
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTheoretical study on molecular mechanism of aerobic oxidation of 5-hydroxymethylfurfural to 2,5-diformyfuran catalyzed by VO2+ with counterpart anion in N,N-dimethylacetamide solution†
Zhen-Bing Si a,
Jin-Shan Xionga,
Ting Qia,
Hong-Mei Yanga,
Han-Yun Mina,
Hua-Qing Yang*a and
Chang-Wei Hub
a,
Jin-Shan Xionga,
Ting Qia,
Hong-Mei Yanga,
Han-Yun Mina,
Hua-Qing Yang*a and
Chang-Wei Hub
aCollege of Chemical Engineering, Sichuan University, Chengdu, Sichuan 610065, P.R. China. E-mail: huaqingyang@scu.edu.cn; Fax: +86 28 85464466; Tel: +86 28 85464466
bKey Laboratory of Green Chemistry and Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu, Sichuan 610064, P.R. China
First published on 15th December 2021
Abstract
Vanadium-containing catalysts exhibit good catalytic activity toward the aerobic oxidation of 5-hydroxymethylfurfural (HMF) to 2,5-diformyfuran (DFF). The aerobic oxidation mechanism of HMF to DFF catalyzed by VO2+ with counterpart anion in N,N-dimethylacetamide (DMA) solution have been theoretically investigated. In DMA solution, the stable VO2+-containing complex is the four-coordinated [V(O)2(DMA)2]+ species. For the gross reaction of 2HMF + O2 → 2DFF + 2H2O, there are three main reaction stages, i.e., the oxidation of the first HMF to DFF with the reduction of [V(O)2(DMA)2]+ to [V(OH)2(DMA)]+, the aerobic oxidation of [V(OH)2(DMA)]+ to the peroxide [V(O)3(DMA)]+, and the oxidation of the second HMF to DFF with the reduction of [V(O)3(DMA)]+ to [V(O)2(DMA)2]+. The rate-determining reaction step is associated with the C–H bond cleavage of –CH2 group of the first HMF molecule. The peroxide [V(O)3(DMA)]+ species exhibits better oxidative activity than the initial [V(O)2(DMA)2]+ species, which originates from its narrower HOMO–LUMO gap. The counteranion Cl− exerts promotive effect on the aerobic oxidation of HMF to DFF catalyzed by [V(O)2(DMA)2]+ species.
1. Introduction
Currently, 5-hydroxymethylfurfural (HMF), as an important platform for connecting biomass and petrochemical industry, has attracted wide attention, due to its sustainable production of high value-added chemicals, such as 2,5-diformylfuran (DFF).1,2 DFF can be used as a multifunctional intermediate for the synthesis of pharmaceuticals and functional polymers.3,4 Especially, DFF can be synthesized via a partial oxidation of the primary hydroxyl group without attacking reactive unsaturated aldehyde group from HMF, using clean O2 as an oxidant.5 At present, however, DFF is still commercially expensive and only available in milligram quantities.In order to improve the yield of selective partial oxidation of HMF to DFF, deep oxidation of HMF must be avoided. Therefore, it is necessary to develop suitable catalytic oxidation system. To this end, various catalytic systems have been explored, including ruthenium-,6–8 silver-,9 copper- ,10 iron-,11 manganese-,12,13 molybdenum-,14–16 and vanadium-containing catalysts.17–23,44 Particularly, V-containing catalysts have been proved to be good efficient and inexpensive. Unfortunately, the instability of HMF limits the application of pure HMF as the raw feedstock, because of the high cost of its downstream purification process. Encouragingly, for the scale synthesis of DFF, a promising approach is the direct production from cheap and accessible glucose, avoiding the separation of unstable HMF intermediate from solution.23,24 Interestingly, our group have previously proposed one-pot-two-steps conversion of glucose into DFF, over both CrCl3/NaBr catalyst for isomerization/dehydration and NaVO3 catalyst for partial oxidation in N,N-dimethylacetamide (DMA) solution.23 Herein, the experimental system includes the water produced from glucose and the Lewis acid Cr3+, which make the solution acidic. Thereupon, when NaVO3 can be readily dissolved in acidic DMA solution, the dissolved V-species may exist as VO2+ cation in acidic DMA solution.25 Moreover, there are some counterpart anions of VO2+, e.g., Cl− and Br−, in actually experimental DMA solution.23 These counterpart anions may affect the catalytic performance of VO2+. Despite the present research achievement, the experimental study of direct synthesis of DFF from glucose still faces some problems, such as low yield of DFF, poor recycling of catalyst, and high cost of product separation and purification. To improve these issues, it is necessary to understand the relevant catalytic molecular mechanism.
In this study, we report a completely molecular mechanism for the aerobic oxidation of HMF to DFF catalyzed by VO2+ with Cl− in DMA solution. The objectives are as following: (a) to ascertain the stable form of VO2+ and solvent DMA coordination, (b) to elucidate the detailed reaction pathways for the aerobic oxidation of HMF to DFF catalyzed by VO2+-containing complex, (c) to acquire the determining intermediate (DI) and the determining transition state (DTS), based on the energetic span,26–29 (d) to gain the catalytic nature of VO2+ species, and (e) to obtain the effects of counterpart anions on the stabilization of both DI and DTS, which may engineer the novel design of green and efficient catalytic system toward the synthesis of DFF from HMF.
2. Computational details
In DMA media, all geometry calculations were performed with Gaussian 09 program.30 To simulate the DMA solvent effect, a polarized continuum model based on solute electron density (PCM-SMD) was employed.31,32 Full geometric optimizations were run to locate all stationary points, using M06 density functional theory method33,34 with the 6-311++G(d, p) basis set35,36 for the main-group elements (H, C, O, N, and Cl), and aug-cc-pVTZ basis set for the 3d transition metal (V) in view of relativistic effect,37 namely, M06/6-311++G(d,p), aug-cc-pVTZ,. Each stationary point was characterized by harmonic vibrational frequency calculation (all real frequencies for intermediate and only one imaginary frequency for transition state), in which the zero-point energy (ZPE) was also gained. Each transition state structure was also verified by intrinsic reaction coordinate (IRC).38,39 The natural atomic charges were analyzed, using the natural bond orbital (NBO) method.40,41 Unless otherwise stated, the Gibbs free energy of formation (ΔG) are relative to the initial catalyst and reactants in DMA solution, under experimental temperature and pressure (383.0 K and 1.0 atm).23Based on the energetic span model,28 the rate-determining step was diagnosed, gaining the determining intermediate (DI) and the determining transition state (DTS). The rate constants were assessed over the 353–433 K temperature range, on the basis of conventional transition state theory, together with tunnelling correction, as described in our previous study.42,43
In DMA solution, DMA molecule coordinates readily to VO2+ species, forming [V(O)2(DMA)x]+ complex. The Gibbs free energy of formation (ΔG) of each species [V(O)2(DMA)x]+ starting from two independent species (VO2+ and DMA) was computed following the reaction
| VO2+ + x(DMA) → [V(O)2(DMA)x]+ | (1) |
And using the following the equation:
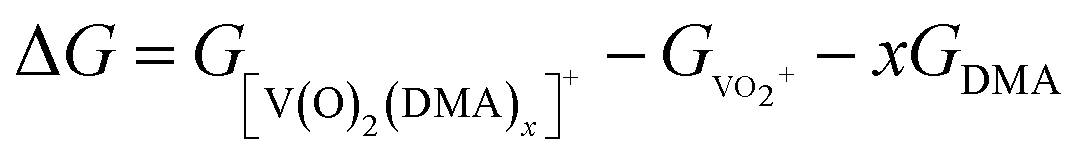 | (2) |
3. Results and discussion
For the reactant HMF molecules, as reported earlier,43 there are eight possible conformers. For eight conformers of HMF in DMA solution, the geometric structures and relative Gibbs free energy are shown in Fig. S3† from ESI. As depicted in Fig. S3,† for the eight conformers of HMF, the relative Gibbs free energies increase as HMF-1 < HMF-2 < HMF-3 < HMF-6 < HMF-4 < HMF-8 < HMF-5 < HMF-7 in DMA solution. It is indicated that the conformer HMF-1 is thermodynamically the most stable in DMA solution.Here, the ground state of O2 molecule is the triplet state with the singlet state as the first excited state. The superscript prefixes “1” and “3” represents the singlet and triplet states, respectively. Unless specified, the default state is the ground state “1”.
For the product DFF molecules, there are three possible conformers. For three conformers of DFF in DMA solution, the geometric structures and relative Gibbs free energy are shown in Fig. S4† from ESI. As depicted in Fig. S4,† the relative Gibbs free energies increase as DFF-2 < DFF-3 < DFF-1. It is inferred that the conformer DFF-2 is thermodynamically the most preferable in DMA solution. Thereupon, the conformer HMF-1 is preferred as the initial reactant in the present study, while the conformer DFF-2 is regarded as the product.
For the aerobic of HMF to DFF, the following gross reaction will be studied:
| 2HMF + O2 → 2DFF + 2H2O | (3) |
The possible reaction pathways are depicted in Scheme 1, based on the relevant literatures.15,16,44
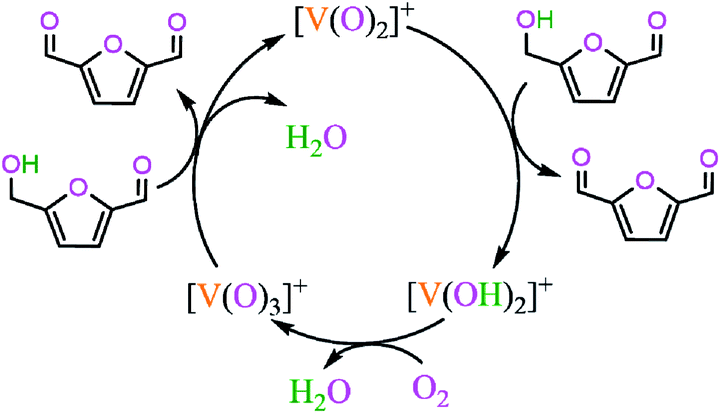 | ||
| Scheme 1 Catalytic oxidation pathways of HMF to DFF. | ||
3.1 Stable coordination species of VO2+ with DMA
In DMA solution, the optimized geometric structures and the formed Gibbs free energies of [V(O)2(DMA)x]+ complexes are shown in Fig. 1.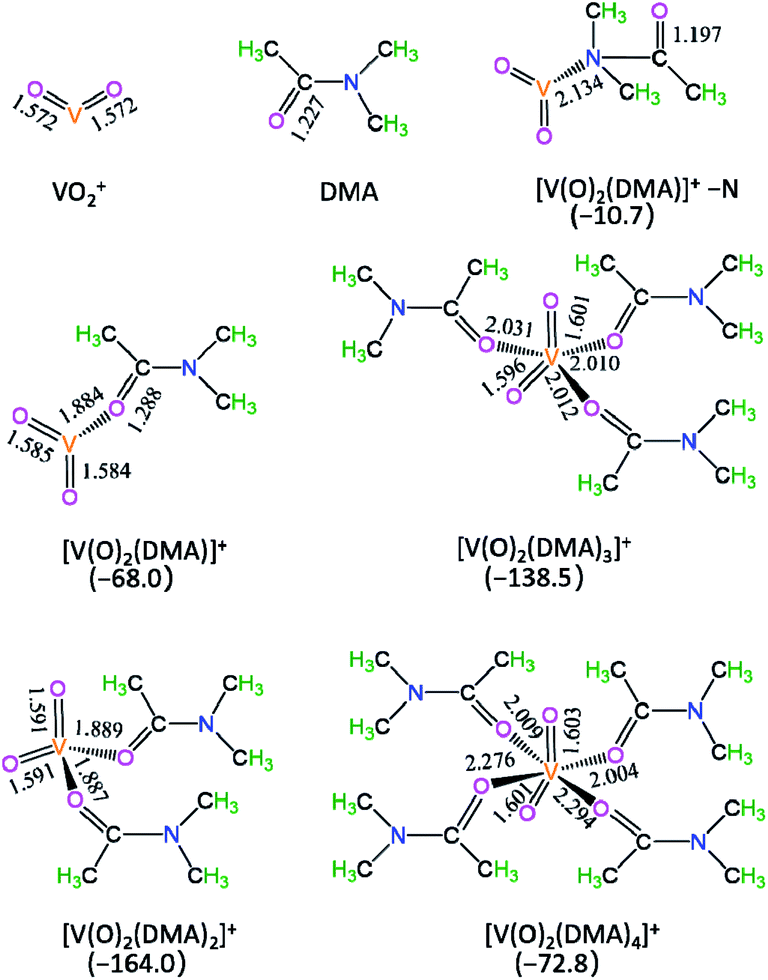 | ||
| Fig. 1 The optimized geometric structures and the formed Gibbs free energies (Gr, kJ mol−1) of [V(O)2(DMA)x]+ complexes in the DMA solution. Bond lengths are reported in Å. | ||
As depicted in Fig. 1, with regard to the DMA coordinating to V-site of VO2+, there are two coordination types, i.e., through N-end and O-end of DMA, i.e., [V(O)2(DMA)]+–N and [V(O)2(DMA)]+. For [V(O)2(DMA)]+–N and [V(O)2(DMA)]+, the ΔG value is −10.7 and −68.0 kJ mol−1, respectively. It is inferred that the O-end coordination [V(O)2(DMA)]+ is 57.3 kJ mol−1 more stable than the N-end coordination [V(O)2(DMA)]+–N. That is to say, for the DMA coordination to V-site of VO2+, the O-end is stronger than the N-end.
Furthermore, for [V(O)2(DMA)x]+ (x = 1–4) species, the ΔG value is −68.0, −164.0, −138.5, and −72.8 kJ mol−1, respectively. It is obvious that the four-coordinated [V(O)2(DMA)2]+ complex is thermodynamically the most preferable. Exceptionally, when the Cl− anion interacts with VO2+, the corresponding complex [V(O)2(Cl)], with the stabilization energy of 147.8 kJ mol−1, as shown in Table S1† from ESI. It is obvious that the [V(O)2(DMA)2]+ complex is more 16.2 kJ mol−1 stable than the [V(O)2(Cl)] complex. Thereupon, the [V(O)2(DMA)2]+ complex is preferred as the initial catalytic species.
3.2 [V(O)2(DMA)2]+ + HMF → [V(OH)2(DMA)]+ + DFF + DMA
The potential energy diagrams and geometric structures for the first HMF oxidation to DFF over [V(O)2(DMA)2]+ cation without/with Cl− anion are shown in Fig. 2, denoted as P-0-1 and P-Cl-1, respectively.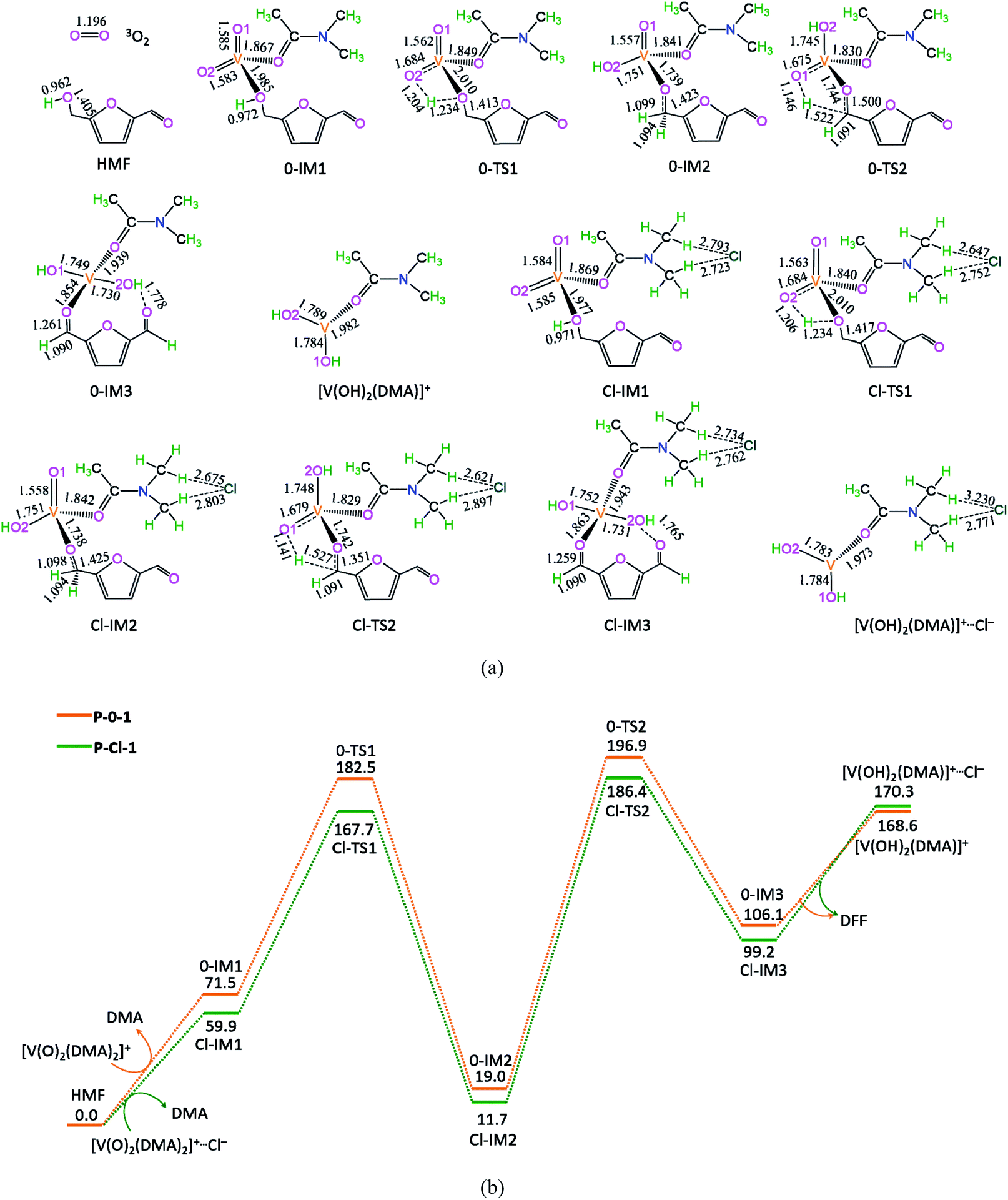 | ||
| Fig. 2 The geometric structures (a) and the schematic energy diagrams (b) with the relative Gibbs free energy (Gr, kJ mol−1) relative to the reactants for the first HMF oxidation to DFF catalyzed by [V(O)2(DMA)2]+ without/with Cl− in the DMA solution. For clarity, the hydrogen atoms on carbon are not shown. Bond lengths are reported in Å. | ||
As depicted in Fig. 2, in the absence of counteranion Cl−, there are four reaction steps, i.e., (1) the formation of molecular complex 0-IM1 from the HMF-1 with exchanging with one DMA molecule from [V(O)2(DMA)2]+, (2) the O–H bond cleavage via a four-membered cyclic 0-TS1 through addition unsaturated the first V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond, (3) the C–H bond cleavage in –CH2OH group via a five-membered 0-TS2 through addition unsaturated the second VO double bond, resulting in the DFF formation, and (4) the release of DFF with leaving the reduced [V(OH)2(DMA)]+ behind.
O double bond, (3) the C–H bond cleavage in –CH2OH group via a five-membered 0-TS2 through addition unsaturated the second VO double bond, resulting in the DFF formation, and (4) the release of DFF with leaving the reduced [V(OH)2(DMA)]+ behind.
The P-0-1 exhibits the energy height of the highest point (EHHP) of 196.9 kJ mol−1 at 0-TS2, the highest energy barrier (HEB) of 177.9 kJ mol−1 at the reaction step of 0-IM2 → 0-TS2 → 0-IM3, and the energy height of the lowest point (EHLP) of 0.0 kJ mol−1 at the initial point. The P-0-1 is concerned with the oxidation of the first HMF molecule to DFF, where the two VO double bonds are reduced by addition to two V–OH group.
Additionally, in the presence of counteranion Cl−, the P-Cl-1 is similar to P-0-1, there are differences in relative Gibbs free energies on the potential energy diagrams. The P-Cl-1 displays the EHHP of 186.4 kJ mol−1 at Cl-TS2, the HEB of 174.7 kJ mol−1 at the reaction step of Cl-IM2 → Cl-TS2 → Cl-IM3, and the EHLP of 0.0 kJ mol−1 at the initial point.
Compared with P-0-1, P-Cl-1 possesses lower EHHP (186.4 vs. 196.9 kJ mol−1) and lower HEB (174.7 vs. 177.9 kJ mol−1). It is indicated that the counteranion Cl− exhibits some degree promotive effect for [V(O)2(DMA)2]+ oxidizing the first HMF to DFF.
3.3 [V(OH)2(DMA)]+ + O2 → [V(O)3(DMA)]+ + H2O
The potential energy diagrams and geometric structures for the oxidative dehydration of [V(OH)2(DMA)]+ to [V(O)3(DMA)]+ without/with Cl− anion are shown in Fig. 3, denoted as P-0-2 and P-Cl-2, respectively.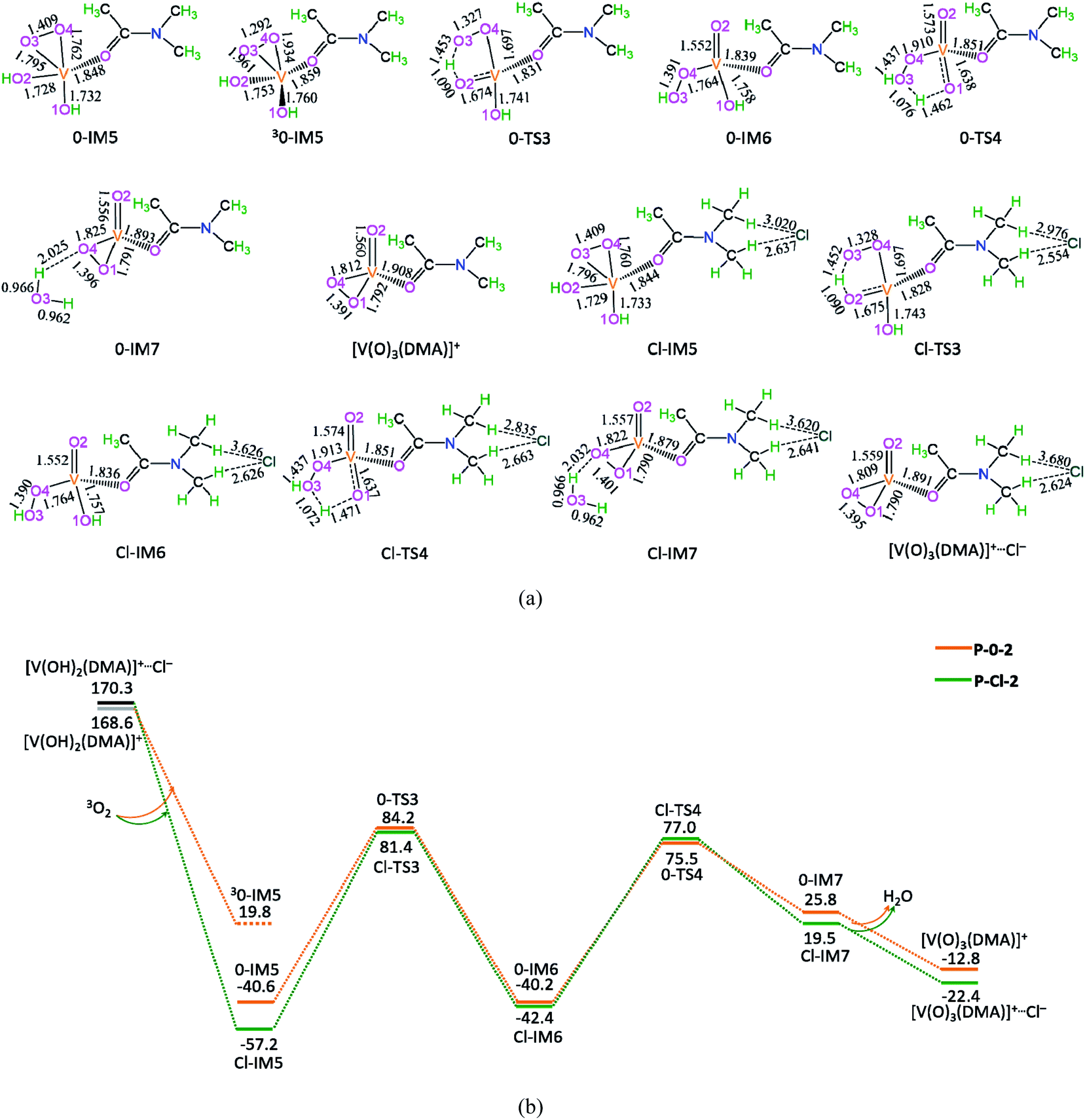 | ||
| Fig. 3 The geometric structures (a) and the schematic energy diagrams (b) with the relative Gibbs free energy (Gr, kJ mol−1) relative to the reactants for the oxidative dehydration of [V(OH)2(DMA)]+ to [V(O)3(DMA)]+ catalyzed by without/with Cl− in the DMA solution. For clarity, the hydrogen atoms on carbon are not shown. Bond lengths are reported in Å. | ||
As depicted in Fig. 3, in the absence of counteranion Cl−, there are four reaction steps, i.e., (1) the formation of molecular complex 0-IM5 between the interaction of O2 with V-site of [V(OH)2(DMA)]+, (2) the O–H bond cleavage through [1,4]-H shift via a five-membered ring 0-TS3 for the formation of hydroxyl peroxide, (3) the cleavage of peroxide bond through [1,4]-H shift via a five-membered 0-TS4, leading to the H2O formation, and (4) the release of H2O molecule and saving the oxidized [V(O)3(DMA)]+ species. Particularly, although the ground state of O2 is the triplet state, the triplet state 30-IM5 lies 60.4 kJ mol−1 above the singlet state 0-IM5. Thereby, a significant triplet–singlet spin inversion may occur once at 0-IM5. Here, for 30-IM5, the Mulliken spin densities are calculated to be 0.56 at O3, 0.48 at O4, and 0.99 at V. Moreover, for 0-IM5, the NBO analysis shows that the occupancies of V–O(3) and V–O(4) are 1.914 and 1.930e, respectively. It is indicated that the single bond of V–O(3) and V–O(4) have been formed. That is to say, in 0-IM5, the O2 molecule is chemically adsorbed on V-site of [V(OH)2(DMA)]+.
The P-0-2 exhibits the EHHP of 168.6 kJ mol−1 at the initial point ([V(OH)2(DMA)]+ + O2), the HEB of 124.8 kJ mol−1 at the reaction step of 0-IM5 → 0-TS3 → 0-IM6, and the EHLP of −40.6 kJ mol−1 at 0-IM5. Besides, the P-Cl-2 displays the EHHP of 170.3 kJ mol−1 at initial point ([V(OH)2(DMA)]+⋯Cl + O2), the HEB of 138.6 kJ mol−1 at the reaction step of Cl-IM5 → Cl-TS3 → Cl-IM6, and the EHLP of −57.2 kJ mol−1 at Cl-IM5.
Compared with P-0-2, P-Cl-2 includes lower EHLP (−57.2 vs. −40.6 kJ mol−1). It is inferred that the counteranion Cl− exhibits some degree stabilization effect toward 0-IM5.
3.4 [V(O)3(DMA)]+ + HMF + DMA → [V(O)2(DMA)2]+ + DFF + H2O
The potential energy diagrams and geometric structures for the oxidative dehydration of the second HMF to DFF by [V(O)3(DMA)]+ species without/with Cl− anion are shown in Fig. 4, denoted as P-0-3 and P-Cl-3, respectively.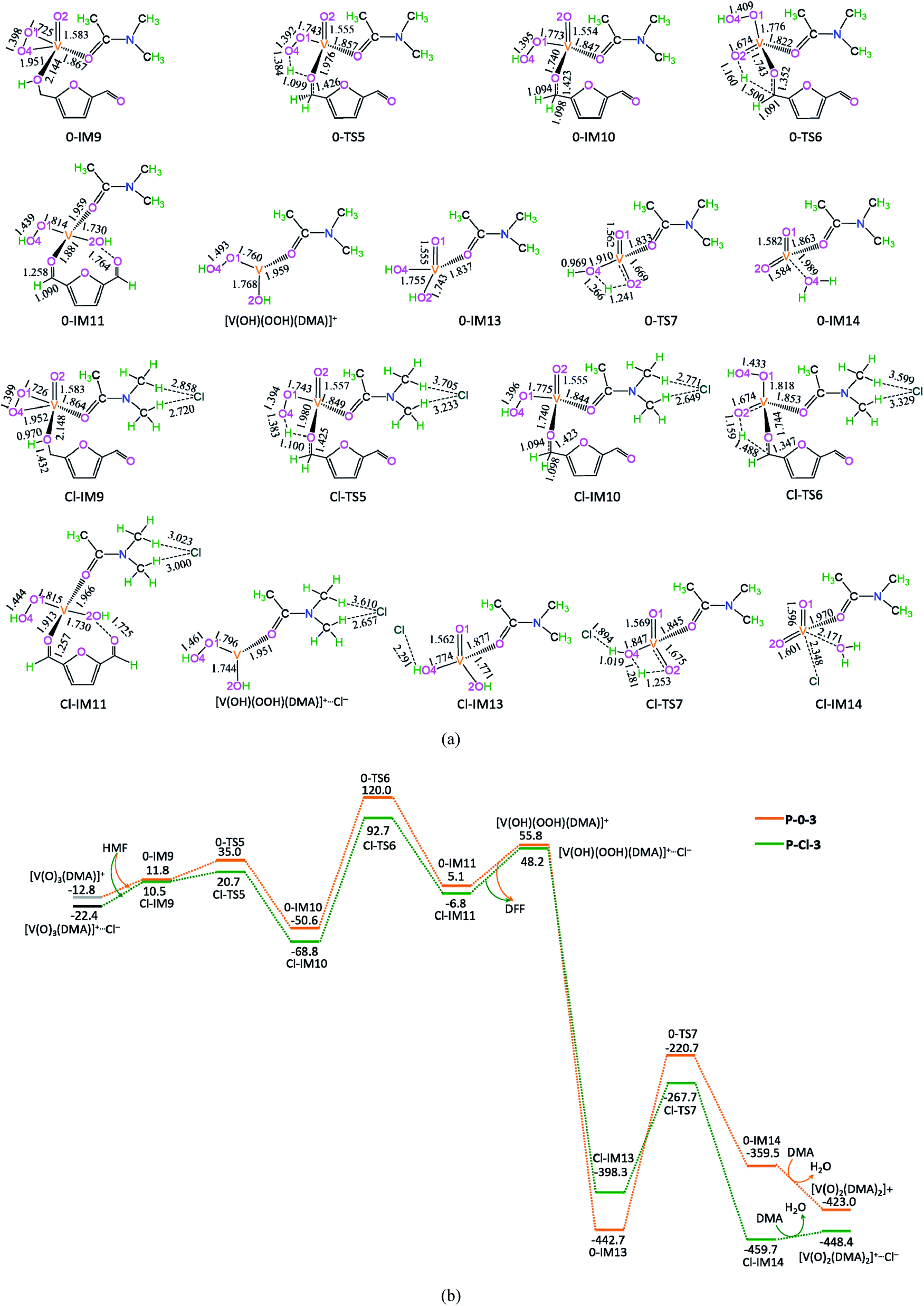 | ||
| Fig. 4 The geometric structures (a) and the schematic energy diagrams (b) with the relative Gibbs free energy (Gr, kJ mol−1) relative to the reactants for the oxidative dehydration of the second HMF to DFF by [V(O)3(DMA)]+ without/with Cl− in the DMA solution. For clarity, the hydrogen atoms on carbon are not shown. Bond lengths are reported in Å. | ||
As depicted in Fig. 4, in the absence of counteranion Cl−, there are seven reaction steps, i.e., (1) the formation of a molecular complex 0-IM9 between the second HMF molecule and [V(O)3(DMA)]+ species through the O-end of –OH in HMF coordinating to V-site, (2) the O–H bond cleavage through [1,4]-H shift via a five-membered cyclic 0-TS5, (3) the C–H bond cleavage in –CH2OH group via a five-membered 0-TS6 through addition unsaturated VO double bond for the DFF formation, (4) the release of DFF, saving the reduced [V(OH)(OOH)(DMA)]+ aside, (5) [V(OH)(OOH)(DMA)]+ readily rearranging to the isomer 0-IM13 with O–OH bond cleavage, (6) the O–H bond cleavage via a four-membered cyclic 0-TS7 through [1,3]-H shift for the H2O formation, and (7) one DMA molecule exchanging with the H2O molecule, regenerating the catalytic species [V(O)2(DMA)2]+.
The P-0-3 displays the EHHP of 120.0 kJ mol−1 at 0-TS6, the HEB of 180.0 kJ mol−1 at the reaction step of 0-IM13 → 0-TS7 → 0-IM14, and the EHLP of −442.7 kJ mol−1 at 0-IM13. In addition, the P-Cl-3 shows the EHHP of 92.7 kJ mol−1 at Cl-TS6, the HEB of 177.6 kJ mol−1 at the reaction step of Cl-IM13 → Cl-TS7 → Cl-IM14, and the EHLP of −459.7 kJ mol−1 at Cl-IM14.
Compared with P-0-3, P-Cl-3 involves lower EHHP (92.7 vs. 120.0 kJ mol−1) and lower EHLP (−459.7 vs. −442.7 kJ mol−1). It is indicated that counteranion Cl− stabilizes in some degree the relevant TS and IM.
3.5 Catalytic global analysis
As mentioned earlier, over [V(O)2(DMA)2]+ species, the global catalytic cycle is made up of the reaction pathways of P-0-1, P-0-2, and P-0-3, denoted as G-0. Using energetic span model analysis, for G-0, the DI and DTS are computed to be the initial point ([V(O)2(DMA)2]+ + HMF) and 0-TS2, respectively. The rate constants kG-0 of ([V(O)2(DMA)2]+ + HMF) → 0-TS2 (in s−1 mol−1 dm3) can be expressed by the following formula:
kG-0 = 1.45 × 108![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−155949/RT) exp(−155949/RT)
| (4) |
Moreover, as mentioned earlier, the crucial reaction step is associated with the C–H bond cleavage of methylene group in HMF. Here, the [V(O)2(DMA)2]+ species exerts oxidative effect for the first HMF oxidation to DFF, while the peroxide [V(O)3(DMA)]+ species behaves as an oxidant for the second HMF oxidation to DFF. The peroxide [V(O)3(DMA)]+ species exhibits higher oxidative activity than the [V(O)2(DMA)2]+ species, toward the C–H bond cleavage of methylene group in HMF. For [V(O)2(DMA)2]+ and [V(O)3(DMA)]+ species, the LUMO and HOMO molecular orbitals are shown in Fig. 5. The LUMO–HOMO gaps are 5.49 and 3.66 eV for [V(O)2(DMA)2]+ and [V(O)3(DMA)]+, respectively. It is obvious that LUMO–HOMO gap for [V(O)3(DMA)]+ is narrower than that for [V(O)2(DMA)2]+. Moreover, the positions of LUMO are relatively concentrated in V–O–O region for [V(O)3(DMA)]+, and in O–V–O region for [V(O)2(DMA)2]+. The V–O–O peroxy functional group in [V(O)3(DMA)]+ shows stronger oxidation than the V–O functional group in [V(O)2(DMA)2]+. Thereupon, [V(O)3(DMA)]+ displays a higher oxidizability than [V(O)2(DMA)2]+, toward the C–H bond cleavage in the methylene group in HMF.
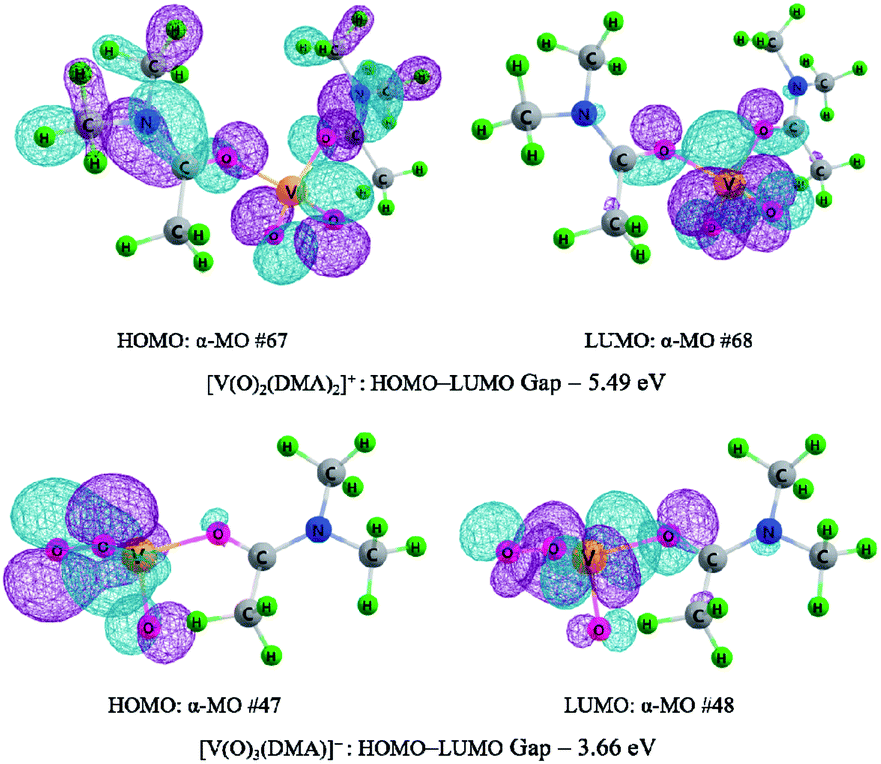 | ||
| Fig. 5 Highest occupied molecular orbital (HOMO), lowest unoccupied molecular orbital (LUMO) and the HOMO–LUMO energy gap for [V(O)2(DMA)2]+ and [V(O)3(DMA)]+. The orbital contour value was 0.03. | ||
In the presence of counteranion Cl−, i.e., over [V(O)2(DMA)2]+⋯Cl−, the global catalytic cycle is composed of P-Cl-1, P-Cl-2, and P-Cl-3, denoted as G-Cl. For G-Cl, the DI and DTS are acquired to be initial point ([V(O)2(DMA)2]+⋯Cl + HMF) and Cl-TS2. The rate constants kG-Cl of ([V(O)2(DMA)2]+⋯Cl− + HMF) → Cl-TS2 (in s−1 mol−1 dm3) can be expressed by the following formula:
|
kG-Cl = 2.74 × 108exp(−155189/RT)
| (5) |
The rate constant of kG-Cl is 1.5–1.3 times larger than that of kG-0, over the 353–433 K temperature range. This result echoes that the counteranion Cl− promotes the catalytic activity of [V(O)2(DMA)2]+ toward the aerobic oxidation of HMF to DFF.
4. Conclusions
The aerobic oxidation mechanism of HMF to DFF catalyzed by VO2+ with counterpart anion in DMA solution have been theoretically studied. The following conclusions can be drawn from the present results.In DMA solution, the stable VO2+-containing complex is the four-coordinated [V(O)2(DMA)2]+ species. For the gross reaction of 2HMF + O2 → 2DFF + 2H2O, there are three main reaction stages, i.e., the oxidation of the first HMF to DFF with the reduction of [V(O)2(DMA)2]+ to [V(OH)2(DMA)]+, the aerobic oxidation of [V(OH)2(DMA)]+ to the peroxide [V(O)3(DMA)]+, and the oxidation of the second HMF to DFF with the reduction of [V(O)3(DMA)]+ to [V(O)2(DMA)2]+. The initial [V(O)2(DMA)2]+ species is responsible for the oxidation of the first HMF, while the peroxide [V(O)3(DMA)]+ species is in charge of the oxidation of the second HMF. The rate-determining reaction step is concerned with the C–H bond cleavage of –CH2 group of the first HMF molecule. The peroxide [V(O)3(DMA)]+ species shows better oxidative activity than the initial [V(O)2(DMA)2]+ species, which stems from its narrower HOMO–LUMO gap. The counteranion Cl− displays promotive effect on the catalytic activity of [V(O)2(DMA)2]+ species, toward the aerobic oxidation of HMF to DFF.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for financial support by the National Natural Science Foundation of China (No: 22073064 and 21573154) and the 111 Project (B17030).Notes and references
- P. Priyanka and S. Shunmugavel, ChemSusChem, 2019, 12, 145–163 CrossRef PubMed
.
- X. Kong, Y. Zhu, Z. Fang, J. A. Kozinski, I. S. Butler, L. Xu, H. Song and X. Wei, Green Chem., 2018, 20, 3657–3682 RSC
- J. Mitra, X. Zhou and T. Rauchfuss, Green Chem., 2015, 17, 307–313 RSC
- Y. Xu, X. Jia, J. Ma, J. Gao, F. Xia, X. Li and J. Xu, Green Chem., 2018, 20, 2697–2701 RSC
- F. W. Lichtenthaler, Acc. Chem. Res., 2002, 35, 728–737 CrossRef CAS PubMed
- C. A. Antonyraj, J. Jeong, B. Kim, S. Shin, S. Kim, K. Lee and J. K. Cho, J. Ind. Eng. Chem., 2013, 19, 1056–1059 CrossRef CAS
- Y. Wang, B. Liu, K. Huang and Z. Zhang, Ind. Eng. Chem. Res., 2014, 53, 1313–1319 CrossRef CAS
- J. Nie, J. Xie and H. Liu, J. Catal., 2013, 301, 83–91 CrossRef CAS
- G. D. Yadav and R. V. Sharma, Appl. Catal., B, 2014, 147, 293–301 CrossRef CAS
- W. Zhang, J. Xie, W. Hou, Y. Liu, Y. Zhou and J. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 23122–23132 CrossRef CAS PubMed
- B. Liu, Z. Zhang, K. Lv, K. Deng and H. Duan, Appl. Catal., A, 2014, 472, 64–71 CrossRef CAS
- J. Nie and H. Liu, J. Catal., 2014, 316, 57–66 CrossRef CAS
- Y. Yao and G. Wang, J. Phys. Chem. C, 2021, 125, 3818–3826 CrossRef CAS
- Y. Liu, L. Zhu, J. Tang, M. Liu, R. Cheng and C. Hu, ChemSusChem, 2014, 7, 3541–3547 CrossRef CAS PubMed
- L. Ren, H. Yang and C. Hu, Catal. Sci. Technol., 2016, 6, 3776–3787 RSC
- Z. Wang, L. Liu, B. Xiang, Y. Wang, Y. Lyu, T. Qi, Z. Si, H. Yang and C. Hu, Catal. Sci. Technol., 2019, 9, 811–821 RSC
- W. Zhang, T. Meng, J. Tang, W. Zhuang, Y. Zhou and J. Wang, ACS Sustainable Chem. Eng., 2017, 5, 10029–10037 CrossRef CAS
- W. Hou, Q. Wang, Z. Guo, J. Li, Y. Zhou and J. Wang, Catal. Sci. Technol., 2017, 7, 1006 RSC
- Y. Zhou, Z. Ma, J. Tang, N. Yan, Y. Du, S. Xi, K. Wang, W. Zhang, H. Wen and J. Wang, Nat. Commun., 2018, 9, 2931 CrossRef PubMed
- I. Sádaba, Y. Y. Gorbanev, S. Kegnaes, S. S. R. Putluru, R. W. Berg and A. Riisager, ChemCatChem, 2013, 5, 284–293 CrossRef
- J. Nie and H. Liu, Pure Appl. Chem., 2012, 84, 765–777 CAS
- N. T. Le, P. Lakshmanan, K. Cho, Y. Han and H. Kim, Appl. Catal., A, 2013, 464–465, 305–312 CrossRef CAS
- X. Xiang, L. He, Y. Yang, B. Guo, D. Tong and C. Hu, Catal. Lett., 2011, 141, 735–741 CrossRef CAS
- A. Takagaki, M. Takahashi, S. Nishimura and K. Ebitani, ACS Catal., 2011, 1, 1562–1565 CrossRef CAS
- L. F. Zhu, B. Guo, D. Y. Tang, X. K. Hu, G. Y. Li and C. W. Hu, J. Catal., 2007, 245, 446–455 CrossRef CAS
- A. Uhe, S. Kozuch and S. Shaik, J. Comput. Chem., 2011, 32, 978–985 CrossRef CAS PubMed
- S. Kozuch and S. Shaik, Acc. Chem. Res., 2011, 44, 101–110 CrossRef CAS PubMed
- S. Kozuch, ACS Catal., 2015, 5, 5242–5255 CrossRef CAS
- W. Wang and G. Wang, RSC Adv., 2015, 5, 83459–83470 RSC
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, et al., Revision C.01, Gaussian, Inc, Wallingford, CT, 2010 Search PubMed
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed
- M. Cossi, G. Scalmani, N. Rega and V. Barone, et al., J. Chem. Phys., 2002, 117, 43–54 CrossRef CAS
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, et al., J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS
- N. B. Balabanov and K. A. Peterson, J. Chem. Phys., 2005, 123, 64107 CrossRef PubMed
- R. Bauernschmitt and R. Ahlrichs, J. Chem. Phys., 1996, 104, 9047–9052 CrossRef CAS
- C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef CAS
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS
- B. Xiang, Y. Wang, T. Qi, H. Yang and C. Hu, J. Catal., 2017, 352, 586–598 CrossRef CAS
- L. Ren, L. Zhu, T. Qi, J. Tang, H. Yang and C. Hu, ACS Catal., 2017, 7, 2199–2212 CrossRef CAS
- L. Liu, Z. Wang, Y. Lyu, J. Zhang, Z. Huang, T. Qi, Z. Si, H. Yang and C. Hu, Catal. Sci. Technol., 2020, 10, 278–290 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Sum of electronic energies, thermal correction to Gibbs free energy and relative Gibbs energies of various species. The MOL files of various species. Arrhenius plots of the calculated rate constants for the crucial reaction steps. See DOI: 10.1039/d1ra07297h |
| This journal is © The Royal Society of Chemistry 2021 |