DOI:
10.1039/D1RA06943H
(Paper)
RSC Adv., 2021,
11, 36257-36264
Theoretical insight into hydroxyl production via H2O2 decomposition over the Fe3O4(311) surface†
Received
16th September 2021
, Accepted 2nd November 2021
First published on 10th November 2021
Abstract
Fenton's reagent provides a method to produce active hydroxyl radicals (˙OH) for chemical oxidation by mixing iron oxide and hydrogen peroxide, which divides into homogeneous and heterogeneous Fenton's reagent. Heterogeneous Fenton's reagent is fabricated from H2O2 and various iron oxide solid materials, such as α-FeOOH, α-Fe2O3, and Fe3O4. Fe3O4 possesses the Fe2+/Fe3+ mixed valence oxidational state and has been reported to have good catalytic activity. However, the reaction mechanism of H2O2 decomposition on Fe3O4 surfaces is still unclear. In this work, we performed DFT calculations to investigate the H2O2 decomposition mechanisms over the Fe3O4(311) surface. There are two iron environments for H2O2 adsorption and decomposition on the Fe3O4(311) surface, a Fe2+/Fe3+ environment and a Fe3+/Fe3+ environment. We found that the H2O2 can adsorb on the Fe2+/Fe3+ environment by molecular adsorption but by dissociative adsorption on the Fe3+/Fe3+ environment. Our results show that both adsorption structures can produce two OH groups on the Fe3O4(311) surface thermodynamically. In addition, based on the electronic property analysis, H2O2 on the Fe2+/Fe3+ environment follows the Haber–Weiss mechanism to form one OH anion and one OH radical. On the other hand, H2O2 on the Fe3+/Fe3+ environment follows the radical mechanism to form two OH radicals. In particular, the OH radical formed on Fe2+/Fe3+ has energy levels on both sides of the Fermi energy level. It can be expected that this OH radical has good redox activity.
1. Introduction
Fenton's reagent is a well-known advanced oxidation process (AOP) that can produce reactive oxygen species (ROS), such as hydroxyl radicals (˙OH), peroxyl radical (˙OOH), and superoxide radical (˙O2).1–4 Fenton's reagent is usually fabricated by mixing two solutions: a reducing transition metal and hydrogen peroxide. The Fenton-type reagent has been reported as an active catalyst for various reactions such as water treatment,5,6 methane partial oxidation, and mercury removal. Most AOPs can damage the organic compounds into H2O, CO2, and other small molecules by means of hydroxyl radicals (˙OH) because of their strong oxidative ability.7 As one of the well-known AOPs, the Fenton reagent shows extraordinary ability for organic decomposition and the least expensive. Moreover, the development of a Fenton reaction can accelerate the application of AOPs for water treatment and purification on an industrial scale.
Conventional homogeneous Fenton reagent composed of hydrogen peroxide (H2O2) and ferrous ion (Fe2+) for the purpose of producing hydroxyl radicals.8 However, there are several drawbacks to the homogeneous Fenton reagent.9 For example, homogeneous Fenton reagent needs to react in the narrow pH range, and the aggregation of iron-containing sludge in the reaction can also limit the recycling of catalysts, which can be considered as secondary pollution in the environment.10,11 Compared to the homogeneous Fenton systems, the heterogeneous iron-based solid catalysts can serve as the other choices for the Fenton reagents, which have been reported in various previous studies.12 Iron oxides-based catalysts, such as α-FeOOH, α-Fe2O3, Fe3O4, and FeO,13–17 possess good catalytic properties in many heterogeneous catalysis reactions, including Fenton reaction18 and Fischer–Tropsch synthesis.19 Theoretically, Song et al. has reported the NO oxidation mechanism over α-Fe2O3 catalyst by H2O2.20 Experimentally, Li et al. have studied the α-FeOOH material as the pCNB degradation catalyst by Fenton-like process.21 Furthermore, several investigations have found that the mixed valence oxidational state of iron atoms possess unique catalytic activity towards many heterogeneous catalysis reactions, including Fenton reaction, methane partial oxidation, and mercury oxidation.10,22–27 Among, magnetite (Fe3O4) is one of the iron oxide material that has a Fe2+/Fe3+ mix valence oxidational state. The bulk magnetite crystal structure was a member of the inverse cubic spinel group with the ferrimagnetic property.28,29 There are many applications of using magnetite as pigment due to its magnetic properties, such as polishing compounds, cosmetics, medicines, polymer, and rubber filler.30–32
There are still some challenges for heterogeneous Fenton reagents, such as that the yield to produce the OH radical is low. In addition, the mechanism to produce the OH radical via heterogeneously mixed valence iron oxide catalysts such as Fe3O4 of the Fenton reaction is unclear. In literature, there have been proposed two possible mechanisms for the traditional Fenton reaction. The first mechanism had been proposed by Haber and Weiss in the 1930s, named hydroxyl free radical mechanism (Haber–Weiss mechanism),8 which involves the formation of hydroxyl radicals (˙OH) in the system with the ferry (Fe2+) oxidation. This radical mechanism for the Fenton reaction with the following elementary reactions.
| Fe2+ + H2O2 → Fe3+ + ˙OH + OH− |
| Fe3+ + H2O2 → Fe2+ + ˙OOH + H+ |
Thereafter, another mechanism was proposed by Bray and Gorin in 1932,33 which generate higher valence Fe complexes such as ferryl (FeIV) ion in the Fenton system, as follows:
| Fe2+ + H2O2 → FeIVO + H2O |
| FeIVO + H2O2 → Fe2+ + H2O + O2 |
As a structure in the spinel family, Fe3O4 also possesses stable low-index surfaces, (100), (110), (111) facets,34–37 and a high-index surface of (311). Some studies have been reported the mercury oxidation mechanism by H2O2 over Fe3O4(001) and Fe3O4(111) surfaces.27,38 However, even though the low-index facets have relatively low surface energies, Xu et al. have successfully synthesized a polyhedral 50-facet Fe3O4 nanocrystal where only one high-index Fe3O4(311) facet can stably exist with other low-index facets jointly.39 In their results, there were even 24 high-index (311) facets in that nanocrystal, which is stable and abundant. Thus, Fe3O4(311) facet is a stable high-index surface experimentally. Because there was no investigation on the mechanism of the Fenton reaction over the Fe3O4(311) surface, in this work, we studied the adsorption and decomposition of H2O2 (Fenton reaction) over Fe3O4(311) surface using DFT calculations. We also investigated the possible reaction mechanisms of the Fenton reaction on the Fe3O4(311) surface. Moreover, we analyzed the electron density difference (EDD) and density of states (DOS) to explore the electronic interaction between H2O2/OH and Fe3O4(311) surface. To clarify the oxidational states of iron atoms between H2O2/OH and Fe3O4(311) surface, we also calculated the Bader charge and magnetic moments for the iron atoms on the Fe3O4(311) surface.
2. Computational details
The spin-polarized periodic DFT calculations were carried out with the Vienna ab initio simulation package (VASP 5.4.1).40–43 The generalized gradient approximation (GGA) with the functional proposed by Perdew, Burke and Enzerhof (PBE) exchange-correlation functional44 was used together with a plane-wave basis set with a kinetic cutoff energy of 500 eV. The electron ion–core interactions were described by the projector augmented wave (PAW) method.45,46 In order to correctly describe the itinerant or localized behavior of the Fe 3d-orbital, it is important to account for the strong on-site Coulomb correlations.47,48 Therefore, in this work, all systems were calculated by using the Hubbard correction term U (GGA + U). We applied the rotationally invariant approach proposed by Dudarev et al. with an effective Hubbard parameter of 4.0 eV for iron, which is the difference between the Coulomb (U) and exchange (J) parameters (Ueff = U − J). The optimized exchange-correlation functional (optB86b-vdW) was considered to determine the effect of van der Waals interactions.49
For the total energy calculations, the Brillouin zone integrations for the pristine and defect system were performed using the 2 × 2 × 1 based on the Monkhorst–Pack k-points scheme50 for all structural configuration relaxations. The convergence threshold was set to be 10−5 eV for electronic optimization and the force convergence was set to be 0.02 eV Å−1 for structural optimization. The adsorption energy (Eads) of adsorbate was defined as following equation:
| Eads = ETotal − Esurf. − EMolecule |
where
ETotal is the total energy of the surface together with the adsorbate,
Esurf. is the total energy of the surface, and
Emolecule is the total energy of the free molecule in gas-phase. Therefore, a negative adsorption energy illustrated a thermodynamically-favored exothermic adsorption process. An 8 × 8 × 1
k-point grid was used to calculate electronic density difference (EDD), Bader charge analysis, and density of state (DOS) with the same cutoff energy. The climbing image nudged elastic band (CI-NEB) method
51 and dimer method was applied to find transition states and minimum energy path of all reactions. The vibrational frequencies analysis was performed for validating the optimized and transition state structures.
The bulk Fe3O4 was adopted the conventional Fe3O4 unit cell comprising 56 atoms (Fe24O32). For the Fe3O4 bulk calculations, 8 × 8 × 8 k-mesh of a grid density was employed. The optimized lattice parameters of bulk Fe3O4 were a = b = c = 8.396 Å and α = β = γ = 90°. The result of the calculation is in good agreement well with the experimental data (a = b = c = 8.394 Å and α = β = γ = 90°).52 The bulk magnetite crystal structure was inverse cubic spinel with ferrimagnet property, the structure comprises the tetrahedral iron site and octahedral iron site with different oxidation number, and the O anions form a close-packed face-centered cubic (fcc) sublattice, as shown in Fig. S1(a).† Moreover, the tetrahedral and octahedral iron sites were performed by two magnetic sub-lattices in the opposite direction to form a typical inverse spinel structure as [Fe3+]tet[Fe3+, Fe2+]octO4. During the calculation, the initial atomic spin magnetic moments were ms = −5 μB and 4 μB for the tetrahedral iron and the octahedral iron sites, respectively. After structure optimization, the magnetic moment per formula unit was calculated to be 4.0 μB, which lies close to experiment data (4.05 μB).32 The magnetic moments of Fe3+ and Fe2+ in the tetrahedral iron and the octahedral iron atoms were with the antiparallel alignment, creating the ferrimagnetic character. Fig. S1(b)† shows the structure of the Fe3O4(311) surface by constructing four molecular layer models with the bottom two layers being fully fixed. The simulated XRD pattern of Fe3O4 bulk structure is drawn in Fig. S1(c),† which is in a good agreement with the experimental observation.53
3. Results and discussion
3.1 Adsorption of H2O2 molecule
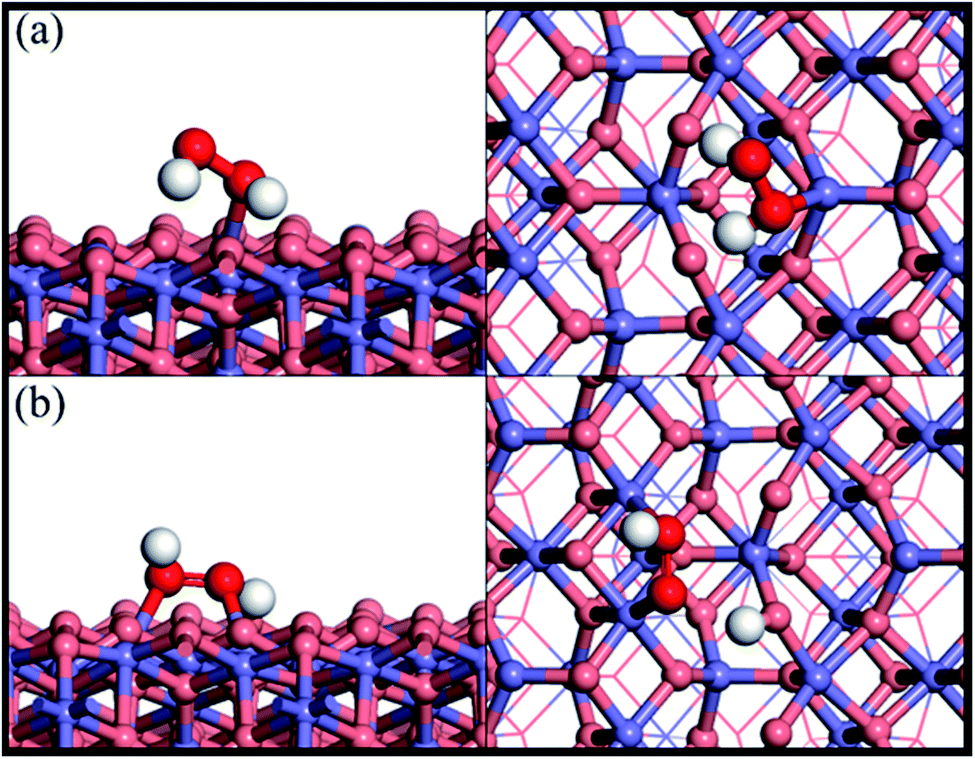
Fig. 1 shows the adsorption configuration of H2O2 on the Fe3O4(311) surface. There are two possible adsorption structures of H2O2 on the Fe3O4(311) surface: (1) molecular adsorption (Fig. 1(a)) and (2) dissociative adsorption (Fig. 1(b)). In the H2O2 molecular adsorption structure, the H2O2 adsorbs on the Fetet site via one OH to form the Fe–O bond, while the other OH end of H2O2 points to the lattice oxygen to form the hydrogen bond instead of forming the Fe–O bond between the adjacent Feoct site, as can be seen in Fig. 1(a). The adsorption energy of the H2O2 molecular adsorption is −1.17 eV. Moreover, the molecule geometry of H2O2 distorts seriously after adsorption that the two OH bonds within the molecule point to the different lattice oxygen atoms and form the hydrogen bonding with the distances of 1.66 Å and 1.47 Å, respectively. It results in the O–H bonds within the molecule elongated from 0.98 Å (free H2O2) to 1.016 and 1.061 Å, respectively. In the dissociative adsorption of H2O2, the H2O2 adsorbs on the two adjacent Feoct sites to form two Fe–O bonds, with the whole O–O bond being paralleled to the bridge of two octahedral iron sites, and one O–H bond directly dissociates to form OOH group, as can be seen in Fig. 1(b). The adsorption energy of H2O2 dissociative adsorption is −1.56 eV. The O–O bond of the OOH is elongated from 1.472 Å (in free H2O2) to 1.483 Å, as listed in Table 1.
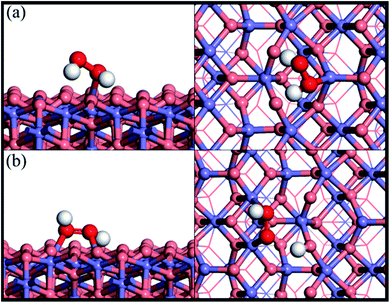 |
| | Fig. 1 Side and top views of optimized adsorption structures of H2O2 on the Fe3O4(311) surface by different adsorption configurations of (a) molecular adsorption and (b) dissociative adsorption. Purple, red, and white spheres represent Fe, O, and H atoms, respectively. The deep red color represents the oxygen of H2O2 while the light red color is the oxygen of Fe3O4(311) surface. | |
Table 1 The adsorption energy and geometrical parameters of H2O2 via different adsorption configurations on the Fe3O4(311) surface
| Adsorption type |
Eads (eV) |
O–O bond length (Å) |
O–H1 bond length (Å) |
O–H2 bond length (Å) |
| Gas phase |
— |
1.472 |
0.98 |
0.98 |
| Molecular adsorption |
−1.17 |
1.475 |
1.016 |
1.061 |
| Dissociate adsorption |
−1.57 |
1.483 |
2.135 |
0.983 |
3.2 The activation of H2O2 on Fe3O4
To understand the reaction mechanism of H2O2 decomposition, we calculated the energy barrier and reaction energy of each reaction step from both adsorption structures of H2O2 on the Fe3O4(311) surface. Two cleave aspects are considered in either molecular adsorption of H2O2 or dissociative adsorption of H2O2: (1) O–O bond-breaking into the OH group; (2) O–H bond-breaking into the OOH group. The following steps show the possible mechanisms for the molecular adsorption of H2O2 on the Fe3O4(311) surface:
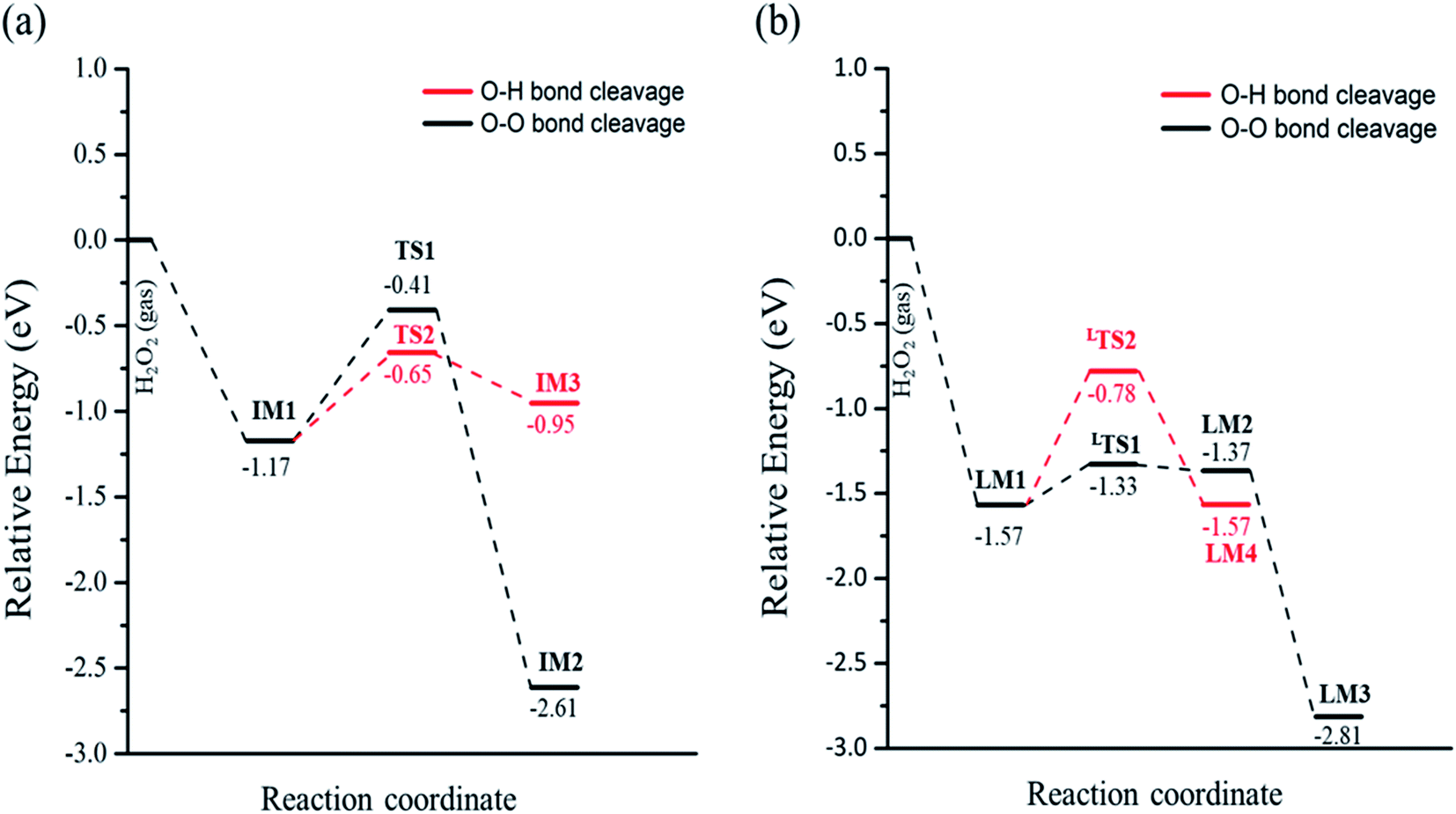
In eqn (1), the reaction barrier is 0.76 eV of that the H2O2 molecule cleaves the O–O bond to form two OH groups on one Fetet site and one Feoct site, and the reaction energy of this reaction is significant exothermic by 1.44 eV. Fig. S2(a)† depicts the structures of the initial state, transition state, and final state of this reaction. In eqn (2), the pathway starts from the deprotonation of H2O2 to deliver one hydrogen atom to the lattice oxygen, then forming an OOH group and the surface hydroxyl group. The oxygen atoms of the OOH group are separately adsorbed on both Fetet and Feoct, as shown in Fig. S2(b).† The calculated energy barrier of this reaction is 0.52 eV by a slight endothermicity of 0.22 eV. Comparing the pathways via O–O bond breaking and deprotonation, the reaction in the former is more thermodynamically favorable to produce the two OH groups, but it has larger activation energy for O–O bond scission, whereas the reaction in the latter possesses a smaller energy barrier to form the OOH group.
When the decomposition reaction starts from the dissociative adsorption of H2O2 (H + OOH), the reaction can also divide into O–O bond breaking and OH deprotonation reactions. The first pathway is the O–O bond decomposition of OOH to form O atom and OH group on two Feoct sites of the surface. The calculated energy barrier and reaction energy of this elementary step (OOH → O + OH) is 0.24 eV and 0.20 eV, respectively. The activated O–O bond length in the transition state structure is 1.70 Å, and the intermediate state will form a hydrogen bonding interaction between the O atom and OH group with a distance of 1.86 Å after dissociation, as shown in Fig. S3(a).† However, the dissociated O atom is unstable so that this O atom can attract the atomic H of the surface hydroxyl group to form another OH group on the Feoct2 site via the hydrogen bonding networks, which results in a large exothermic reaction by 1.44 eV due to the stability of forming two OH groups. The second pathway is that deprotonation from OOH to transfer one H atom to the lattice oxygen to form dioxygen on the two octahedral iron sites, as shown in Fig. S3(b).† This process is a thermal neutral process with an activation energy of 0.79 eV. This result demonstrates that the most favorable pathway from the decomposition reaction of the dissociative adsorption of H2O2 is to produce two OH groups on the Fe3O4(311) surface. Table 2 summarizes the calculated reaction energy and activated barriers of the elementary steps of the O–O bond and O–H bond dissociation of H2O2 on Fe3O4(311) surface. Fig. 2 depicts the potential energy profiles for H2O2 decomposition on different sites over Fe3O4(311) surface. Our results show that it is thermodynamically favorable to produce the two hydroxyl groups on the Fe3O4(311) surface from both molecular adsorption and dissociative adsorption.
Table 2 Calculated reaction barriers (Ea in eV), reaction energies (ΔE in eV), and imaginary frequencies (IMF, cm−1) for elementary reactions of H2O2 decomposition on the Fe3O4(311) surface
| Elementary steps |
Ea |
ΔE |
IMF (cm−1) |
| Molecular adsorption |
| H2O2(gas) → H2O2* |
— |
−1.17 |
|
| H2O2* → 2OH* |
0.76 |
−1.44 |
i424 |
| H2O2* → H* + OOH* |
0.52 |
0.22 |
i493 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
| Dissociate adsorption |
| H2O2*(gas) → H* + OOH* |
— |
−1.57 |
|
| H* + OOH* → H* + O* + OH* |
0.24 |
0.20 |
i641 |
| H* + O* + OH*→ 2OH* |
— |
−1.44 |
|
| H* + OOH* → OO* + 2H* |
0.79 |
0.002 |
i943 |
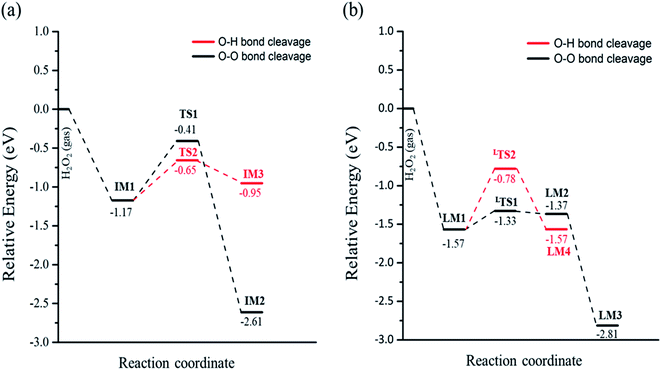 |
| | Fig. 2 The potential energy profiles of the decomposition of H2O2 on the Fe3O4(311) surface via (a) H2O2 molecular adsorption and (b) H2O2 dissociative adsorption. | |
3.3 Solvent effect
To realistically simulate the effect of the catalytic environment on Fenton's reaction, we carried out the implicit solvent model by using VASPsol.54,55 The implicit solvent effect of water was adopted in this work, where the corresponding dielectric constant of water was 78.3553. As listed in Table S1,† the reaction barriers of either O–O bond breaking or O–H bond breaking of the H2O2 molecule in an aqueous solution become smaller than that of the H2O2 molecule in the vacuum, which is contributed by the solvent effects. Besides, the OH groups production is still the most thermodynamically favorable pathway via O–O bond breaking of the H2O2 on the Fe3O4(311) surface, as shown in Fig. S4.† Thus, with/without the solvent effect, the reaction trend of H2O2 decomposition on the Fe3O4(311) surface is similar.
3.4 Electronic property analysis
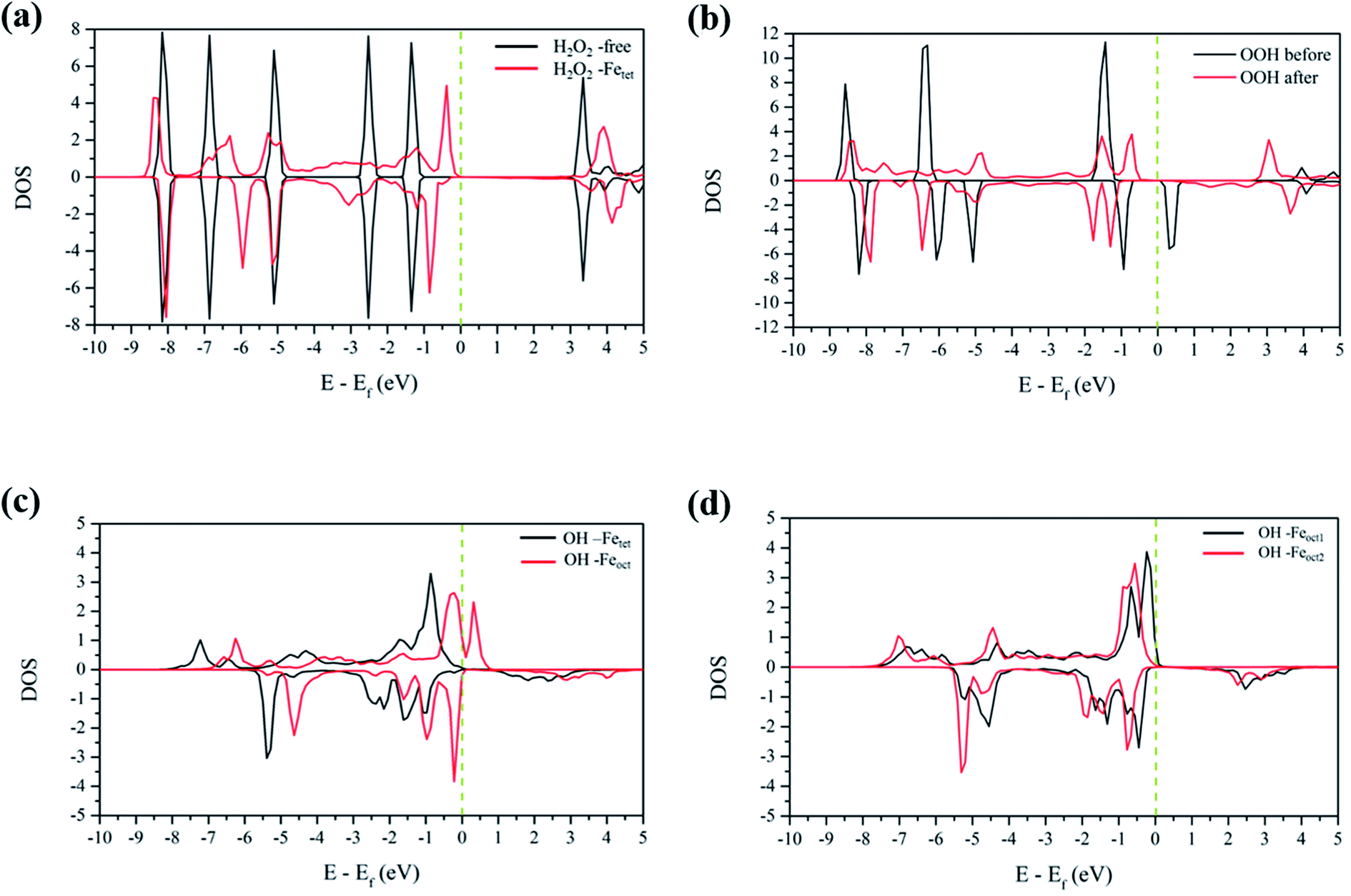
3.4.1 H2O2 molecular adsorption. To analyze the interactions between H2O2 and Fe atoms upon adsorption on Fe3O4(311) surface, we calculate the electron density difference (EDD) and density of states (DOS). Fig. 3(a) and (b)† show the different views of 2D EDD contour plots of H2O2 molecular adsorption. Fig. 3(a) illustrates the accumulation of electron region between the OH group and Fetet site, representing that the oxygen donates electrons to the iron atom to form a dative bond between oxygen and iron atom. On the other hand, Fig. 3(b) shows the cutting plane along with the other H atom and the O–O bond (Ht–O–O), which displays the electron density accumulation (red contour) between the OH group and the lattice O. This EDD plot in Fig. 3(b) indicates that the H2O2 donates the electron transfer from the lattice O to the OH group via the hydrogen bonding interaction. In addition, Fig. 4(a) shows the PDOS diagram of the H2O2 molecule before and after its adsorption. One can observe that the spin up and spin down of the peaks of H2O2 is getting splitting after adsorption. Moreover, as can be seen in Fig. S5(a) and (b),† the projected d orbitals of Fetet and Feoct atoms show that the d orbitals of Fetet have obviously changed, and the peak is getting broad. In comparison, the peaks of the Feoct changed slightly after H2O2 adsorption. Additionally, the d orbitals of the Fetet atom have an overlap with the p orbitals of H2O2 around −0.50 eV, reflecting the dative bond between H2O2 and Fetet atom.
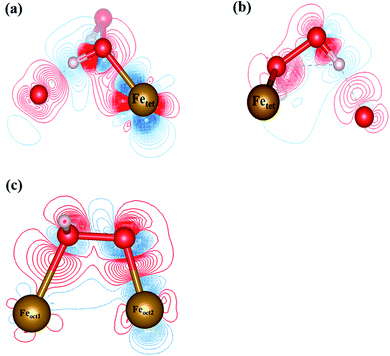 |
| | Fig. 3 The 2D electron density difference diagrams of (a) Fetet–OH–O plane, and (b) Fetet–O–O plane of H2O2 molecular adsorption, and (c) H2O2 dissociative adsorption. The isosurface level is 0.002|e|/bohr3. Brown, red, and white spheres represent Fe, O, and H atoms, respectively. | |
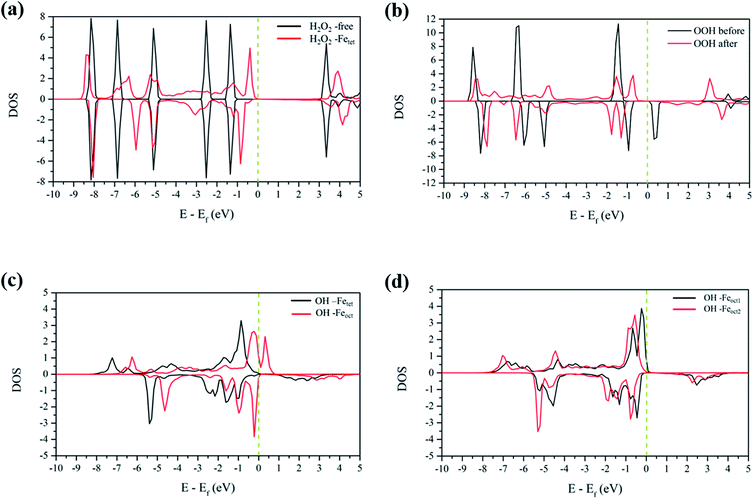 |
| | Fig. 4 The PDOS plots of (a) H2O2 before and after molecular adsorption, (b) H2O2 before and after dissociative adsorption, (c) OH groups from the decomposition of H2O2 molecular adsorption, and (d) OH groups from the decomposition of H2O2 dissociative adsorption. The dotted line is the Fermi level. | |
3.4.2 H2O2 dissociative adsorption. Fig. 3(c) shows the 2D EDD plot of the OOH group of the H2O2 dissociative adsorption configuration, and the cutting plane is along the bridge between two Feoct atoms and the O–O bond (Feoct–O–O–Feoct). The blue contour around the OOH group reveals that the OOH group loses electrons electron. There is also a broad electron density accumulation (red contour) around the bound OOH and two Feoct atoms, and the region around the OOH group, which indicates the strong electron interactions between Fe atoms and the OOH group. The strong electronic interactions mean that the OOH can form a dative chemical bond with the Fe atoms.Further, the PDOS plots, as shown in Fig. 4(b), demonstrated that the peaks of the OOH (from the dissociative adsorption of H2O2) has a significant splitting on both spin up and spin down, reflecting the strong radical property of the OOH group. The peaks around −1.50 eV and −1.00 eV belong to α-HOMO and β-HOMO, respectively, as well as the peak around 0.50 eV represents the LUMO of the OOH group before adsorption (black line in Fig. 4(b)). For the dissociative adsorption configuration of H2O2, the peaks of the OOH group (red line in Fig. 4(a)) display a broad region above Fermi level energy to 3.00 eV, but the peaks around 0.50 eV disappear. It reflects that the OOH group can gain electrons from the Fe atoms. After dissociative adsorption configuration of H2O2, the peaks still show a notable splitting on both spin up and spin down, representing the strong radical property of the OOH group. Besides, as can be seen in Fig. S5(c) and (d),† the d orbital of the Feoct2 atom shows a more remarkable change than that of the Feoct1 atom before and after OOH group formation, indicating the strong interaction between the Feoct2 atom and the OOH group.
3.4.3 OH on the Fetet/Feoct of the Fe3O4(311) surface. According to the calculated results, both molecular adsorption of H2O2 and dissociative adsorption of H2O2 can produce two OH groups on either Feoct or Fetet atoms of the Fe3O4(311) surface. Because Fenton's reaction involves the redox reaction, we did the electronic property analysis to understand the electronic and magnetic properties after H2O2 decomposition. Fig. S6(a) and (b)† show the EDD plots of the adsorption of the OH group on the Feoct and Fetet sites after H2O2 decomposition, respectively. It shows wide regions of electron density accumulation (red lines) between the OH group and either Fetet or Feoct atoms, which shows the electron transferred from the OH group to the Fe atoms. Fig. 4(c) represents the PDOS of the OH groups on the Fetet and Feoct sites after H2O2 decomposition. For the OH on the Feoct atom, there are peaks with similar intensity on both sides of the Fermi level, indicating that the oxygen atom on OH has unpaired electrons after H2O2 decomposition. Since the peaks are close to the Fermi level, we can expect that the OH group on the Feoct atom should be very active. Besides, it can be observed from Fig. S7(a)† that the DOS distribution of Fetet changes significantly before and after adsorption, whereas from Fig. S7(b)† that the DOS distribution of Feoct is very similar after H2O2 decomposition. The changes of the DOS distribution of Fetet implies that Fetet2+ might have been oxidized to Fetet3+.To further understand the redox reaction before and after H2O2 decomposition on the Fe3O4(311) surface, we calculated the Bader charge and magnetic moments of iron ions and the fragments of H2O2 during the reaction. The calculated Bader charge values and magnetic moment values of iron ions are similar before and after the adsorption of H2O2. After the adsorption of H2O2, the calculated Bader charge values of Fetet and Feoct are around 1.36 and 1.67|e|, respectively, and the calculated magnetic moment values of both Fetet and Feoct atoms are around 3.804 and 4.243 μB, respectively. The results indicate that the oxidational state of Fetet and Feoct atoms upon H2O2 adsorption is Fe2+ and Fe3+, respectively. Furthermore, after the H2O2 decomposition to OH groups, the calculated Bader charge value of both Fetet and Feoct atoms become 1.69 and 1.71|e|, respectively, and the calculated magnetic moment of both Fetet and Feoct atoms become 4.249 and 4.234 μB, respectively, as listed in Table 3. The changes in Bader charge and the magnetic moment suggest that the Fetet is oxidized from Fe2+ to Fe3+, while the Feoct is maintained at Fe3+. The calculated results indicate that the OH group on the Fetet will be reduced to the OH− anion, while the one on the Feoct will be an OH radical, which is consistent with the prediction of DOS analysis. Therefore, we predict that the Fetet and Feoct atoms may be the ferrous ion (Fe2+) and ferric ion (Fe3+) initially and then the Fetet can be oxidized to Fe3+ after H2O2 decomposition, in agreement with the prediction from PDOS analysis. As a result, we predict that this redox mechanism follows the Haber–Weiss mechanism of the Fenton's reaction as the following equations:
| Fetet2+ + ˙OH → Fetet3+ + OH− |
| Feoct3+ + ˙OH → Feoct3+ + ˙OH |
Table 3 Calculated Bader charge and magnetization for Fe atoms, H2O2, and OH groups before and after H2O2 decomposition. The positive and negative values of the Bader charge represent the loss and gain electrons, respectively
| Species |
Bader charge |
Magnetization |
Species |
Bader charge |
magnetization |
| Free surface |
|
|
Free surface |
|
|
| Fetet |
1.32 |
3.782 |
Feoct1 |
1.68 |
4.248 |
| Feoct |
1.67 |
4.240 |
Feoct2 |
1.69 |
4.262 |
| Molecular adsorption |
|
|
Dissociative adsorption |
|
|
| Fetet |
1.36 |
3.804 |
Feoct1 |
1.73 |
4.258 |
| Feoct |
1.67 |
4.243 |
Feoct2 |
1.71 |
4.276 |
| H2O2 |
−0.012 |
— |
H–OOH |
−0.09 |
— |
| OH on the Fetet/Feoct |
|
|
OH on the Feoct1/Feoct2 |
|
|
| Fetet |
1.69 |
4.249 |
Feoct1 |
1.69 |
4.258 |
| Feoct |
1.71 |
4.234 |
Feoct2 |
1.70 |
4.278 |
| OH–Fetet |
−0.47 |
— |
OH–Feoct1 |
−0.50 |
— |
| OH–Feoct |
−0.51 |
— |
OH–Feoct2 |
−0.45 |
— |
3.4.4 OH on the Feoct1/Feoct2 of the Fe3O4(311) surface. After the decomposition of the H2O2 to two OH groups on the Feoct1 and Feoct2 atoms, we also investigated the electronic interaction between these OH groups and the Feoct atoms via EDD, PDOS, Bader charge and magnetization analyses. Fig. S6(c) and (d)† show the EDD contour plots from the different perspective cutting planes of these two OH groups. These EDD plots display a wide region with electron density accumulation (red color) between the OH group and either Feoct1 or Feoct2 atoms, demonstrating the OH group has a strong interaction with either Feoct1 or Feoct2 atoms. In Fig. 4(d), the peaks of OH groups show a little split, which also denotes the radical character for these OH groups. Besides, the PDOS of the d orbital of either Feoct1 atom or Feoct2 atom display only slightly changes before and after H2O2 decomposition, as shown in Fig. S7(c) and (d).† This result implies that the redox reactivity between OH groups and Feoct1/Feoct2 atoms is weak.The calculated Bader charge values have slight changes on both Feoct1 and Feoct2 atoms before and after O–O decomposition, where the value of Feoct1 atom changes from 1.68|e| to 1.73|e|, and the value of Feoct2 atom from 1.69|e| becomes 1.71|e|. The calculated magnetic moment values of both Feoct1 and Feoct2 atoms remain around 4.25 μB before and after O–O decomposition. These results indicate that both Feoct1 and Feoct2 atoms are the ferric ions (Fe3+) initially and the Fe atoms will get hardly further oxidized after forming the OH group on these Fe atoms. Thus, the OH groups on these ferric ions (Fe3+) also possess a radical character after H2O2 decomposition. The calculated Bader charge and magnetic moment values are listed in Table 3.
Finally, in a comparison of two mechanisms of H2O2 decomposition on the Fe3O4(311) surface, we found that it can occur a redox reaction between H2O2 and Fe3O4(311) surface when H2O2 adsorbed on both Fetet and Feoct sites. The mechanism obeys the Haber–Weiss mechanism to produce one OH anion and one OH radical, which has a strong redox activity. On the other hand, while H2O2 adsorbed on two Feoct sites, H2O2 can dissociate into two OH radicals without oxidizing the Feoct atoms, which is the radical mechanism of H2O2 decomposition. Based on these results, we could predict that the active OH radicals would be the surface abundant species on the Fe3O4(311) surface. Moreover, the calculated desorption energies of OH radicals are 2.84 eV and 2.94 eV at Fetet2+/Feoct3+ and Feoct3+/Feoct3+ sites, respectively. The desorption energy indicated that the hydroxyl radicals might be challenging to desorb from the Fe3O4(311) surface. Therefore, in our study, we predict that the surface OH groups might go through surface heterogeneous oxidation reactions.
4. Conclusions
In this work, we have employed DFT calculations to study the H2O2 adsorption and the decomposition mechanism on the Fe3O4(311) surface. Fe3O4(311) surface possesses a mixed valence (Fe2+/Fe3+) state so that there are two possible adsorption sites for H2O2 adsorption. When H2O2 adsorbs on the Fetet2+/Feoct3+ sites, the adsorption structure is molecular adsorption, whereas the adsorption structure of H2O2 on Feoct3+/Feoct3+ sites becomes dissociative adsorption (OOH + H). We found that the molecular adsorption of H2O2 can proceed O–H or O–O bond scission to form OOH or 2OH on the Fe3O4(311) surface, respectively. The O–H and O–O bond dissociation is kinetic control and thermodynamic control of the decomposition reaction for the molecular adsorption of H2O2, respectively. On the contrary, the dissociative adsorption of H2O2 only prefers to go through the O–O bond scission to produce 2OH on the Fe3O4(311) surface with a small energy barrier of 0.24 eV. In addition, according to various electronic property calculations, DOS, EDD, Bader charge, and magnetic moments, we observed that the reaction from H2O2 to 2OH on the Fetet2+/Feoct3+ sites obeys the Haber–Weiss mechanism of the Fenton reaction. The Fetet2+ site can be oxidized to Fetet3+ while the other Feoct3+ site maintains its original oxidation state. Thus, the 2OH formed from the molecular adsorption of H2O2 is a composition of OH anion and OH radical. On the contrary, the 2OH produced from the dissociative adsorption of H2O2 is two radicals because the Feoct3+/Feoct3+ sites did not change their oxidation states, which belongs to the radical mechanism. Compared to two mechanisms, it can produce an active OH radical on the surface via either the Haber–Weiss mechanism or radical mechanism, where the oxidation state of Fe on the Fe3O4(311) surface can stay at Fe3+ instead of further oxidation. As a result, our results predict that these OH radicals from the Fenton-like reaction of H2O2 on the Fe3O4(311) surface might be a candidate of oxidant for other heterogeneous catalysis reactions.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully acknowledge the financial support for this work from the grant 106-0210-02-11-05 of Executive Yuan, Republic of China, Taiwan; 107-2113-M-011-003-MY3 and 109-2113-M-035-003-MY3 of Ministry of Science and Technology (MOST). The National Center of High-Performance Computing (NCHC) contributed to this project by allowing access to their computer facilities and donating computer time.
References
- R.-S. Juang, S.-H. Lin and T.-Y. Wang, Chemosphere, 2003, 53, 1221–1228 CrossRef CAS.
- B. Durham, M. M. Bourbigot and T. Pankratz, Desalination, 2001, 138, 83–90 CrossRef CAS.
- F. Fu and Q. Wang, J. Environ. Manage., 2011, 92, 407–418 CrossRef CAS PubMed.
- I. Zinicovscaia, in Cyanobacteria for Bioremediation of Wastewaters, ed. I. Zinicovscaia and L. Cepoi, Springer International Publishing, Cham, 2016, pp. 17–25, DOI:10.1007/978-3-319-26751-7_3.
- W. H. Glaze, Environ. Sci. Technol., 1987, 21, 224–230 CrossRef CAS PubMed.
- C. P. Huang, C. Dong and Z. Tang, J. Waste Manage., 1993, 13, 361–377 CrossRef CAS.
- K. Keyer, A. S. Gort and J. A. Imlay, J. Bacteriol., 1995, 177, 6782 CrossRef CAS PubMed.
- F. Haber and J. Weiss, Naturwissenschaften, 1932, 20, 948–950 CrossRef CAS.
- Y. F. Huang and Y. H. Huang, J. Hazard. Mater., 2009, 162, 1211–1216 CrossRef CAS PubMed.
- Y. Yao, L. Wang, L. Sun, S. Zhu, Z. Huang, Y. Mao, W. Lu and W. Chen, Chem. Eng. Sci., 2013, 101, 424–431 CrossRef CAS.
- M. Zhou, Q. Yu, L. Lei and G. Barton, Sep. Purif. Technol., 2007, 57, 380–387 CrossRef CAS.
- S. Rahim Pouran, A. A. Abdul Raman and W. M. A. Wan Daud, J. Cleaner Prod., 2014, 64, 24–35 CrossRef CAS.
- Z. Ai, Y. Cheng, L. Zhang and J. Qiu, Environ. Sci. Technol., 2008, 42, 6955–6960 CrossRef CAS PubMed.
- R. Huang, Z. Fang, X. Yan and W. Cheng, Chem. Eng. J., 2012, 197, 242–249 CrossRef CAS.
- K. Rajeshwar, M. E. Osugi, W. Chanmanee, C. R. Chenthamarakshan, M. V. B. Zanoni, P. Kajitvichyanukul and R. Krishnan-Ayer, J. Photochem. Photobiol., C, 2008, 9, 171–192 CrossRef CAS.
- T. Shahwan, S. Abu Sirriah, M. Nairat, E. Boyacı, A. E. Eroğlu, T. B. Scott and K. R. Hallam, Chem. Eng. J., 2011, 172, 258–266 CrossRef CAS.
- Y. Zhao, H. Jiangyong and H. Chen, J. Photochem. Photobiol., A, 2010, 212, 94–100 CrossRef CAS.
- M. C. Pereira, L. C. A. Oliveira and E. Murad, Clay Miner., 2018, 47, 285–302 CrossRef.
- H. Almkhelfe, X. Li, P. Thapa, K. L. Hohn and P. B. Amama, J. Catal., 2018, 361, 278–289 CrossRef CAS.
- Z. Song, B. Wang, J. Yu, C. Ma, C. Zhou, T. Chen, Q. Yan, K. Wang and L. Sun, Appl. Surf. Sci., 2017, 413, 292–301 CrossRef CAS.
- X. Li, Y. Huang, C. Li, J. Shen and Y. Deng, Chem. Eng. J., 2015, 260, 28–36 CrossRef CAS.
- X.-X. Ji, H.-F. Wang and P.-J. Hu, Rare Met., 2018, 38, 783–792 CrossRef.
- M. I. Litter and M. Slodowicz, J. Adv. Oxid. Technol., 2017, 20, 20160164 CAS.
- J. I. Nieto-Juarez and T. Kohn, Photochem. Photobiol. Sci., 2013, 12, 1596–1605 CrossRef CAS PubMed.
- A. Szecsenyi, G. Li, J. Gascon and E. A. Pidko, ACS Catal., 2018, 8, 7961–7972 CrossRef CAS PubMed.
- R. B. Wang and A. Hellman, J. Chem. Phys., 2018, 148, 094705 CrossRef.
- C. Zhou, H. Yang, D. Qi, J. Sun, J. Chen, Z. Zhang, L. Mao, Z. Song and L. Sun, Fuel, 2018, 216, 513–520 CrossRef CAS.
- M. B. Robin and P. Day, in Advances in Inorganic Chemistry and Radiochemistry, ed. H. J. Emeléus and A. G. Sharpe, Academic Press, 1968, vol. 10, pp. 247–422 Search PubMed.
- J. M. D. Coey, M. Viret and S. von Molnár, Adv. Phys., 1999, 48, 167–293 CrossRef CAS.
- H. Wei and E. Wang, Anal. Chem., 2008, 80, 2250–2254 CrossRef CAS PubMed.
- J. Ahdjoudj, C. Martinsky, C. Minot, M. A. Van Hove and G. A. Somorjai, Surf. Sci., 1999, 443, 133–153 CrossRef CAS.
- D. Santos-Carballal, A. Roldan, R. Grau-Crespo and N. H. de Leeuw, Phys. Chem. Chem. Phys., 2014, 16, 21082–21097 RSC.
- W. C. Bray and M. H. Gorin, J. Am. Chem. Soc., 1932, 54, 2124–2125 CrossRef CAS.
- R. Gargallo-Caballero, L. Martin-Garcia, A. Quesada, C. Granados-Miralles, M. Foerster, L. Aballe, R. Bliem, G. S. Parkinson, P. Blaha, J. F. Marco and J. de la Figuera, J. Chem. Phys., 2016, 144, 094704 CrossRef PubMed.
- X. Li and J. Paier, J. Phys. Chem. C, 2016, 120, 1056–1065 CrossRef CAS.
- D. Santos-Carballal, A. Roldan, R. Grau-Crespo and N. H. de Leeuw, Phys. Chem. Chem. Phys., 2014, 16, 21082–21097 RSC.
- T. Yang, X.-d. Wen, J. Ren, Y.-w. Li, J.-g. Wang and C.-f. Huo, J. Fuel Chem. Technol., 2010, 38, 121–128 CrossRef CAS.
- C. Zhou, B. Wang, Z. Song, C. Ma, J. Yu, Z. Zhang, H. Yang and L. Sun, Fuel, 2017, 202, 318–327 CrossRef CAS.
- Y. Xu, H. Hao, P. Liu, Q. Wang, Y. Sun and G. Zhang, CrystEngComm, 2014, 16, 10451–10459 RSC.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- T. K. Ng and P. A. Lee, Phys. Rev. Lett., 1988, 61, 1768–1771 CrossRef PubMed.
- R. Zimmermann, P. Steiner, R. Claessen, F. Reinert, S. Hüfner, P. Blaha and P. Dufek, J. Phys.: Condens. Matter, 1999, 11, 1657–1682 CrossRef CAS.
- J. Klimeš, D. R. Bowler and A. Michaelides, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 195131 CrossRef.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- N. T. H. Kim-Ngan, W. Soszka and A. Kozłowski, J. Magn. Magn. Mater., 2004, 279, 125–133 CrossRef CAS.
- H. Shagholani, S. M. Ghoreishi and M. Mousazadeh, Int. J. Biol. Macromol., 2015, 78, 130–136 CrossRef CAS PubMed.
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. A. Arias and R. G. Hennig, J. Chem. Phys., 2014, 140, 084106 CrossRef PubMed.
- K. Mathew, V. S. C. Kolluru, S. Mula, S. N. Steinmann and R. G. Hennig, J. Chem. Phys., 2019, 151, 234101 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra06943h |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b and
Jyh-Chiang Jiang
*b and
Jyh-Chiang Jiang