
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBis(imino)-6,7-dihydro-5H-quinoline-cobalt complexes as highly active catalysts for the formation of vinyl-terminated PE waxes; steps towards inhibiting deactivation pathways through targeted ligand design†
Mingyang Han ab,
Zheng Zuoab,
Yanping Maa,
Gregory A. Solan*ac,
Xinquan Hu*d,
Tongling Lianga and
Wen-Hua Sun*abe
ab,
Zheng Zuoab,
Yanping Maa,
Gregory A. Solan*ac,
Xinquan Hu*d,
Tongling Lianga and
Wen-Hua Sun*abe
aKey Laboratory of Engineering Plastics and Beijing National Laboratory for Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: whsun@iccas.ac.cn
bCAS Research/Education Center for Excellence in Molecular Sciences, University of Chinese Academy of Sciences, Beijing 100049, China
cDepartment of Chemistry, University of Leicester, University Road, Leicester LE1 7RH, UK. E-mail: gas8@leicester.ac.uk
dCollege of Chemical Engineering, Zhejiang University of Technology, Hangzhou 310014, China. E-mail: xinquan@zjut.edu.cn
eState Key Laboratory for Oxo Synthesis and Selective Oxidation, Lanzhou Institute of Chemical Physics Chinese Academy of Sciences, Lanzhou 730000, China
First published on 15th December 2021
Abstract
A set of five related bis(imino)-6,7-dihydro-5H-quinoline-cobalt(II) complexes, [2-(ArN = CPh)-8-(NAr)-C9H8N]CoCl2 (Ar = 2,6-Me2C6H3 Co1, 2,6-Et2C6H3 Co2, 2,6-i-Pr2C6H3 Co3, 2,4,6-Me3C6H2 Co4, 2,6-Et2-4-MeC6H2 Co5), have been synthesized in reasonable yield by the template reaction of cobalt(II) chloride hexahydrate, 2-benzoyl-6,7-dihydro-5H-quinolin-8-one and the corresponding aniline. The molecular structures of Co1 and Co4 highlight both the differences in the two imino-carbon environments (phenyl-capped chain vs. cyclic) and also the steric properties exerted by the bulky Nimine-aryl groups. On pre-treatment with either modified methylaluminoxane (MMAO) or methylaluminoxane (MAO), all complexes proved productive catalysts for the polymerization of ethylene. In particular, Co1/MAO was the most active reaching a very high level of 1.62 × 107 g PE per mol (Co) per h over a 30 minute run time. Owing to the presence of the imino-phenyl substituent, Co1–Co5 were able to exhibit good thermal stability by displaying appreciable catalytic activity at temperatures between 50 and 80 °C, generating polyethylenes with narrow dispersities (Mw/Mn range: 1.66–3.28). In particular, the least sterically bulky precatalysts, Co1 and Co4 formed polyethylene waxes (Mw range: 1.94–5.69 kg per mol) with high levels of vinyl unsaturation as confirmed by high temperature 1H/13C NMR spectroscopy and by IR spectroscopy.
Introduction
Late transition metal catalyzed polymerization of ethylene has been the subject of widespread interest1–5 ever since the groundbreaking discoveries that α-diimino-Ni(II)/Pd(II)1a and bis(imino)pyridyl-Fe(II)/Co(II) complexes, under suitable activation, can exhibit very high catalytic activities. With particular regard to bis(imino)pyridyl-cobalt(II) halides (A, Chart 1),1b,2a it is often considered that the highly electrophilic nature of the active catalyst facilitates a low barrier towards ethylene insertion while bulky ortho-substituted N-aryl groups inhibit chain transfer resulting in the formation of high molecular weight polyethylene. As a result, a wide array of substituent variations has over the years been made to the bis(imino)pyridine ligand skeleton.6,7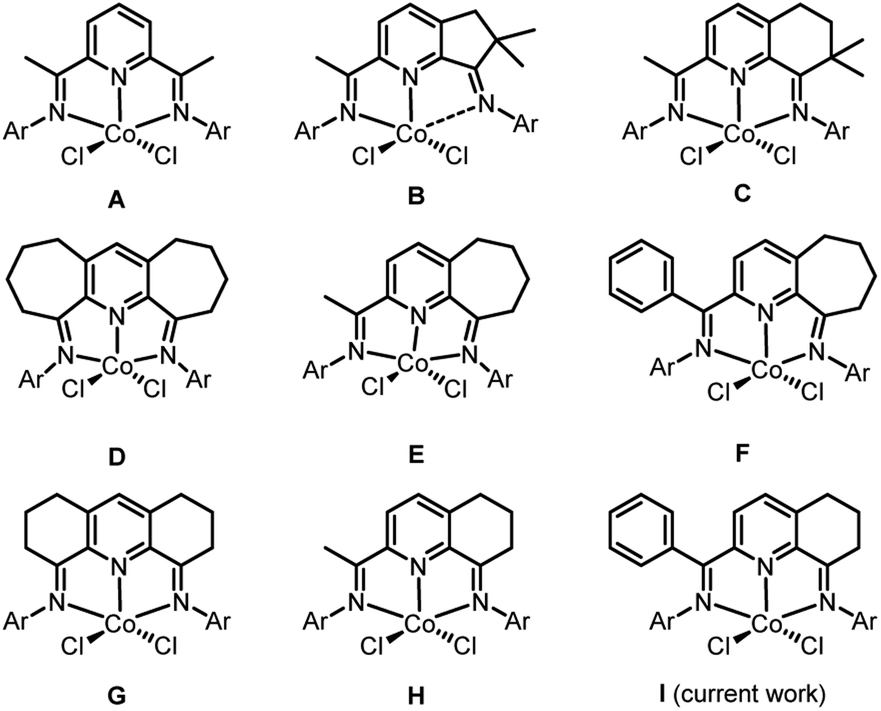 | ||
| Chart 1 2,6-Bis(arylimino)pyridine-cobalt(II) halide A and its fused counterparts B–I. | ||
Besides high molecular weight polyethylene products, linear polyethylene waxes of narrow dispersity and incorporating vinyl chain ends have gained increasing attention owing to their various applications such as lubricants and colorants for plastics processes.8 In view of this, alternative iron and cobalt ethylene polymerization catalysts have been investigated as a means to target such low molecular weight polymers (typical Mw range: 1–5 kg per mol). In particular, cobalt catalysts bearing bis(imino)pyridines fused with carbocyclic rings have shown a propensity towards the formation of such materials; broader polymer dispersities can be a drawback with their iron counterparts.6 However, the specific ring size of the fused carbocycle and its substitution pattern can be influential on not only the polymer specification but also the performance of the catalyst. For example, precatalysts B9a and C9b (Chart 1), that contain dimethyl-substituted five and six-membered fused rings respectively, exhibit quite different productivities while the molecular weight of their polyethylenes fall outside the desirable range for a polyethylene wax. On the other hand, precatalysts containing doubly or singly fused seven-membered rings (D–F,9c,9d,9e Chart 1) or six-membered rings devoid of methyl substituents (G,9f H,9g,9h Chart 1) are able to form the target waxes whilst maintaining, in general, good catalytic performance. As an additional benefit of the fused ring is its ability to influence the thermal stability of the catalyst allowing the polymerization process to operate at a higher temperature. In a similar way, modifications to the imino-C substituent in the N,N,N-ligand can be influential on thermostability. For instance, E exhibits its optimal activity for ethylene polymerization at 50 °C,9d while its phenyl-substituted counterpart F operates most effectively at 70 °C.9e
In this article, we return to the cyclohexyl-fused H-type family of precatalysts with a view to replacing the imino C-methyl substituent with a phenyl group. It was envisaged that this change could impede deactivation pathways and in-turn increase the thermostability of the catalysts.9e,9g Furthermore, it was of interest to examine how this modification would impact on catalytic activity and its ability to form polyethylene waxes. In particular, we target five examples of 2-(phenyl-(arylimino)methyl)-8-arylimino-6,7-dihydro-5H-quinoline-cobalt(II) chloride (I, Chart 1) in which the steric and electronic properties of the N-aryl groups have been varied. In addition, a full catalytic evaluation for the polymerization of ethylene is reported that explores not only the effect of precatalyst structure but also the co-catalyst type, precatalyst/co-catalyst ratio, run time, temperature and pressure. The intrinsic properties of the polyethylenes will also be discussed (e.g., molecular weight, dispersity, melting temperatures, vinyl content) as will the synthetic and characterization data for the new cobalt complexes.
Results and discussion
Synthesis and characterization of Co1–Co5
The 2-(phenyl(arylimino)methyl)-8-arylimino-6,7-dihydro-5H-quinoline-cobalt(II) chloride complexes, [2-(ArN = CPh)-8-(NAr)-C9H8N]CoCl2 (Ar = 2,6-Me2C6H3 Co1, 2,6-Et2C6H3 Co2, 2,6-i-Pr2C6H3 Co3, 2,4,6-Me3C6H2 Co4, 2,6-Et2-4-MeC6H2 Co5), were prepared in reasonable yield (37–52%) by the template reaction of 2-benzoyl-6,7-dihydro-5H-quinolin-8-one, cobalt dichloride hexahydrate and the corresponding aniline in acetic acid at reflux for 6 hours (Scheme 1). Attempts at trying to isolate the free bis(imino)-6,7-dihydro-5H-quinoline compounds by condensation of 2-benzoyl-6,7-dihydro-5H-quinolin-8-one with the aniline, proved unsuccessful.10 Complexes Co1–Co5 have been characterized by a combination of IR spectroscopy, microanalysis and single crystal X-ray diffraction.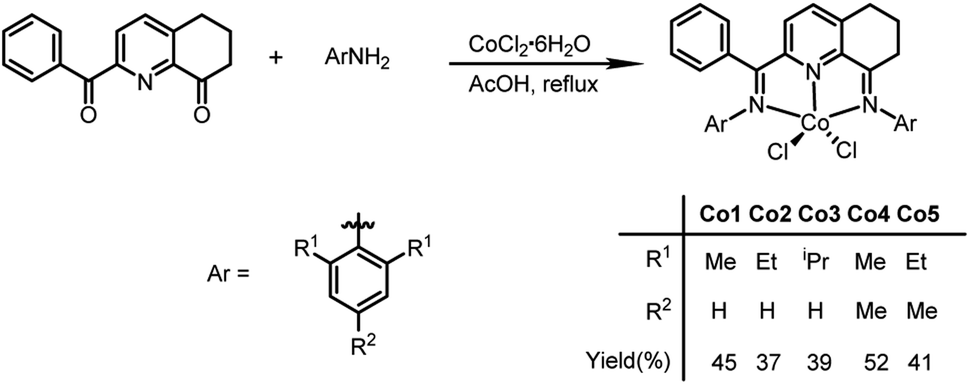 | ||
| Scheme 1 Synthesis of complexes Co1–Co5 via a one-pot route. | ||
For the purposes of the X-ray determination, single crystals of Co1 and Co4 were grown by diffusing diethyl ether into dichloromethane solutions of the corresponding complex. Views of Co1 and Co4 are shown in Fig. 1 and 2; selected bond lengths and angles are gathered in Table 1.
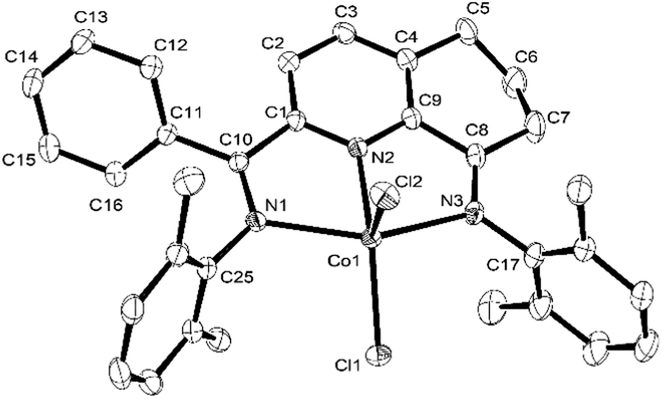 | ||
| Fig. 1 OLEX2 representation of Co1. The thermal ellipsoids are shown at the 30% probability level, while all hydrogen atoms and a molecule of CH2Cl2 have been removed for clarity. | ||
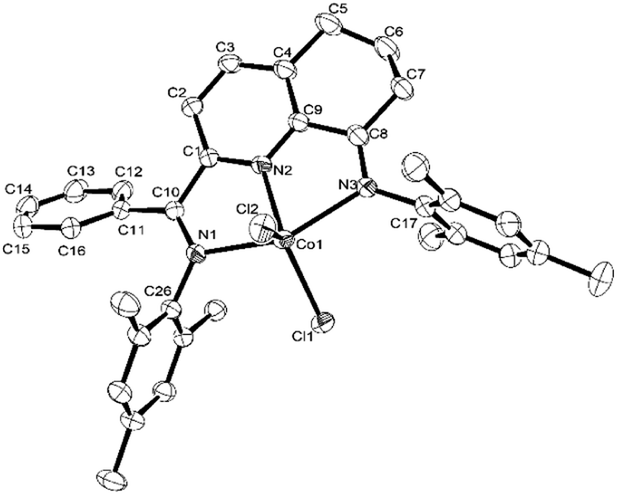 | ||
| Fig. 2 OLEX2 representation of Co4. The thermal ellipsoids are shown at the 30% probability level, while all hydrogen atoms and two molecules of CH2Cl2 have been removed for clarity. | ||
| Co1 | Co4 | |
|---|---|---|
| Bond lengths (Å) | ||
| Co(1)–N(1) | 2.238(2) | 2.213(3) |
| Co(1)–N(2) | 2.040(2) | 2.024(3) |
| Co(1)–N(3) | 2.209(2) | 2.263(3) |
| Co(1)–Cl(1) | 2.2579(8) | 2.2475(10) |
| Co(1)–Cl(2) | 2.2775(8) | 2.2808(11) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||
| Bond angles (deg) | ||
| N(1)–Co(1)–N(2) | 74.06(9) | 75.31(11) |
| N(1)–Co(1)–N(3) | 148.11(9) | 148.73(10) |
| N(2)–Co(1)–N(3) | 75.80(9) | 75.15(11) |
| N(1)–Co(1)–Cl(2) | 95.66(6) | 99.48(8) |
| N(2)–Co(1)–Cl(2) | 100.14(7) | 105.69(9) |
| N(3)–Co(1)–Cl(2) | 99.65(7) | 98.14(9) |
| N(1)–Co(1)–Cl(1) | 101.54(6) | 98.24(8) |
| N(2)–Co(1)–Cl(1) | 146.28(7) | 136.48(10) |
| N(3)–Co(1)–Cl(1) | 97.69(7) | 96.08(9) |
| Cl(2)–Co(1)–Cl(1) | 113.58(4) | 117.79(4) |
Owing to the structural similarity, both Co1 and Co4 will be discussed together. In common to both structures, a cobalt center is surrounded by three nitrogen donors belonging to the tridentate 2-(phenyl(arylimino)-methyl)-8-arylimino-6,7-dihydro-5H-quinoline chelating ligand (aryl = 2,6-dimethylphenyl Co1, 2,4,6-trimethylphenyl Co4) and two chlorides to complete a geometry best described as pseudo-square pyramidal. The three nitrogen atoms N1, N2, N3 and Cl1 fill the basal plane while Cl2 occupies the apical position. The cobalt atom sits above the square base by 0.600 Å for Co4 and 0.496 Å for Co1 in a manner akin to that seen with related (N,N,N)CoCl2 complexes.9g The exterior Co–Nimine bond lengths [2.213, 2.263 Å (Co4), 2.238, 2.209 Å (Co1)] are longer than the central Co–Npyridine bond length [2.024 Å (Co4), 2.040 Å (Co1)] in line with the stronger donor ability of the pyridine. As with its analogues,11,12 the N-aryl groups are inclined towards perpendicular with respect to the N,N,N-Co coordination plane with dihedral angles of 71.06°, 76.73° Co4 and 72.59°, 65.67° Co1. On account of the sp3-hybrization of carbon atoms, C5, C6 and C7, there is some puckering of this section of the fused 6-membered ring that folds away from the apical Cl2 ligand in both structures. There are no intermolecular contacts of significance.
The FT-IR spectra of Co1–Co5 exhibited absorption bands for the two C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Nimine groups around 1610 cm−1, values that are characteristic of cobalt(II) bound imines.9b,9e Further examination of the spectra revealed the absence of any absorptions corresponding to complexed CO groups or free diketone. In addition, the elemental analysis data were in good agreement with the proposed formulae for all five cobalt complexes.
Nimine groups around 1610 cm−1, values that are characteristic of cobalt(II) bound imines.9b,9e Further examination of the spectra revealed the absence of any absorptions corresponding to complexed CO groups or free diketone. In addition, the elemental analysis data were in good agreement with the proposed formulae for all five cobalt complexes.
Exploring the scope of Co1–Co5 as precatalysts in ethylene polymerization
To facilitate the ethylene polymerization study, a direct in situ activation process was followed whereby the cobalt precatalyst (Co1–Co5) was pre-activated by the addition of co-catalyst and the resulting active species used to mediate the polymerization. In view of previous studies, most bis(imino)pyridine-cobalt(II) halide complexes have been shown to exhibit their optimal performance for ethylene polymerization when activated with either modified methyl aluminoxane (MMAO) or methylaluminoxane (MAO).13,14 With this in mind, the current work focuses on using these two types of co-catalyst with Co4 selected as the test precatalyst to develop a suitable set of conditions that could then be used to screen the remaining cobalt complexes. For each Co4/co-catalyst combination, variations in reaction temperature, Al:Co molar ratio, run time and pressure will be independently conducted; the results are compiled in Tables 2 and 3. As a matter of routine, the polyethylene generated was characterized by gel permeation chromatography (GPC) and differential scanning calorimetry (DSC). In addition, high temperature NMR spectroscopy as well as IR spectroscopy was used to explore the microstructural properties of selected samples of polyethylene.
| Entry | Precat. | Al:Co |
T (°C) | t (min) | Activityb | Mwc | Mw/Mnc | Tmd (°C) |
|---|---|---|---|---|---|---|---|---|
| a Conditions: 2.0 μmol of cobalt precatalyst, 10 atm of ethylene, 100 mL of toluene.b Activity: 106 g PE per mol (Co) per h.c Mw in kg per mol. Mw and Mw/Mn measured by GPC.d Measured by DSC.e 5 atm. | ||||||||
| 1 | Co4 | 2000 | 30 | 30 | 2.20 | 5.69 | 2.27 | 126.6 |
| 2 | Co4 | 2000 | 40 | 30 | 3.25 | 4.39 | 2.28 | 125.9 |
| 3 | Co4 | 2000 | 50 | 30 | 5.80 | 3.64 | 2.93 | 125.0 |
| 4 | Co4 | 2000 | 60 | 30 | 4.97 | 3.06 | 2.03 | 124.5 |
| 5 | Co4 | 2000 | 70 | 30 | 4.23 | 2.93 | 2.34 | 124.1 |
| 6 | Co4 | 1750 | 50 | 30 | 3.84 | 3.77 | 2.05 | 125.1 |
| 7 | Co4 | 2250 | 50 | 30 | 8.65 | 3.17 | 2.05 | 124.2 |
| 8 | Co4 | 2500 | 50 | 30 | 6.79 | 2.92 | 1.66 | 125.0 |
| 9 | Co4 | 2750 | 50 | 30 | 5.16 | 2.77 | 1.95 | 123.3 |
| 10 | Co4 | 2250 | 50 | 5 | 28.2 | 3.47 | 2.00 | 124.8 |
| 11 | Co4 | 2250 | 50 | 15 | 10.4 | 3.48 | 2.19 | 124.9 |
| 12 | Co4 | 2250 | 50 | 45 | 4.71 | 3.30 | 1.95 | 124.7 |
| 13 | Co4 | 2250 | 50 | 60 | 3.78 | 3.20 | 1.95 | 124.7 |
| 14e | Co4 | 2250 | 50 | 30 | 4.52 | 3.68 | 3.28 | 125.0 |
| 15 | Co1 | 2250 | 50 | 30 | 16.18 | 2.67 | 2.43 | 123.3 |
| 16 | Co2 | 2250 | 50 | 30 | 2.42 | 9.91 | 2.30 | 129.6 |
| 17 | Co3 | 2250 | 50 | 30 | 0.16 | 146.43 | 1.67 | 131.2 |
| 18 | Co5 | 2250 | 50 | 30 | 1.48 | 11.38 | 2.08 | 129.3 |
| Entry | Precat. | Al:Co |
T (°C) | t (min) | Activityb | Mwc | Mw/Mnc | Tmd (°C) |
|---|---|---|---|---|---|---|---|---|
| a Conditions: 2.0 μmol of cobalt precatalyst, 10 atm of ethylene, 100 mL of toluene.b Activity: 106 PE per mol (Co) per h.c Mw in kg per mol. Mw and Mw/Mn measured by GPC.d Measured by DSC.e 5 atm. | ||||||||
| 1 | Co4 | 2000 | 40 | 30 | 2.27 | 4.11 | 2.28 | 126.3 |
| 2 | Co4 | 2000 | 50 | 30 | 3.50 | 3.44 | 2.96 | 124.5 |
| 3 | Co4 | 2000 | 60 | 30 | 3.98 | 2.87 | 1.67 | 124.7 |
| 4 | Co4 | 2000 | 70 | 30 | 3.13 | 2.67 | 1.89 | 124.1 |
| 5 | Co4 | 2000 | 80 | 30 | 2.45 | 2.59 | 2.05 | 124.3 |
| 6 | Co4 | 1500 | 60 | 30 | 3.00 | 2.81 | 1.77 | 124.4 |
| 7 | Co4 | 1750 | 60 | 30 | 3.25 | 2.89 | 1.98 | 124.8 |
| 8 | Co4 | 2250 | 60 | 30 | 3.35 | 2.90 | 1.87 | 124.5 |
| 9 | Co4 | 2500 | 60 | 30 | 2.34 | 2.43 | 1.92 | 124.6 |
| 10 | Co4 | 2000 | 60 | 5 | 14.52 | 2.23 | 1.95 | 124.6 |
| 11 | Co4 | 2000 | 60 | 15 | 6.30 | 1.94 | 1.96 | 124.6 |
| 12 | Co4 | 2000 | 60 | 45 | 2.13 | 2.52 | 2.28 | 124.6 |
| 13 | Co4 | 2000 | 60 | 60 | 2.05 | 2.89 | 2.35 | 124.7 |
| 14e | Co4 | 2000 | 60 | 30 | 2.44 | 3.58 | 1.82 | 124.8 |
| 15 | Co1 | 2000 | 60 | 30 | 7.14 | 2.43 | 1.96 | 123.2 |
| 16 | Co2 | 2000 | 60 | 30 | 1.72 | 10.01 | 2.24 | 129.4 |
| 17 | Co3 | 2000 | 60 | 30 | 0.18 | 166.57 | 2.67 | 132.7 |
| 18 | Co5 | 2000 | 60 | 30 | 1.20 | 9.87 | 2.31 | 129.5 |
As the temperature of the polymerization run can play a key role in catalytic performance,11b,15 we first explored its effect on the catalytic activity of Co4/MAO. Specifically, five runs were conducted at fixed temperatures between 30 °C and 70 °C with the Al:Co molar ratio at 2000:1 (entries 1–5, Table 2). A peak of 5.80 × 106 g PE per mol (Co) per h was observed with the run temperature set at 50 °C. Even at 70 °C, the catalyst still performed well with only a 37% loss of activity. With respect to the molecular weights of the resulting polyethylenes, these gradually declined from 5.69 kg per mol to 2.93 kg per mol as the temperature was increased from 30 °C to 70 °C in keeping with an increased rate of chain transfer as the temperature was raised (Fig. 3).15b,16 Nevertheless, the distributions of the polyethylenes obtained across the temperature range remained reasonably narrow (Mw/Mn = 2.09–2.93) highlighting the good control and single site-like behavior of the cobalt active species.
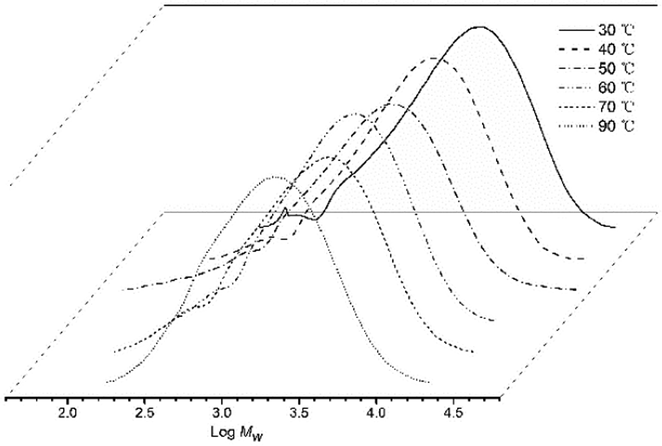 | ||
| Fig. 3 GPC traces of the polyethylene produced using Co4/MAO at different run temperatures (entries 1–5, Table 2). | ||
With the polymerization temperature retained at 50 °C, the impact of the Al:Co molar ratio was investigated by performing the runs at ratios of between 1750:1 and 2750:1 (entries 3 and 6–9, Table 2). Inspection of the data for Co4/MAO revealed the highest activity of 8.65 × 106 g PE per mol (Co) per h was achieved at a molar ratio of 2250:1. In terms of molecular weight of the polymer, it was noted that this steadily dropped as the Al:Co ratio was increased, which may suggest that chain transfer from the active cobalt species to aluminum co-catalyst is operative.9d,9e
To explore the profile of Co4/MAO with respect to time, the polymerizations were undertaken at five distinct run times namely, 5, 15, 30, 45 and 60 minutes (entries 7 and 10–13, Table 2 and Fig. S1†). After 5 minutes, the catalyst reached peak activity of 28.2 × 106 g PE per mol (Co) per h before steadily losing performance and reaching its lowest level of 3.78 × 106 g PE per mol (Co) per h after 60 minutes. Such a downward trend in activity over time would suggest some gradual deactivation of the catalyst. With the ethylene pressure lowered from 10 to 5 atm, the catalytic activity dropped by nearly a half (entries 14 vs. 7, Table 2), while the molecular weight of the polymer increased from 3.17 kg per mol to 3.68 kg per mol. Moreover, the dispersity of the polyethylene generated was broader at the lower ethylene pressure, a finding that has been reported elsewhere for a cobalt analogue.17
By using the favored set of conditions established with Co4/MAO, the remaining cobalt precatalysts, Co1–Co3 and Co5, were additionally evaluated for ethylene polymerization (entries 15–18, Table 2). In general, all the cobalt complexes showed good catalytic activities across a wide range (0.16–16.18 × 106 g PE per mol (Co) per h) with the relative levels following the order: Co1[2,6-di(Me)] > Co4[2,4,6-tri(Me)] > Co2[2,6-di(Et)] > Co5[2,6-di(Et)-4-Me] > Co3[2,6-di(i-Pr)]. It is noteworthy that the activity of Co1[2,6-di(Me)] (16.18 × 106 g PE per mol (Co) per h) exceeded that for Co3[2,6-di(i-Pr)](0.16 × 106 g PE per mol (Co) per h) by an order of magnitude (Fig. 4).
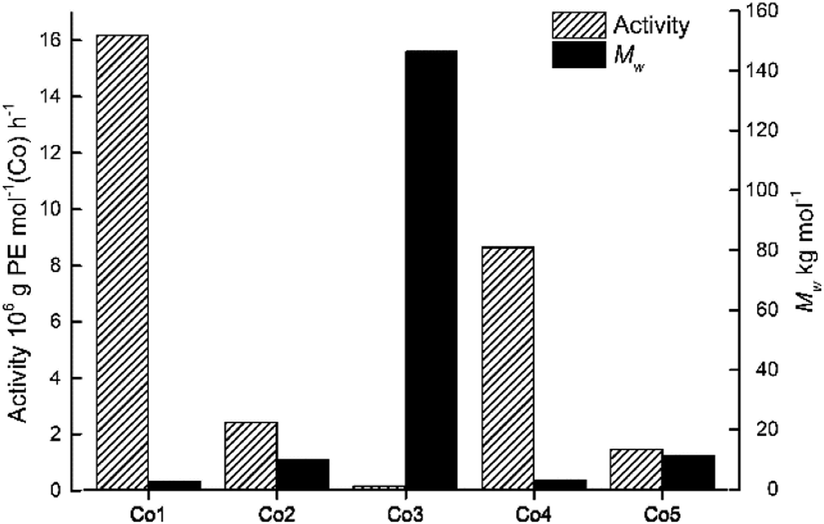 | ||
| Fig. 4 Bar chart showing catalytic activity and molecular weight of the polyethylene as a function of the precatalyst employed (entries 7 and 15–18, Table 2); MAO used as co-catalyst in each case. | ||
It would seem that the steric properties exerted by the ortho-substituents are influential with increased hindrance generally leading to lower activity, a finding that can be credited to inefficient coordination of the monomer. On the other hand, the molecular weight of the polymers (Mw range: 2.67–146.43 kg per mol) followed the reverse order with respect to the precatalyst: Co3[2,6-di(i-Pr)] > Co5[2,6-di(Et)-4-Me] > Co2[2,6-di(Et)] > Co4[2,4,6-tri(Me)] > Co1[2,6-di(Me)]. Indeed, the molecular weight of the polymer generated by Co1 (Mw = 2.67 kg per mol) was substantially less than that seen with Co3 (Mw = 146.43 kg per mol) which highlights how steric effects can be beneficial to chain propagation. Furthermore, the dispersity (Mw/Mn) of the polyethylene generated using Co1–Co5 fell in the range 1.67–2.96, which again underlines the good control of the polymerizations in accord with single-site active species.
On increasing the run temperature from 40 °C to 80 °C, the topmost level of activity for Co4/MMAO (3.98 × 106 g PE per mol (Co) per h) was seen at 60 °C (entries 1–5, Table 3). By comparison with Co4/MAO, this optimal temperature was 10 degrees lower albeit with a higher catalytic activity (5.80 × 106 g PE per mol (Co) per h, entry 3, Table 2). By raising the temperature to 70 °C, a 27% loss of activity was noted with Co4/MMAO, while at 80 °C a further 28% loss was seen. With regard to the molecular weight of the polymer, this decreased steadily from 4.11 kg per mol at 40 °C to 2.59 kg per mol at 80 °C. Indeed, this downward trend in molecular weight is similar to that seen for Co4/MAO, with temperature-induced chain-transfer likely responsible (Fig. 5). Likewise, a relatively narrow dispersity of the polymers was again observed (Mw/Mn range: 1.67–2.96) which highlights the effective control of the polymerizations as well as single-site nature of the active species.
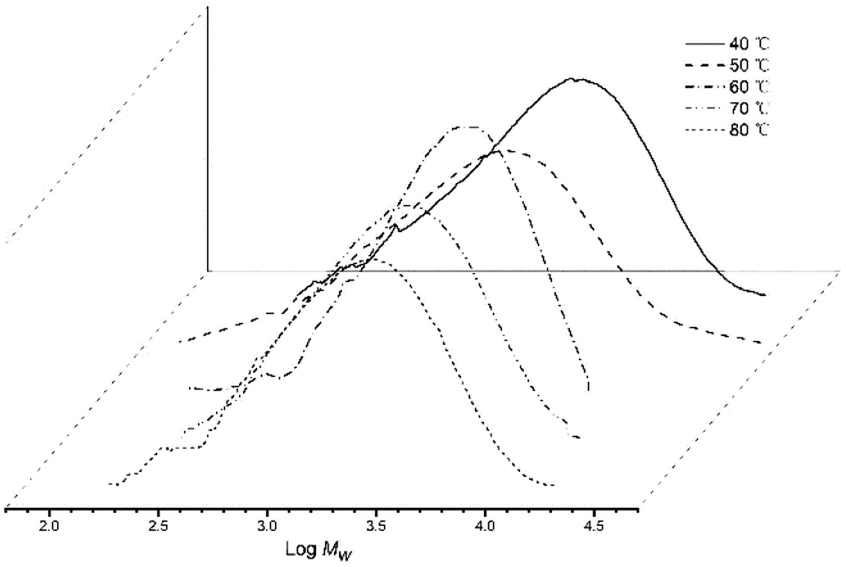 | ||
| Fig. 5 GPC traces of the polyethylene produced using Co4/MMAO at different run temperatures (entries 1–5, Table 3). | ||
With the polymerization temperature kept at 60 °C, a series of runs were conducted by varying the amount of MAO with respect to Co4. In particular, with the Al:Co molar ratio adjusted between 1500:1 and 2500:1 (entries 3, 6–9, Table 3), a maximum level of catalytic activity was reached with 2000 molar equivalents. Notably, when the Al:Co molar ratio was increased to 2500:1, a noticeable drop in both catalytic activity and the molecular weight was observed which would suggest the onset of chain transfer to aluminum (Fig. S2†).9d,9e In all cases the dispersity of polyethylene remained fairly narrow between 2.43 and 2.90.
To explore the time–activity profile for Co4/MMAO, the polymerizations runs were conducted at intervals between 5 and 60 minutes with the temperature and Al:Co molar ratio maintained at 60 °C and 2500:1, respectively (entries 3, 10–13, Table 3). The highest activity of 14.52 × 106 g PE per mol (Co) per h was seen in the 5 first minutes which compares with 28.2 × 106 g PE per mol (Co) per h with Co4/MAO (entry 10, Table 2). Upon prolonging the reaction time, the polymerization activity smoothly decreased, underlining the gradual deactivation of the cobalt active species; similar observations were seen for Co4/MAO and for their imino-C methyl counterparts (H, Chart 1). Furthermore, the activity of Co4/MMAO was observed to dramatically decrease when the ethylene pressure was reduced from 10 atm to 5 atm. Conversely, the molecular weight of the polyethylene generated at 5 atm was higher than that at 10 atm. Similar observations were noted with Co4/MAO and indeed the molecular weight of the polymer at 5 atm was comparable (3.68 kg per mol (MAO) vs. 3.58 kg per mol (MMAO)).
To probe the effect of precatalyst structure, the remaining cobalt catalysts, Co1–Co3 and Co5 were also screened for ethylene polymerization under the optimal conditions ascertained for Co4/MMAO. Collectively, the activities fell in the range 0.18–7.14 × 106 g PE per mol (Co) per h) which is narrower when compared to 0.18–16.18 × 106 g PE per mol (Co) per h with MAO. Specifically, the activities decreased in the order, Co1[2,6-di(Me)] > Co4[2,4,6-tri(Me)] > Co2[2,6-di(Et)] > Co5[2,6-di(Et)-4-Me] > Co3[2,6-di(i-Pr)], which is identical to that seen with MAO and indeed similar to that reported elsewhere.18 Once again steric properties played a key role with greater hindrance resulting in lower activity. As for the molecular weight, a broad range in values from 2.43 kg per mol to 166.57 kg per mol were seen with bulky diisopropyl-containing Co3 falling at the very top end, while dimethyl-containing Co1 occupying the bottom; trends that are the inverse of that found for activity (Fig. 6).
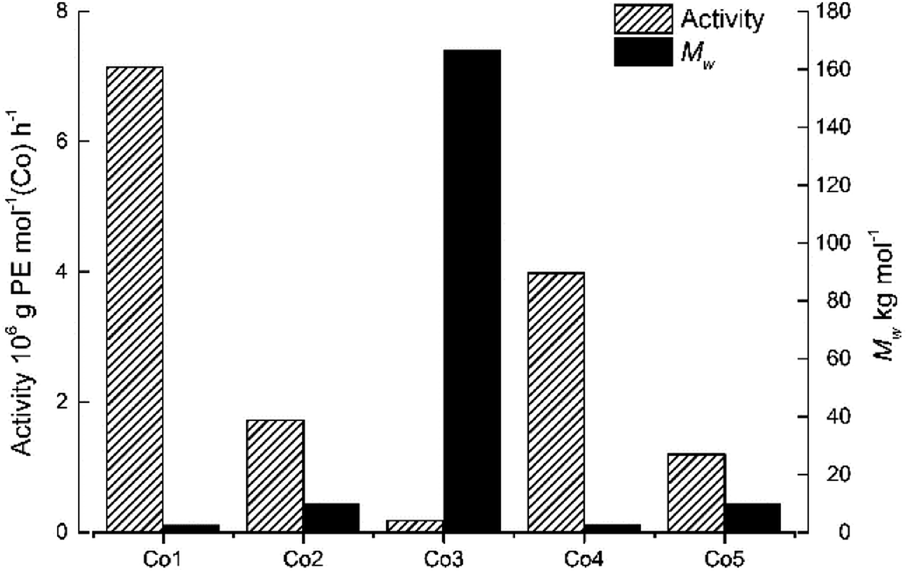 | ||
| Fig. 6 Bar chart showing catalytic activity and molecular weight of the polyethylene as a function of the precatalyst employed (entries 3 and 15–18, Table 3); MMAO used as co-catalyst in each case. | ||
In general, the catalytic activities to Co1–Co5 with MAO as co-catalyst were higher than that with MMAO as has been noted with related cobalt catalysts.9d,9e,9g Likewise, the molecular weights of the polyethylenes produced using MAO exceeded that found using MMAO with the exception of Co2 and Co3. To allow a comparison to be made with previously reported bis(imino)pyridine-Co(II) precatalysts that contain six- and seven-membered fused rings,9d,9e,9g the molecular weight and activity data for the 2,4,6-trimethylphenyl-containing precatalysts H, E and F (Chart 1) are displayed along with that for Co4 in Fig. 7; all precatalysts have been screened with MAO as the co-catalyst at 10 atm C2H4.
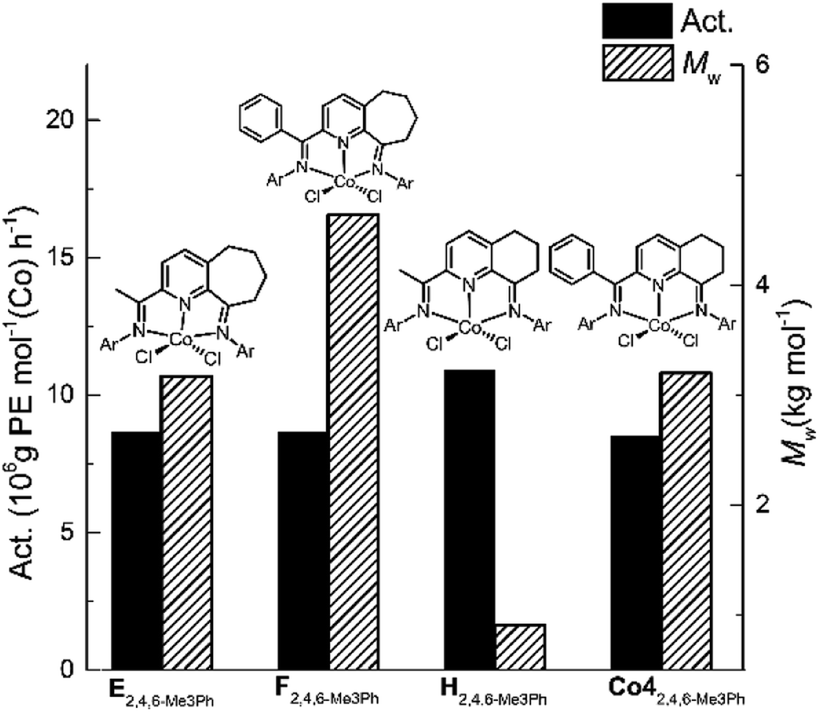 | ||
| Fig. 7 Comparison of catalytic activity and polymer molecular weight for Co4 (entry 7, Table 2) with that produced by E, F and H (Ar = 2,4,6-Me3Ph); all precatalysts screened with MAO as the co-catalyst at 10 atm C2H4. | ||
Analysis of the data reveals that the introduction of a phenyl group at the imino-C had a beneficial effect on the molecular weight of the resulting polyethylene. For instance, the molecular weight obtained using F (Mw = 4.6 kg per mol)9e was clearly higher than for E (Mw = 3.2 kg per mol),9d while their activity was comparable. In a similar manner, the polyethylene produced using Co4 (Mw = 3.2 kg per mol) was notably higher than that formed by H (Mw = 0.91 kg per mol).9g This observation may be due to the protection imparted by the phenyl group on the active center.
Microstructural properties of the polyethylene waxes
On inspection of Tables 2 and 3, it evident that the Tm values for all the polyethylenes produced with either MAO or MMAO exceeded 123 °C. Such a temperature range would suggest high linearity for these polymers.19 To gather more information about their microstructural properties, representative samples of polyethylene wax obtained from the separate MAO and MMAO investigations were selected, namely that formed using Co4/MAO (Mw = 3.17 kg per mol, entry 7, Table 2) and Co4/MMAO (Mw = 2.87 kg per mol, entry 3, Table 3), were characterized by 1H and 13C NMR spectroscopy as well as by inverse-gated decoupled 13C NMR spectroscopy. Typically, these spectra were obtained at 100 °C in 1,1,2,2-tetrachloroethane-d2 so as to engender suitable solubility of the polymers.9,16In the case of the sample obtained using Co4/MAO, the 1H NMR spectrum displayed downfield multiplets at around δ 5.87 ppm (He: integration = 1) and δ 5.04 ppm (Hd: integration = 2) that are characteristic of a –CHCH2 end group (Fig. S3†). Corroboration of this assignment was provided by the 13C NMR spectrum which showed the corresponding unsaturated carbon signals at δ 114.4 ppm and 139.6 ppm (Ce and Cd in Fig. S4†).7c,19d The methyl chain end protons (Ha) were visible at δ 0.96 ppm in the 1H NMR spectrum with a relative integration of 4.58 which, on account of being >3H, suggested the presence of both vinyl-terminated polyethylene and some fully saturated polymer.7c In an attempt to quantify the relative proportion of vinyl-terminated polyethylene to fully saturated polyethylene, we determined the molar fraction X (where X = 1 refers to 100% vinyl-terminated polyethylene) based on the integrations of the signals for Ha and Hd in the 1H NMR spectrum (Fig. S5†).
A value of 0.79 was determined which implied a clear bias towards a vinyl-terminated polymer in line with β-H elimination constituting the key termination pathway.16,19d As supporting evidence, the inverse-gated decoupled 13C NMR spectrum of the sample displayed an integral ratio for Cd, Ce and Ca was 0.98:1.00:1.24 (Fig. S5†). Likewise, the sample of polyethylene wax obtained using Co4/MMAO revealed similar features with a sharp singlet at δ 30.0 ppm in its 13C NMR spectrum and an intense signal at δ 1.35 ppm in its 1H NMR spectrum in support of linear polymer (Fig. 8). Once again, the value of X (0.81) highlighted the predominance of β-H elimination in chain termination with vinyl-terminated polyethylenes being the major type of chain end. Similarly, the inverse-gated decoupled 13C NMR spectrum for the polymer sample supported this finding by revealing a Cd:Ce:Ca integral ratio of 1.00:1.02:1.24 (Fig. 9).
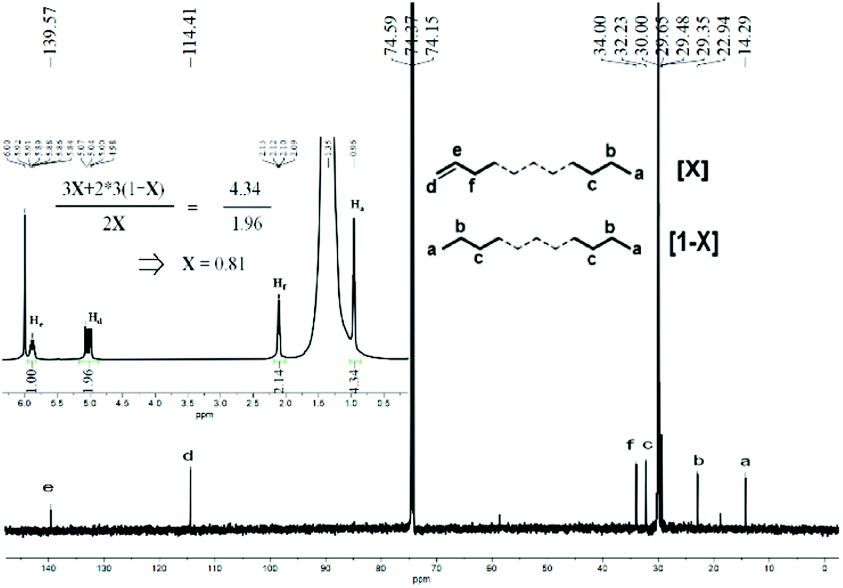 | ||
| Fig. 8 13C NMR spectrum of the polyethylene wax generated using Co4/MMAO along with an inset showing its 1H NMR spectrum (entry 3, Table 3); recorded in tetrachloroethane-d2 (δC 74.37, δH 6.0). | ||
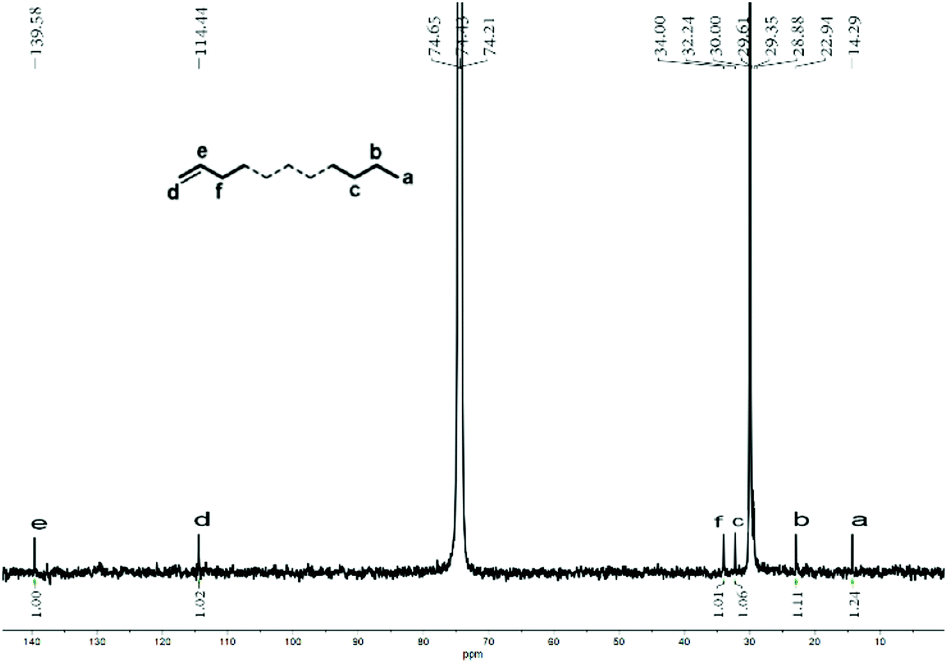 | ||
| Fig. 9 Inverse-gated decoupled 13C NMR spectrum of the polyethylene wax generated using Co4/MMAO (entry 3, Table 3); recorded in tetrachloroethane-d2 (δC 74.43). | ||
The two polymer wax samples formed using Co4/MAO (Mw = 3.17 kg per mol, entry 7, Table 2) and Co4/MMAO (Mw = 2.87 kg per mol, entry 3, Table 3), were also studied by FT-IR spectroscopy. As shown in Fig. 10 (middle) and Fig. S6 (middle),† weak bands at 1642 cm−1, 990 cm−1 and 909 cm−1 could be assigned to the vinyl group in the form of a vibration for the CC stretching mode, the in-phase plane vibration and the out of phase plane vibration, respectively. On the other hand, the stronger peaks at 700–750 cm−1 (CH2 rocking vibration) and at 1440–1490 cm−1 (CH2 wagging vibration) were characteristic of linear polyethylenes.20 In the same way, polyethylene samples prepared using higher Al:Co molar ratios of Co4/MAO (Mw = 2.77 kg per mol, entry 9, Table 2) and Co4/MMAO (Mw = 2.43 kg per mol, entry 9, Table 3) showed related features through the intensity of the vinyl vibrations was visibly less.
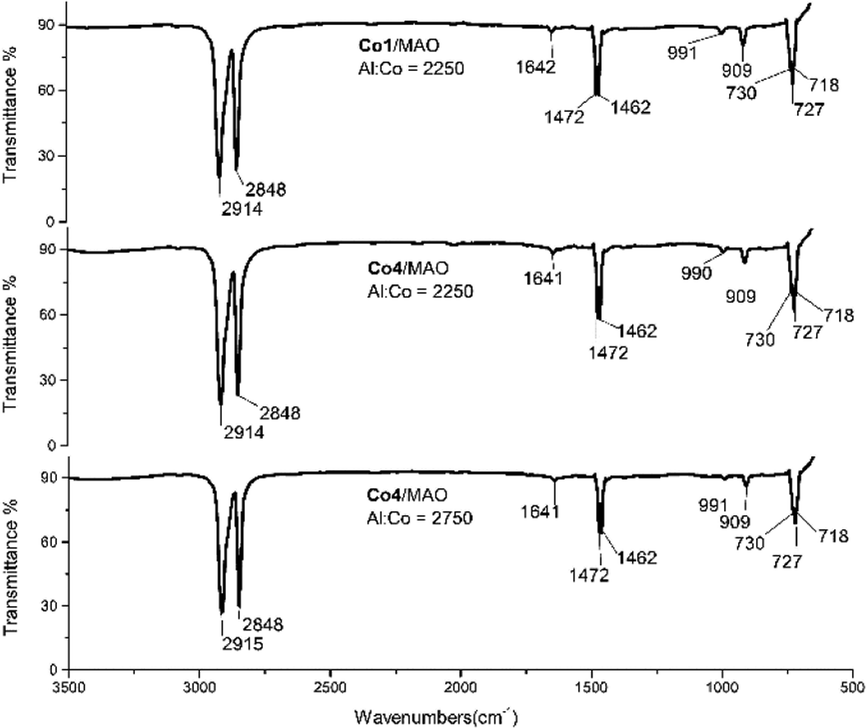 | ||
| Fig. 10 FT-IR spectra of the polyethylene samples generated using Co1/MAO (top, entry 15, Table 2) and Co4/MAO (middle, entry 7, Table 2 and bottom, entry 9, Table 2). | ||
In addition, samples of the polyethylene waxes generated using the more active catalysts, Co1/MAO (Mw = 2.67 kg per mol, entry 15, Table 2) and Co1/MMAO (Mw = 2.43 kg per mol, entry 15, Table 3), were also studied using IR spectroscopy. Once more, the spectra revealed the presence of vinyl-terminated polyethylenes (Fig. 10 (top) and Fig. S6 (top)†). Clearly, chain termination via β-H elimination represents a key pathway for all these polymerizations.
Conclusions
Five examples of I-type (Chart 1) cobalt precatalysts namely, Co1–Co5, each incorporating an imino C-phenyl group but differing in the imino N-aryl substitution pattern, have been successfully synthesized and characterized. Confirmation of the structural type by single crystal X-ray diffraction was also undertaken in the cases of Co1 and Co4. On activation with MMAO or MAO, all cobalt complexes exhibited high activity with Co1/MAO reaching the maximum level of 1.62 × 107 g PE per mol (Co) per h generating strictly linear polyethylenes with narrow dispersity. In addition, all five cobalt(II) precatalysts displayed good thermal stability with Co4/MMAO achieving an appreciable activity of 2.45 × 106 g PE per mol (Co) per h when the polymerization run was conducted at a more industrially relevant temperature of 80 °C. It is believed that the introduction of the imino C-phenyl substituent improved this thermal stability by inhibiting deactivation pathways. In addition, the molecular weights of the polyethylene were found to fall in an exceptionally broad range (2.43–166.57 kg per mol) which is controlled by the steric hindrance imparted by the ortho-substituents on the N-aryl groups. Notably the least sterically hindered Co1 and Co4 were able to form narrowly dispersed polyethylene waxes (Mw range: 1.94–5.69 kg per mol) with high levels of vinyl unsaturation. Subsequent investigations will focus on using these ligand frameworks and their derivatives as supports for other polymerization-active late transition metals.Experimental
General procedures
All operations of air- and/or moisture-sensitive compounds were carried out using standard Schlenk techniques under an atmosphere of nitrogen. Toluene was purified by initially heating it to reflux over sodium (containing a small amount of benzophenone) and then distilled under a nitrogen atmosphere prior to use. Methylaluminoxane (MAO) (1.46 M solution in toluene) and modified methylaluminoxane (MMAO) (1.93 M in n-heptane) were purchased from Albemarle Corporation, while high purity ethylene was purchased from Beijing Yansan Petrochemical Company. Other reagents were obtained from Aldrich or local suppliers. 1H and 13C NMR spectroscopic measurements on the organic compounds were performed on Bruker DMX 400 MHz instruments at room temperature using TMS as an internal standard; δ values are given in ppm. A PerkinElmer System 2000 FT-IR spectrometer was used to collect spectra from 4000 cm−1 to 400 cm−1 at room temperature with a data interval of 1 cm−1 when resolution was set at 4 cm−1. Elemental analysis was carried out with a Flash EA 1112 microanalyzer. Gel permeation chromatography (GPC) was undertaken with an Agilent PLGPC 220 GPC system using triple detection and with a polystyrene standard to determine molecular weights (Mw) and molecular weight distributions (Mw/Mn). Typically, polyethylene samples were dissolved in 1,2,4-trichlorobenzene at 150 °C and heated for at least two hours to dissolve. The melting temperatures (Tm) of the polyethylene samples were measured by differential scanning calorimeter (DSC) under a nitrogen atmosphere on a PerkinElmer DSC-7 instrument. As a matter of course, polyethylene samples were performed in crimped aluminum pans and data were collected with heat/cool cycles (−20 to 160 °C and 160 to −20 °C) at a heating rate of 10 °C min−1. All reported melting temperatures (Tm) were recorded from the second heating cycle. 1H and 13C NMR spectra of the polyethylene samples was recorded on a Bruker AVIII 500WB instrument. Typically, a weighed amount of polyethylene (20–40 mg) was first dissolved in 1,1,2,2-tetrachloroethane-d2 (2 mL) with TMS as an internal standard. Spectra were then recorded at 100 °C on a Bruker AVANCE III 500 MHz instrument in 5 mm standard glass tubes. Operating conditions used for the 13C NMR spectra: number of scans between 2000 and 3000, spectral width 31250.0 Hz, acquisition time 0.5243 s, relaxation delay 1.5000 s. Inverse-gated decoupled 13C NMR spectra were recorded with 6 mg of the relaxation reagent chromium(III) acetylacetonate. The operating conditions were set as follows: spectral frequency 125.70 MHz, pulse width 10.0 μs, spectral width 21.3675 kHz, acquisition time 0.7668 s, relaxation delay 5.0000 s.
Synthesis of 2-benzoyl-6,7-dihydro-5H-quinolin-8-one
The compound, 2-benzoyl-6,7-dihydro-5H-quinolin-8-one, was prepared in an acceptable yield (ca. 30%) from 2-chloro-6,7-dihydro-5H-quinolin-8-one according to a literature method.9e 1H NMR (400 MHz, CDCl3, TMS): δ 8.27 (m, 2H, Py), 8.12 (d, J = 8.0 Hz, 1H, Ph), 7.86 (d, J = 8.0 Hz, 1H, Ph), 7.60 (m, 1H, Ph), 7.57 (m, 2H, Ph), 3.13 (d, J = 6.0 Hz, 2H, Ph), 2.86 (d, J = 6.6 Hz, 2H, Ph), 2.27 (m, 2H, Ph). 13C NMR (100 MHz, CDCl3, TMS): δ 195.79, 192.35, 154.58, 147.00, 143.26, 138.95, 135.88, 133.39, 131.69, 128.39, 127.51, 39.98, 29.57, 22.61.Synthesis of [2-(ArN = CPh)-8-(NAr)-C9H8N]CoCl2 (Co1–Co5)
General procedure for ethylene polymerization
A stainless autoclave (250 mL), equipped with an ethylene pressure control system, a mechanical stirrer and a temperature controller, was firstly evacuated and backfilled with ethylene three times. Once the desired temperature was reached the precatalyst (2.0 μmol), pre-dissolved in freshly distilled toluene (25 mL), was injected into the autoclave under an atmosphere of ethylene (ca. 1 atm). More toluene (25 mL) was injected followed by the required amount of co-catalyst (MAO, MMAO) and then a final portion of toluene (50 mL) to complete the addition. The autoclave was immediately pressurized to the desired pressure (5 or 10 atm) and the stirring commenced. Once the designated time (5, 10, 30, 45 and 60 min) was reached, the reactor was cooled to room temperature with an external ice/water bath. The autoclave was then vented and the reaction quenched by the addition of 10% HCl in ethanol (200 mL). The polyethylene was filtered, washed with ethanol, and then dried overnight under reduced pressure in an oven at 60 °C.Crystal data and structure refinement
Single crystal X-ray diffraction studies were conducted on a XtaLAB Synergy-R single-crystal diffractometer with mirror-monochromatic Cu-Kα radiation (λ = 1.54184 Å) for Co1 at 170(10) K and Co4 at 220(13) K; the cell parameters were obtained by global refinement of the positions of all collected reflections. Structure solution was performed by using the SHELXT program (Sheldrick, 2015)21a and structure refinement was performed by using the SHELXL program (Sheldrick, 2015).21b All hydrogen atoms were placed in calculated positions. Crystallographic parameters for Co1 and Co4 are summarized in Table S1.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21871275). G.A.S. thanks the Chinese Academy of Sciences for a President's International Fellowship for Visiting Scientists.References
- (a) L. Johnson, C. Killian and M. Brookhart, J. Am. Chem. Soc., 1995, 117, 6414 CrossRef CAS; (b) G. J. P. Britovsek, V. C. Gibson, B. S. Kimberley, P. J. Maddox, S. J. McTavish, G. A. Solan, A. J. P. White and D. J. Williams, Chem. Commun., 1998, 7, 849 RSC.
- (a) B. L. Small and M. Brookhart, J. Am. Chem. Soc., 1998, 120, 4049 CrossRef CAS; (b) G. J. P. Britovsek, S. Mastroianni, G. A. Solan, S. P. D. Baugh, C. Redshaw, V. C. Gibson, A. J. P. White, D. J. Williams and M. R. J. Elsegood, Chem.–Eur. J., 2000, 6, 2221 CrossRef CAS PubMed.
- (a) A. M. F. Phillips, H. Suo, M. F. Silva, A. Pombeiro and W.-H. Sun, Coord. Chem. Rev., 2020, 416, 213332 CrossRef CAS; (b) S. D. Ittel, L. K. Johnson and M. Brookhart, Chem. Rev., 2000, 100, 1169 CrossRef CAS PubMed.
- (a) B. L. Small, Acc. Chem. Res., 2015, 48, 2599 CrossRef CAS PubMed; (b) B. L. Small and M. Brookhart, J. Am. Chem. Soc., 1998, 120, 7143 CrossRef CAS.
- Z. Flisak and W.-H. Sun, ACS Catal., 2015, 5, 4713 CrossRef CAS.
- Z. Wang, G. A. Solan, W. Zhang and W.-H. Sun, Coord. Chem. Rev., 2018, 363, 92 CrossRef CAS.
- (a) C. Bariashir, Z. Wang, Y. Ma, A. Vignesh, X. Hao and W.-H. Sun, Organometallics, 2019, 38, 4455 CrossRef CAS; (b) W.-H. Sun, S. Kong, W. Chai, T. Shiono, C. Redshaw, X. Hu, C. Guo and X. Hao, Appl. Catal., A, 2012, 448, 67 CrossRef; (c) M. Han, Q. Zhang, I. I. Oleynik, H. Suo, G. A. Solan, I. V. Oleynik, Y. Ma, T. Liang and W.-H. Sun, Dalton Trans., 2020, 49, 4774 RSC.
- K. Wang, K. Wedeking, W. Zuo, D. Zhang and W.-H. Sun, J. Organomet. Chem., 2008, 693, 1073 CrossRef CAS.
- (a) J. Ba, S. Du, E. Yue, X. Hu, Z. Flisak and W.-H. Sun, RSC Adv., 2015, 5, 32720 RSC; (b) R. Zhang, Y. Huang, G. A. Solan, W. Zhang, X. Hu, X. Hao and W.-H. Sun, Dalton Trans., 2019, 48, 8175 RSC; (c) S. Du, Z. Wang, E. Yue, F. Huang, T. Liang and W.-H. Sun, Eur. J. Inorg. Chem., 2016, 11, 1748 CrossRef; (d) F. Huang, W. Zhang, E. Yue, T. Liang, X. Hu and W.-H. Sun, Dalton Trans., 2016, 45, 657 RSC; (e) F. Huang, W. Zhang, Y. Sun, X. Hu, G. A. Solan and W.-H. Sun, New J. Chem., 2016, 40, 8012 RSC; (f) V. K. Appukuttan, Y. Liu, B. C. Son, C.-S. Ha, H. Suh and I. Kim, Organometallics, 2011, 30, 2285 CrossRef CAS; (g) W.-H. Sun, S. Kong, W. Chai, T. Shiono, C. Redshaw, X. Hu, C. Guo and X. Hao, Appl. Catal., A, 2012, 447–448, 67 CrossRef CAS; (h) W. Zhang, W. Chai, W.-H. Sun, X. Hu, C. Redshaw and X. Hao, Organometallics, 2012, 31, 5039 CrossRef CAS.
- (a) Z. Wang, Y. Ma, J. J. Guo, Q. Liu, G. A. Solan, T. Liang and W.-H. Sun, Dalton Trans., 2019, 48, 2582 RSC; (b) A. M. Archer, M. W. Bouwkamp, M. P. Cortez, E. Lobkovsky and P. J. Chirik, Organometallics, 2006, 25, 4269 CrossRef CAS.
- (a) Q. Zhang, Y. Ma, H. Suo, G. A. Solan, T. Liang and W.-H. Sun, Appl. Organomet. Chem., 2019, 33, e5134 Search PubMed; (b) H. Suo, I. I. Oleynik, C. Bariashir, I. V. Oleynik, Z. Wang, G. A. Solan, Y. Ma, T. Liang and W.-H. Sun, Polymer, 2018, 149, 45 CrossRef CAS.
- Q. Zhang, N. Wu, J. Xiang, G. A. Solan, H. Suo, Y. Ma, T. Liang and W.-H. Sun, Dalton Trans., 2020, 49, 9425 RSC.
- J. Guo, W. Zhang, I. I. Oleynik, G. A. Solan, I. V. Oleynik, T. Liang and W.-H. Sun, Dalton Trans., 2020, 49, 136 RSC.
- F. Huang, Q. Xing, T. Liang, Z. Flisak, B. Ye, X. Hu, W. Yang and W.-H. Sun, Dalton Trans., 2014, 43, 16818 RSC.
- (a) Z. Chen, M. Mesgar, P. White, O. Dauguils and M. Brookhart, ACS Catal., 2015, 5, 631 CrossRef CAS; (b) M. Han, Q. Zhang, I. I. Oleynik, H. Suo, I. V. Oleynik, G. A. Solan, Y. Ma, T. Liang and W.-H. Sun, Catalysts, 2020, 10, 1002 CrossRef CAS.
- (a) Q. Zhang, Z. Li, M. Han, J. Xiang, G. A. Solan, Y. Ma, T. Liang and W.-H. Sun, Catal. Sci. Technol., 2021, 11, 656 RSC; (b) Q. Zhang, W. Yang, Z. Wang, G. A. Solan, T. Liang and W.-H. Sun, Catal. Sci. Technol., 2021, 11, 4605 RSC.
- (a) S.-H. Yuan, Z. Fan, M. Han, Y. Yan, Z. Flisak, Y. Ma, T. Liang and W.-H. Sun, Eur. J. Inorg. Chem., 2021, 1571 CrossRef CAS; (b) H. Suo, I. I. Oleynik, I. V. Oleynik, G. A. Solan, Y. Ma, T. Liang and W.-H. Sun, Polymer, 2021, 213, 123294 CrossRef CAS.
- T. Liu, Y. Ma, G. A. Solan, T. Liang and W.-H. Sun, Appl. Organomet. Chem., 2021, 35, e6259 CAS.
- (a) L. Guo, M. Zada, W. Zhang, A. Vignesh, D. Zhu, Y. Ma, T. Liang and W.-H. Sun, Dalton Trans., 2019, 48, 5604 RSC; (b) R. Zhang, M. Han, I. V. Oleynik, G. A. Solan, I. I. Oleynik, Y. Ma, T. Liang and W.-H. Sun, Appl. Organomet. Chem., 2021, e6376 CAS; (c) L. Guo, W. Zhang, F. Cao, Y. Jiang, R. Zhang, Y. Ma, G. A. Solan, Y. Sun and W.-H. Sun, Polym. Chem., 2021, 12, 4214 RSC; (d) R. Zhang, Y. Huang, Y. Ma, G. A. Solan, X. Hu, T. Liang and W.-H. Sun, Polymer, 2021, 222, 123684 CrossRef CAS.
- (a) A. Martínez-Romo, R. González-Mota, J. J. Soto-Bernal and I. Rosales-Candelas, J. Spectrosc., 2015, 2015, 1 CrossRef; (b) B. M. Fanconi, K. L. DeVries and R. H. Smith, Polymer, 1982, 23, 1027 CrossRef CAS; (c) W. Glenz and A. Peterl, Die Makromolekulare Chemie, 1971, 150, 163 CrossRef CAS; (d) M. R. Jung, F. David Horgen, S. V. Orski, V. Rodriguez, K. L. Beers, G. H. Balazs, T. Todd Jones, T. M. Work, K. C. Brignace, S.-J. Royer, K. David Hyrenbach, B. A. Jensen and J. M. Lynch, Mar. Pollut. Bull., 2018, 127, 704 CrossRef CAS PubMed; (e) T. Masa-hide, T. Kohji and A. Shigetoshi, Macromolecules, 1999, 32, 5860 CrossRef; (f) M. Han, I. I. Oleynik, Y. Ma, I. V. Oleynik, G. A. Solan, T. Liang and W.-H. Sun, Appl. Organomet. Chem., 2021, e6429 CAS.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic data. CCDC 2105336 (Co1) and 2105337 (Co4). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra07279j |
| This journal is © The Royal Society of Chemistry 2021 |