen, G. Boschloo and A. Hagfeldt, J. Solid State Electrochem., 2015, 19, 3139–3144 CrossRef CAS.
- J. Zhang, N. Vlachopoulos, Y. Hao, T. W. Holcombe, G. Boschloo, E. M. J. Johansson, M. Grätzel and A. Hagfeldt, ChemPhysChem, 2016, 17, 1441–1445 CrossRef CAS PubMed.
- N. Sakmeche, E. a. Bazzaoui, M. Fall, S. Aeiyach, M. Jouini, J. C. Lacroix, J. J. Aaron and P. C. Lacaze, Synth. Met., 1997, 84, 191–192 CrossRef CAS.
- N. Sakmeche, S. Aeiyach, J. J. Aaron, M. Jouini, J. C. Lacroix and P. C. Lacaze, Langmuir, 1999, 15, 2566–2574 CrossRef CAS.
- N. Sakmeche, J. J. Aaron, M. Fall, S. Aeiyach, M. Jouini, J. C. Lacroix and P. C. Lacaze, Chem. Commun., 1996, 2723 RSC.
- M. Fall, J. J. Aaron, N. Sakmeche, M. M. Dieng, M. Jouini, S. Aeiyach, J. C. Lacroix and P. C. Lacaze, Synth. Met., 1998, 93, 175–179 CrossRef CAS.
- A. Delices, J. Zhang, M. P. Santoni, C. Z. Dong, F. Maurel, N. Vlachopoulos, A. Hagfeldt and M. Jouini, Electrochim. Acta, 2018, 269, 163–171 CrossRef CAS.
- J. H. J. Kim, J. K. Koh, B. Kim, S. H. Ahn, H. Ahn, D. Y. Ryu, J. H. J. Kim and E. Kim, Adv. Funct. Mater., 2011, 21, 4633–4639 CrossRef CAS.
- B. Kim, J. K. Koh, J. H. J. Kim, W. S. Chi, J. H. J. Kim and E. Kim, ChemSusChem, 2012, 5, 2173–2180 CrossRef CAS PubMed.
Footnote |
| † Dedicated to Professor Seth Marder, Georgia Institute of Technology, for his 65th birthday. |
| This journal is © The Royal Society of Chemistry 2021 |