
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolyfluoroalkylated antipyrines in Pd-catalyzed transformations†
Evgeny V. Shchegolkov a,
Yanina V. Burgarta,
Daria A. Matsnevaa,
Sophia S. Borisevichb,
Renata A. Kadyrovac,
Iana R. Orshanskayac,
Vladimir V. Zarubaevc and
Victor I. Saloutin*a
a,
Yanina V. Burgarta,
Daria A. Matsnevaa,
Sophia S. Borisevichb,
Renata A. Kadyrovac,
Iana R. Orshanskayac,
Vladimir V. Zarubaevc and
Victor I. Saloutin*a
aPostovsky Institute of Organic Synthesis, Ural Branch of the Russian Academy of Sciences, S. Kovalevskoi St., 22, Ekaterinburg 620990, Russia. E-mail: saloutin@ios.uran.ru
bUfa Institute of Chemistry, Russian Academy of Sciences, 71 October Ave., Ufa, 450054, Russia
cSaint Petersburg Pasteur Research Institute of Epidemiology and Microbiology, 14 Mira St., Saint-Petersburg, 197101, Russia
First published on 31st October 2021
Abstract
In the direct C–H arylation with arylhalogenides in the presence of Pd(OAc)2, trifluoromethyl-containing antipyrine reacts very slowly and incompletely owing to the low nucleophilicity of its C4 center. However, it was effective in modifying polyfluoroalkyl-substituted 4-bromo- and 4-iodo antipyrines by the Suzuki and Sonogashira reactions. It was established that using Pd2(dba)3 as catalyst and XPhos as phosphine ligand was the optimal catalytic system for the synthesis of 4-aryl- and 4-phenylethynyl-3-polyfluoroalkyl-antipyrines. Moreover, iodo-derivatives as the initial reagents were found to be more advantageous compared to bromo-containing analogs. It was found that 4-phenylethynyl-5-CF3-antipyrine has a moderate activity against the influenza virus A/Puerto Rico/8/34 (H1N1) and 4-iodo-5-CF3-antipyrine reveals a weak activity against the vaccine virus (strain Copenhagen) and bovine diarrhea virus (strain VC-1).
Introduction
The pyrazole core is an acknowledged privileged scaffold in medicinal chemistry.1–10 In this regard, pyrazolone derivatives are of particular interest because, based on them, an entire class of analgesic-antipyretics has been developed to be successfully applied in clinical practice for a long time.11,12 The predecessor of this pyrazolone family is phenazone (antipyrine) (1) (Fig. 1) that is still part of some combination drugs, e.g., A/B otic drops (ear drops).13 | ||
| Fig. 1 Drugs based on pyrazolone core. | ||
Research in recent years has shown that the pyrazolone core is a universal structure14 to develop substances with various types of biological action including antimicrobial,11 antitubercular,15,16 antiviral,17 anticancer,18–21 analgesic,22 anti-inflammatory,23 antioxidant,24 and anti-diabetic25 activities as well as action on CNS,26,27 etc.
The several drugs containing pyrazolone scaffold is presently approved for medical use (Fig. 1). These are edaravone (2) with antiradical properties for the treatment of amyotrophic lateral sclerosis, aminophenazone (3) with antipyretic and anti-inflammatory activity, eltrombopag (4) for the treatment of low blood platelet counts, dichloralphenazone (5) for relieving tension and vascular headaches, metamizole (6) to stop severe pain and fever, sulfamazone (7) – sulfanilamide antibiotic with antipyretic properties, and analgesics propyphenazone (8) and nifenazone (9) (please see, DrugBank).
Note that fluorine-containing pyrazolone derivatives are promising because of their potential use in the pharmaceutical industry.28–33 For example, the medicines containing trifluoromethyl–pyrazole cycle are celecoxib,34 mavacoxib35 (COX-2 selective inhibitors), and razaxaban36 (inhibitor of coagulation blood factor Xa).
Recently, we have developed the methods for synthesis of polyfluoroalkyl-containing antipyrines.37 CF3-antipyrine (1-methyl-2-phenyl-5-trifluoromethyl-1,2-dihydro-3H-pyrazolon-3-one) was found to show an anti-inflammatory and analgesic activity at the level of diclofenac and metamizole or significantly higher, and its antipyretic effect was higher than that of paracetamol.38 Therefore, the promising challenge is the development of methods for functionalization of polyfluoroalkyl-antipyrines.
The modification of organic compounds via the generation of new carbon–carbon bond is known to be one of the most used approaches to generate new molecules. In the mid-seventies of the 20th century, a wide range of C–C bond formation reactions catalyzed by metal complexes was discovered, which made it possible to put these transformations in a row of the most effective and successful tools of organic synthesis. The great research interest in these transformations is caused primarily by their synthetic capabilities, which allow a wide range of organic substrates to be involved in the reactions, as well as by high selectivity in the formation of target products and by the comparatively low requirements to purity of the reagents.39,40
The cross-coupling reactions allow C–C bonds not only to be generated between aryl components but also to involve various hetaryl fragments in the process, which significantly extends their synthetic potential to modify the natural substances and pharmacologically active compounds.41,42 Recently, the methods have been enthusiastically investigated for modifying pyrazoles, including polyfluoroalkyl-containing ones, at the position 4. For example, the Suzuki cross-coupling reaction of 3-chloro-4-iodo-1-methyl-5-(trifluoromethyl)-1H-pyrazole with phenylboronic acid in the presence of Pd(PPh3)4 led to 3-chloro-1-methyl-4-phenyl-5-(trifluoromethyl)-1H-pyrazole.43 We used an analogous catalyst to modify 4-bromo-1,5-diphenyl-3-(polyfluoroalkyl)-1H-pyrazoles in the conditions of microwave irradiation to obtain 4-(het)aryl-substituted pyrazoles.44 The 4-iodo-derivatives of a celecoxib and its analogs were used in the cross-coupling reactions with copper(I) cyanide to give 4-cyano-3-CF3-pyrazoles, with phenylboronic acid in the presence of Pd(PPh3)4 and K2CO3 – triaryl-substituted 3-CF3-pyrazoles, with 4-methoxythiophenol in the presence of the catalytic system of Pd(dba)2-Xantphos – 4-arylthio-3-CF3-pyrazoles, and in the conditions of Sonogashira reaction – 4-phenylethynyl-3-CF3-pyrazoles in.45
A great research attention has also been paid to the modification of antipyrine and its analogs in the cross-coupling reactions. A convenient and ligand-free method has been described for the synthesis of a series of 4-aryl-antipyrines based on 4-unsubstituted derivatives and arylhalogenides via an activation of C–H bond by palladium(II) acetate in the presence of Ag2CO3.46 Reactivity of para-, meta- and ortho-substituted arylbromides has been investigated in the reaction with antipyrine by Pd(OAc)2 and K2CO3.47 A method has been suggested for the direct introduction of arylthiol and arylselenium residues into antipyrines by di-p-tolyldisulfide or diphenyldiselenide using catalyst system AgOTf/AgOAc.48 An effective way of carrying out the cross-coupling reaction of 4-iodo-antipyrine with arylboronic acids has been found on Pd/nanoglobular carbon support.49 The reactions have been described for Sonogashira cross-coupling of antipyrine with electrophile alkynylation reagent – 1-[(triisopropylsilyl)ethynyl]-1,2-benziodoxol-3(1H)-one (TIPS-EBX).50
In this report, we investigated and compared the capabilities of polyfluoroalkyl-containing antipyrines in Pd-catalyzed C–H arylations and Suzuki and Sonogashira cross-coupling reactions, and studied antiviral activity of the synthesized compounds.
Results and discussion
The literature data analysis has shown that antipyrine and its non-fluorinated analogs readily undergo the Pd-catalyzed direct arylation with the different arylhalogenides.46,47 However, our numerous efforts to involve 5-trifluoromethyl-antipyrine 1a into the direct arylation with arylhalogenides were ineffective, despite the use of various palladium catalysts {Pd(OAc)2, Pd2(dba)3}, bases (AcOK, K2CO3, Cs2CO3, K3PO4), ligands (ligand-free, XPhos), solvents (CO(OEt)2, EtOH–H2O, DMA, toluene, 1,4-dioxane), temperature modes (100–160 °C) and ratios of reagents in these reactions. The extended experiments are presented in Table S1 (please see, ESI†).The attempts to introduce phenyl iodide 2a and 3-cyanophenylbromide 2b in the reaction with CF3-antipyrine 1a (entry 1–4, Table S1†) may be recognized as practically unsuccessful, since the content of the expected 4-aryl-antipyrines 3a, b in accordance with GLC-mass spectrometry was only 5 and 12%, respectively. The conversion of CF3-antipyrine 1a in the reaction with 4-cyanophenyl halides 2b, c and with 4-nitrophenyl iodide 2e into the corresponding products 3c–d was no more than 21% (entries 5–23, Table S1†). However, the use of aryl bromides 2f, h, i in the reaction with antipyrine 1a led to the higher content of the target products 3d–f in a reaction mixture in the range of 32–42% (entries 24, 26 and 27, Table S1†). We were successful in isolating the substances 3d–f in a pure form and the compound 3c in a mixture with the initial antipyrine 1a (the 3c![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1a ratio was 83:17) (Scheme 1).
:1a ratio was 83:17) (Scheme 1).
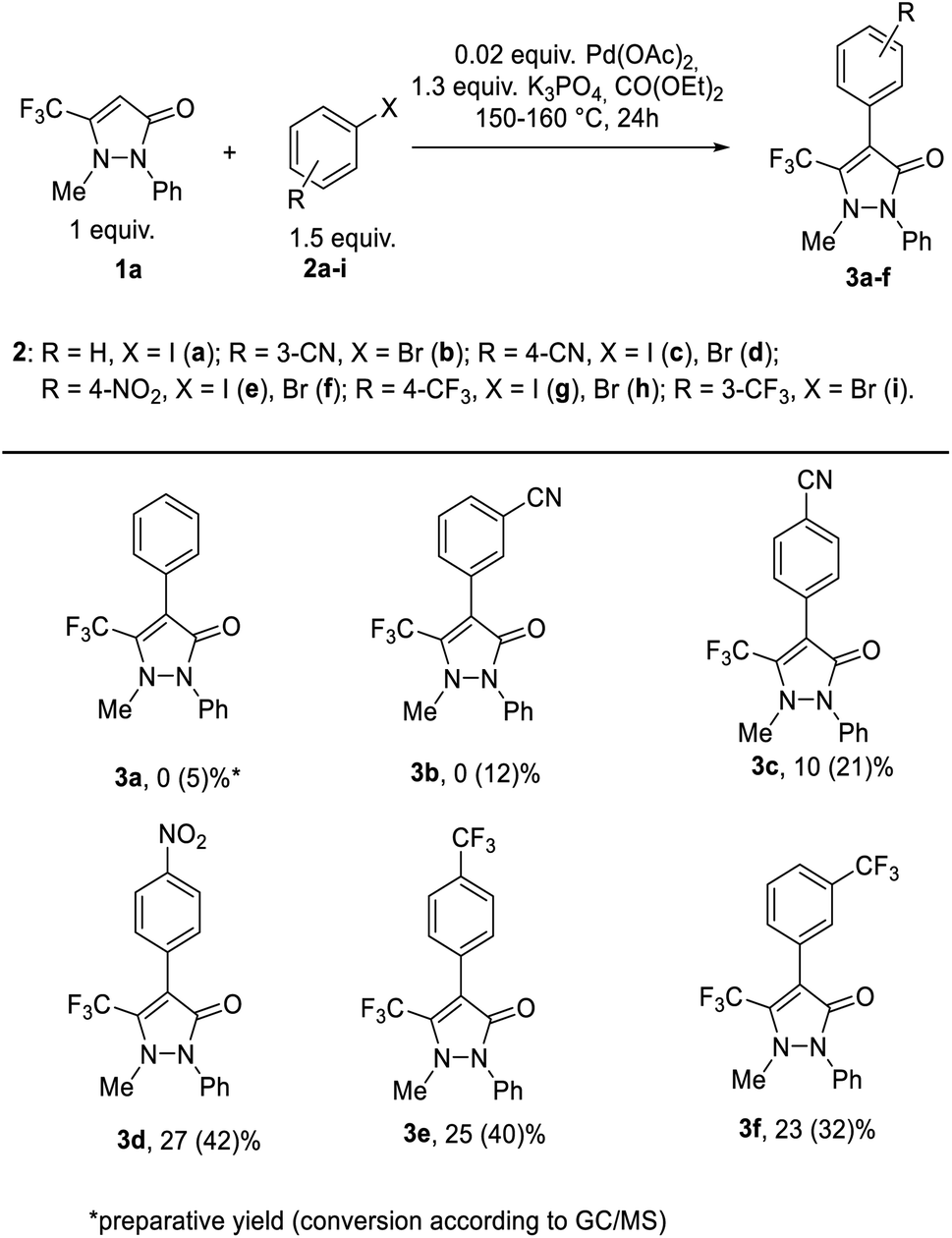 | ||
| Scheme 1 Direct C–H arylation of antipyrine 1a. | ||
The analysis of all the reaction conditions showed that the catalyst Pd(OAc)2 was more effective than Pd2(dba)3 in the direct arylation, and using the base K3PO4 was more successful. Among all the solvents, the definitely best one was eco-friendly CO(OEt)2 (ref. 51–54) at 150–160 °C. In addition, all transformations did not require a phosphonium ligand to be used. Using aryl bromides was found to be more effective than aryl iodide.
Apparently, in the reactions with electrophile reagents compared to non-fluorinated analogs, the reduced reactivity of 5-trifluoromethyl-antipyrine 1a was assumed to be caused by the deactivation of carbon C4 due to the influence of the neighboring electron-withdrawing fluorinated substituent. To explain this assumption, we evaluated the Fukui dual descriptors for centers C4 of antipyrine (Ant) and CF3-antipyrine 1a (Fig. 2) using quantum-chemical calculations.
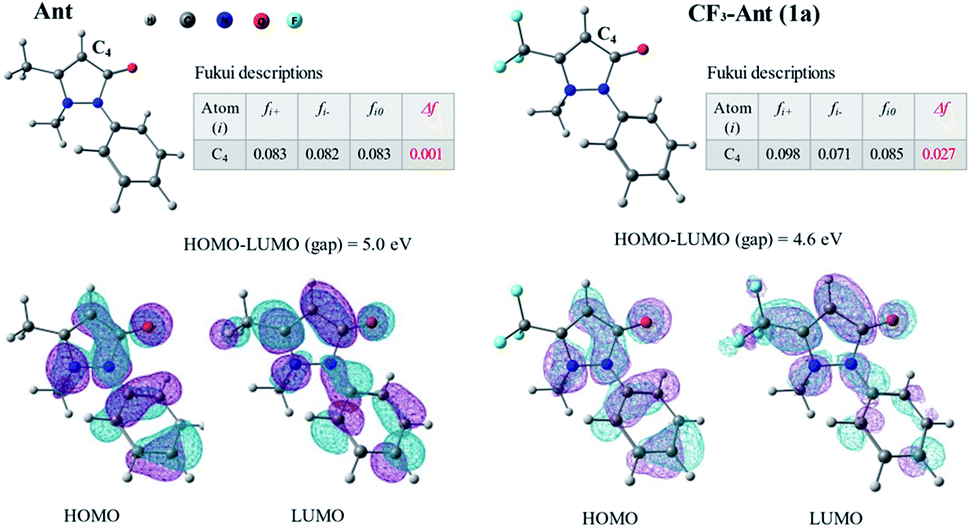 | ||
| Fig. 2 The dual descriptor of Fukui functions for antipyrine (Ant) and CF3-antipyrine 1a, HOMO–LUMO gap and visualization. | ||
According to previous report,55 the dual descriptor is able to unambiguously expose truly nucleophilic and electrophilic regions on a molecule. The reaction center C4 of CF3-antipyrine 1a was found to be characterized with more positive values of the dual descriptor of Fukui function Δf, which points to its higher electrophilicity in comparison with the same center of Ant.56 It explains the weak reactivity of compound 1a in the reactions of electrophile arylation comparing to the non-fluorinated antipyrine in analogous transformations.46,47
Then, we investigated the possibility of modifying polyfluoroalkyl-antipyrines 1 at the center C4 in the Pd-catalyzed Suzuki and Sonogashira cross-coupling reactions. At first, 4-bromo-5-polyfluoroalkyl-antipyrines 4a–d and 4-iodo-5-polyfluoroalkyl-antipyrines 5a, b were synthesized by the treatment of the initial heterocycles 1a–d with N-bromo- or N-iodosuccinimide (NXS) (Scheme 2).
 | ||
| Scheme 2 Synthesis of 4-bromo-antipyrines 4a–d and 4-iodo-antipyrines 5a, b. | ||
The reactions were running readily at room temperature with the good yields. The structure of the obtained halogen-containing antipyrines 4a–d and 5a, b was confirmed by IR and NMR spectroscopy and the elemental analysis. The halogenation reaction is confirmed by the absence of the singlet signal of ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH proton at δ ∼ 5–6 ppm in 1H NMR spectra of the compounds 4a–d and 5a, b.
CH proton at δ ∼ 5–6 ppm in 1H NMR spectra of the compounds 4a–d and 5a, b.
Further, we studied the Suzuki reaction of halogen-substituted antipyrines 4a–d and 5a, b. To search for optimal conditions, we chose the synthesis of 4-phenyl-5-trifluoromethyl-antipyrine 3a via cross-coupling reaction of 1 equiv. of 4-bromo-5-trifluoromethyl-antipyrine 4a with 1.2 equiv. of phenylboronic acid in the presence of 2.5 equiv. of base K2CO3, varying the solvent, palladium catalyst, and phosphine ligand. The reactions were performed at 100 °C in the closed vials under inert gas. A ratio of the reaction products was determined by GLC-mass spectrometry. The data on selecting the reaction conditions are given in Table S2.†
Initially, a mixture of THF–H2O (3:4) was applied as solvent, and Pd(PPh3)4 as catalyst/ligand system. There was a partial conversion of the initial bromo-antipyrine 4a under these conditions (entry 1, Table S2†). The replacement of THF by aqueous ethanol led to full conversion of the initial heterocycle 4a, but the reaction was accompanied by debromination of bromo-antipyrine 4a into the parent antipyrine 1a and the by-products formation (entry 2, Table S2†).
With aqueous ethanol chosen as a solvent, we investigated the possibility to apply Pd2(dba)3 as a catalyst in combination with various phosphine ligands: P(o-Tol)3, P(p-Tol)3, XPhos – 2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl, Xantphos – 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene and BINAP – (±)-2,2′-bis(diphenylphosphino)-1,1′-binaphthalene, [1,1′-Binaphthalene]-2,2′-diylbis[diphenylphosphine]. It was found that in all the cases the reaction proceeded with almost complete conversion of the starting bromo-antipyrine 4a (entries 3–7, Table S2†).
However, the catalyst system Pd2(dba)3/XPhos (entry 5, Table S2†) was found to be the most effective. In these conditions the content of targeted compound 4a reached 86%, and the contents of debromination product and impurities were 6% and 8%, respectively. Note that the reaction in the same catalyst system under the microwave irradiation (entry 8, Table S2†) led to the formation of a large number of by-products, and as a result the product 3a yield decreased to 62%.
It is known that (het)aryl-containing iodides should often enter into cross-coupling reactions easier.57 Thus, we carried out a series of reactions of 4-iodo-5-trifluoromethyl-antipyrines 5a with phenylboronic acid to give 4-phenyl-5-trifluoromethyl-antipyrine 3a using similar catalytic systems as those used in the reactions of bromo-substituted analog 4a. The performed experiments showed, regardless of the use of various phosphonium ligands and Pd catalysts, the main process in these reactions at high temperature was the deiodation of 4-iodo-antipyrine 5a into antipyrine 1a. The yield of the desired product 3a was reduced significantly (entries 1–4, Table S3†). The replacement of base K2CO3 by Cs2CO3 did not have influence on the reaction (entry 5, Table S3†). However, the reaction under the optimal conditions for transformations of bromo-antipyrine 4a (1.2 equiv. of phenylboronic acid in the presence of 2.5 equiv. of base K2CO3, 0.02 equiv. of Pd2(dba)3 and 0.04 equiv. of XPhos), carried out at room temperature, allowed us to realize 100% conversion of the initial iodo-antipyrine 5a into 4-phenyl-5-trifluoromethyl-antipyrine 3a (entry 6, Table S3†). The yield of compound 3a was 89% after purification. Therefore, these conditions may be considered as the most effective, although the reaction at room temperature proceeded for longer time but with almost quantitative yield. The comparative reaction with 0.05 equiv. of Pd(PPh3)4 was found to be ineffective (entry 7, Table S3†).
Using the selected optimal conditions, a series of 4-phenyl-5-polyfluoroalkyl-antipyrines 3a, h, j, l was synthesized (Scheme 3). The yields of heterocycles 3a, h obtained from iodo-antipyrines 5a, b were slightly higher than in the case of bromo-substituted analogs 4a, b, because the reaction at room temperature allowed dehalogention of the initial compounds 5a, b to be avoided. Note that bromo-antipyrine 4a practically did not react at room temperature.
 | ||
| Scheme 3 Synthesis of 4-aryl-5-polyfluoroalkyl-antipyrines 3a, g–l. | ||
In addition, the optimal conditions were successfully applied to the reactions of halogen antipyrines 4a–d and 5a, b with [4-(methylthio)phenyl]boronic acid to generate new 4-[4-(methylthio)phenyl]-5-polyfluoroalkyl-antipyrines 3g, i, k (Scheme 3).
The structure of the obtained 4-aryl-5-polyfluoroalkyl-antipyrines 3a, d–f, g–l was confirmed by the elemental analysis, IR and NMR spectroscopy. The IR spectra of compounds 3a, d–f, g–l had a similar character. For example, the absorption bands of the carbonyl groups of compounds 3a, d–f, g–l were observed at νCO 1660–1680 cm−1. The 1H NMR spectra of antipyrines 3a, d–f, g–l were characterized with the presence of methyl and additional aryl protons.
We also performed the X-ray diffraction analysis (XRD) for the compound 3e to determine its exact structure (Fig. 3).
 | ||
| Fig. 3 The structure of 1-methyl-2-phenyl-5-(trifluoromethyl)-4-[4-(trifluoromethyl)phenyl]-1,2-dihydro-3H-pyrazol-3-one 3e according to XRD (CCDC 2110385†). | ||
Then, we studied the Sonogashira cross-coupling reactions of 4-halogen-antipyrines 4a–d, 5a, b with phenylacetylene. We also began the investigation of these reactions with the search for the optimal conditions, varying the phosphonium ligands and palladium catalyst.
In contrast to the Suzuki reactions, the Sonogashira cross-coupling reactions are commonly carried out in acetonitrile in the presence of copper(I) iodide as co-catalyst and DIPEA as a base. Therefore, we performed a series of the reactions of 1 equiv. of 4-bromo-5-trifluoromethyl-antipyrine 4a with 1.5 equiv. of phenylacetylene in the presence of 2.0 equiv. of DIPEA and 0.1 equiv. of co-catalyst CuI, using various pallidum catalyst and phosphine ligands in acetonitrile at 80 °C in the closed vials under argon for 12 h (entries 1, 3–5, Table S4†). However, these conditions did not lead to satisfactory results owing to incomplete conversion of the starting bromo-antipyrine 4a and the formation of a large number of by-products (mainly phenylacetylene crosslinking product – 1,4-diphenyl-1,3-butadiyne). According to GLC-mass spectrometry data, the content of the targeted 1-methyl-2-phenyl-4-(phenylethynyl)-5-(trifluoromethyl)-1,2-dihydro-3H-pyrazol-3-one 6a in the reactions mixtures was recorded in the range of 8 to 30%. The reaction with 0.05 equiv. of Pd(PPh3)4 (entry 2) or with 0.02 equiv. of Pd2(dba)3 and 0.04 equiv. of XPhos (entry 6) at 100 °C for 6 h resulted in a slight increase of product 6a to 35 and 36%, respectively. The yield of the targeted heterocycle 6a was 26% after purification (entry 6, Table S4†).
Further, we used 4-iodo-5-trifluoromethyl-antipyrine 5a as the initial reagent in the reaction with phenylacetylene (Table S5†). To perform a series of experiments, the already proven catalytic system Pd2(dba)3-XPhos was chosen. The reaction of antipyrine 5a with 1.5 equiv. of phenylacetylene was carried out in the closed vials under argon in the presence of 0.02 equiv. of Pd2(dba)3, 0.04 equiv. of XPhos, 2.0 equiv. of DIPEA and 0.1 equiv. of CuI in different solvents at heating or at room temperature (Table S5†).
The reaction in toluene at 100 °C was found to result in a mixture of by-products, and the content of the targeted 4-(phenylethynyl)-5-(trifluoromethyl)-antipyrine 6a was only 2% (entry 1, Table S5†). Using 1,4-dioxane (entry 2) or acetonitrile (entry 3) at the same temperature led to significant deiodation of the initial iodo-antipyrine 5a, but the content of the desired heterocycle 6a was slightly higher (25%) in acetonitrile than in 1,4-dioxane (13%). At a lower reaction temperature (50 °C) in acetonitrile the content of targeted compound 6a increased up to 35% (entry 4, Table S5†).
The most effective conditions for the Sonogashira reaction of iodo-antipyrine 5a with phenylacetylene was found to hold the reaction mass in acetonitrile at room temperature for 72 h (entry 6, Table S5†). Here, the full conversion of the initial iodo-antipyrine 5a occurred but this transformation was also accompanied by a side process: self-condensation of phenylacetylene to form 1,4-diphenyl-1,3-butadiyne. The content of the desired product 6a in the reaction mixture was 75% and the yield after purification was 60%. The found optimal conditions were used to generate pentafluoroethyl substituted analog 6b (Scheme 4).
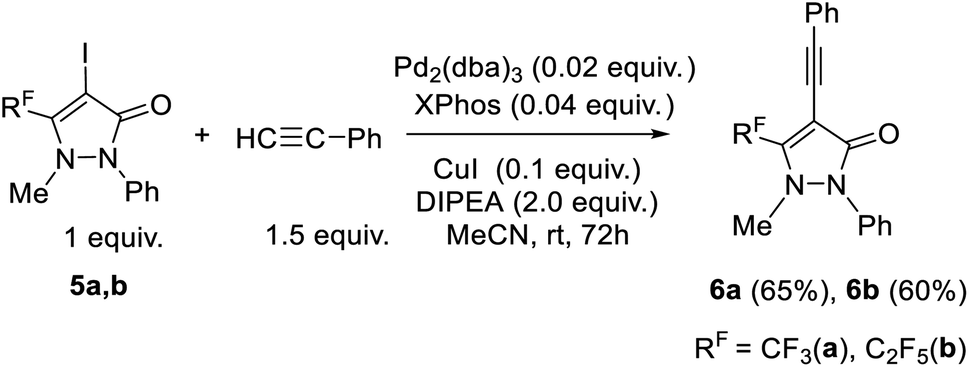 | ||
| Scheme 4 Synthesis of 4-(phenylethynyl)-5-(polyfluoroalkyl)-antipyrines 6a, b. | ||
The structure of 4-(phenylethynyl)-5-(polyfluoroalkyl)-antipyrines 6a, b was confirmed by the elemental analysis, IR and NMR spectroscopy. The existence of triple bond was affirmed by the presence in the 13C NMR spectra of the chemical shifts at δ 76.4–76.6 and 97.8–98.0 ppm, which are typical of two sp-hybridized carbon atoms.45,58
Biological testing
Iodoantipyrine has been known to be applied in the medicinal practice to treat and prevent tick-borne encephalitis,59,60 influenza and infections caused by nonpoliomyelitis enteroviruses of Coxsackie and ECHO groups.60 We evaluated an antiviral activity of the synthesized polyfluoroalkyl-containing iodo-antipyrines 5a, b, and aryl-3a, g and phenylethynyl-substituted 6a antipyrines against the influenza virus A/Puerto Rico/8/34 (H1N1) on MDCK cell line (Table 1) using ribavirin as a reference drug according the published procedure.61| No. | Compound | CC50, μM | IC50, μM | SI |
|---|---|---|---|---|
| a CC50 – 50% cytotoxic concentration, the concentration resulting in death of 50% of cells in culture; IC50 – 50% inhibiting concentration, the concentration resulting in a 50% decrease of virus production as compared to the control; SI – selectivity index (CC50/IC50). | ||||
| 1 | 5a | >815 | 177 ± 21 | 5 |
| 2 | 5b | 378 ± 26 | 205 ± 19 | 2 |
| 3 | 3a | 789 ± 57 | >314 | 3 |
| 4 | 3g | 263 ± 19 | >91 | 3 |
| 5 | 6a | >877 | 58 ± 8 | 15 |
| 6 | Ribavirin | >2130 | 36 ± 5 | 59 |
Studying the cytotoxicity of compounds on the MDCK cell line, it was found that compounds 5a, 6a have lower cytotoxicity (CC50 > 815 μM) compared to the derivatives 5b, 3a and 3g. The elongation of polyfluoroalkyl chain in iodo-antipyrines 5a, b led to the increase of cytotoxicity (5a CC50 > 815 μM vs. 5b CC50 378 μM). The introduction of phenyl substituent resulted in the increased cytotoxic properties of compound 3a (CC50 789 μM) in comparison with the methylthiophenyl analog 3g (CC50 263 μM). Phenylethyl derivative 6a (CC50 > 877 μM) showed the lowest cytotoxicity.
The virus-inhibiting activity of iodo-antipyrines 5a, b and phenyl-antipyrines 3a was low (IC50 > 314–177 μM), therefore their selectivity index did not exceed 5. The activity of methylthiophenyl-antipyrine 3g increased up to IC50 91 μM, but it has a low SI = 3 owing to the raised cytotoxicity. The best ratio of cytotoxic and inhibitory properties was shown by phenylethynyl antipyrine 6a with SI = 15.
The evaluation of antiviral activity of CF3-iodo-antipyrine 5a was carried out in vitro against the vaccine virus (VV, strain Copenhagen), herpes simplex virus type 1 (HSV-1, strain VR-3) and bovine diarrhea virus (BDV) (strain VC-1), a surrogate of the hepatitis C virus on the cells of the calf coronary vessels (CCV). It was found that heterocycle 5a was inactive against HSV-1, but it showed a weak activity against strains of SVV and VDC with SI 2.9 and 6.4, respectively (Table 2).
| CC50, μM | IC50, μM | SIBOB | IC50, μM | SIHSV-1 | CC50, μM | IC50, μM | SIVDC |
|---|---|---|---|---|---|---|---|
| Vero | VV | HSV-1 | CCV | BDV | |||
| a CC50 – 50% cytotoxic concentration, the concentration resulting in death of 50% of cells in culture; IC50 – 50% inhibiting concentration, the concentration resulting in a 50% decrease of the virus production as compared to control; SI – selectivity index. | |||||||
| 298 ± 50 | 103 ± 10 | 2.9 | Inactive | — | 298 ± 50 | 46.88 ± 2 | 6.4 |
Conclusions
In summary, we found a weak reactivity of CF3-antipyrine in Pd-catalyzed reactions of the direct arylation owing to a decrease in the nucleophilicity of its C4 site compared to antipyrine. For the purpose of modification of polyfluoroalkyl-substituted antipyrines at the C4 center, the Suzuki and Sonogashira reactions via the preliminary synthesis of 4-bromo- and 4-iodo-derivatives were found to be effective. Using Pd2(dba)3 as catalyst and XPhos as phosphine ligand were found to be the optimal conditions for the preparation of 4-aryl- and 4-phenylethynyl-3-RF-antipyrines. In addition, the application of iodo-RF-antipyrines as the initial reagents was more advantageous compared to bromo-containing analogs, since they were easily and more efficiently transformed into the target products at room temperatures. The synthesis under such mild conditions allowed us to avoid dehalogenation of the starting halogen-RF-antipyrines. Note that this process was the main side reaction in the studied transformations, which reduces significantly the yield of the target products.The investigation of antiviral activity of the new derivatives of polyfluoroalkyl-containing antipyrines showed a moderate activity of 4-phenylethynyl-5-CF3-antipyrine against the influenza virus A/Puerto Rico/8/34 (H1N1) and a weak activity of iodo-5-CF3-antipyrines against vaccine virus (strain Copenhagen) and bovine diarrhea virus (strain VC-1), a surrogate of the hepatitis C virus.
Author contributions
E. V. Shchegolkov: conceptualization, methodology, validation, investigation, data curation, writing – original draft, writing – review & editing, visualization. Y. V. Burgart: conceptualization, methodology, validation, writing – original draft, writing – review & editing, visualization. D. A. Matsneva: investigation, formal analysis. S. S. Borisevich: formal analysis, visualization. R. A. Kadyrova: investigation, formal analysis. I. R. Orshanskaya: investigation, formal analysis. V. V. Zarubaev: data curation, validation, resources, writing – original draft. V. I. Saloutin: conceptualization, resources, writing – review & editing, funding acquisition.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the Russian Science Foundation (grant no. 21-13-00390). Analytical studies were carried out using equipment of the Center for Joint Use “Spectroscopy and Analysis of Organic Compounds” at the Postovsky Institute of Organic Synthesis of UB RAS. We are grateful to Natalia Yu. Pomortseva for her assistance in preparing the manuscript.Notes and references
- J. Akhtar, A. A. Khan, Z. Ali, R. Haider and M. Shahar Yar, Eur. J. Med. Chem., 2017, 125, 143–189 CrossRef CAS PubMed.
- D. Havrylyuk, O. Roman and R. Lesyk, Eur. J. Med. Chem., 2016, 113, 145–166 CrossRef CAS PubMed.
- M. F. Khan, M. M. Alam, G. Verma, W. Akhtar, M. Akhter and M. Shaquiquzzaman, Eur. J. Med. Chem., 2016, 120, 170–201 CrossRef CAS PubMed.
- K. Karrouchi, S. Radi, Y. Ramli, J. Taoufik, Y. Mabkhot, F. Al-aizari and M. Ansar, Molecules, 2018, 23, 134 CrossRef PubMed.
- K. Kaur, V. Kumar and G. K. Gupta, J. Fluorine Chem., 2015, 178, 306–326 CrossRef CAS.
- Ş. G. Küçükgüzel and S. Şenkardeş, Eur. J. Med. Chem., 2015, 97, 786–815 CrossRef PubMed.
- R. S. Keri, K. Chand, T. Ramakrishnappa and B. M. Nagaraja, Arch. Pharm., 2015, 348, 299–314 CrossRef CAS PubMed.
- M. Aghazadeh Tabrizi, P. G. Baraldi, P. A. Borea and K. Varani, Chem. Rev., 2016, 116, 519–560 CrossRef CAS PubMed.
- L. Pizzuti, A. G. Barschak, F. M. Stefanello, M. D. Farias, C. Lencina, M. Roesch-Ely, W. Cunico, S. Moura and C. M. P. Pereira, Curr. Org. Chem., 2014, 18, 115–126 CrossRef CAS.
- D. Dias, B. Pacheco, W. Cunico, L. Pizzuti and C. Pereira, Mini-Rev. Med. Chem., 2015, 14, 1078–1092 CrossRef PubMed.
- Z. Zhao, X. Dai, C. Li, X. Wang, J. Tian, Y. Feng, J. Xie, C. Ma, Z. Nie, P. Fan, M. Qian, X. He, S. Wu, Y. Zhang and X. Zheng, Eur. J. Med. Chem., 2020, 186, 111893 CrossRef CAS PubMed.
- P. Chauhan, S. Mahajan and D. Enders, Chem. Commun., 2015, 51, 12890–12907 RSC.
- J. Sahoo, C. R. Sahoo, P. K. Nandini Sarangi, S. K. Prusty, R. N. Padhy and S. K. Paidesetty, Eur. J. Med. Chem., 2020, 186, 111911 CrossRef CAS PubMed.
- L. Yet, Five-Membered Ring Systems: With More than One N Atom, in Progress in Heterocyclic Chemistry, 2020, Ch. 5.4, pp. 325–361 Search PubMed.
- P. Gunasekaran, S. Perumal, P. Yogeeswari and D. Sriram, Eur. J. Med. Chem., 2011, 46, 4530–4536 CrossRef CAS PubMed.
- C. Soares de Melo, V. Singh, A. Myrick, S. B. Simelane, D. Taylor, C. Brunschwig, N. Lawrence, D. Schnappinger, C. A. Engelhart, A. Kumar, T. Parish, Q. Su, T. G. Myers, H. I. M. Boshoff, C. E. Barry, F. A. Sirgel, P. D. van Helden, K. I. Buchanan, T. Bayliss, S. R. Green, P. C. Ray, P. G. Wyatt, G. S. Basarab, C. J. Eyermann, K. Chibale and S. R. Ghorpade, J. Med. Chem., 2021, 64, 719–740 CrossRef CAS PubMed.
- T. Pillaiyar, M. Manickam, V. Namasivayam, Y. Hayashi and S.-H. Jung, J. Med. Chem., 2016, 59, 6595–6628 CrossRef CAS PubMed.
- Y. Mostinski, G. J. J. E. Heynen, M. P. López-Alberca, J. Paul, S. Miksche, S. Radetzki, D. Schaller, E. Shanina, C. Seyffarth, Y. Kolomeets, N. Ziebart, J. de Schryver, S. Oestreich, M. Neuenschwander, Y. Roske, U. Heinemann, C. Rademacher, A. Volkamer, J. P. von Kries, W. Birchmeier and M. Nazaré, J. Med. Chem., 2020, 63, 14780–14804 CrossRef CAS PubMed.
- G. Saidachary, K. Veera Prasad, D. Divya, A. Singh, U. Ramesh, B. Sridhar and B. China Raju, Eur. J. Med. Chem., 2014, 76, 460–469 CrossRef CAS PubMed.
- N. S. Gavande, P. VanderVere-Carozza, A. K. Mishra, T. L. Vernon, K. S. Pawelczak and J. J. Turchi, J. Med. Chem., 2017, 60, 8055–8070 CrossRef CAS PubMed.
- A. L. Smith, K. L. Andrews, H. Beckmann, S. F. Bellon, P. J. Beltran, S. Booker, H. Chen, Y.-A. Chung, N. D. D'Angelo, J. Dao, K. R. Dellamaggiore, P. Jaeckel, R. Kendall, K. Labitzke, A. M. Long, S. Materna-Reichelt, P. Mitchell, M. H. Norman, D. Powers, M. Rose, P. L. Shaffer, M. M. Wu and J. R. Lipford, J. Med. Chem., 2015, 58, 1426–1441 CrossRef CAS PubMed.
- N. Uramaru, H. Shigematsu, A. Toda, R. Eyanagi, S. Kitamura and S. Ohta, J. Med. Chem., 2010, 53, 8727–8733 CrossRef CAS PubMed.
- O. Corminboeuf and X. Leroy, J. Med. Chem., 2015, 58, 537–559 CrossRef CAS PubMed.
- P. M. P. Santos, A. M. M. Antunes, J. Noronha, E. Fernandes and A. J. S. C. Vieira, Eur. J. Med. Chem., 2010, 45, 2258–2264 CrossRef CAS PubMed.
- S. Yousuf, K. M. Khan, U. Salar, S. Chigurupati, M. T. Muhammad, A. Wadood, M. Aldubayan, V. Vijayan, M. Riaz and S. Perveen, Eur. J. Med. Chem., 2018, 159, 47–58 CrossRef CAS PubMed.
- D. Zimmermann, Y. L. Janin, L. Brehm, H. Bräuner-Osborne, B. Ebert, T. N. Johansen, U. Madsen and P. Krogsgaard-Larsen, Eur. J. Med. Chem., 1999, 34, 967–976 CrossRef CAS PubMed.
- Y. Zhang, K. T. Zhao, S. G. Fox, J. Kim, D. R. Kirsch, R. J. Ferrante, R. I. Morimoto and R. B. Silverman, J. Med. Chem., 2015, 58, 5942–5949 CrossRef CAS PubMed.
- W. R. Dolbier, J. Fluorine Chem., 2005, 126, 157–163 CrossRef CAS.
- P. K. Mykhailiuk, Chem. Rev., 2021, 121, 1670–1715 CrossRef CAS PubMed.
- J. C. Sloop, C. Holder and M. Henary, Eur. J. Org. Chem., 2015, 2015, 3405–3422 CrossRef CAS.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- E. Prchalová, O. Štěpánek, S. Smrček and M. Kotora, Future Med. Chem., 2014, 6, 1201–1229 CrossRef PubMed.
- J. Han, L. Kiss, H. Mei, A. M. Remete, M. Ponikvar-Svet, D. M. Sedgwick, R. Roman, S. Fustero, H. Moriwaki and V. A. Soloshonok, Chem. Rev., 2021, 121, 4678–4742 CrossRef CAS PubMed.
- T. D. Penning, J. J. Talley, S. R. Bertenshaw, J. S. Carter, P. W. Collins, S. Docter, M. J. Graneto, L. F. Lee, J. W. Malecha, J. M. Miyashiro, R. S. Rogers, D. J. Rogier, S. S. Yu, G. D. Anderson, E. G. Burton, J. N. Cogburn, S. A. Gregory, C. M. Koboldt, W. E. Perkins, K. Seibert, A. W. Veenhuizen, Y. Y. Zhang and P. C. Isakson, J. Med. Chem., 1997, 40, 1347–1365 CrossRef CAS PubMed.
- S. R. Cox, S. P. Lesman, J. F. Boucher, M. J. Krautmann, B. D. Hummel, M. Savides, S. Marsh, A. Fielder and M. R. Stegemann, J. Vet. Pharmacol. Ther., 2010, 33, 461–470 CrossRef CAS PubMed.
- M. L. Quan, P. Y. S. Lam, Q. Han, D. J. P. Pinto, M. Y. He, R. Li, C. D. Ellis, C. G. Clark, C. A. Teleha, J.-H. Sun, R. S. Alexander, S. Bai, J. M. Luettgen, R. M. Knabb, P. C. Wong and R. R. Wexler, J. Med. Chem., 2005, 48, 1729–1744 CrossRef CAS PubMed.
- N. A. Nemytova, E. V. Shchegol’kov, Y. V. Burgart, P. A. Slepukhin, S. S. Borisevich, S. L. Khursan and V. I. Saloutin, J. Fluorine Chem., 2018, 206, 72–81 CrossRef CAS.
- N. Agafonova, E. Shchegolkov, Y. Burgart, V. Saloutin, A. Trefilova, G. Triandafilova, S. Solodnikov, V. Maslova, S. Borisevich, O. Krasnykh and S. Khursan, Med. Chem., 2019, 15, 521–536 CrossRef CAS PubMed.
- A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722–6737 CrossRef CAS PubMed.
- C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed.
- K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442–4489 CrossRef CAS PubMed.
- D. Wang and S. Gao, Org. Chem. Front., 2014, 1, 556–566 RSC.
- J. P. Chupp, J. Heterocycl. Chem., 1994, 31, 1377–1380 CrossRef CAS.
- A. E. Ivanova, Y. V Burgart and V. I. Saloutin, Russ. J. Org. Chem., 2018, 54, 1265–1267 CrossRef CAS.
- V. M. Muzalevskiy and V. G. Nenajdenko, Org. Biomol. Chem., 2018, 16, 7935–7946 RSC.
- H. Gong, Y. Yang, Z. Wang and C. Kuang, Beilstein J. Org. Chem., 2013, 9, 2033–2039 CrossRef PubMed.
- A. Sasmal, J. K. Bera, H. Doucet and J.-F. Soulé, Tetrahedron Lett., 2020, 61, 151798 CrossRef CAS.
- W. Ma, H. Dong, D. Wang and L. Ackermann, Adv. Synth. Catal., 2017, 359, 966–973 CrossRef CAS.
- S. A. Yakukhnov, E. O. Pentsak, K. I. Galkin, R. M. Mironenko, V. A. Drozdov, V. A. Likholobov and V. P. Ananikov, ChemCatChem, 2018, 10, 1869–1873 CrossRef CAS.
- X. Wang, X. Li, Y. Zhang and L. Xia, Org. Biomol. Chem., 2018, 16, 2860–2864 RSC.
- S. Mao, X. Shi, J.-F. Soulé and H. Doucet, Adv. Synth. Catal., 2018, 360, 3306–3317 CrossRef CAS.
- P. Arockiam, V. Poirier, C. Fischmeister, C. Bruneau and P. H. Dixneuf, Green Chem., 2009, 11, 1871 RSC.
- J. J. Dong, J. Roger, C. Verrier, T. Martin, R. Le Goff, C. Hoarau and H. Doucet, Green Chem., 2010, 12, 2053 RSC.
- A. Hfaiedh, K. Yuan, H. Ben Ammar, B. Ben Hassine, J.-F. Soulé and H. Doucet, ChemSusChem, 2015, 8, 1794–1804 CrossRef CAS PubMed.
- J. I. Martínez-Araya, J. Math. Chem., 2015, 53, 451–465 CrossRef.
- J. Oláh, C. Van Alsenoy and A. B. Sannigrahi, J. Phys. Chem. A, 2002, 106, 3885–3890 CrossRef.
- In Name Reactions for Homologations, Part I, ed. J. J. Li, Wiley & Sons, Inc., 2009, pp. 1–333 Search PubMed.
- E. Pretsch, P. Bulmann and C. Affolter, Structure determination of organic compounds, Springer-Verlag, Berlin, Heidelberg, 2000 Search PubMed.
- V. E. Iavorovskaia, A. S. Saratikov, I. V. Fedorov, A. N. Evstropov, R. G. Solianik, G. V. Anosova, E. V. Lepekhin and A. V. Portniagina, Exp. Clin. Pharmacol., 1998, 61, 51–53 CAS.
- A. S. Saratikov and V. E. Yavorovskaya, Pharm. Chem. J., 1997, 31, 333–334 CrossRef.
- N. A. Elkina, Y. V. Burgart, E. V. Shchegolkov, O. P. Krasnykh, V. V. Maslova, G. A. Triandafilova, S. S. Solodnikov, A. A. Muryleva, M. A. Misiurina, A. V. Slita, V. V. Zarubaev and V. I. Saloutin, J. Fluorine Chem., 2020, 240, 109648 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, Tables S1–S5, copies of NMR spectra. CCDC 2110385. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra06967e |
| This journal is © The Royal Society of Chemistry 2021 |