
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolid-state nitrogen-doped carbon nanoparticles with tunable emission prepared by a microwave-assisted method†
Fitri Aulia Permatasari a,
Fitriyanti Nakulb,
Tirta Rona Mayangsaric,
Akfiny Hasdi Aimona,
Bebeh Wahid Nuryadind,
Satria Zulkarnaen Bisrie,
Takashi Ogif and
Ferry Iskandar*ag
a,
Fitriyanti Nakulb,
Tirta Rona Mayangsaric,
Akfiny Hasdi Aimona,
Bebeh Wahid Nuryadind,
Satria Zulkarnaen Bisrie,
Takashi Ogif and
Ferry Iskandar*ag
aDepartment of Electrical Engineering, Politeknik Negeri Batam, Jalan Ahmad Yani, Batam, Riau 29461, Indonesia
bDepartment of Electrical Engineering, Politeknik Negeri Batam, Batam, Indonesia
cDepartment of Chemistry, Universitas Pertamina, Jakarta 12220, Indonesia
dDepartment of Physics, Faculty of Science and Technology, UIN Sunan Gunung Djati Bandung, Jl. A. H. Nasution 105, Bandung, Indonesia 40614
eRIKEN Center for Emergent Matter Science, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan
fChemical Engineering Program, Department of Advanced Science and Engineering, Graduate School of Advanced Science and Engineering, Hiroshima University, 1-4-1 Kagamiyama, Higashihiroshima 739-8527, Japan
gResearch Center for Nanoscience and Nanotechnology, Institut Teknologi Bandung, Jalan Ganesha 10, Bandung, Indonesia 40132. E-mail: ferry@fi.itb.ac.id
First published on 16th December 2021
Abstract
Tunable emissive solid-state carbon nanoparticles (CNPs) have been successfully synthesized by a facile synthesis through microwave irradiation. Modulating microwave interaction with the sample to generate abrupt localized heating is a long-term challenge to tailor the photoluminescence properties of CNPs. This study systematically revealed that the sample temperature through microwave irradiation plays a crucial role in controlling the photoluminescence properties over other reaction conditions, such as irradiation time and microwave duty cycle. When the sample temperature reached 155 °C in less than three minutes, the CNP sample exhibited a green-yellowish emission with the highest quantum yield (QY) of 14.6%. Time-dependent density functional theory (TD-DFT) study revealed that the tunable photoluminescence properties of the CNPs can possibly be ascribed to their nitrogen concentrations, which were dictated by the sample temperature during irradiation. This study opens up a promising route for the well-controlled synthesis of luminescent CNPs through microwave irradiation.
Introduction
Carbon-based phosphors, such as carbon nanoparticles (CNPs), carbon dots, graphene quantum dots, and boron carbon oxynitride (BCNO), have gained scientific interest in various semiconductor applications, such as biosensing, bioimaging, nanomedicine, solar cells, and catalysis.1–5 Owing to their superior properties (tunable photoluminescence emission, high quantum efficiency, low cytotoxicity, facile production, cost-effective preparation, photostability, chemical inertness, and excellent biocompatibility), carbon-based phosphors are expected to be excellent potential substitutes for semiconductor quantum dots and rare earth element-based phosphors.6–8 Among other carbon-based phosphors, carbon nanoparticles (CNPs) are the most promising materials for large-scale production due to their scalable and facile synthesis with superior properties.In the last decade, much research has been devoted to developing high-performance CNPS with tunable emission. Most of these methods involve several syntheses and purification steps that are unpreferable for up-scaled synthesis. In addition, the solution-based synthesis process hinders the optoelectronic application of CNPs due to their self-quenching phenomenon.9,10 Similar to many organic molecules, fluorescence quenching occurs in the aggregate state, ascribed to direct π–π interactions or excessive resonance energy transfer (RET).11,12 To overcome this problem, many scientists have made some efforts to develop solid-state CNPs.13–16 Therefore, the challenge of developing a synthesis method for solid-state CNPs has aroused great interest.
Among the various facile syntheses, microwave treatment is the most popular method for synthesizing and modifying carbon-based phosphors due to the environmentally friendly process and high conductivity of carbon, leading to a strong interaction with microwave radiation. Wang et al. reported the tunable emission of a solid-state CNP prepared from citric acid and piperazine as precursors via microwave heating.14 They suggested a significant influence of the mass ratio of the precursors on CNP size and PL properties. The tunable emission PL properties of these CNPs are mainly dictated by the size effect, controlled by the precursors' mass ratio. A higher mass ratio between citric acid and piperazine leads to a red-shifted emission peak that was preferable for the tunable emission of the CNP. However, the PL intensity was significantly reduced due to the rich surface defects preserved by excessive piperazine precursors. Wei et al. successfully synthesized solid CNPs that exhibited strong PL using citric acid and urea in the presence of NaOH by a microwave heating process.16 The prepared CNP exhibited a green emission with a maximum quantum yield (QY) of 75.9%, higher than other reports. However, the NaOH presence that serves as a matrix to prevent the self-quenching phenomenon also impeded the tuning of the emission wavelength of the CNP. Thus, even after several experimental condition optimization, the PL emission of the CNP is limited in the blue until green color range. These two reports suggest the possibility of several emission pathways on CNPs that can be controlled by experimental conditions to obtain tunable emission with high QY.
Several explanations have been proposed to explain the optical nature of CNPs. They are classified into three categories. First, the quantum confinement effect that takes place from the graphitic core, involving the presence of different π-conjugated islands inside the core.17 Second, the surface-mediated optical property that arises from the surface defect states.18 Third, the doping atoms that originate from the precursors and the configuration of their bonds with carbon atoms.19 The experimental conditions may lead to one of these pathways, selectively enabling control of the PL properties of the CNPs. Therefore, in this work, we developed solid-state CNPs by microwave heating and elucidated the emission pathways of the as-synthesized CNPs based on experimental and time-dependent density functional theory (TD-DFT) studies.
This study selected citric acid and urea as raw materials due to their simple structures and known reaction mechanisms through the heating process based on our previous research.20,21 A commercial microwave was modified to monitor the sample temperature through irradiation. A solid phase of the precursor was used in microwave irradiation to avoid water molecule resonance. Then, the microwave irradiation conditions were systematically investigated, i.e., microwave duty cycle, irradiation time, and sample temperature. This study revealed that the nitrogen content firmly ascribed to the reaction temperature is crucial to controlling the photoluminescence properties of the solid-state CNPs. This study opens up a new understanding of the well-controlled synthesis of solid CNPs through microwave treatment. Furthermore, the tunable emission properties of the CNPs induced by the nitrogen content were elucidated based on comprehensive experimental and TD-DFT studies.
Experimental
Material and synthesis
The CNPs were prepared from citric acid and urea through microwave irradiation, as reported in our previous work.21 In brief, 0.18 mmol citric acid (Merck) and 50 mmol urea (Sigma Aldrich) were dissolved in 6 ml distilled water. Then, the solution was stirred for 5 minutes at room temperature and heated in an oven at 100 °C for an hour. The obtained white powder was irradiated by a modified microwave with 800 W of power for several minutes (±2 minutes). This process increases the powder temperature to 95–235 °C. The powder colour changed to light yellowish-brown after microwave irradiation. To characterize the temperature dependence of the sample, we developed an experimental microwave synthesis apparatus, as shown in the ESI (Fig. S1†). The sample temperature was monitored during microwave irradiation by an infrared thermometer mounted on top of the microwave. The infrared thermometer was calibrated with a thermocouple to maintain measurement accuracy, as shown in the ESI (Fig. S2†).Characterization
The photoluminescence of the sample was measured using a spectrofluorophotometer (PL Spectra, RF-5300 PC, Shimadzu Corp., Kyoto, Japan), which was equipped with a xenon lamp that emitted a continuous spectrum with a wavelength of 200–800 nm. Meanwhile, the internal QY measurements were recorded using an absolute-PLQY measurement system (C992002, Hamamatsu Photonics, Shizuoka, Japan) with a BaSO4-coated integrating sphere and a xenon lamp as the light source. To investigate their chemical bonds, all samples were characterized by Fourier transform infrared spectrometry (FTIR, Bruker ALPHA). Thermal analyses of the sample were conducted by differential scanning calorimetry (DSC) and thermal gravimetric analysis (TGA, LINSEIS STA Platinum Series) under flowing air in a 25 °C to 600 °C temperature range. The sample morphology was characterized by transmission electron microscopy (TEM, JEM-3000F JEOL, Japan) with an operating voltage of 200 kV. In addition, the crystal structure of the sample was characterized by powder X-ray diffraction (XRD, Bruker, Advance, Germany) with Cu Kα radiation.TD-DFT calculation method
The solid CNP was modelled as a C54 aromatic carbon ring. Then, the geometry and ground-state calculations were optimized by density functional theory (DFT) using the Becke three-parameter Lee–Yang–Parr hybrid functional (B3LYP) and 6-31G (dp) basis set in Gaussian 09. In this work, the calculations for each model were conducted in the gas phase to represent the solid-state structure. The visualizations of the CNP model and the energy level diagram were generated using the Chemcraft software.22Results and discussion
The resonance between the microwave irradiation and precursors induced an elevated sample temperature in the range of 95–235 °C. It is worth noting that the photoluminescence (PL) emission of the samples did not shift significantly under different molar ratios of citric acid and urea (Fig. S3 and S4†). 0.18 mmol citric acid and 50 mmol urea are the optimum concentrations that produce the best sample with intense PL intensity. Thus, the effects of sample temperature on the PL properties were investigated at a fixed concentration ratio of citric acid and urea. Fig. 1a shows the colour change of the powder samples, which were produced using the microwave-assisted reaction with various final sample temperatures. The initial sample powder has a white colour, and it turned to light yellow when the sample temperature reached 125 °C. The sample colour then became deep yellow as the final sample temperature increased. Finally, the powder sample became brown upon reaching a temperature of more than 215 °C. Under UV irradiation (365 nm), a bright green-yellowish emission could be observed from the samples prepared at 155–175 °C.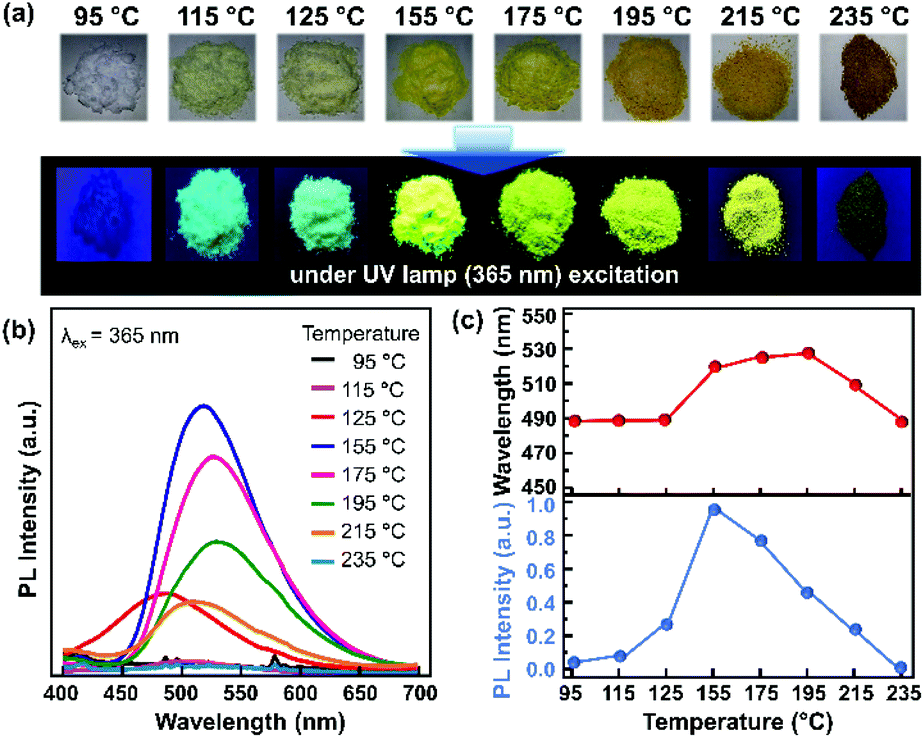 | ||
| Fig. 1 CNP samples synthesized through 80% DC microwave irradiation. (a) Digital micrographs of the sample without UV excitation (top) and under UV excitation (bottom). (b) PL spectra of the CNP samples under 365 nm excitation. (c) PL peak wavelengths and PL intensities for each sample with varying synthesis temperature. | ||
Fig. 1b shows the corresponding PL spectra of the as-synthesized CNPs at different temperatures. When the temperature reached 115 °C, the sample started to exhibit emission in the wavelength range of 400–650 nm. The PL intensity was enhanced with increasing sample temperature and reached its maximum intensity at 155 °C with a 520 nm emission peak. Then, the PL intensity decreased with increasing temperature and no emission was observed for the sample with the temperature of 235 °C. A red-shift of the PL emission was observed from 487 nm to 528 nm in the samples that were prepared until the temperature reached 115–195 °C (Fig. 1c). It has been reported that a red-shifted emission may originate from the increase of the π-conjugated graphitic core or the presence of heteroatoms, such as nitrogen and oxygen.23–25
Furthermore, microwave irradiation can also be modulated based on the duty cycle (Dc) of the irradiation pulse, i.e., Dc 17%, Dc 80%, and Dc 100%. At a fixed microwave irradiation time, the sample temperature profiles of the different Dc modes were monitored, as shown in Fig. 2a. After two minutes of irradiation at 17% Dc, the sample temperature only reached 70 °C. This temperature remains constant, although the irradiation time was prolonged to 5 minutes. On the other hand, using 80% Dc and 100% Dc, the sample temperature increased abruptly, reaching 200 °C and 300 °C, respectively. 100% Dc is more suitable for the rapid synthesis process since it provides faster heating and is more energy efficient. Meanwhile, 80% Dc provides a relatively moderate constant heating rate, preferable for controlling the reaction mechanism.
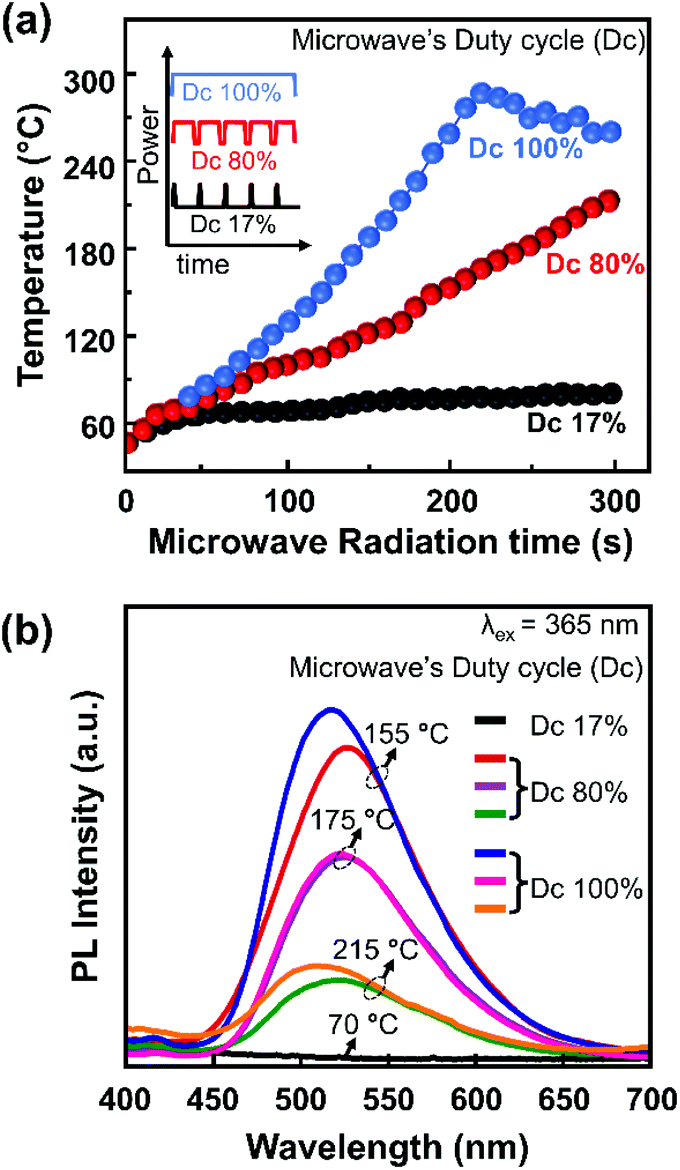 | ||
| Fig. 2 (a) Temperature profiles of a sample through microwave irradiation for different Dc modes. (b) PL spectra of a representative sample under 365 nm excitation. | ||
Several samples were selected as representative samples for investigating the effects of temperature and DC mode on the PL spectra, as shown in Fig. 2b. A consistent red-shift in the emission from Dc 100% to Dc 80% was observed for the samples prepared at 155 °C and 215 °C. In contrast, a subtle change was observed for the sample prepared at 175 °C. Interestingly, the PL spectra were almost similar for all samples prepared at the same temperature, although the irradiation time and Dc modes differed. This finding suggested that the sample temperature is a more influential parameter on the PL spectra than the other reaction conditions, i.e., microwave irradiation time and Dc mode. Another implication of this finding is that the conventional heating process in ambient atmosphere to prepare these CNPs is more crucial than the molecular resonance of the sample with the microwave.
Microwave irradiation can generate heat rapidly and shorten the heating time due to its penetration ability, leading to a uniform sample temperature. This is vital to achieve in CNP production since uniform temperature will produce both a homogeneous morphology and surface functional groups. The importance of the temperature of the sample for controlling the carbon structure has also been reported in our previous works in the case of the hydrothermal route.20,21 In these studies, the formation mechanism of carbon nanoparticle structures from citric acid and urea as raw materials were investigated comprehensively. It was revealed that the molecular reaction between citric acid and urea naturally began at 130 °C. Then, the emission of the as-synthesized carbon nanostructure reached the highest QY at a reaction temperature of 160 °C. Consistent with our previous study, the as-synthesized CNPs also began to exhibit emission at 125 °C and reached the maximum QY at a temperature of 155 °C.
To understand the above occurrence, the molecular structure and its possible formation mechanism were investigated using FTIR spectroscopy, DFT calculations, TGA, and DSC. The FTIR spectra of the samples exhibiting different PL spectra, such as those prepared at 95 °C, 155 °C, and 235 °C, showed strikingly distinctive features, as shown in Fig. 3a. A sample prepared at 95 °C showed a strong presence of N–H bond vibration (3330 cm−1 and 1586 cm−1) and O–H bonds (3420 cm−1 and 1150 cm−1). The signature of the carbon graphitic core of this sample could not be observed clearly, indicating that the graphitic core of the CNP had not formed yet. This sample did not generate any PL (Fig. 1b), suggesting that the conjugated graphitic core might be responsible for the CNP fluorescence mechanism.
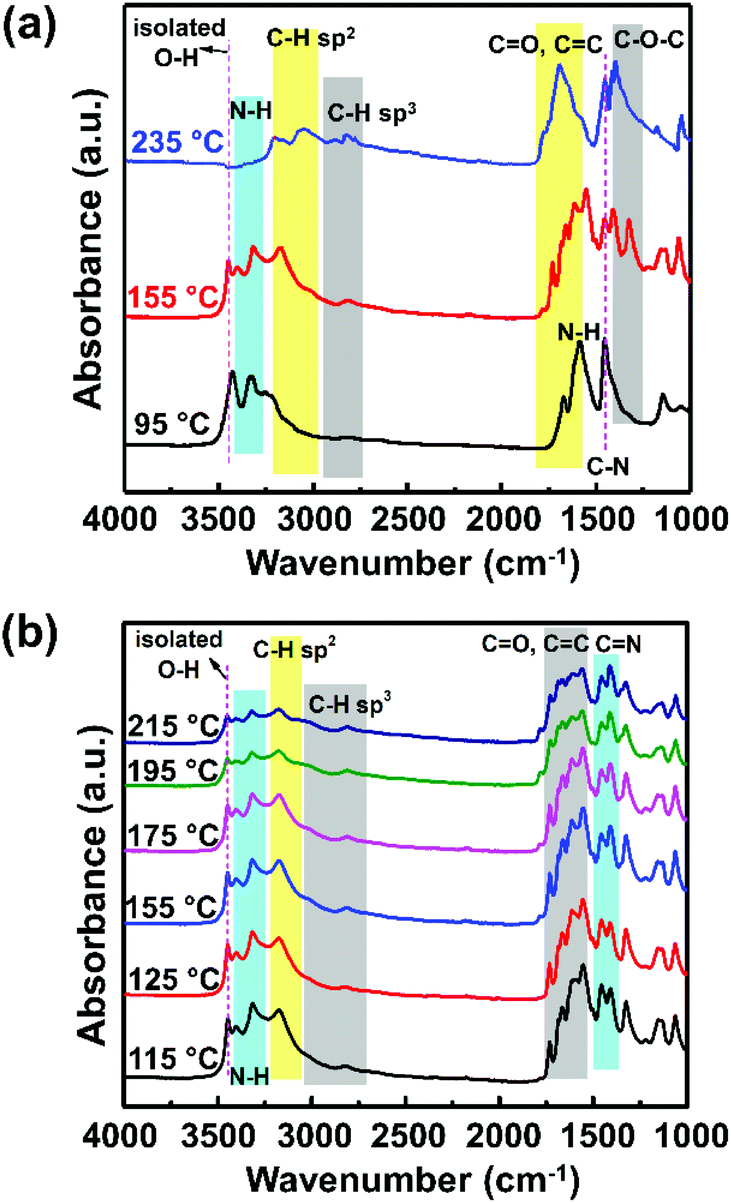 | ||
| Fig. 3 (a) FTIR spectra of CNPs synthesized until the reaction temperature reached 95 °C, 155 °C, and 235 °C, and (b) FTIR spectra of a CNP sample that exhibits PL emission (reaction temperature sample of 115 °C to 215 °C). | ||
Furthermore, the sample prepared at 155 °C showed a slight blue-shift of the O–H bond to 3450 cm−1 relative to the signature observed in the sample prepared at 95 °C. The observation of this blue-shift corresponds to the increase of the isolated hydroxyl groups. Absorbance at around 3170 cm−1 was observed, which corresponds to the C–H sp2 bond vibration, indicating the formation of the graphitic core. The other FTIR absorbance peaks related to the graphitic core of the CNP were also observed at lower wavenumbers of 1000–1800 cm−1, which correspond to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, CO, and C–O–C bonds. The presence of the C–H sp2 and CC bond signatures indicates the formation of the π-conjugated system as a core of the CNP structure.17,26,27 These π-conjugated systems play a crucial role in the energy level diagram of the CNP, which dictates their PL properties.25,26 On the other hand, the sample prepared at 235 °C showed a significant decrease of N–H bonds (3200–3400 cm−1), accompanied by increase of the C–H sp3 and CO bonds at 3060 cm−1 and 1680 cm−1, respectively. The observation of the signatures of these functional groups suggests the carbonization of the sample. This observation can explain the physical appearance of the sample in Fig. 1a.
C, CO, and C–O–C bonds. The presence of the C–H sp2 and CC bond signatures indicates the formation of the π-conjugated system as a core of the CNP structure.17,26,27 These π-conjugated systems play a crucial role in the energy level diagram of the CNP, which dictates their PL properties.25,26 On the other hand, the sample prepared at 235 °C showed a significant decrease of N–H bonds (3200–3400 cm−1), accompanied by increase of the C–H sp3 and CO bonds at 3060 cm−1 and 1680 cm−1, respectively. The observation of the signatures of these functional groups suggests the carbonization of the sample. This observation can explain the physical appearance of the sample in Fig. 1a.
To scrutinize the carbon structure that might cause the PL shift, we compared the FTIR spectra of the samples prepared at 115–215 °C (Fig. 3b). To accurately observe the absorbance tendency, the integrated peak height ratios for the C–H sp2, CC, CN, N–H, and C–OH bonds should be determined (Table 1). From these ratio comparisons, we obtained several caveats. First, the absorption band at 1150 cm−1, assigned to the C–OH stretching vibration, is steadily weakened by the increase of the sample preparation temperature, as indicated by the ratio of ICC/IC–OH (Table 1). This tendency indicates the loss of the hydroxyl groups due to a nucleophilic reaction with the amino groups from urea.28
| Temperature (°C) | 115 | 125 | 155 | 175 | 195 | 215 |
|---|---|---|---|---|---|---|
| IC–H/IN–H | 0.91 | 0.90 | 0.98 | 0.98 | 1.04 | 1.06 |
| ICC/IC–N |
0.82 | 0.79 | 0.78 | 0.79 | 0.85 | 0.91 |
| ICC/IC–OH |
1.22 | 1.31 | 1.33 | 1.32 | 1.61 | 1.56 |
Second, the ratio between the two absorption bands at around 1500 cm−1 and 1660 cm−1, respectively indicating the presence of C–N amide groups24 and CC bonds,25 is enhanced when the sample preparation temperature increases from 115 °C to 155 °C. The presence of C–N amide groups might be correlated with the existence of free electrons, explaining the increased probability of having more excited electrons, which results in enhanced PL intensity.29 On the other hand, an increase in CC bonds would indicate the increase of the π-conjugated graphitic core. Incorporating heteroatoms such as nitrogen and oxygen as both surface defects and in the core structure would provide a nonradiative relaxation mechanism that leads to red-shifted PL (Fig. 5a).30 However, at higher temperatures, the heteroatom incorporations were reduced, as seen from the ratios of IC–H/IN–H, ICC/IC–N, and ICC/IC–OH. The loss of surface defects would reduce nonradiative emission, causing an increase of the bandgap and the PL blue-shift.
Third, an absorption band at 1720 cm−1, which indicates the presence of CO (carbonyl) groups, started to appear at 155 °C. In addition, a broad absorption band is present at 3150–3500 cm−1 in all samples. This band can be assigned to the O–H and N–H stretching vibrations, which are the hydrophilic groups that ensure the excellent solubility of the species in water.31,32
We modelled and simulated the gap energy of each sample with different synthesis temperatures assuming different nitrogen concentrations on simplified CNP structures (Fig. 4). It is worth pointing out that the different nitrogen concentrations could also influence other physicochemical properties of the CNPs, such as the absorption feature, carrier mobility and structural stability, as reported elsewhere.33–35 In this TD-DFT study, we only focus on bandgap energy calculations to indicate the different nitrogen concentration effects on the CNP PL emission. Even though the nitrogen content could not be indicated precisely, the CNP model presented in Fig. 4a was constructed based on the C/N bond ratios from the FTIR spectra. Thus, these TD-DFT calculations could emphasize the tendency of the gap energy depending on the C/N ratios. This simulation assumed that the surface functional groups on the CNPs did not alter their optical properties. Therefore, the optical and electronic properties of the CNP are mainly attributed to the C/N ratio. To simulate the lowest formation energy of those configurations, nitrogen atoms were incorporated in the CNP structure in pyridinic-N and graphitic-N.36–38 The calculations showed that the bandgap of the CNP was reduced from 1.47 eV to 1.23 eV by increasing the nitrogen concentration from 5.6% to 9.9%, as can be seen in Fig. 4b. These results agree with the PL spectra, which showed a red-shift of the emissions emitted from the samples prepared from 115 °C to 195 °C. It also further confirms that the presence of heteroatom species provides a different fluorescence mechanism. Surprisingly, the high nitrogen concentration induces red-shifts of both the PL emission and absorption spectra (Fig. S5†). Broadening the UV-vis absorption spectra in the same wavelength emission range leads to re-absorption of the emission, suppressing the quantum yield. As observed in this study, the high nitrogen concentration induces a red-shift phenomenon with low PL intensity (Fig. 1c).
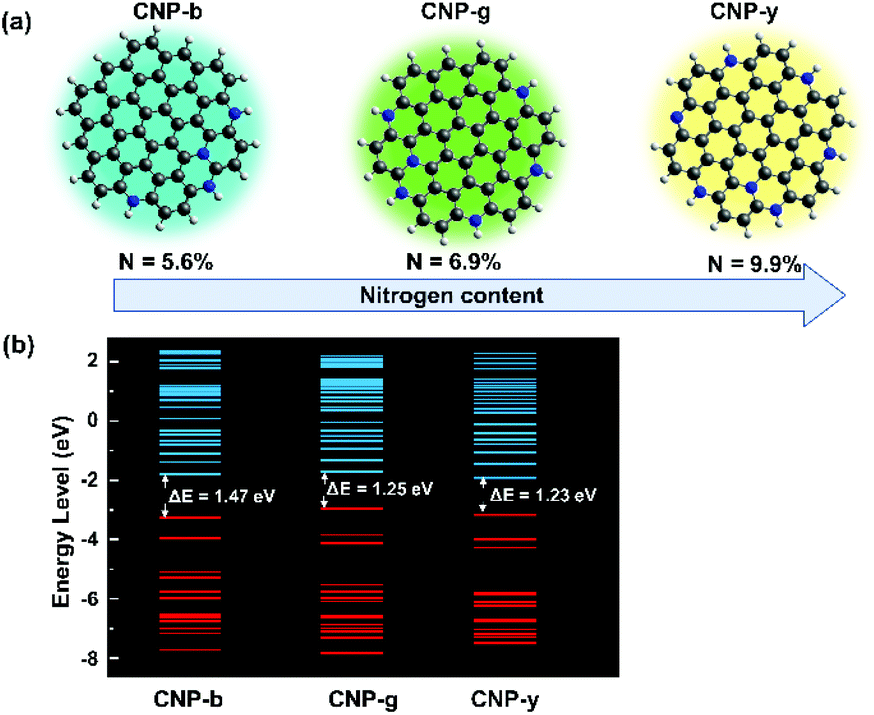 | ||
| Fig. 4 (a) CNP models with different nitrogen concentrations for different sample temperatures assumed from the FTIR discussion, and (b) the occupied and unoccupied energy levels of each model generated from the DFT calculations. ΔE was calculated from the difference in the HOMO and LUMO energy levels. | ||
Fig. 5 shows the TGA and DSC graphs of the solid precursor obtained after drying the liquid precursor using an oven at 100 °C for an hour. The TGA curve showed weight loss over the temperature range of 122–400 °C, with a prior weight increase that started from 45 °C. The initial weight increase results from water vapour adsorption by the hydrophilic functional groups of citric acid.39–41 The sharp drop to 30 wt% that happened from 122 °C until 224 °C can be attributed to the solid precursor, i.e., a mixture of citric acid and urea. The weight loss corresponds to the DSC graph, which shows an endothermic peak at 125 °C. Since citric acid starts to decompose at 180 °C and its concentration in the mixture is lower than that of urea, it is reasonable to expect that the weight loss can be attributed to the melting process of solid urea.42 This melting process resulted in ammonium gas and isocyanic acid gas. Note that the solid urea started to melt into ammonium gas and isocyanic acid gas at 133 °C.43 The melting temperature of urea could also be decreasing as the heating rate increases. However, the weight loss occurred until 224 °C, while the DSC graph shows three endothermic peaks in that range. This indicates that other reactions probably take place in that temperature range instead of only the melting of urea. One of the possible reactions is the formation of citric acid amide due to the nucleophilic reaction between citric acid and ammonia gas,44,45 indicated by the loss of hydroxyl groups shown in the FTIR spectra (Fig. 3a).
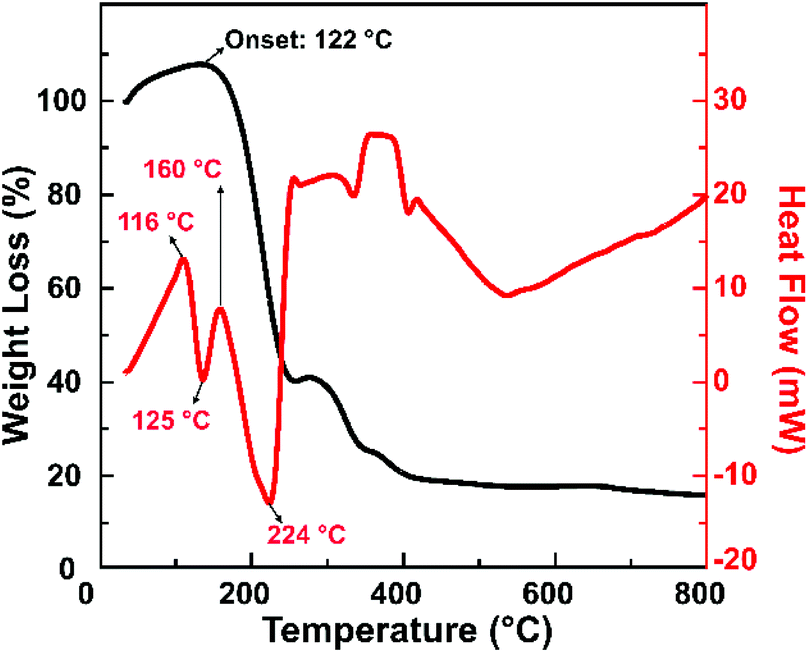 | ||
| Fig. 5 TGA and DSC curves of a sample that was synthesized by microwave heating until the reaction temperature reached 155 °C. | ||
This observation is consistent with our previous findings, revealing that citric acid and urea are condensed into a citric acid amide. They would self-assemble to become carbon sheets, then the sheets would be cut into carbon nanoparticles under heating treatment.20 We also confirmed that upon heating, the citric acid amide generation started from 130 °C, and the self-assembling process that initiates the carbon nanoparticle generation takes place at this temperature up to 200 °C.21
The PL measurement indicates that the PL intensity of the CNP is strongly affected by the sample temperature rather than the microwave irradiation power. With the elaboration of the FTIR, DSC and TGA curves, this fact suggests a high possibility that the CNP formation mechanism in a microwave-irradiation-assisted synthesis is similar to a hydrothermal synthesis route. Achieving a specific temperature in microwave-assisted synthesis is also crucial in obtaining highly luminescent CNPs. An intense PL emission was observed for the samples that have final temperatures in the range of 125–215 °C, suggesting the importance of this range. Specifically, the optimum temperature to obtain the brightest CNP is 155 °C. The synthesis temperature control enables the tuning of nitrogen concentration management, thus tuning the PL characteristics of the produced carbon nanoparticles.
We further analysed the sample that was synthesised at 155 °C, which exhibits the highest PLQY. This sample structure was characterized using TEM and XRD, as shown in Fig. 6. Fig. 6a shows that the solid sample exhibits blue to green-yellowish emission, with the highest QY of 14.6%. The TEM image (Fig. 6b) reveals that the sample was dominated by spherical particles in the diameter range of 40–100 nm, with an average diameter of 66.9 nm (Fig. 6c). This indicates that the as-synthesized CNP is out of the quantum confinement range, implying that the size effect did not significantly affect the electronic and optical properties of the CNPs. To evaluate the possible reaction mechanism of CNP formation, XRD characterization was conducted on a sample that was synthesized by microwave heating until the reaction temperature reached 155 °C. The XRD pattern (Fig. 6d) indicates that these spherical particles were polycrystalline, with predominantly graphitic carbon corresponding to JCPDS 01-075-0444.46 It also indicates that this sample possibly still contained some residues of unreacted raw materials, i.e. urea and citric acid.47,48 In line with the TGA and DSC measurements (Fig. 5), the urea started to melt at a temperature of 133 °C. Thus, at a temperature of 155 °C, the melting of urea and citric acid formation compete with each other, dictating the nitrogen concentration on the CNP.
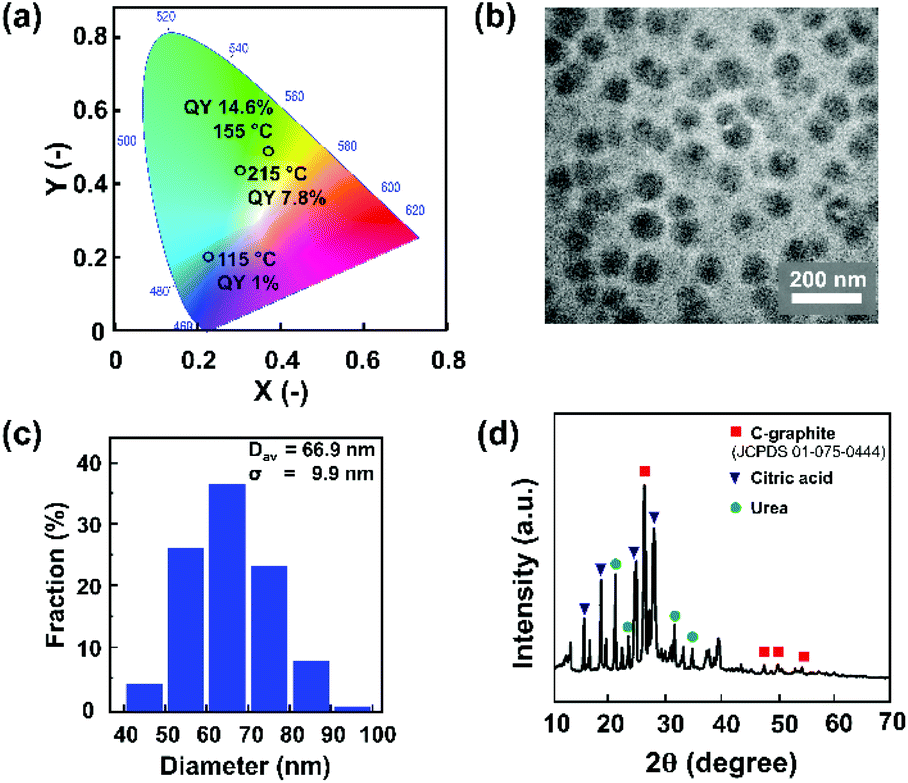 | ||
| Fig. 6 The CNP sample that was synthesized by microwave heating until the reaction temperature reached 155 °C: (a) CIE diagram; (b) TEM image. (c) Size distribution determined by measuring one hundred particles. (d) XRD pattern. | ||
Conclusions
In conclusion, the PL properties of the CNPs prepared by a microwave-assisted method strongly depend on the temperature reached in the samples. Our systematic studies showed that the generated sample temperature is the most important parameter affecting the PL spectral peak and intensity. This factor eclipses the other parameters controlling the reaction conditions, i.e., the microwave irradiation time and Dc mode. Comprehensive measurements of the physical characteristics of the produced CNPs suggest that 125–155 °C is the critical temperature range to establish highly luminescent nanoparticles. This study opens a new avenue to the facile and well-controlled synthesis of CNPs through microwave heating. Furthermore, a synergetic study of TD-DFT and experimental characterization suggest an efficient way to tune the CNP emission from blue to yellow, which is preferable for any optoelectronic application.Author contributions
Fitri Aulia Permatasari: conceptualization, methodology, investigation, and writing-original draft. Fitriyanti Nakul: methodology and investigation. Rona Tirta Mayangsari: visualization and writing-review & editing. Bebeh Wahid Nuryadin: characterization. Akfiny Hasdi Aimon: writing-review & editing. Satria Zulkarnaen Bisri: writing-review & editing. Takashi Ogi: writing-review & editing. Ferry Iskandar: conceptualization, supervision, visualization and writing-review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was fully supported by the Indonesian Endowment Fund for Education (LPDP) and the Indonesian Science Fund (DIPI) through the International Collaboration RISPRO Funding Program Grant No. RISPRO/KI/B1/KOM/11/4542/2/2020. FAP would like to thank the Ministry of Fiscal Indonesia Endowment Fund for Education (LPDP) for her doctoral scholarship. S. Z. B. acknowledges Grant-in-Aid for Scientific Research (C) (JP21K04815) from The Japan Society for the Promotion of Science (JSPS). T. O acknowledges JSPS KAKENHI Grant Number 19H02500.References
- L. Sinclair, J. Brown, M. G. Salim, D. May, B. Guilvaiee, A. Hawkins and L. Cathles, Carbon, 2020, 169, 395–402 CrossRef CAS.
- M. Taspika, F. Aulia Permatasari, B. Wahid Nuryadin, T. Rona Mayangsari, A. H. Aimon and F. Iskandar, RSC Adv., 2019, 9, 7375–7381 RSC.
- B. W. Nuryadin, F. A. Permatasari, A. Y. Nuryantini, I. D. Faryuni, M. Abdullah and F. Iskandar, RSC Adv., 2017, 7, 4161–4166 RSC.
- T. Ogi, Y. Kaihatsu, F. Iskandar, W.-N. Wang and K. Okuyama, Adv. Mater., 2008, 20, 3235–3238 CrossRef CAS.
- W.-N. Wang, T. Ogi, Y. Kaihatsu, F. Iskandar and K. Okuyama, J. Mater. Chem., 2011, 21, 5183–5189 RSC.
- L. Yin, J. Zhou, W. Li, J. Zhang and L. Wang, RSC Adv., 2019, 9, 9301–9307 RSC.
- S. Paulo-Mirasol, E. Martínez-Ferrero and E. Palomares, Nanoscale, 2019, 11, 11315–11321 RSC.
- T. V. de Medeiros, J. Manioudakis, F. Noun, J.-R. Macairan, F. Victoria and R. Naccache, J. Mater. Chem. C, 2019, 7, 7175–7195 RSC.
- D. Xu, F. Lei, H. Chen, L. Yin, Y. Shi and J. Xie, RSC Adv., 2019, 9, 8290–8299 RSC.
- H. J. Yoo, B. E. Kwak and D. H. Kim, J. Phys. Chem. C, 2019, 123, 27124–27131 CrossRef CAS.
- Y. Song, S. Zhu, S. Xiang, X. Zhao, J. Zhang, H. Zhang, Y. Fu and B. Yang, Nanoscale, 2014, 6, 4676–4682 RSC.
- F. Zu, F. Yan, Z. Bai, J. Xu, Y. Wang, Y. Huang and X. Zhou, Microchim. Acta, 2017, 184(7), 1899–1914 CrossRef CAS.
- J. Ren, L. Stagi and P. Innocenzi, Prog. Solid State Chem., 2021, 62, 100295 CrossRef CAS.
- H. J. Wang, T. T. Yu, H. L. Chen, W. Bin Nan, L. Q. Xie and Q. Q. Zhang, Dyes Pigm., 2018, 159, 245–251 CrossRef CAS.
- J. He, Y. He, Y. Chen, B. Lei, J. Zhuang, Y. Xiao, Y. Liang, M. Zheng, H. Zhang and Y. Liu, Small, 2017, 13, 1700075 CrossRef PubMed.
- J.-Y. Wei, Q. Lou, J.-H. Zang, Z.-Y. Liu, Y.-L. Ye, C.-L. Shen, W.-B. Zhao, L. Dong and C.-X. Shan, Adv. Opt. Mater., 2020, 8, 1901938 CrossRef CAS.
- S. Zhu, Y. Song, J. Wang, H. Wan, Y. Zhang, Y. Ning and B. Yang, Nano Today, 2017, 13, 10–14 CrossRef CAS.
- S. Zhu, X. Zhao, Y. Song, S. Lu and B. Yang, Nano Today, 2016, 11, 128–132 CrossRef CAS.
- S. D. Dsouza, M. Buerkle, P. Brunet, C. Maddi, D. B. Padmanaban, A. Morelli, A. F. Payam, P. Maguire, D. Mariotti and V. Svrcek, Carbon, 2021, 183, 1–11 CrossRef CAS.
- T. Ogi, K. Aishima, F. A. Permatasari, F. Iskandar, E. Tanabe and K. Okuyama, New J. Chem., 2016, 40, 5555–5561 RSC.
- T. Ogi, H. Iwasaki, K. Aishima, F. Iskandar, W.-N. Wang, K. Takimiya and K. Okuyama, RSC Adv., 2014, 4, 55709–55715 RSC.
- Chemcraft – Graphical program for visualization of quantum chemistry computations.
- B. Wang, J. Yu, L. Sui, S. Zhu, Z. Tang, B. Yang and S. Lu, Adv. Sci., 2021, 8, 2001453 CrossRef CAS PubMed.
- L. Wang, W. Li, L. Yin, Y. Liu, H. Guo, J. Lai, Y. Han, G. Li, M. Li, J. Zhang, R. Vajtai, P. M. Ajayan and M. Wu, Sci. Adv., 2020, 6, eabb6772 CrossRef CAS PubMed.
- E. V. Kundelev, N. V. Tepliakov, M. Y. Leonov, V. G. Maslov, A. V. Baranov, A. V. Fedorov, I. D. Rukhlenko and A. L. Rogach, J. Phys. Chem. Lett., 2019, 10, 5111–5116 CrossRef CAS PubMed.
- A. Diac, M. Focsan, C. Socaci, A.-M. Gabudean, C. Farcau, D. Maniu, E. Vasile, A. Terec, L. M. Veca and S. Astilean, RSC Adv., 2015, 5, 77662–77669 RSC.
- S. D. Hettiarachchi, R. M. Graham, K. J. Mintz, Y. Zhou, S. Vanni, Z. Peng and R. M. Leblanc, Nanoscale, 2019, 11, 6192–6205 RSC.
- H. Tetsuka, A. Nagoya, T. Fukusumi and T. Matsui, Adv. Mater., 2016, 28, 4632–4638 CrossRef CAS PubMed.
- F. A. Permatasari, A. H. Aimon, F. Iskandar, T. Ogi and K. Okuyama, Sci. Rep., 2016, 6, 1–8 CrossRef PubMed.
- H. Ding, S.-B. Yu, J.-S. Wei and H.-M. Xiong, ACS Nano, 2015, 10, 484–491 CrossRef PubMed.
- J. Peng, W. Gao, B. K. Gupta, Z. Liu, R. Romero-Aburto, L. Ge, L. Song, L. B. Alemany, X. Zhan, G. Gao, S. A. Vithayathil, B. A. Kaipparettu, A. A. Marti, T. Hayashi, J.-J. Zhu and P. M. Ajayan, Nano Lett., 2012, 12, 844–849 CrossRef CAS PubMed.
- H. Ding, S.-B. Yu, J.-S. Wei and H.-M. Xiong, ACS Nano, 2016, 10, 484–491 CrossRef CAS PubMed.
- F. A. Permatasari, H. Fukazawa, T. Ogi, F. Iskandar and K. Okuyama, ACS Appl. Nano Mater., 2018, 1, 2368–2375 CrossRef CAS.
- J. Feng, H. Dong, B. Pang, F. Shao, C. Zhang, L. Yu and L. Dong, Phys. Chem. Chem. Phys., 2018, 20, 15244–15252 RSC.
- F. Chen, L. L. Liu, Y. J. Zhang, J. H. Wu, G. X. Huang, Q. Yang, J. J. Chen and H. Q. Yu, Appl. Catal., B, 2020, 277, 119218 CrossRef CAS.
- E. B. Yutomo, F. A. Noor and T. Winata, RSC Adv., 2021, 11, 18371–18380 RSC.
- H. Yan, L. Wang, Y. Chen, L. Jiao, Y. Wu, W. Xu, W. Gu, W. Song, D. Du and C. Zhu, Research, 2020, 1, 1–11 Search PubMed.
- P. Lazar, R. Mach and M. Otyepka, J. Phys. Chem. C, 2019, 123, 10695–10702 CrossRef CAS.
- C. Peng, A. H. L. Chow and C. K. Chan, Aerosol Sci. Technol., 2010, 35, 753–758 CrossRef.
- H. Abdillahi, A. Rouilly, E. Chabrat and L. Rigal, Ind. Crops Prod., 2013, 50, 104–111 CrossRef CAS.
- M. M. Chim, C. Y. Lim, J. H. Kroll and M. N. Chan, ACS Earth Space Chem., 2018, 2, 1323–1329 CrossRef CAS.
- D. Wyrzykowski, E. Hebanowska, G. Nowak-Wiczk, M. Makowski and L. Chmurzyński, J. Therm. Anal. Calorim., 2010, 104, 731–735 CrossRef.
- M. Koebel and E. O. Strutz, Ind. Eng. Chem. Res., 2003, 42, 2093–2100 CrossRef.
- M. J. Milewska, Z. Chem., 1988, 28, 204–211 CrossRef CAS.
- W. Kasprzyk, T. Świergosz, S. Bednarz, K. Walas, N. V. Bashmakova and D. Bogdał, Nanoscale, 2018, 10, 13889–13894 RSC.
- V. Duraisamy, S. Palanivel, R. Thangamuthu and S. M. S. Kumar, ChemistrySelect, 2018, 3, 11864–11874 CrossRef CAS.
- Y. Liao, S. Zhu, J. Ma, Z. Sun, C. Yin, C. Zhu, X. Lou and D. Zhang, ChemCatChem, 2014, 6, 3419–3425 CrossRef CAS.
- U. Baruah, M. J. Deka and D. Chowdhury, RSC Adv., 2014, 4, 36917–36922 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra07290k |
| This journal is © The Royal Society of Chemistry 2021 |