
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of quinolines via sequential addition and I2-mediated desulfurative cyclization†
Mingming Yanga,
Yajun Jian a,
Weiqiang Zhanga,
Huaming Suna,
Guofang Zhanga,
Yanyan Wang*a and
Ziwei Gao*ab
a,
Weiqiang Zhanga,
Huaming Suna,
Guofang Zhanga,
Yanyan Wang*a and
Ziwei Gao*ab
aKey Laboratory of Applied Surface and Colloid Chemistry, Xi'an Key Laboratory of Organometallic Material Chemistry, School of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi'an 710119, P. R. China. E-mail: zwgao@snnu.edu.cn; yyw@snnu.edu.cn
bA School of Chemistry & Chemical Engineering, Xinjiang Normal University, Urumqi 830054, P. R. China
First published on 6th December 2021
Abstract
An efficient one-pot approach for the synthesis of quinolines from o-aminothiophenol and 1,3-ynone under mild conditions is disclosed. With the aid of ESI-MS analysis and parallel experiments, a three-step mechanism is proposed—a two-step Michael addition–cyclization condensation step leading to intermediate 1,5-benzothiazepine catalyzed by zirconocene amino acid complex Cp2Zr(η1-C9H10NO2)2, followed by I2-mediated desulfurative step.
Introduction
Quinoline is a nitrogen containing heterocyclic aromatic compound1–5 which was first extracted from coal tar by Friedlieb Ferdinand Runge in 1834.6 Since that, extensive attention has been paid to the synthesis of quinolines and their derivatives for their broad range of pharmaceutical activity, such as anti-viral, anti-cancer and anti-HIV.7–9 Various synthetic transformations have been developed to obtain quinolines, including Combes synthesis,10–12 Skraup reaction,13 Conrad–Limpach synthesis,14–16 Povarov reaction,17,18 Doebner reaction,19,20 Doebner–Miller reaction,21–23 Gould–Jacobs reaction,24,25 Friedländer reaction,26–29 and so on. However, considering the extreme significance of quinoline derivatives, the development of mild and economic approaches is still attractive.Recently, the ynones and amine synthons' combination toward the synthesis of quinolines have been verified as effective strategies,30,31 started by using preconnected starting materials containing both amine and ynone parts. For example, β-(2-aminophenyl)-α,β-ynones32–36 can undergo intramolecular cyclization to generate quinoline at room temperature (Scheme 1, entry a).37 In contrast, the reaction between anilines and ynones via 3 + 3 cycloaddition is more direct and step-economical, thus became more popular. Although C–H activation of aniline has less demanding for starting material, the reaction usually needs to be driven by noble metal catalysts, such as AgOTf, under high temperature (entry b).38 Therefore, using α-position preactivated aniline is more practical as cheap metal catalyst could be adopted. For example, 2-iodoanilines and ynones can be catalyzed by a nickel-catalyzed cyclization to generate quinolines (entry c).39 o-Sulfhydryl-aniline also proven to effective as preactivated 3-atom synthons,40,41 due to both the soft basic nature of sulfur atom for the ease to carry out 1,4-Michael addition reactions, and the viability to go through desulfurization via 7-membered ring's42–44 contraction from 1,5-benzothiazoles (entry d). However, such one-pot procedures are still suffering from negligible drawbacks, like incompatible solvent system for the thiol Michael addition and desulfurization processes, and high temperatures for desulfurization step. Therefore, it is still challenging to synthesize quinoline in one solvent under mild temperature.
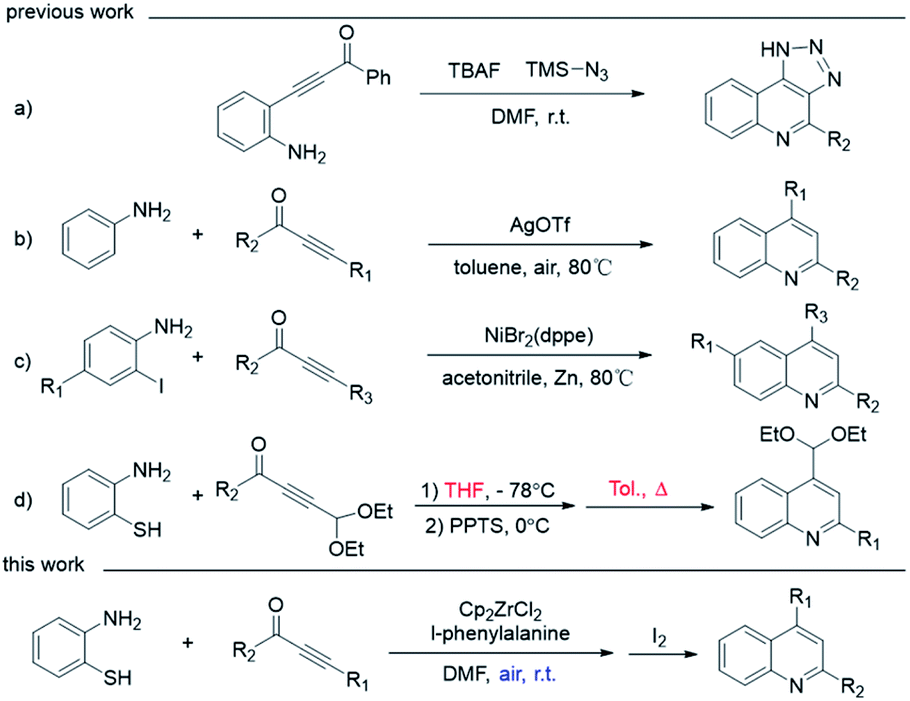 | ||
| Scheme 1 The synthesis of quinolines. | ||
In our previous work, by using titanocone complex as catalyst, 7-membered ring contained 1,5-benzodiazepines were successfully synthesized from o-phenylenediamine and 1,3-ynones through the [4 + 3] cyclization reaction under mild conditions, indicating such organometallic Lewis acid catalyst can facilitate the Michael addition process and cyclization reaction at room temperature.45 Additionally, the Lewis acidic catalytic activity46,47 and selectivity48 of such metallocene could be conveniently adjusted by changing carboxylic acid ligand bearing different electronic characteristic,49 based on our group's investigation accumulation of IVB metal based complex. In particular, the –NH2 group as a Brønsted base is potential to activate o-aminothiophenol through hydrogen bonding. In this work, we disclosed a one-pot approach for the synthesis of quinolines from o-aminothiophenol and 1,3-ynone, catalyzed and mediated by Cp2ZrCl2 and I2 at room temperature, via sequential thio-Michael addition, cyclization and desulfurization by ESI-MS and control experiments.
Results and discussion
It was proven in our previous study that solvents play important role for the synthesis of nitrogen containing cyclic products, so the solvent effect for the reaction were investigated initially. From the screening results shown in Table 1, we can see that aprotic polar solvent are helpful for the proceeding of the reaction, for example, 83% yield product could be produced when DMF was chosen (entry 6). What need to mention is that protic solvent, like EtOH, is extremely harmful for the reaction, as only 17% yield quinolines was afforded accompanied with a lot other byproducts, which might be attributed to the inhibition effect from the possible complexation between EtOH with zirconocene catalyst (entry 7). Next, we examined the ligand effect systematically by using various Brønsted acid ligand. In general, compared to aryl(heteroaryl) carboxylic acids (entries 13–15) and sulfonic acids (entry 16), amino acids ligands (entries 9–12) are more beneficial for the reaction. We tentatively speculate that the amino group on the ligand might be involved in the catalysis. At last, some other representative oxidant besides TBHP was evaluated, including H2O2, DMP, and I2. It turns out that I2 is the best option, as 92% yield product was obtained (entries 17–19). The yield of desired product declined to 60% without an oxidant (entry 20). Base on this optimized catalytic condition, a new one-pot method for the synthesis of quinolines have been developed.| Run | Solvent | Ligand | Oxidant | Yieldb (%) |
|---|---|---|---|---|
| a Reaction conditions: 0.5 mmol of 1,3-ynone, 0.6 mmol of o-aminothiophenol, 5 mol% of Cp2ZrCl2 and 10 mol% of ligand in 1 mL DMF at room temperature for 5 h, then 0.6 mmol oxidant was added for 1 h.b Yield of the GC. | ||||
| 1 | Toluene | L-Phenylalanine | TBHP | 56 |
| 2 | CH2Cl2 | L-Phenylalanine | TBHP | 56 |
| 3 | THF | L-Phenylalanine | TBHP | 43 |
| 4 | 1,4-Dioxane | L-Phenylalanine | TBHP | 65 |
| 5 | CH3CN | L-Phenylalanine | TBHP | 48 |
| 6 | DMF | L-Phenylalanine | TBHP | 83 |
| 7 | DMSO | L-Phenylalanine | TBHP | 67 |
| 7 | EtOH | L-Phenylalanine | TBHP | 17 |
| 9 | DMF | Alanine | TBHP | 78 |
| 10 | DMF | Tyrosine | TBHP | 80 |
| 11 | DMF | Serine | TBHP | 77 |
| 12 | DMF | L-Proline | TBHP | 77 |
| 13 | DMF | Salicylic acid | TBHP | 62 |
| 14 | DMF | Benzoic acid | TBHP | 60 |
| 15 | DMF | 2-Hydroxynicotinic acid | TBHP | 52 |
| 16 | DMF | Camphorsulfonic acid | TBHP | 24 |
| 17 | DMF | L-Phenylalanine | H2O2 | 80 |
| 18 | DMF | L-Phenylalanine | DMP | 83 |
| 19 | DMF | L-Phenylalanine | I2 | 92 |
| 20 | DMF | L-Phenylalanine | — | 60 |
To further expand the substrate scope of this one-pot method, the utilization of 1,3-ynone derivatives was then explored (Table 2). The scope of this one-pot method concerning the substituents of the substrates was proven to be very wide. 1,3-Ynone bearing electron-donating and electron-withdrawing groups on the phenyl ring at the carbonyl group side were applied to the reaction. The introduction of electron-donating groups such as –Me and –OMe in different positions of the ring, affording quinolines in the yield range of 62–93% (3b, 3c, 3d and 3e). Electron-withdrawing halogen groups like p-fluoro, p-chloro and p-bromo led to the desired products in good yields of 90%, 84% and 80% respectively (3f–3h). The reaction of o-chloro and m-chloro substituted 1,3-ynones proceeded to afford quinolines in good yields of 64% (3i) and 81% (3j), respectively. Fortunately, heterocyclic thiophene- methylthiophen- and naphthyl-substituted 1,3-ynone were also compatible with this reaction, giving products in yield of 73%, 63%, and 50%, respectively (3k–3m). Then we turned attention to test the toleration with aliphatic 1,3-ynones. Ethoxy-substituted 1,3-ynone afforded the product in 84% yield (3n). Methyl-substituted 1,3-ynone assured a yield in 52% (3x).
| a Reaction conditions: 0.5 mmol of 1,3-ynone, 0.6 mmol of o-aminothiophenol in the presence of 5 mol% Cp2ZrCl2, 10 mol% of L-phenylalanine and 1 mL DMF at room temperature for 5 h, then 0.6 mmol I2 was added for 1 h. Isolated yields were obtained after purification by column chromatography. |
|---|
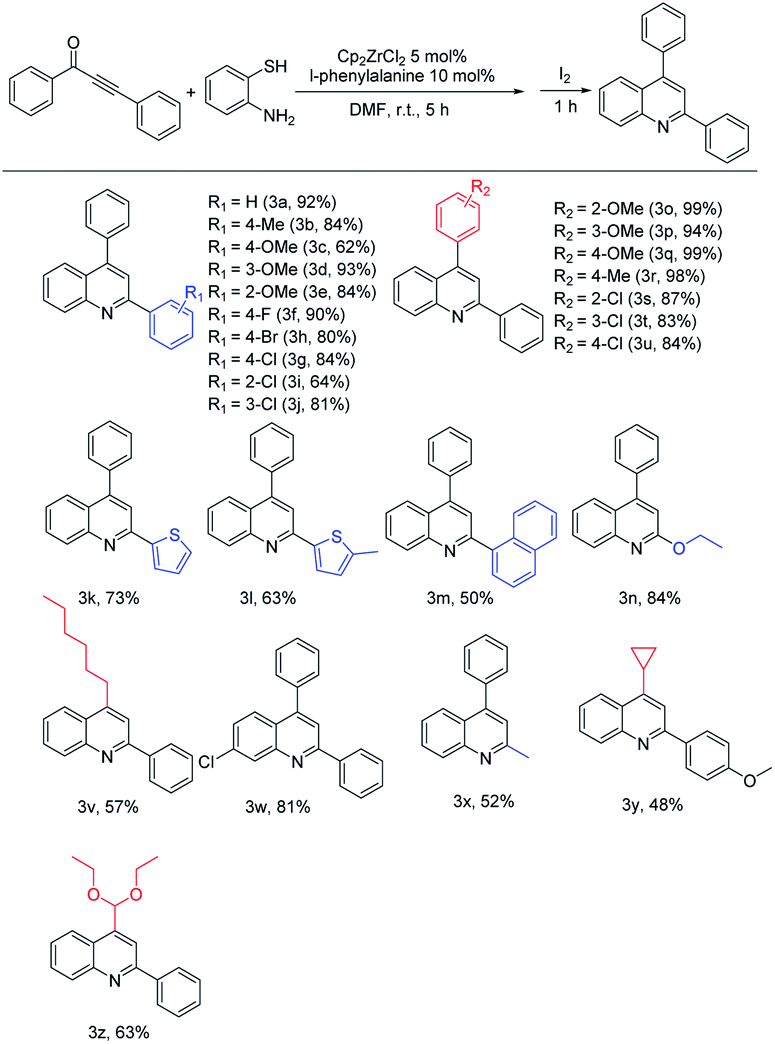 |
Secondly, we investigated the scope of this method concerning the impact of substitution of the alkyne side on 1,3-ynones. For p-methoxy, m-methoxy and o-methoxy substituents, the products were afforded in excellent yields of 99%, 94% and 99% (3o–3q), respectively. And p-methyl group also assured an excellent yield in 98% (3r). When electron-withdrawing substrates were subjected to this transformation, halogen groups like o-chloro, m-chloro and p-chloro also led to the product in good yields of 87%, 83% and 84% respectively (3s–3u). These results indicate that 1,3-ynones derivatives with whether electron-donating or electron-withdrawing groups can be used in this method to give excellent yields. Even for 1,3-ynone derivatives with aliphatic substituents, such as hexyl-, cyclopropyl- and diethoxymethyl-, were successfully applied to the reaction and gave the corresponding products in good yields (3v, 3y and 3z). Moreover, the p-chloro derivative of o-aminothiophenol provided the corresponding quinoline 3w in an excellent yield of 81%.
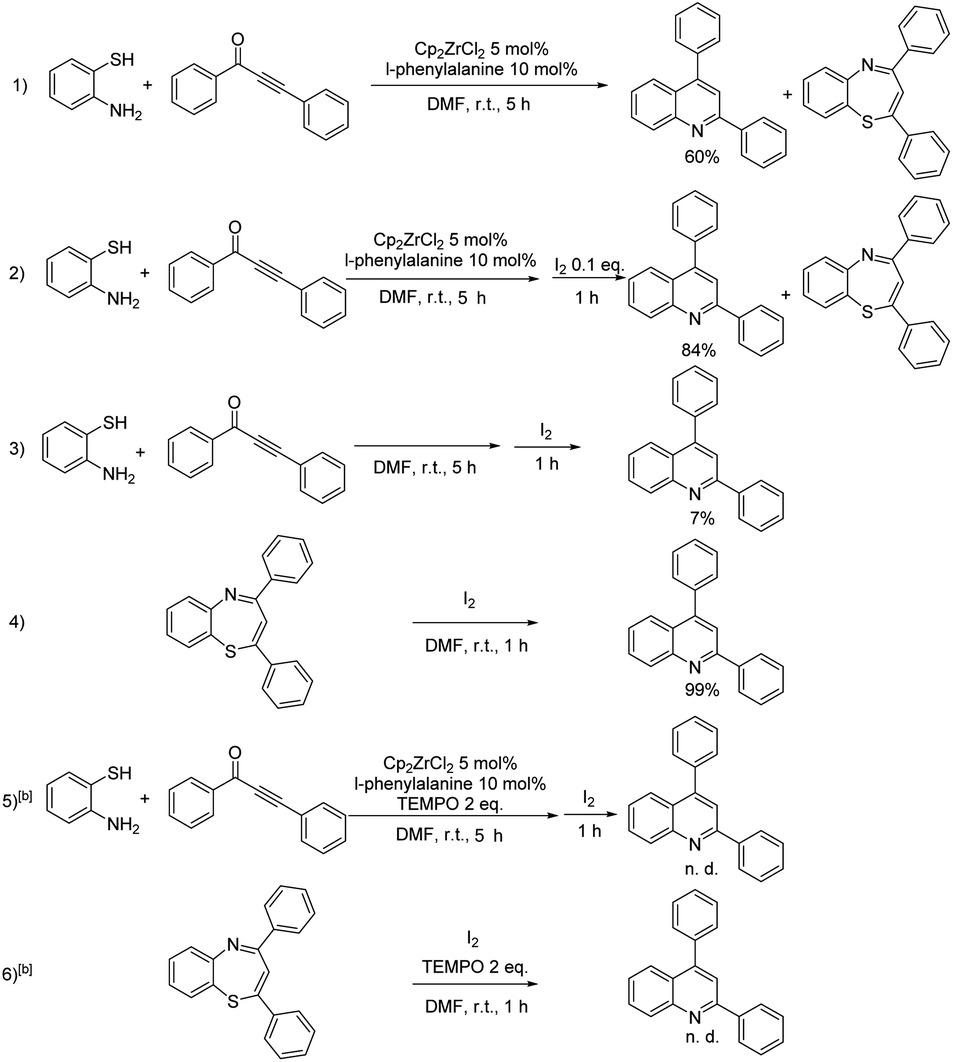
The next task is to map the route for the reaction. To elucidate the action mode of this one-pot tandem reaction, some reactions were performed (Table 3). Under the oxidant-free condition, the conversion of 1,3-ynone was almost 100%, but the yield of the target product was only 60%, and 1,5-benzothiazepines in 40% yield was obtained (Table 3, entry 1). This result indicates that both 1,5-benzothiazepines containing 7-membered ring and quinolines containing 6-membered ring could be formed from 1,3-ynones and o-aminothiophenol without I2. 0.1 equiv. of I2 prompted the reaction to give the quinoline in yield of 84% (entry 2), which indicated that a small amount of oxidant benefits the conversion from 1,5-benzothiazepine to quinoline. Meanwhile, the reaction couldn't be catalyzed by the oxidant alone, for that only 7% yield of quinoline was detected in Cp2ZrCl2-free and ligand-free condition (entry 3). However, 1,5-benzothiazepine could be almost completely converted to quinoline by I2, which further suggested the positive effect of I2 on the desulfurization of 1,5-benzothiazepine (entry 4). Regardless of the starting material, quinoline was not detectable after adding the radical scavenger TEMPO in the experiment (entries 5, 6). These results suggested that the iodine-mediated desulfurization is most likely a radical process. We also carried out ESI-MS experiment for the real reaction system, with important intermediates detected (proposed structures shown in Scheme 2), including zirconocene catalytic species Cp2Zr(η1-C9H10NO2)2 (II), and intermediate II′.
| a Normal reaction conditions: 0.5 mmol of 1,3-ynone, 0.6 mmol of o-aminothiophenol in the presence of 5 mol% of Cp2ZrCl2, 10 mol% of L-phenylalanine and 1 mL DMF at room temperature for 5 h, then 0.6 mmol I2 was added and reaction for 1 h, yields were obtained after separation and purification of the product by column chromatography.b Adding 2 equiv. of TEMPO. |
|---|
 |
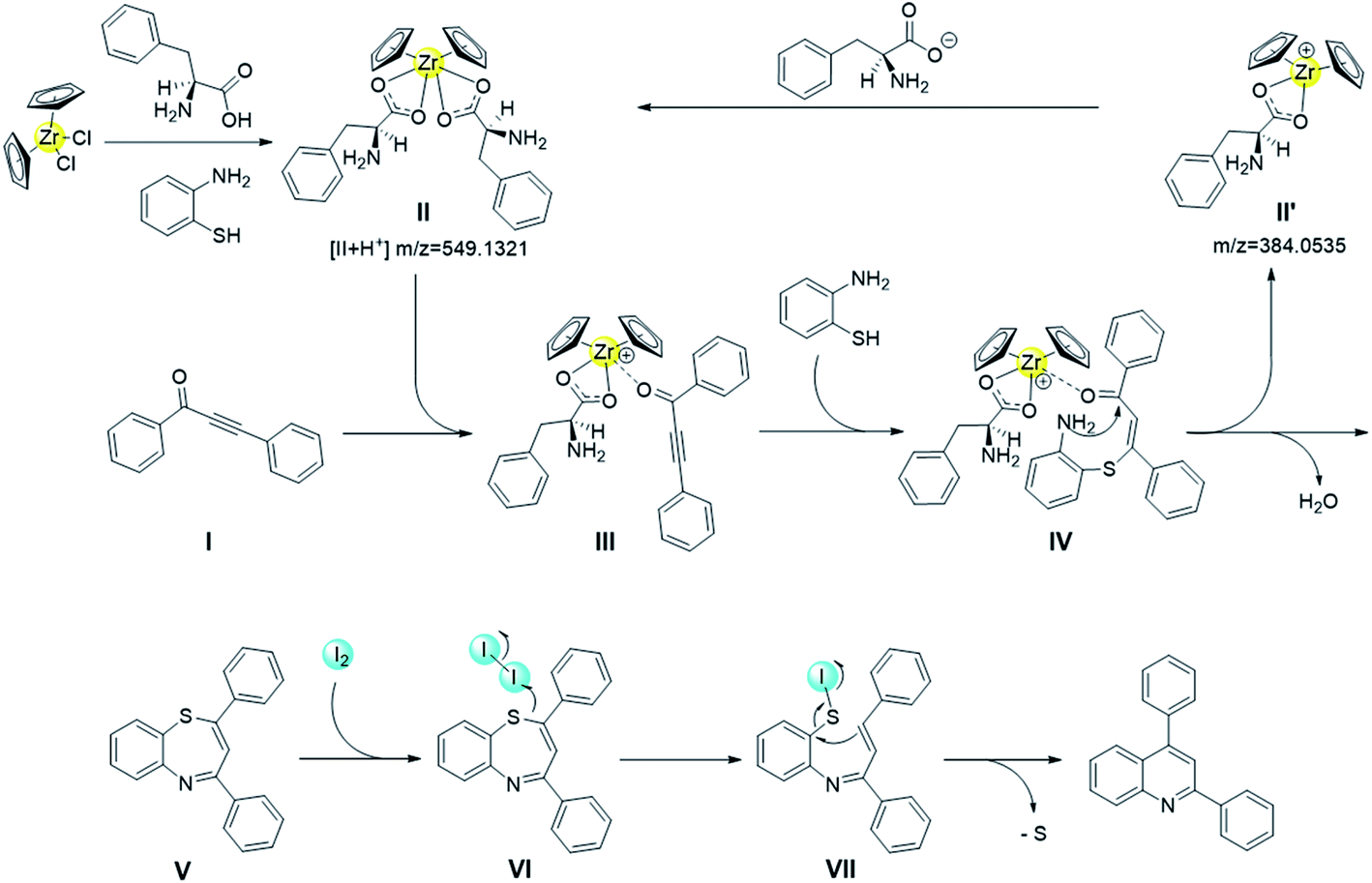 | ||
| Scheme 2 Plausible mechanism for the synthesis of quinolines. | ||
Taking all the aforementioned control experiment results and ESI-MS analysis together, a plausible reaction mechanism is proposed in Scheme 2. Initially, the zirconocene catalyst Cp2Zr(η1-C9H10NO2)2 is very likely to be formed in situ by the pre-catalyst and ligand in the presence of o-aminothiophenol.50,51 As one amino acid ligand is labile, the 1,3-ynone readily coordinated to the generated Cp2Zr(η1-C9H10NO2)2 to form the catalyst–substrate intermediate III, which facilitates a Michael addition with o-phenylenediamine to form an intermediate IV.45 Then the intermediate IV experiences an intramolecular cyclization condensation, followed by the removal of one molecule of H2O to form a seven-membered ring 1,5-benzothiazepine V, thus a catalytic cycle furnished. Finally, intermediate V experiences a desulfurization process mediated by I2.52–54 An intermediate VI is formed from the V and iodine molecular via I–S coordination, followed by a cleavage of I–I bond that generates an intermediate VII. Finally, the subsequent intramolecular cycloaddition followed by cleavage of the C–S bond affords the quinoline framework.
Conclusions
In summary, a one-pot, tandem process has been developed for the synthesis of quinolines. The procedure for the synthesis of quinolines is composed of the Michael addition–cyclization condensation catalyzed by a Lewis acid catalyst, Cp2Zr(η1-C9H10NO2)2, generated in situ, and the desulfuration process promoted by I2. ESI-MS analysis verified the tandem formation of quinolines underwent the less stable 1,5-benzothiazepine intermediates and designed control experiments clarified how the desulfurization conducted. This one-pot method is illuminated efficient for broad substrate scope that provides a new approach for the synthesis of quinolines.Experimental
General procedure for the synthesis of the product 3a
A representative example for preparation of 2,4-diphenylquinoline (3a) is as following: a reactor was charged with 1,3-ynone (103 mg, 0.5 mmol), o-phenylenediamine (64.8 mg, 0.6 mmol), Cp2ZrCl2 (7.31 mg, 0.025 mmol), L-phenylalanine (8.26 mg, 0.05 mmol) and DMF (1 mL) to form a reaction system. The reaction was then carried out under magnetic stirring at room temperature for 5 hours, followed by adding I2 (152.28 mg, 0.6 mmol) and continuing for another 1 hour. TLC analysis indicated the completion of the reaction. Finally, 4 mL of ethyl acetate was added to the reagent to form a mixture, in which the DMF and iodine were washed away for three times with saturated sodium thiosulfate solution by extraction and separation. The organic phase was carried out via silica gel flash column chromatography (eluent![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :petroleum ether:EtOAc = 10:1) to obtain 129 mg of the target product, 3a (white solid, yield: 92%).
:petroleum ether:EtOAc = 10:1) to obtain 129 mg of the target product, 3a (white solid, yield: 92%).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the National Natural Science Foundation of China (21771122), the Natural Science Foundation of Shaanxi Province (2020JM291), the Fundamental Research Funds for the Central Universities (GK202103046, GK202103027), the 111 Project (B14041) and the Key Research and Development Program in Shaanxi Province of China (2021GY-308).References
- V. Sridharan, P. A. Suryavanshi and J. C. Menéndez, Chem. Rev., 2011, 111, 7157–7259 CrossRef CAS PubMed.
- Y. Wang, C. Chen, J. Peng and M. Li, Angew. Chem., Int. Ed., 2013, 52, 5323–5327 CrossRef CAS PubMed.
- M. A. Gouda, A. A. Abu-Hashem and A. A. M. Abdelgawad, J. Heterocycl. Chem., 2021, 58, 908–927 CrossRef CAS.
- K. Ilina and M. Henary, Chem.–Eur. J., 2021, 27, 4230–4248 CrossRef CAS PubMed.
- A. Weyesa and E. Mulugeta, RSC Adv., 2020, 10, 20784–20793 RSC.
- M. Akhil and C. Tejpal Singh, Mini-Rev. Org. Chem., 2019, 16, 631–652 CrossRef.
- J. P. Michael, Nat. Prod. Rep., 2008, 25, 166–187 RSC.
- R. Kaur and K. Kumar, Eur. J. Med. Chem., 2021, 215, 113220 CrossRef CAS PubMed.
- F. A. Saddique, M. Farhad, S. Aslam and M. Ahmad, Synth. Commun., 2021, 51, 13–36 CrossRef CAS.
- S. Yuan, K. Zhang and J. Xia, Asian J. Chem., 2013, 25, 5535–5538 CrossRef CAS.
- S. E. Kharrat, P. Laurent and H. Blancou, Tetrahedron, 2014, 70, 1252–1266 CrossRef.
- S. Banerjee and R. N. Zare, J. Phys. Chem. A, 2019, 123, 7704–7709 CrossRef CAS PubMed.
- J. Jin, S. Guidi, Z. Abada, Z. Amara, M. Selva, M. W. George and M. Poliakoff, Green Chem., 2017, 19, 2439–2447 RSC.
- A. Monastyrskyi, N. K. Namelikonda and R. Manetsch, J. Org. Chem., 2015, 80, 2513–2520 CrossRef CAS PubMed.
- B. Lőrinczi, A. Csámpai, F. Fülöp and I. Szatmári, RSC Adv., 2021, 11, 543–554 RSC.
- C. Tan, H. Xiang, Q. He and C. Yang, Eur. J. Org. Chem., 2015, 2015, 3656–3660 CrossRef CAS.
- S. Yamamoto, Z. Y. Zhou, G. Hiruta, K. Takeuchi, J.-C. Choi, T. Yasuda, T. Kanbara and J. Kuwabara, J. Org. Chem., 2021, 86, 7920–7927 CrossRef CAS PubMed.
- X.-X. Yu, P. Zhao, Y. Zhou, C. Huang, L.-S. Wang, Y.-D. Wu and A.-X. Wu, J. Org. Chem., 2021, 86, 8381–8388 CrossRef CAS PubMed.
- T. T. H. Hajalsiddig and A. E. M. Saeed, Eur. J. Chem., 2019, 10, 57–63 CrossRef CAS.
- N. Omidkhah and R. Ghodsi, Synth. Commun., 2021, 51, 1947–1955 CAS.
- G. A. Ramann and B. J. Cowen, Tetrahedron Lett., 2015, 56, 6436–6439 CrossRef CAS.
- S. K. Ghosh and R. Nagarajan, Tetrahedron Lett., 2016, 57, 4009–4011 CrossRef CAS.
- M. N. Shahi, N. Perveen, M. N. Khan, S. Khakwani and M. A. Khan, Asian J. Chem., 2016, 28, 2665–2669 CrossRef CAS.
- N. M. Shah, V. C. Ramani and R. D. Shah, Asian J. Chem., 2017, 29, 2750–2754 CrossRef CAS.
- M. Wernik, P. E. Hartmann, G. Sipos, F. Darvas, A. D. Boese, D. Dallinger and C. O. Kappe, Eur. J. Org. Chem., 2020, 2020, 7051–7061 CrossRef CAS.
- N. Anand, S. Koley, B. J. Ramulu and M. S. Singh, Org. Biomol. Chem., 2015, 13, 9570–9574 RSC.
- L. Guy, M. Mosser, D. Pitrat, J.-C. Mulatier, M. Kukułka, M. Srebro-Hooper, E. Jeanneau, A. Bensalah-Ledoux, B. Baguenard and S. Guy, J. Org. Chem., 2019, 84, 10870–10876 CrossRef CAS PubMed.
- J. Xu, Q. Chen, Z. Luo, X. Tang and J. Zhao, RSC Adv., 2019, 9, 28764–28767 RSC.
- S. S. Choudhury, S. Jena, D. K. Sahoo, S. Shekh, R. K. Kar, A. Dhakad, K. H. Gowd and H. S. Biswal, ACS Omega, 2021, 6, 19304–19313 CrossRef CAS PubMed.
- Y. Zhang, Y. Sun, Y. Wei and M. Shi, Adv. Synth. Catal., 2019, 361, 2129–2135 CrossRef CAS.
- B. Ganesan, K. Govindan, G. C. Senadi, M. Kandasamy and W.-Y. Lin, Chem. Commun., 2020, 56, 6488–6491 RSC.
- G. Abbiati, A. Arcadi, F. Marinelli and E. Rossi, Synthesis, 2014, 46, 0687–0721 CrossRef.
- A. Arcadi, F. Marinelli and E. Rossi, Tetrahedron, 1999, 55, 13233–13250 CrossRef CAS.
- G. Abbiati, A. Arcadi, F. Marinelli and E. Rossi, Eur. J. Org. Chem., 2003, 1423–1427 CrossRef CAS.
- E. Rossi, G. Abbiati, V. Canevari, D. Nava and A. Arcadi, Tetrahedron, 2004, 60, 11391–11398 CrossRef CAS.
- G. Abbiati, A. Arcadi, F. Marinelli, E. Rossi and M. Verdecchia, Eur. J. Org. Chem., 2009, 1027–1031 CrossRef CAS.
- N. Sun, H. Yang, K. Zheng, L. Jin, B. Hu, Z. Shen and X. Hu, Eur. J. Org. Chem., 2020, 2020, 6805–6812 CrossRef CAS.
- X. Zhang and X. Xu, Chem.–Asian J., 2014, 9, 3089–3093 CrossRef CAS PubMed.
- R. P. Korivi and C.-H. Cheng, J. Org. Chem., 2006, 71, 7079–7082 CrossRef CAS PubMed.
- T. Masquelin and D. Obrecht, Terrahedron, 1997, 53, 641–646 CrossRef CAS.
- G. Cabarrocas, M. Ventura, M. Maestro, J. Mahía and J. M. Villalgordoa, Tetrahedron: Asymmetry, 2001, 12, 1851–1863 CrossRef CAS.
- G. Cabarrocas, S. Rafel, M. Ventura and J. M. Villalgordo, Synlett, 2000, 595–598 CAS.
- T. Blitzke, D. Sicker and H. Wilde, Synthesis, 1995, 236–238 CrossRef CAS.
- L. Nagarapu, N. Ravirala and D. Akkewar, Heterocycl. Commun., 2001, 7, 237–242 CAS.
- M. Yang, Y. Wang, Y. Jian, D. Leng, W. Zhang, G. Zhang, H. Sun and Z. Gao, Mol. Catal., 2020, 498, 111247 CrossRef CAS.
- Y. Luo, H. Sun, W. Zhang, X. Wang, S. Xu, G. Zhang, Y. Jian and Z. Gao, RSC Adv., 2017, 7, 28616–28625 RSC.
- Y. Wu, C. Chen, G. Jia, X. Zhu, H. Sun, G. Zhang, W. Zhang and Z. Gao, Chem.–Eur. J., 2014, 20, 8530–8535 CrossRef CAS PubMed.
- X. Wang, Z. Wang, G. Zhang, W. Zhang, Y. Wu and Z. Gao, Eur. J. Org. Chem., 2016, 2016, 502–507 CrossRef.
- Y. Wang, Y. Jian, Y. Wu, H. Sun, G. Zhang, W. Zhang and Z. Gao, Appl. Organomet. Chem., 2019, 33, e4925 Search PubMed.
- R. L. Webster, RSC Adv., 2014, 4, 5254–5260 RSC.
- T. M. Klapötke, H. Köpf, I. C. Tornieporth-Oetting and P. S. White, Angew. Chem., Int. Ed. Engl., 1994, 33, 1518–1519 CrossRef.
- J. Nath, B. K. Patel, L. Jamir, U. B. Sinha and K. V. V. V. Satyanarayana, Green Chem., 2009, 11, 1503–1506 RSC.
- M. S. Chernov'yants, I. V. Burykin, Z. A. Starikova, A. Y. Tereznikov and T. S. Kolesnikova, J. Mol. Struct., 2013, 1047, 204–208 CrossRef.
- S. Zhang, Q. Zhao, Y. Zhao, W. Yu and J. Chang, Adv. Synth. Catal., 2020, 362, 1993–1997 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR data and ESI(+)-MS spectra. See DOI: 10.1039/d1ra06976d |
| This journal is © The Royal Society of Chemistry 2021 |