
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper-mediated construction of benzothieno[3,2-b]benzofurans by intramolecular dehydrogenative C–O coupling reaction†
Liankun Ai,
Ibrahim Yusuf Ajibola and
Baolin Li*
and
Baolin Li*
School of Chemical Sciences, University of Chinese Academy of Sciences, Beijing 100049, P. R. China. E-mail: libl@ucas.ac.cn
First published on 10th November 2021
Abstract
An efficient method to synthesize benzothieno[3,2-b]benzofurans via intramolecular dehydrogenative C–H/O–H coupling has been developed. Good to excellent yields (64–91%) could be obtained no matter if the substituted group is electron-donating or electron-withdrawing. Notably, three-to-six fused ring thienofuran compounds could be constructed using this method. A reaction mechanism study showed that 1,1-diphenylethylene can completely inhibit the reaction. Therefore, it is a radical pathway initiated by single electron transfer between the hydroxyl of the substrate and the copper catalyst.
Introduction
The research on polycyclic aromatic hydrocarbons has achieved substantial advances in the past decades, which have led to the development of various materials for organic light-emitting diodes (OLEDs), organic field-effect transistors (OFETs) and photovoltaics (OPVs).1–10 Ladder-type thienoacenes such as benzothieno[3,2-b]benzothiophene (BTBT) have played a key role in the development of high-performance optoelectronic devices such as OFETs and OPVs.11 2,7-Dioctyl[1]benzothieno[3,2-b][1]benzothiophene (C8-BTBT) showed an ultra-high maximum hole mobility of 43 cm2 V−1 s−1 in thin film transistors,12 which is among the highest values reported to date for all organic molecules. Inspired by the great success of BTBT derivatives in OFETs, we have recently designed and synthesized benzo[4,5]thieno[3,2-b]benzofuran (BTBF) derivatives13,14 and benzofuro[3,2-b]benzofuran (BFBF) derivatives15 with either one or two furan rings substituting for thiophene rings, and investigated their packing mode, photophysical and charge transport properties.As we all know, an oxygen atom and a sulfur atom have a similar valence electron configuration; the difference is that the oxygen atom has a smaller atomic radius and a larger electronegativity. The smaller atomic radius of the oxygen atom may decrease the intermolecular distance and increase the intermolecular π-orbital overlap, which facilitates the charge transport property in the solid state. Thus, furan-fused π-conjugated molecules are being viewed as very promising materials in organic electronics.9,15–21 BTBF motif is believed to be a promising candidate for material applications as it possesses a similar structure and energy profile with those of BTBT according to the density functional theory (DFT) calculations.13 However, there are only a few examples for BTBFs used in optoelectronic devices.13,22–24 One major problem restraining the applications of BTBFs can be attributed to the lack of facile synthetic routes toward these oxygen-containing heteroacenes as oxygen atom has only one valence state of −2 to construct the furan ring. Thus, developing a new and efficient synthetic method toward BTBFs is paramount for their material applications.
The synthesis of BTBF was firstly realized by Aitken group using flash vacuum pyrolysis of 2-methylthiophenyl substituted phosphorus ylides in 1995 (Scheme 1a).25,26 By the reaction of benzothieno[3,2-b]furan with substituted dienes, Svoboda group synthesized tetrahydro[1]benzothieno[3,2-b][1]benzofuran derivatives which were transformed into BTBFs by dehydrogenative aromatization (Scheme 1b).27 A Pd-catalyzed intramolecular oxidative C–H/C–H coupling of 3-aryloxybenzo[b]thiophenes was independently reported by Kuninobu28 and Miura group29 in 2015 (Scheme 1c). Then a Cu-catalyzed Ullmann-type intramolecular C–O bond coupling reaction toward BTBFs was developed by our group and coworkers (Scheme 1d).13 Later on, You group used a similar strategy to synthesize these compounds.30 Recently, Mitsudo group have reported an electrochemical synthesis of this type of compounds from 2-(benzo[b]furan-2-yl)benzenethiol via a dehydrogenative C–H/S–H coupling (Scheme 1e).31 Most recently, John group have developed a mild metal-free synthetic route toward BTBFs by the annulation of 3-nitrobenzothiophene with phenols (Scheme 1f).32 However, all these methods required metal catalysts or harsh conditions, and multistep transformations for the synthesis of BTBFs with limited scopes.
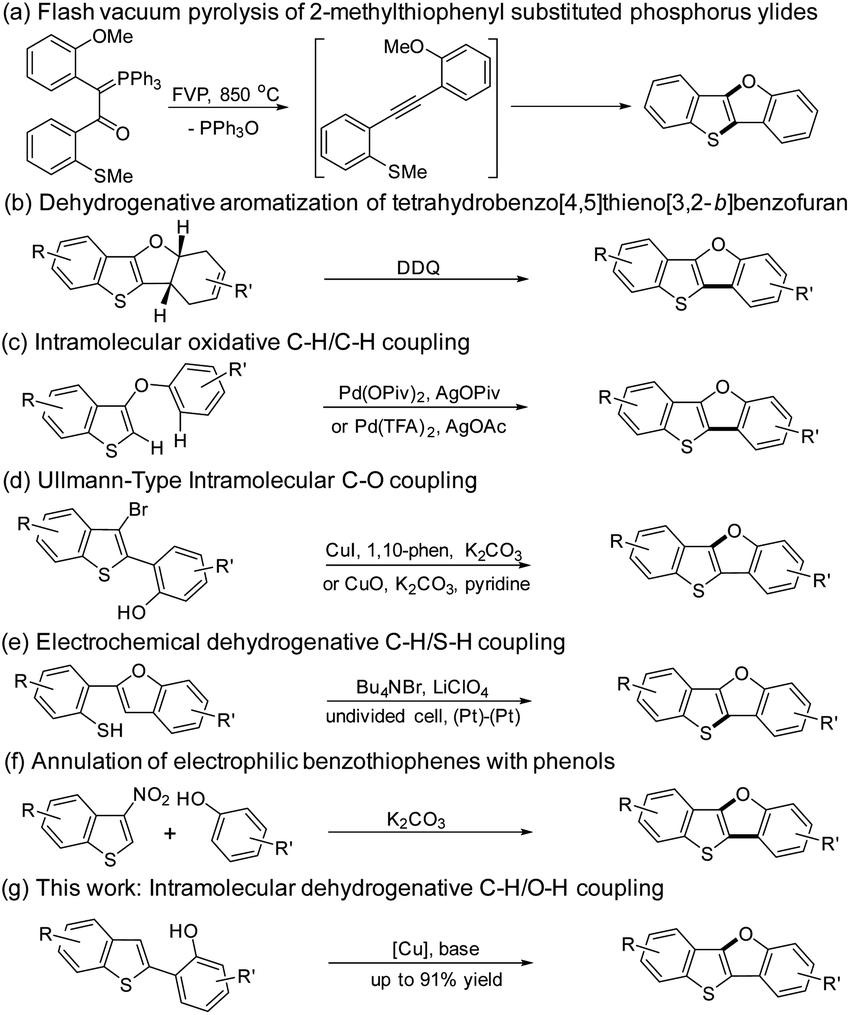 | ||
| Scheme 1 Synthetic strategies toward BTBFs. | ||
The construction of furan-fused π-conjugated molecules through dehydrogenative C–H/O–H coupling has been studied in the past decade. Liu33 and Yoshikai group34 independently reported the Pd-catalyzed synthesis of dibenzofuran derivatives through dehydrogenative C–H/O–H coupling. Zhu group discovered that copper catalysts could also promote dehydrogenative etherification to form dibenzofurans.35,36 Hong et al. reported the synthesis of heterocyclic-fused benzofurans via dehydrogenative C–H/O–H coupling of flavones and coumarins.37 Shimada and coworkers reported that dual C–H/O–H coupling of binaphthols occurred to furnish peri-xanthenoxanthenes.38 To be noted, the products by the methods mentioned above have been limited to aryl ethers and lactones.
Inspired by the results mentioned above, we hypothesized that BTBFs could be constructed via intramolecular dehydrogenative C–H/O–H coupling reaction. Herein, due to our continuing interest in Cu-catalyzed synthesis of furan-fused π-conjugated molecules,13–15 we have been motivated to investigate the construction of BTBFs via dehydrogenative C–H/O–H coupling reaction as shown in Scheme 1g. The advantages of this novel approach compared with our previous strategy (Scheme 1d) are as followed: (1) the bromination of β-position of thiophene is unnecessary which simplifies the reaction; (2) the debromination side reaction which was found in our previous strategy could be avoided which helps to improve the reaction yield.
Results and discussion
Our investigations commenced with the synthesis of 2-(benzo-[b]thiophen-2-yl)phenol derivatives in Suzuki coupling reaction, and the yields were very high (75–98%, see ESI†). With the above compounds in hand, we started to investigate the construction of BTBFs via intramolecular dehydrogenative C–H/O–H coupling reaction. Initially, intramolecular dehydrogenative C–H/O–H coupling reaction of 2-(benzo[b]thiophen-2-yl)phenol (1a) was selected as the model reaction. We first tried Pd(OAc)2 as the catalyst, 1,3-bis(2,6-diisopropylphenyl)-2,3-dihydro-1H-imidazole (IPr) as the ligand, Cs2CO3 as the base, toluene as the solvent, and the reaction was performed under air (i.e., air as the oxidant), but no target product BTBF (2a) was observed (see Table 1, entry 1). When the oxidant was replaced by 3 equivalent of Cu(OAc)2, the target product was produced in 8% nuclear magnetic resonance (NMR) yield (see Table 1, entry 2). Then the ligand was replaced by pyridine (Table 1, entry 3), the yield of BTBF (2a) increased to 11%; if pyridine was used as the ligand and solvent, the yield was further increased to 21% (Table 1, entry 4). However, if a catalytic amount of Cu(OAc)2 (0.2 equivalent) was used as the catalyst instead of Pd(OAc)2, and the reaction was performed under air, only a trace amount of target compound was observed either with or without ligands (Table 1, entries 5–7). To our delight, when the amount of Cu(OAc)2 was gradually increased to 0.5, 1 and 3 equivalent, the yield increased to 32%, 47%, 51% respectively (Table 1, entries 8–10). When the reaction was performed under pure oxygen, the yield dropped to 35% (Table 1, entry 11), which is probably due to oxidation of starting material by oxygen. Intriguingly, when 3 equivalent of Cu(OAc)2 was used, and the reaction was performed under nitrogen, the NMR yield increased to 90% (86% isolated yield) (Table 1, entry 12). Actually, Cu(OAc)2 was used as a catalyst and oxidant under this condition. Afterward, we screened a variety of copper oxidants (entries 13–20), bases (entries 21–23) and solvents (entries 24–28), the yields were much lower than the above yield (Table 1, entry 12).
|
||||||
|---|---|---|---|---|---|---|
| Entry | [Pd]/[Cu] | Ligand | Base | Oxidant | Solvent | Yieldb (%) |
| a Reaction conditions: 1a (0.20 mmol), Pd(OAc)2 or Cu catalyst (0.2–3 equiv., 0.04–0.6 mmol), ligand (0.4 equiv., 0.08 mmol), base (1 equiv., 0.20 mmol), oxidant (for entries 2–4, 3 equiv., 0.60 mmol), solvent (4 mL), 110 °C, the reactions were performed under nitrogen if the oxidant is neither air nor oxygen.b Determined by 1 H NMR with 1,3,5-trimethoxybenzene as an internal standard.c Isolated yields. | ||||||
| 1 | Pd(OAc)2, 0.2 equiv. | IPr | Cs2CO3 | Air | Toluene | 0 |
| 2 | Pd(OAc)2, 0.2 equiv. | IPr | Cs2CO3 | Cu(OAc)2 | Toluene | 8 |
| 3 | Pd(OAc)2, 0.2 equiv. | Pyridine | Cs2CO3 | Cu(OAc)2 | Toluene | 11 |
| 4 | Pd(OAc)2, 0.2 equiv. | Pyridine | Cs2CO3 | Cu(OAc)2 | Pyridine | 21 |
| 5 | Cu(OAc)2, 0.2 equiv. | o-Phen | Cs2CO3 | Air | Pyridine | Trace |
| 6 | Cu(OAc)2, 0.2 equiv. | 2,2′-Bipyridine | Cs2CO3 | Air | Pyridine | Trace |
| 7 | Cu(OAc)2, 0.2 equiv. | — | Cs2CO3 | Air | Pyridine | Trace |
| 8 | Cu(OAc)2, 0.5 equiv. | — | Cs2CO3 | Air | Pyridine | 32 |
| 9 | Cu(OAc)2, 1 equiv. | — | Cs2CO3 | Air | Pyridine | 47 |
| 10 | Cu(OAc)2, 3 equiv. | — | Cs2CO3 | Air | Pyridine | 51 |
| 11 | Cu(OAc)2, 3 equiv. | — | Cs2CO3 | O2 | Pyridine | 35 |
| 12 | Cu(OAc)2, 3 equiv. | — | Cs2CO3 | — | Pyridine | 90 (86)c |
| 13 | Cu(OAc)2·H2O, 3 equiv. | — | Cs2CO3 | — | Pyridine | 64 |
| 14 | CuOAc, 3 equiv. | — | Cs2CO3 | — | Pyridine | 51 |
| 15 | Cu2S, 3 equiv. | — | Cs2CO3 | — | Pyridine | 7 |
| 16 | CuS, 3 equiv. | — | Cs2CO3 | — | Pyridine | Trace |
| 17 | CuCl, 3 equiv. | — | Cs2CO3 | — | Pyridine | 0 |
| 18 | CuCl2, 3 equiv. | — | Cs2CO3 | — | Pyridine | 0 |
| 19 | CuBr2, 3 equiv. | — | Cs2CO3 | — | Pyridine | 0 |
| 20 | CuI, 3 equiv. | — | Cs2CO3 | — | Pyridine | 0 |
| 21 | Cu(OAc)2, 3 equiv. | — | K2CO3 | — | Pyridine | 34 |
| 22 | Cu(OAc)2, 3 equiv. | — | NaOAc | — | Pyridine | 59 |
| 23 | Cu(OAc)2, 3 equiv. | — | — | — | Pyridine | 20 |
| 24 | Cu(OAc)2, 3 equiv. | Pyridine | Cs2CO3 | — | Toluene | 30 |
| 25 | Cu(OAc)2, 3 equiv. | — | Cs2CO3 | — | DMSO | 44 |
| 26 | Cu(OAc)2, 3 equiv. | — | Cs2CO3 | — | DMF | Trace |
| 27 | Cu(OAc)2, 3 equiv. | — | Cs2CO3 | — | Isopropanol | 0 |
| 28 | Cu(OAc)2, 3 equiv. | — | Cs2CO3 | — | t-Butanol | 0 |

To clarify the reaction scope, we next devoted our efforts to test a variety of 2-(benzo-[b]thiophen-2-yl)phenol derivatives 1 under the optimized reaction conditions (Scheme 2). Firstly, we selected o-, m- or p-substituted 2-(benzo[b]-thiophen-2-yl)phenols as the substrates, and found that all the yields (2b–k, 64–91%) were good to excellent no matter the substituted group is electron-donating or electron-withdrawing. The substrates with weak electron-donating groups such as alkyl chains afforded very high yields (2b–d, 85–91%), however, the substrate with a strong electron-donating group such as methoxy group gave comparably low yield (2e, 64%). For a specific substituent, the substituting position affects the yield: p-fluoro substituted BTBF (2f, 85%) > m-substituted BTBF (2g, 78%) > o-substituted BTBF (2h, 68%), probably due to the different steric hindrance of fluoro substituent in different position.
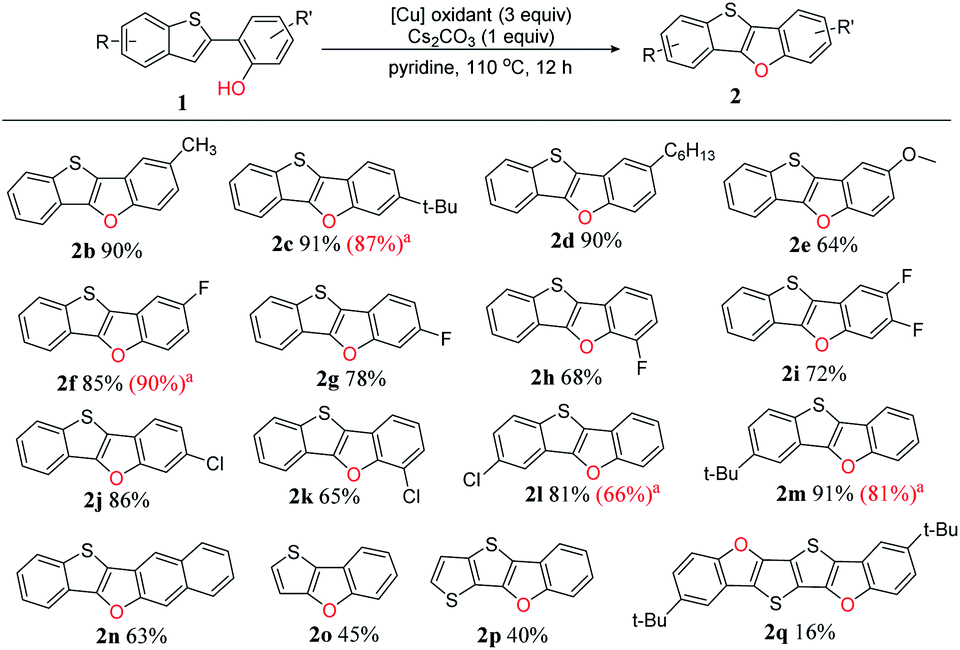 | ||
| Scheme 2 Reaction scope for the intramolecular dehydrogenative C–O coupling reaction under optimized conditions. a The yields in the parenthesis reported in our previous paper are shown for comparison with this work.13 | ||
Next, we tested phenols with substituted benzo[b]thiophene moieties, and also got excellent yields (2l–2m, 81–91%). For more π-extended 3-(benzo[b]thiophen-2-yl)naphthalen-2-ol as the substrate, the yield only slightly dropped, affording benzo[4,5]thieno[3,2-b]naphtho[2,3-d]furan (2n, BTNF, 63%). Although substrates having a thiophene unit (1o) or bithiophene unit (1p) could also be used in this reaction, the yields significantly dropped (2o, 45%; 2p, 40%), probably because the cation intermediate could not be effectively stabilized without the resonance with phenyl group (see proposed mechanism, vide infra). Thus, not surprisingly, the yield of 2q is even lower (16%) because it is obtained via two subsequent intramolecular dehydrogenative C–H/O–H couplings. Thus, three-to-six fused ring thienofuran compounds could be constructed. Notably, the yields of BTBFs are comparable or even higher than those reported in our previous paper (2c 91% vs. 87%, 2f 85% vs. 90%, 2l 81% vs. 66%, and 2m 91% vs. 81%),13 indicating advantage of the new synthetic approach.
Subsequently, we investigated the gram-scale synthesis of copper-mediated construction of benzothieno[3,2-b]benzofurans by intramolecular dehydrogenative C–O coupling reaction. 2-(Benzo-[b]thiophen-2-yl)phenol derivatives 1a and 1f (6 mmol) reacted under the optimised reaction conditions, affording their corresponding benzothieno[3,2-b]benzofurans 2a and 2f in high yields which are comparable with those obtained on a 0.2 mmol scale (Fig. 1).
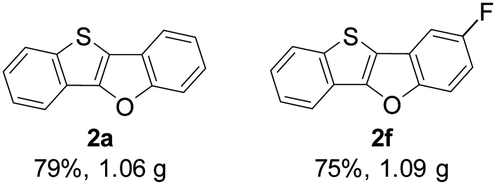 | ||
| Fig. 1 Gram-scale synthesis. | ||
To gain insight into the reaction mechanism, some deuterium-labeling experiments were conducted with deuterated 2-(benzo[b]thiophen-2-yl-3-d)phenol (1a-D) (Scheme 3). The rate measurements revealed kinetic isotope effects (KIEs) of 1.1, indicating that the C3–H bond cleavage is not necessarily involved in the turnover limiting step of the reactions. The deuterated percentage in residual compounds did not change obviously, which indicated that cleavage of C3–D or C3–H bond is irreversible and thus phenolic hydroxyl played an important role in C–H functionalization.37
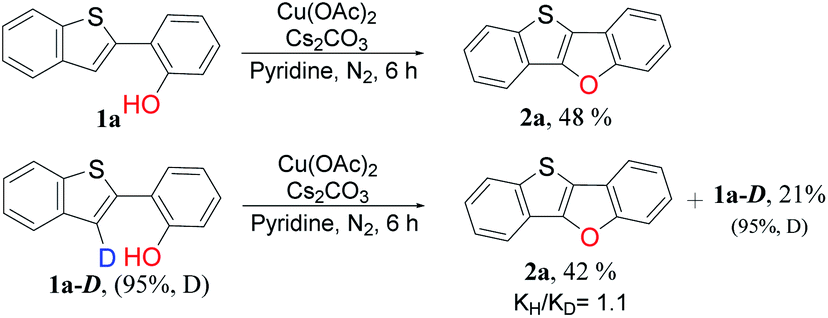 | ||
| Scheme 3 Kinetic isotope effect study. Reaction conditions: 1a and 1a-D (0.20 mmol), Cu catalyst (3 equiv., 0.6 mmol), base (1 equiv., 0.20 mmol), solvent (4 mL), 110 °C, 6 h under nitrogen. | ||
A radical inhibition test was carried out in order to gain insight into whether the reaction proceeded through radical intermediates (Scheme 4). We selected 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO), butylated hydroxytoluene (BHT) and 1,1-diphenylethylene as the radical scavenger which are widely used in free radical capture experiments.39–43 It is found that the NMR yield dropped to 10% in the presence of TEMPO, and 55% of the starting material was still left. In the presence of BHT, the yield of the target product dropped to 20%. While the desired cyclization reaction was completely inhibited by 1,1-diphenylethylene. These results clearly indicate that the reaction pathway is a radical mechanism. Unfortunately, all the efforts to detect the adducts between the substrate and radical intermediates failed.
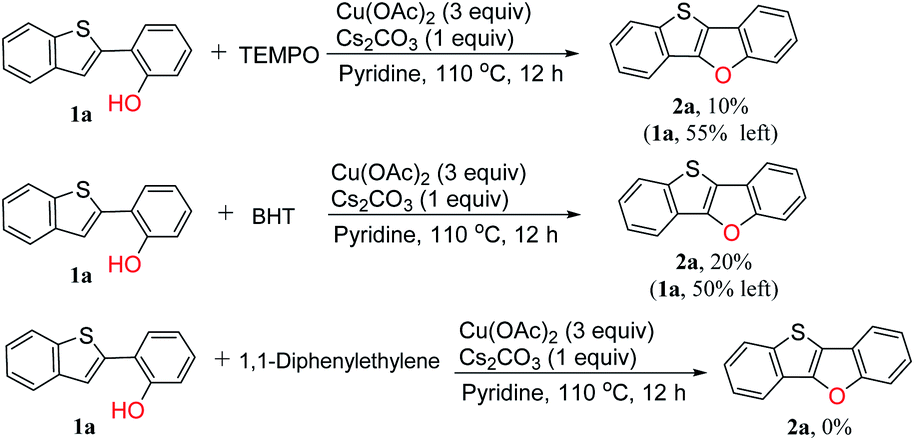 | ||
| Scheme 4 Free radical capture experiment. | ||
Although the detailed mechanism remains to be elucidated, a possible reaction pathway is proposed based on the above observations (Scheme 5). At first, 1a reacts with a base, generating anionic intermediate A. Then phenoxy radical B is produced via single electron transfer with Cu(OAc)2, followed by intramolecular C–O cyclization to generate radical intermediate C. Afterward, cationic intermediate D is formed by oxidation with Cu(OAc)2. Finally, the target product 2a is obtained by the abstraction of a proton with the base.
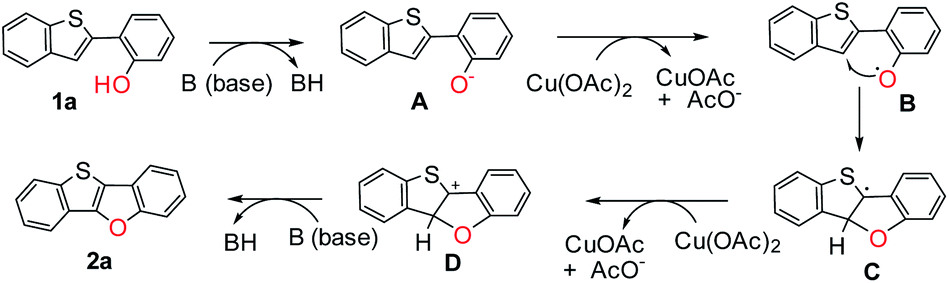 | ||
| Scheme 5 Proposed mechanism. | ||
Conclusions
In summary, we have developed an efficient method to synthesize benzothieno[3,2-b]benzofurans via intramolecular dehydrogenative C–H/O–H coupling in up to 91% yield. Notably, three-to-six fused ring thienofuran compounds could be constructed. The advantages of this novel approach include unnecessary bromination of β-position of thiophene and thus inhibition of the debromination side reaction and high yield. Reaction mechanism study showed that it is a radical pathway initiated by single electron transfer between substrate and copper catalyst.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We appreciate the financial support from the National Natural Science Foundation of China (21402194), Fundamental Research Funds for the Central Universities, and University of Chinese Academy of Sciences.Notes and references
- J. E. Anthony, Chem. Rev., 2006, 106, 5028–5048 CrossRef CAS PubMed.
- J. Wu, W. Pisula and K. Mullen, Chem. Rev., 2007, 107, 718–747 CrossRef CAS PubMed.
- A. Pron, P. Gawrys, M. Zagorska, D. Djurado and R. Demadrille, Chem. Soc. Rev., 2010, 39, 2577–2632 RSC.
- W. Wu, Y. Liu and D. Zhu, Chem. Soc. Rev., 2010, 39, 1489–1502 RSC.
- A. Chaskar, H. F. Chen and K. T. Wong, Adv. Mater., 2011, 23, 3876–3895 CrossRef CAS PubMed.
- J. Jung, N. J. Tremblay, M. L. Yeh and H. E. Katz, Chem. Mater., 2011, 23, 568–582 CrossRef.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed.
- J. Lee, A. J. Kalin, T. Yuan, M. Al. Hashimi and L. Fang, Chem. Sci., 2017, 8, 2503–2521 RSC.
- H. Tsuji and E. Nakamura, Acc. Chem. Res., 2017, 50, 396–406 CrossRef CAS PubMed.
- Y. Yan, X. Liu and T. Wang, Adv. Mater., 2017, 29, 1601074 Search PubMed.
- For recent representative reports, see: (a) A. Y. Amin, A. Khassanov, K. Reuter, T. M. Friedrichsen and M. Halik, J. Am. Chem. Soc., 2012, 134, 16548–16550 CrossRef CAS PubMed; (b) K. Takimiya, I. Osaka, T. Mori and M. Nakano, Acc. Chem. Res., 2014, 47, 1493–1502 CrossRef CAS PubMed; (c) G. Schweicher, V. Lemaur, C. Niebel, C. Ruzié, Y. Diao, O. Goto, W. Y. Lee, Y. Kim, J. B. Arlin, J. Karpinska, A. R. Kennedy, S. R. Parkin, Y. Olivier, S. C. B. Mannsfeld, J. Cornil, Y. H. Geerts and Z. Bao, Adv. Mater., 2015, 27, 3066–3072 CrossRef CAS PubMed; (d) A. L. Capodilupo, E. Fabiano, L. De Marco, G. Ciccarella, G. Gigli, C. Martinelli and A. Cardone, J. Org. Chem., 2016, 81, 3235–3245 CrossRef CAS PubMed.
- Y. Yuan, G. Giri, A. L. Ayzner, A. P. Zoombelt, S. C. Mannsfeld, J. Chen, D. Nordlund, M. F. Toney, J. Huang and Z. Bao, Nat. Commun., 2014, 5, 3005 CrossRef PubMed.
- D. Chen, D. Yuan, C. Zhang, H. Wu, J. Zhang, B. Li and X. Zhu, J. Org. Chem., 2017, 82, 10920–10927 CrossRef CAS PubMed.
- W. Ma, J. Huang, C. Li, Y. Jiang, B. Li, T. Qi and X. Zhu, RSC Adv., 2019, 9, 7123–7127 RSC.
- D. Chen, J. Li, W. Ma, B. Li, Y. Zhen, X. Zhu, W. Hu, H. Tsuji and E. Nakamura, Asian J. Org. Chem., 2018, 7, 2228–2232 CrossRef CAS.
- K. Niimi, H. Mori, E. Miyazaki, I. Osaka, H. Kakizoe, K. Takimiya and C. Adachi, Chem. Commun., 2012, 48, 5892–5894 RSC.
- B. Li, Chin. J. Org. Chem., 2015, 35, 2487 CrossRef CAS.
- C. Mitsui, J. Soeda, K. Miwa, K. Shoyama, Y. Ota, H. Tsuji, J. Takeya and E. Nakamura, Bull. Chem. Soc. Jpn., 2015, 88, 776–783 CrossRef CAS.
- C. Mitsui, M. Yamagishi, R. Shikata, H. Ishii, T. Matsushita, K. Nakahara, M. Yano, H. Sato, A. Yamano, J. Takeya and T. Okamoto, Bull. Chem. Soc. Jpn., 2017, 90, 931–938 CrossRef CAS.
- Y. Zhao, L. Yan, I. Murtaza, X. Liang, H. Meng and W. Huang, Org. Electron., 2017, 43, 105–111 CrossRef CAS.
- X. Zeng, D. Zhang, Y. Zhu, M. Chen, H. Chen, S. Kasai, H. Meng and O. Goto, J. Mater. Chem. C, 2019, 7, 14275–14283 RSC.
- Y. S. Yang, T. Yasuda and C. Adachi, Bull. Chem. Soc. Jpn., 2012, 85, 1186–1191 CrossRef CAS.
- W. Ma, G. Liu, L. Zhou, B. Li and Y. Wang, J. Mater. Chem. C, 2020, 8, 8796–8803 RSC.
- M. Shi, Y. He, Y. Sun, D. Fang, J. Miao, M. U. Ali, T. Wang, Y. Wang, T. Zhang and H. Meng, Org. Electron., 2020, 84, 105793 CrossRef CAS.
- (a) R. A. Aitken, C. K. Bradbury, G. Burns and J. J. Morrison, Synlett, 1995, 53–54 CrossRef CAS; (b) R. A. Aitken, G. Burns and J. J. Morrison, J. Chem. Soc., Perkin Trans. 1, 1998, 3937–3941 RSC.
- R. A. Aitken and A. N. Garnett, Synthesis, 2017, 49, 4955–4977 CrossRef CAS.
- P. Pihera, J. Paleček and J. Svoboda, Collect. Czech. Chem. Commun., 1998, 63, 681 CrossRef CAS.
- K. Saito, P. K. Chikkade, M. Kanai and Y. Kuninobu, Chemistry, 2015, 21, 8365–8368 CrossRef CAS PubMed.
- H. Kaida, T. Satoh, K. Hirano and M. Miura, Chem. Lett., 2015, 44, 1125–1127 CrossRef CAS.
- Y. M. Wu, W. Li, L. F. Jiang, L. Q. Zhang, J. B. Lan and J. S. You, Chem. Sci., 2018, 9, 6878–6882 RSC.
- K. Mitsudo, R. Matsuo, T. Yonezawa, H. Inoue, H. Mandai and S. Suga, Angew. Chem., Int. Ed., 2020, 59, 7803–7807 CrossRef CAS PubMed.
- R. A. Krishnan, S. A. Babu, P. R. Nitha, J. Krishnan and J. John, Org. Lett., 2021, 23, 1814–1819 CrossRef PubMed.
- B. Xiao, T. J. Gong and L. Liu, J. Am. Chem. Soc., 2011, 133, 9250–9253 CrossRef CAS PubMed.
- Y. Wei and N. Yoshikai, Org. Lett., 2011, 13, 5504–55047 CrossRef CAS PubMed.
- Y. W. Jiaji Zhao, Y. He, L. Liu and Q. Zhu, Org. Lett., 2012, 14, 1078–1081 CrossRef PubMed.
- J. Zhao, Q. Zhang and Q. Zhu, Org. Lett., 2012, 14, 5362–5365 CrossRef CAS PubMed.
- Y. Moon, Y. Kim, H. Hong and S. Hong, Chem. Commun., 2013, 49, 8323–8325 RSC.
- T. Kamei, M. Uryu and T. Shimada, Org. Lett., 2017, 19, 2714–2717 CrossRef CAS PubMed.
- H. J. Zhang, F. Su and T. B. Wen, J. Org. Chem., 2015, 80, 11322–11329 CrossRef CAS PubMed.
- R. H. Liu, D. Wei, B. Han and W. Yu, ACS Catal., 2016, 6, 6525–6530 CrossRef CAS.
- C. Theunissen, J. Wang and G. Evano, Chem. Sci., 2017, 8, 3465–3470 RSC.
- M. Wang, J. Wei, Q. Fan and X. Jiang, Chem. Commun., 2017, 53, 2918–2921 RSC.
- A. Cai, W. Yan and W. Liu, J. Am. Chem. Soc., 2021, 143, 9952–9960 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, UV-vis absorption spectra, emission spectra, crystal structure data, and other additional information. See DOI: 10.1039/d1ra06985c |
| This journal is © The Royal Society of Chemistry 2021 |