
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExperimental and theoretical study on converting myoglobin into a stable domain-swapped dimer by utilizing a tight hydrogen bond network at the hinge region†
Cheng Xiea,
Hiromitsu Shimoyama b,
Masaru Yamanakaa,
Satoshi Nagao‡
a,
Hirofumi Komoric,
Naoki Shibatad,
Yoshiki Higuchid,
Yasuteru Shigeta*b and
Shun Hirota*a
b,
Masaru Yamanakaa,
Satoshi Nagao‡
a,
Hirofumi Komoric,
Naoki Shibatad,
Yoshiki Higuchid,
Yasuteru Shigeta*b and
Shun Hirota*a
aDivision of Materials Science, Graduate School of Science and Technology, Nara Institute of Science and Technology, 8916-5 Takayama, Ikoma, Nara 630-0192, Japan. E-mail: hirota@ms.naist.jp
bDivision of Life Science, Center for Computational Sciences, University of Tsukuba, 1-1-1, Tennodai, Ibaraki, 305-8577, Japan. E-mail: shigeta@ccs.tsukuba.ac.jp
cFaculty of Education, Kagawa University, 1-1 Saiwai-cho, Takamatsu, Kagawa 760-8522, Japan
dGraduate School of Science, University of Hyogo, 3-2-1 Koto, Kamigori-cho, Ako-gun, Hyogo 678-1297, Japan
First published on 23rd November 2021
Abstract
Various factors, such as helical propensity and hydrogen bonds, control protein structures. A frequently used model protein, myoglobin (Mb), can perform 3D domain swapping, in which the loop at the hinge region is converted to a helical structure in the dimer. We have previously succeeded in obtaining monomer–dimer equilibrium in the native state by introducing a high α-helical propensity residue, Ala, to the hinge region. In this study, we focused on another factor that governs the protein structure, hydrogen bonding. X-ray crystal structures and thermodynamic studies showed that the myoglobin dimer was stabilized over the monomer when keeping His82 to interact with Lys79 and Asp141 through water moleclues and mutating Leu137, which was located close to the H-bond network at the dimer hinge region, to a hydrophilic amino acid (Glu or Asp). Molecular dynamics simulation studies confirmed that the number of H-bonds increased and the α-helices at the hinge region became more rigid for mutants with a tighter H-bond network, supporting the hypothesis that the myoglobin dimer is stabilized when the H-bond network at the hinge region is enhanced. This demonstrates the importance and utility of hydrogen bonds for designing a protein dimer from its monomer with 3D domain swapping.
1 Introduction
In nature, many proteins adopt oligomeric forms for biological functions. Research activity on oligomeric protein design has increased; for example, various shaped oligomeric proteins have been constructed using fusion proteins1–3 and metal coordination.4–6 Proteins with a specific structural motif have been constructed with the assistance of computational design.7–9 Moreover, the H-bond network helps drive the protein to the folded state10,11 and determines its mechanical and thermodynamic properties.12 Many experimental approaches, including molecular dynamics (MD) simulation, have revealed the dynamic and structural properties of biological water at protein interfaces.13–19 A better understanding of the effect of H-bonds on protein structures will provide deeper understanding of the principles of protein structure and will be useful for the design of oligomeric proteins.Three-dimensional domain swapping (3D-DS) was described in diphtheria toxin by Eisenberg and coworkers in 1994.20 In 3D-DS, one or more identical structural elements of a protein molecule exchange between molecules, forming dimers or oligomers.20–22 Some 3D-DS proteins are relevant to biologically related species23 and disease-related aggregation.24,25 3D-DS structures are characterized by no specific properties, such as chain length, structural class, or amino acid composition.21,26 This suggests that almost any protein has the potential to form a 3D-DS structure. The insertion of three amino acids into the middle of a 3D-DS homodimeric archaeal histone fold motif resulted in a soluble and stable monomer.27 Several attempts have also been made to construct a stable 3D-DS dimer from a monomer by shortening the hinge region.28,29
We have previously shown that horse myoglobin (Mb) can form a 3D-DS dimer, in which the hinge region K77–H82 (KKKGHH; K3GH2) is converted from a loop to a helical structure connecting the neighboring E and F α-helices (Fig. S1†).30 A heterodimer with different active sites can be obtained by controlling the electrostatic interaction between the protomers in the dimer.31 To increase the helical propensity at the hinge region, we mutated residues 80, 81, and 82 of wild type (WT) Mb to Ala (K3A3 Mb), which resulted in an increase in its 3D-DS tendency.32,33 However, H82 was involved in the H-bond network with K79 and D141 in the WT Mb 3D-DS dimer (Fig. 1), whereas this H-bond network was not observed in the 3D-DS dimer of K3A3 Mb. It has been deduced that the H-bond at the hinge region stabilizes the monomer structure against the 3D-DS dimer in Human Cellular Retinol Binding Protein II.34 On the other hand, the 3D-DS dimer structure has been stabilized by forming an anti-parallel β-sheet at the hinge region between protomers with insertion of a QVVAG motif,35 whereas loop deletion and insertion of a polyproline rod to the outer-surface protein A from Borrelia stabilized the 3D-DS structure by formation of the polyproline II structure at the hinge region.36 In this study, to investigate the effect of the H-bond network constructed at the hinge loop containing amino acid residues and water molecules on 3D-DS, we introduced two Ala residues at G80 and H81 but retained H82 to interact with K79 and D141 through water molecules at the hinge region (G80A/H81A (K3A2H) Mb). Additionally, L137 is located relatively close to the H-bond network in the WT Mb 3D-DS dimer; thus, L137 was additionally mutated to a hydrophilic amino acid: Glu and Asp (G80A/H81A/L137E (K3A2H-L137E) Mb and G80A/H81A/L137D (K3A2H-L137D) Mb). The dimer-to-monomer ratio increased in the order WT Mb < K3A2H Mb < K3A2H-L137E Mb < K3A2H-L137D Mb, according to the enhancement of the H-bond network at the hinge region, demonstrating the importance of the H-bond network for the stabilization of the α-helices at the hinge region and the 3D-DS dimer.
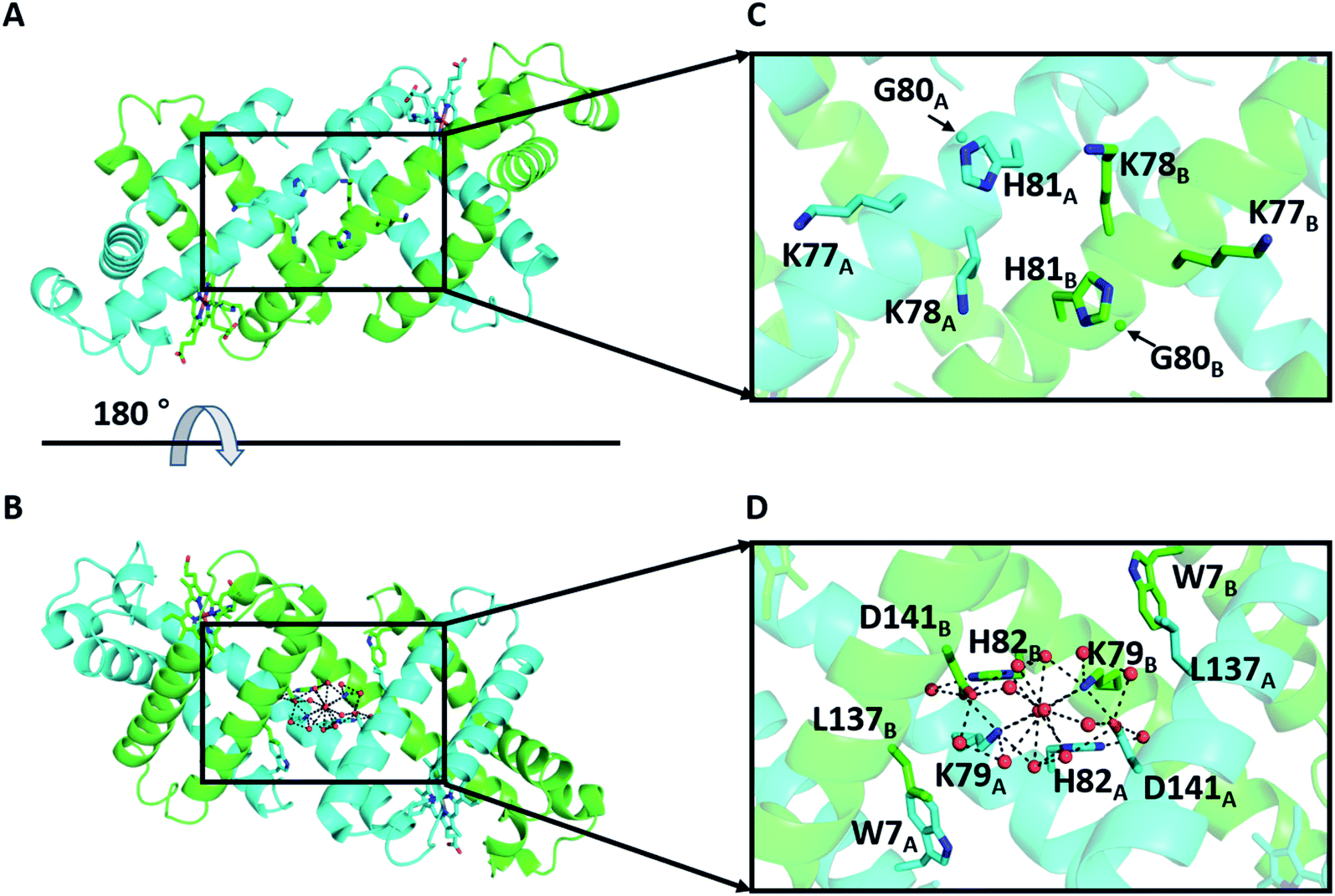 | ||
| Fig. 1 X-ray crystal structure of the WT Mb 3D-DS dimer (PDB ID: 3VM9): (A and B) overall structure and (C and D) enlarged view of the hinge region K77–H82. (B) is a 180°-rotated view of (A). The two protomers are shown in green and cyan. The side-chain atoms of residues W7, K77, K78, K79, H81, H82, and L137 and the hemes are shown as stick models. The Cα atoms of G80 are depicted as spheres. Water molecules are depicted as red spheres. The H-bond network involving K79, H82, D141, and water molecules in the hinge region is shown as dotted lines. The nitrogen and oxygen atoms of the hemes and the side chains of the stick-model residues are depicted in blue and red, respectively. | ||
2 Materials and methods
2.1 Preparation of Mb mutants
WT horse Mb was purchased from Sigma-Aldrich (Saint Louis, MO, USA). Its K3A2H, K3A2H-L137E, and K3A2H-L137D Mb mutant genes were constructed using primers (Eurofins Genomics) with the KOD Plus Mutagenesis Kit (Toyobo, Japan) and confirmed by DNA sequencing (ABI PRISM 310 genetic analyzer sequencing system, Applied Biosystems, Inc.). The primers used to generate the mutations are listed in Table S1†. Mb mutants were expressed in Escherichia coli LE392 cells. All Mb mutants were obtained as a mixture of monomer and dimer using a procedure similar to that described previously.37 The monomers and dimers of K3A2H, K3A2H-L137E, and K3A2H-L137D Mb were purified by size exclusion chromatography (SEC; HiLoad 26/60 Superdex 75 pg, GE Healthcare) using a fast protein liquid chromatography (FPLC) system (Biologic DuoFlow 10, Bio-Rad) with 50 mM potassium phosphate buffer, pH 7.0, at 4 °C. The Mb concentration was adjusted by the Soret band intensity at 408 nm.2.2 Analysis of the monomer and dimer ratio of Mb mutants
Monomer and dimer solutions (100 μM, in heme unit) of WT and mutant Mbs were heated at 70 °C for 30 min. The K3A2H Mb monomer solution (100 μM) was also heated at 63.5 °C, 65.4 °C, 67.5 °C, and 69.5 °C for 930 min, 750 min, 120 min, and 90 min, respectively, and the K3A2H-L137E and K3A2H-L137D Mb monomer solutions (5 μM) were heated at 67.2 °C, 69.5 °C, 71.3 °C, and 73.1 °C for 480 min, 150 min, 60 min, and 30 min, respectively. No precipitation was observed during heating for all mutants. The amount of the monomers and dimers in the heated Mb protein solution was analyzed by SEC (HiLoad 16/60 Superdex 75 pg, GE Healthcare) using the FPLC system (Biologic DuoFlow 10) with 50 mM potassium phosphate buffer, pH 7.0, at 4 °C. The ratios of Mb monomer and dimer at various temperatures were determined from the peak areas in the elution curves of SEC. The areas were obtained by least-square fitting of the peaks with Gaussian curves using the Igor Pro 6.0 software (WaveMetrics, Portland).2.3 Optical absorption and circular dichroism measurements
The absorption spectra of Mb monomers and dimers in their met forms were measured with a UV-2450 spectrophotometer (Shimadzu, Japan) using a 10 mm path-length quartz cell at 25 °C. Circular dichroism (CD) spectra of monomers and dimers of WT and mutant Mbs in their met forms were measured with a J-820 circular dichroism spectropolarimeter (Jasco, Japan) using a 1 mm path-length quartz cell at 25 °C. CD ellipticity changes with temperature at 222 nm for monomers and dimers of WT and mutant Mbs in their met forms were measured with a J-820 circular dichroism spectropolarimeter (Jasco) using a 1 mm path-length quartz cell. The scan rate was 0.25, 0.5, or 1 °C min−1.2.4 X-ray crystallography
Crystallization was carried out at 277 K using the sitting drop vapor diffusion method with crystal plates (CrystalEX Second Generation Corning 3552, Hampton Research, CA, USA). The protein concentration was adjusted to 17 mg mL−1 in 50 mM potassium phosphate buffer, pH 7.0. Droplets prepared by mixing 1 μL of protein solution with 1 μL of reservoir solution were equilibrated. The best reservoir solutions were found to be 0.1 M Tris–HCl buffer, pH 7.0, containing 0.1 M sodium acetate and 10% (w/v) PEG 6000 for the K3A2H Mb dimer; 0.1 M Tris–HCl buffer, pH 7.0, containing 0.1 M sodium acetate, 10% (w/v) PEG 6000, and 5% (w/v) PEG 8000 for K3A2H-L137E Mb dimer; and 0.1 M Tris–HCl buffer, pH 7.0, containing 0.1 M sodium acetate, 10% (w/v) PEG 6000, and 5% (v/v) PEG 200 for the K3A2H-L137D Mb dimer.The diffraction data of K3A2H, K3A2H-L137E, and K3A2H-L137D Mb dimers were collected at the BL38B1, BL41XU, and BL45XU beamlines, respectively, at SPring-8, Japan. The crystals were mounted on cryo-loops and flash-frozen at 100 K in a nitrogen cryo system. The diffraction data were collected automatically using the ZOO system,38 and an automatic data process was performed using the KAMO system.39 The preliminary structure was obtained by a molecular replacement method (MOLREP)40 using the atomic coordinates of the dimer structure of WT horse Mb (PDB code: 3VM9) for all Mb dimers. The structure refinements were performed using the program REFMAC5.41 The molecular model was manually corrected, and water molecules were picked up in the electron density map using the program COOT.42 The data collection and refinement statistics are summarized in Table S2†.
2.5 Molecular dynamics simulations of Mb
MD simulations were performed for WT, K3A2H, K3A2H-L137E, and K3A2H-L137D Mb dimers. The standard force fields of the proteins, the heme, and the water molecule (TIP3P43) were obtained from CHARMM-GUI.44–46 The box size was taken to be 1203 Å3, and the periodic boundary condition was used. The particle mesh Ewald method47,48 with cutoff = 10 Å was used for Coulomb interaction calculations. The total charge of the system was neutralized by adding Na+ ions (Na+ parameter was σ = 1.9623 Å and ε = 0.24299 kJ mol−1). The Lennard-Jones potential with cutoff = 12 Å was used for van der Waals interactions. After energy minimizations and 1.25 ns (Δt = 1 fs) of equilibration simulations with position-restraints, 10 ns (Δt = 2 fs) of production simulations were performed for the production run. Each simulation was started at the corresponding crystal structure. To accelerate the simulation, all chemical bonds including hydrogens were constrained by the LINCS49 method. The temperature of these simulations was maintained by Nose–Hoover thermostat50 so that T = 300 K. The pressure was controlled at 1 bar using a Parrinello–Rahman barostat.51 The trajectory after 3 ns was used for statistical analyses. The trajectory was divided every 1 ns, and the error bar of a physical quantity X was estimated from the eight ensembles as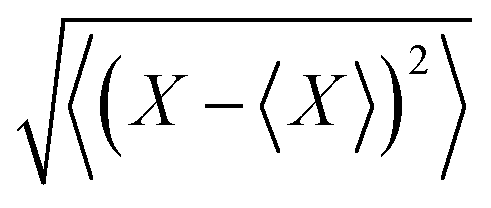 (〈⋯〉 indicates the ensemble average).
(〈⋯〉 indicates the ensemble average).
The distance between the center-of-mass coordinates of residues 77–82 in chains A and B were calculated; the centroid was defined as follows:
 | (1) |
3 Results and discussion
K3A2H, K3A2H-L137E, and K3A2H-L137D Mb were purified, and the molar extinction coefficients at 408 nm of their monomers and dimers in the met forms were determined by the hemochrome method,52 where the values were similar between the monomer and dimer for each species (Table S3†). The stability of the dimer was compared among WT, K3A2H, K3A2H-L137E, and K3A2H-L137D Mb by heating each monomer and dimer solution at 70 °C for 30 min and subsequently analyzing the solution with SEC. All of the dimers of WT Mb dissociated to monomers upon heating, whereas no dimer formed from its monomer (Fig. S2†). The dimer-to-monomer ratios after heating the monomer and dimer solutions were similar for K3A2H Mb, as well as for K3A2H-L137E and K3A2H-L137D Mb, demonstrating that the Mb mutants were under monomer–dimer equilibrium after heating. Dimer-to-monomer ratios of 92, 97, and 98% were obtained after heating K3A2H, K3A2H-L137E, and K3A2H-L137D Mb, respectively, indicating stabilization of the dimer by the mutation. Although the helical propensity of Ala is higher than that of His,32 the dimer-to-monomer ratio of K3A2H Mb was higher than that of K3A3 Mb (Fig. S2†), demonstrating the importance of other factors on the stabilization of the dimer.The dimer-to-monomer ratios at equilibriums for K3A2H, K3A2H-L137E, and K3A2H-L137D Mb were obtained at various temperatures from the monomer and dimer peak areas in the elution curves of SEC (Table. S4†). The equilibrium constant Keq and the Gibbs free energy change ΔGM–D for the monomer-to-dimer conversion of K3A2H, K3A2H-L137E, and K3A2H-L137D Mb were obtained at various temperatures using eqn (2) and (3),
| Keq = [dimer]eq/[monomer]eq2 | (2) |
ΔGM–D = −RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnKeq lnKeq
| (3) |
Keq vs. 1/T) (Fig. 2). The ΔHM–D and ΔSM–D values were both negative, indicating that the 3D-DS dimerization of the mutant Mbs was enthalpically favorable but entropically unfavorable, similar to the characteristics of the G80A (K3AH2) and K3A3 Mb mutants.33 The enthalpy gain and entropy loss on the 3D-DS dimerization of these mutants can be explained by the increased stability of the α-helical structure and decreased flexibility of the dimer structure with the enhanced H-bond network in the hinge region (see text below). Concerning the heat capacity change ΔCp between the monomer and dimer, we could not obtained accurate values due to the limited temperature range. However, ΔCp may be small, since the three dimensional structures are similar between the monomer and dimer of mutant Mbs except for the hinge region.
| K3A2H | K3A2H-L137E | K3A2H-L137D | |
|---|---|---|---|
| a Values at 342.7 K.b The temperature range for the calculation of ΔHM–D and ΔSM–D is 336.7–342.7 K for K3A2H Mb and 340.4–346.3 K for K3A2H-L137E and K3A2H-L137D Mb. | |||
| Keqa (M−1) | (9.2 ± 0.8) × 105 | (9.4 ± 0.8) × 106 | (2.3 ± 0.2) × 107 |
| ΔGM–Da (kcal mol−1) | −9.3 ± 0.1 | −10.9 ± 0.1 | −11.5 ± 0.1 |
| ΔHM–Db (kcal mol−1) | −30.8 ± 1.1 | −63.2 ± 5.1 | −100.5 ± 4.0 |
| ΔSM–Db (cal mol−1) | −62.7 ± 3.1 | −152.8 ± 14.9 | −259.4 ± 11.7 |
 | ||
| Fig. 2 van't Hoff plot of mutant Mbs. Experimental conditions: Mb concentration (heme unit), 100 μM for K3A2H Mb, and 5 μM for K3A2H-L137E and K3A2H-L137D Mb; 50 mM potassium phosphate buffer, pH 7.0; temperature, 63.5, 65.4, 67.5, and 69.5 °C for K3A2H Mb, and 67.2, 69.5, 71.3, and 73.1 °C for K3A2H-L137E and K3A2H-L137D Mb. | ||
The optical absorption spectra of the monomer and dimer of K3A2H, K3A2H-L137E, and K3A2H-L137D Mb in the met form were all similar to each other and similar to those of the WT Mb monomer and dimer (Fig. S3†), indicating that the mutation and dimerization did not significantly alter the heme coordination structure. The overall CD spectra of the Mb monomer and dimer of the mutants in the met form were also similar to the corresponding spectra of WT Mb (Fig. S4†), indicating that the mutations did not perturb the structure of the Mb monomer and dimer significantly. However, the difference in the CD ellipticity between the monomer and dimer was slightly larger for K3A2H-L137E and K3A2H-L137D Mb than for WT and K3A2H Mb, indicating that the helicity increased in solution slightly more for the conversion of the monomer to dimer in K3A2H-L137E and K3A2H-L137D Mb than in WT and K3A2H Mb.
The denaturation temperature was 84, 81, 82, and 81 °C for WT, K3A2H, K3A2H-L137E, and K3A2H-L137D Mb in the met form, respectively, according to the CD ellipticity changes at 222 nm with increasing temperature (Fig. 3), indicating similar stabilities between WT and mutant Mbs. The intensity of the CD ellipticity increased relatively rapidly from ∼65 °C for the WT Mb dimer until it overlapped with the ellipticity of the WT Mb monomer at ∼75 °C, which we attributed to the dissociation of the dimers to monomers by heating (Fig. 3A). A slight ellipticity change was observed for the K3A2H Mb dimer at 76–78 °C, which may correspond to the conversion of the dimers to monomers (Fig. 3B), owing to the stabilization of the dimer (Fig. 3). Dissociation of the dimer was not observed for K3A2H-L137E and K3A2H-L137D Mb; instead, a clear conversion of monomers to dimers was observed at 68 and 67 °C, respectively (Fig. 3C and D), corresponding to the high stability of the dimers (Fig. S2† and Table 1). When we decreased the temperature increase rate in the measurement for K3A2H-L137E and K3A2H-L137D Mb, more monomers converted to dimers at lower temperatures, indicating that the monomer-to-dimer conversion is a relatively slow process (Fig. S5†).
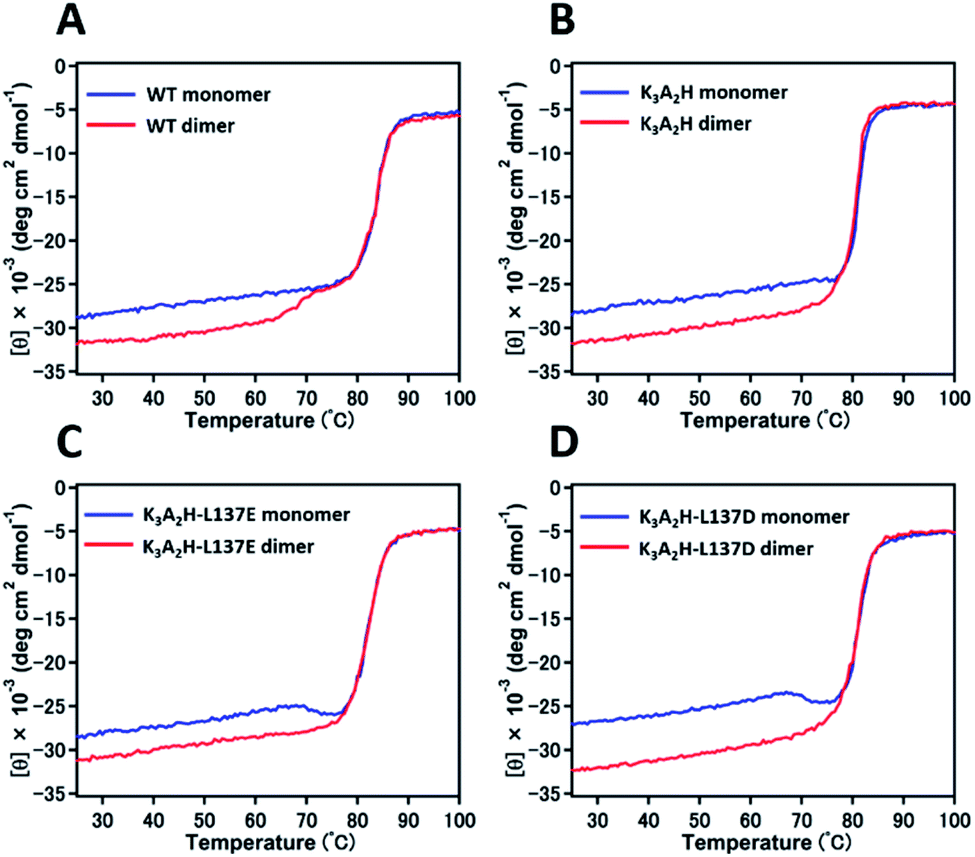 | ||
| Fig. 3 Changes in CD ellipticity at 222 nm with temperature increase for WT and mutant Mbs in the met form: (A) WT, (B) K3A2H, (C) K3A2H-L137E, and (D) K3A2H-L137D Mb. The changes in the Mb monomer (blue) and dimer (red) are shown. Measurement conditions: Mb concentration (heme unit), 30–33 μM; 50 mM potassium phosphate buffer, pH 7.0; path-length, 1 mm; temperature, 25–100 °C; scan rate, 1 °C min−1. | ||
The X-ray crystal structures were determined at 1.16 Å (PDB ID: 7V5P), 1.38 Å (PDB ID: 7V5Q), and 1.39 Å (PDB ID: 7V5R) resolution, respectively (Fig. 4 and Table S2†). All dimers exhibited similar overall structures to that of the WT Mb 3D-DS dimer (PDB: 3VM9, Fig. S6†), and all structures contained an H-bond network at the hinge region involving K79, H82, D141, and water molecules, with most H-bond distances in the range 2.5–3.2 Å (Fig. S7†). E137 interacted with W7 via water molecules in the K3A2H-L137E Mb dimer (Fig. 4E), whereas a new hydrogen bond was formed between K79 and D137 in the K3A2H-L137D Mb dimer, extending the H-bond network from that of WT and K3A2H Mb (Fig. 4F and S8†). The extended H-bond network with more water molecules may stabilize the helical structure at the hinge region and thereby stabilizes the 3D-DS dimer.
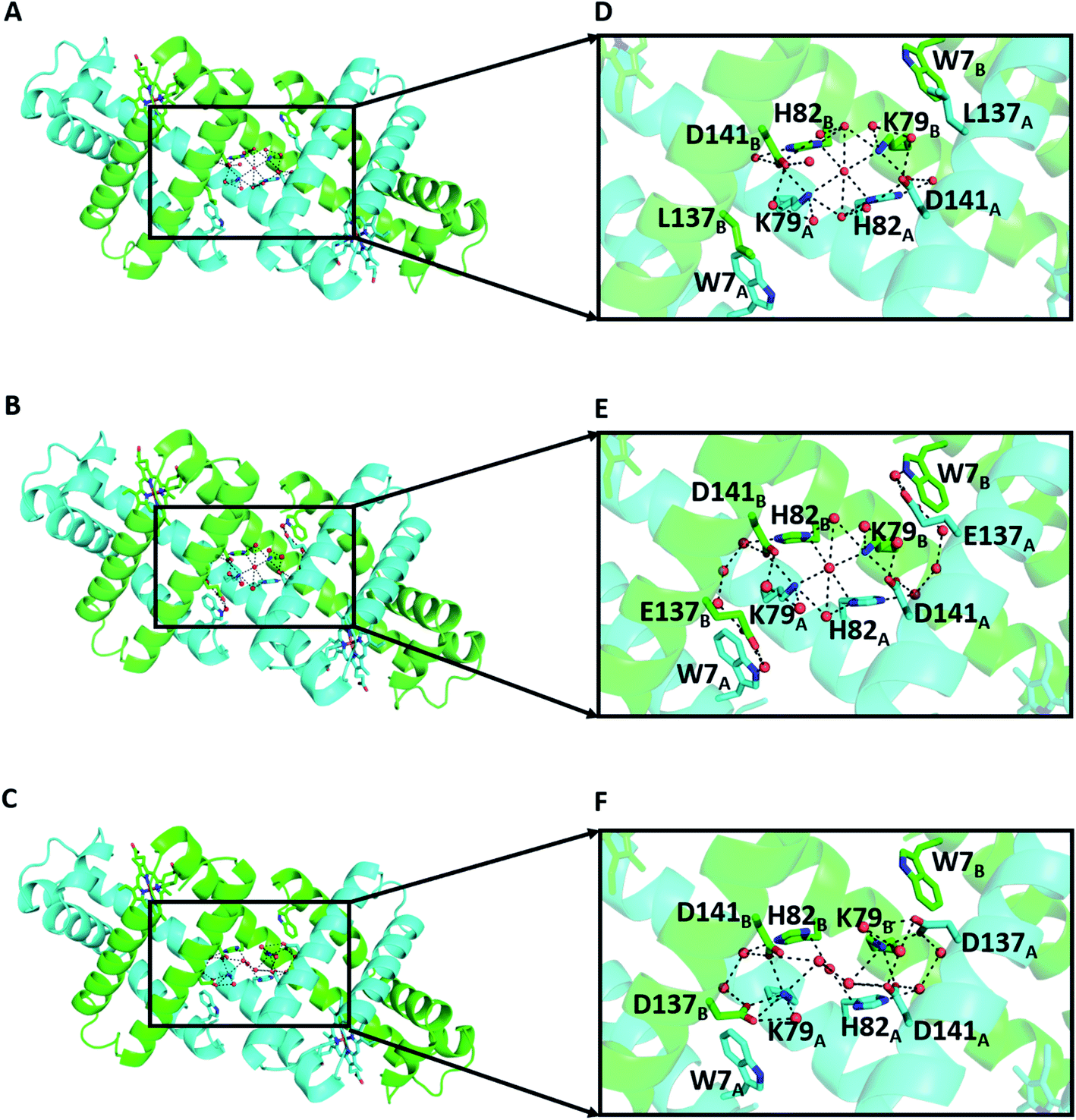 | ||
| Fig. 4 X-ray crystal structures of the mutant Mb 3D-DS dimers: (A) K3A2H (PDB ID: 7V5P), (B) K3A2H-L137E (PDB ID: 7V5Q), and (C) K3A2H-L137D Mb (PDB ID: 7V5R). (D), (E), and (F) are enlarged views of the hinge regions of (A), (B), and (C), respectively. The representations of the amino acids are the same as those in Fig. 1. | ||
The obtained X-ray crystal structures are snapshots of energetically stable structures, and the observation of water molecules may depend on the resolution of the crystal structure. To verify the structures and investigate the H-bonds in solution, we analyzed the number of H-bonds formed for the specific amino acid residues in Fig. 4, i.e., W7, K79, H82, L/D/E137, and D141, by MD simulation (Fig. S9†). The H-bonds of these amino acid residues with H-bond mediating water molecules within cutoff = 7 Å were included. WT, K3A2H, K3A2H-L137E, and K3A2H-L137D Mb 3D-DS dimers contained approximately 72, 85, 94, and 97 H-bonds, respectively, at the hinge region (Fig. 5A). The stability of the Mb dimer increased as the number of H-bonds at the hinge region increased, owing to the inhibition of the dimer-to-monomer conversion by the tight H-bond network. The order of the dimer-to-monomer ratio (Fig. S2†) correlated well with that of the number of H-bonds (WT Mb < K3A2H Mb < K3A2H-L137E Mb < K3A2H-L137D Mb). From the experiments, thermal conversion of the dimer to monomers was observed for WT and K3A2H Mb dimers, whereas the conversion of the monomers to dimer was observed for K3A2H-L137E and K3A2H-L137D Mb (Fig. 3). This difference could be attributed to the difference in the monomer and dimer stability at approximately 65–75 °C. Since the K3A2H-L137E(D) Mb dimer apparently contains more H-bonds than the WT and K3A2H Mb dimers, the K3A2H-L137E(D) Mb dimer may be stabilized over the monomer though the H-bond network but not much for the WT and K3A2H Mb dimers.
 | ||
| Fig. 5 MD simulation for WT (black), K3A2H (red), K3A2H -L137E (blue), and K3A2H -L137D Mb dimers (magenta). (A) Population of the number of H-bonds among W7, K79, H82, L/D/E137, D141, and surrounding water molecules. The H-bonds within 7 Å measured from these amino acid residues were counted considering the tight H-bond network. (B) Population of the centroid distance. The definition of the centroid is described in the Materials and methods section. | ||
To investigate the effect of the H-bonds on the α-helical structure at the hinge region, we calculated the histogram of the distance between the center-of-mass coordinates of residues 77–82 in chains A and B in the dimer during MD simulation, which is referred to as the centroid distance (Fig. 5B). The centroid distances in WT, K3A2H, K3H2A-L137E, and K3H2A-D137 Mb dimers in the crystal structures were 7.6, 7.9, 7.9, and 7.8 Å, respectively. These values were in good agreement with the distances of the (shorter distance) peaks obtained by MD simulation except for the WT Mb dimer (Fig. 5B). For the WT Mb dimer, the distance was about 9 Å, which was longer by 2 Å than that obtained from the X-ray crystal structure. The shorter distance in the crystal structure may be attributed to the stabilization of the crystal structure to the most stable structure, while the protein structure fluctuates in solution, making the centroid distances shorter and similar among WT and mutant Mbs in the crystals. In the MD simulation, the same tendency as the number of H-bonds was found for the centroid distance. The WT Mb dimer showed longer centroid distances than the K3A2H Mb dimer, indicating greater compactness of the K3A2H Mb dimer at the hinge region with approximately 15 more H-bonds (Fig. 5A). For the K3A2H-L137E(D) Mb dimer, the centroid distance was very stable with shorter distances: 8.08 Å (K3A2H-L137E Mb) and 7.95 Å (K3A2H-L137D Mb), supporting the hypothesis of a tight H-bond network in the K3A2H-L137E(D) Mb dimer. For the WT and K3A2H Mb dimers that have broader distribution, the centroid distance negatively correlated with the number of H-bonds; correlation coefficient, WT Mb: −0.81; K3A2H Mb: −0.70 (Fig. S10†). The centroid distance was distributed in the range of 8–11.5 Å and 7.5–11 Å for the WT and K3A2H Mb dimers, respectively, both with an ∼3.5 Å distribution, which was approximately half of the cutoff = 7 Å. The decrease in the centroid distance in the K3A2H Mb dimer could be attributed to the increase in the number of H-bonds included in the tight H-bond network, supporting the hypothesis that the H-bond network is an important driving force for the 3D-DS dimerization of Mb.
4 Conclusions
Experimental and theoretical analyses of WT and mutant Mbs revealed that the 3D-DS dimer is stabilized when the H-bond network at the hinge region is enhanced. Thermodynamic and CD studies showed that the WT Mb dimer dissociates to monomers upon heating at ∼70 °C, whereas K3A2H-L137E and K3A2H-L137D Mb monomers are converted to dimers by the heating. X-ray crystal structures revealed that the H-bond network is enhanced in the order WT < K3A2H < K3A2H-L137E ≈ K3A2H-L137D Mb. In the crystal structure of the K3A2H-L137E Mb dimer, the mutated E137 interacted with W7, whereas the mutated D137 interacted with K79 in the K3A2H-L137D Mb dimer, both enhancing the H-bond network. The MD simulations revealed that the number of H-bonds increases in the order WT < K3A2H < K3A2H-L137E < K3A2H-L137D Mb. It also indicated that the distance between the centroids of the two helices in the hinge region decreases in the same order. These results provide additional evidence for the hypothesis that the 3D-DS dimer becomes more stable compared to the monomer as the H-bond network at the hinge region of the dimer is enhanced in Mb. This study provides a new perspective on utilizing hydrogen bonds in the design of stable domain-swapped oligomers.Author contributions
S. H. designed the project. C. X. performed the experiments with the help of M. Y. H. S. performed the MD simulations. C. X. and S. H. analyzed the spectroscopic and kinetic data. C. X., S. N., H. K., N. S., Y. H., and S. H. analyzed the X-ray crystal structures. H. S and Y. S. analyzed the MD simulation data. C. X., H. S., Y. S., and S. H. wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by Grants-in-Aid from JSPS for Scientific Research (B) (No. JP26288080 and No. JP21H02060 (S. H.)), Scientific Research (C) (No. JP19K05695 (S. N.)), and Innovative Researches “Hydrogeomics” and “Biometal Sciences” (No. JP19H05047 and No. JP20H05497 (S. Y.)), and by JST CREST, Japan (No. JP20338388 (S. H.)). C. X. acknowledges the support from the China Scholarship Council (201708350126) for his PhD studies. We are grateful to the staff at beamlines BL38B1, BL41XU, and BL45XU, SPring-8, Japan (Proposal No. 2018A2722, 2019B2719, and 2021A2732). Numerical calculations were performed using Cygnus at the CCS, University of Tsukuba.Notes and references
- Y. T. Lai, D. Cascio and T. O. Yeates, Science, 2012, 336, 1129 CrossRef CAS PubMed.
- N. Kobayashi, K. Yanase, T. Sato, S. Unzai, M. H. Hecht and R. Arai, J. Am. Chem. Soc., 2015, 137, 11285–11293 CrossRef CAS.
- A. Sciore, M. Su, P. Koldewey, J. D. Eschweiler, K. A. Diffley, B. M. Linhares, B. T. Ruotolo, J. C. Bardwell, G. Skiniotis and E. N. Marsh, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 8681–8686 CrossRef CAS PubMed.
- S. Biswas, K. Kinbara, T. Niwa, H. Taguchi, N. Ishii, S. Watanabe, K. Miyata, K. Kataoka and T. Aida, Nat. Chem., 2013, 5, 613–620 CrossRef CAS PubMed.
- Y. Bai, Q. Luo, W. Zhang, L. Miao, J. Xu, H. Li and J. Liu, J. Am. Chem. Soc., 2013, 135, 10966–10969 CrossRef CAS PubMed.
- J. B. Bailey, R. H. Subramanian, L. A. Churchfield and F. A. Tezcan, Methods Enzymol., 2016, 580, 223–250 CAS.
- N. P. King, J. B. Bale, W. Sheffler, D. E. McNamara, S. Gonen, T. Gonen, T. O. Yeates and D. Baker, Nature, 2014, 510, 103–108 CrossRef CAS.
- P. Lu, D. Min, F. DiMaio, K. Y. Wei, M. D. Vahey, S. E. Boyken, Z. Chen, J. A. Fallas, G. Ueda, W. Sheffler, V. K. Mulligan, W. Xu, J. U. Bowie and D. Baker, Science, 2018, 359, 1042–1046 CrossRef CAS.
- F. Pirro, N. Schmidt, J. Lincoff, Z. X. Widel, N. F. Polizzi, L. Liu, M. J. Therien, M. Grabe, M. Chino, A. Lombardi and W. F. DeGrado, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 33246–33253 CrossRef CAS.
- D. F. Stickle, L. G. Presta, K. A. Dill and G. D. Rose, J. Mol. Biol., 1992, 226, 1143–1159 CrossRef CAS.
- G. D. Rose and R. Wolfenden, Annu. Rev. Biophys. Biomol. Struct., 1993, 22, 381–415 CrossRef CAS PubMed.
- D. R. Livesay, D. H. Huynh, S. Dallakyan and D. J. Jacobs, Chem. Cent. J., 2008, 2, 17 CrossRef PubMed.
- Y. Levy and J. N. Onuchic, Annu. Rev. Biophys. Biomol. Struct., 2006, 35, 389–415 CrossRef PubMed.
- M. C. Bellissent-Funel, A. Hassanali, M. Havenith, R. Henchman, P. Pohl, F. Sterpone, D. van der Spoel, Y. Xu and A. E. Garcia, Chem. Rev., 2016, 116, 7673–7697 CrossRef.
- D. Laage, T. Elsaesser and J. T. Hynes, Chem. Rev., 2017, 117, 10694–10725 CrossRef.
- S. Bandyopadhyay, S. Chakraborty and B. Bagchi, J. Am. Chem. Soc., 2005, 127, 16660–16667 CrossRef.
- K. Shiraga, Y. Ogawa and N. Kondo, Biophys. J., 2016, 111, 2629–2641 CrossRef.
- Y. Qin, L. Wang and D. Zhong, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 8424–8429 CrossRef.
- W. W. Park, K. M. Lee, B. S. Lee, Y. J. Kim, S. H. Joo, S. K. Kwak, T. H. Yoo and O. H. Kwon, Angew. Chem., Int. Ed. Engl., 2020, 59, 7089–7096 CrossRef CAS PubMed.
- M. J. Bennett, S. Choe and D. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 3127–3131 CrossRef PubMed.
- Y. Liu and D. Eisenberg, Protein Sci., 2002, 11, 1285–1299 CrossRef.
- M. P. Schlunegger, M. J. Bennett and D. Eisenberg, Adv. Protein Chem., 1997, 50, 61–122 CrossRef.
- V. Cafaro, C. De Lorenzo, R. Piccoli, A. Bracale, M. R. Mastronicola, A. Di Donato and G. D'Alessio, FEBS Lett., 1995, 359, 31–34 CrossRef.
- A. Sanders, C. J. Craven, L. D. Higgins, S. Giannini, M. J. Conroy, A. M. Hounslow, J. P. Waltho and R. A. Staniforth, J. Mol. Biol., 2004, 336, 165–178 CrossRef PubMed.
- M. Nilsson, X. Wang, S. Rodziewicz-Motowidlo, R. Janowski, V. Lindstrom, P. Onnerfjord, G. Westermark, Z. Grzonka, M. Jaskolski and A. Grubb, J. Biol. Chem., 2004, 279, 24236–24245 CrossRef.
- L. Liu, I. J. Byeon, I. Bahar and A. M. Gronenborn, J. Am. Chem. Soc., 2012, 134, 4229–4235 CrossRef CAS PubMed.
- M. Hadjithomas and E. N. Moudrianakis, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 13462–13467 CrossRef CAS.
- N. Nandwani, P. Surana, J. B. Udgaonkar, R. Das and S. Gosavi, Protein Sci., 2017, 26, 1994–2002 CrossRef PubMed.
- F. Rousseau, J. W. Schymkowitz and L. S. Itzhaki, Structure, 2003, 11, 243–251 CrossRef.
- S. Nagao, H. Osuka, T. Yamada, T. Uni, Y. Shomura, K. Imai, Y. Higuchi and S. Hirota, Dalton Trans., 2012, 41, 11378–11385 RSC.
- Y. W. Lin, S. Nagao, M. Zhang, Y. Shomura, Y. Higuchi and S. Hirota, Angew. Chem., Int. Ed., 2015, 54, 511–515 Search PubMed.
- J. W. Bryson, S. F. Betz, H. S. Lu, D. J. Suich, H. X. Zhou, K. T. O'Neil and W. F. DeGrado, Science, 1995, 270, 935–941 CrossRef.
- S. Nagao, A. Suda, H. Kobayashi, N. Shibata, Y. Higuchi and S. Hirota, Chem. Asian. J., 2020, 15, 1743–1749 CrossRef PubMed.
- Z. Assar, Z. Nossoni, W. Wang, E. M. Santos, K. Kramer, C. McCornack, C. Vasileiou, B. Borhan and J. H. Geiger, Structure, 2016, 24, 1590–1598 CrossRef CAS PubMed.
- N. Nandwani, P. Surana, H. Negi, N. M. Mascarenhas, J. B. Udgaonkar, R. Das and S. Gosavi, Nat. Commun., 2019, 10, 452 CrossRef.
- S. Shiga, M. Yamanaka, W. Fujiwara, S. Hirota, S. Goda and K. Makabe, Chembiochem, 2019, 20, 2454–2457 CrossRef PubMed.
- T. E. Carver, R. E. Brantley Jr, E. W. Singleton, R. M. Arduini, M. L. Quillin, G. N. Phillips Jr and J. S. Olson, J. Biol. Chem., 1992, 267, 14443–14450 CrossRef.
- K. Hirata, K. Yamashita, G. Ueno, Y. Kawano, K. Hasegawa, T. Kumasaka and M. Yamamoto, Acta Crystallogr. D Struct. Biol., 2019, 75, 138–150 CrossRef PubMed.
- K. Yamashita, K. Hirata and M. Yamamoto, Acta Crystallogr. D Struct. Biol., 2018, 74, 441–449 CrossRef PubMed.
- A. Vagin and A. Teplyakov, J. Appl. Crystallogr., 1997, 30, 1022–1025 CrossRef.
- A. T. Brunger, P. D. Adams, G. M. Clore, W. L. DeLano, P. Gros, R. W. Grosse-Kunstleve, J. S. Jiang, J. Kuszewski, M. Nilges, N. S. Pannu, R. J. Read, L. M. Rice, T. Simonson and G. L. Warren, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1998, 54, 905–921 CrossRef CAS PubMed.
- P. Emsley and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 2126–2132 CrossRef PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- S. Jo, T. Kim, V. G. Iyer and W. Im, J. Comput. Chem., 2008, 29, 1859–1865 CrossRef CAS.
- B. R. Brooks, C. L. Brooks III, A. D. Mackerell Jr., L. Nilsson, R. J. Petrella, B. Roux, Y. Won, G. Archontis, C. Bartels, S. Boresch, A. Caflisch, L. Caves, Q. Cui, A. R. Dinner, M. Feig, S. Fischer, J. Gao, M. Hodoscek, W. Im, K. Kuczera, T. Lazaridis, J. Ma, V. Ovchinnikov, E. Paci, R. W. Pastor, C. B. Post, J. Z. Pu, M. Schaefer, B. Tidor, R. M. Venable, H. L. Woodcock, X. Wu, W. Yang, D. M. York and M. Karplus, J. Comput. Chem., 2009, 30, 1545–1614 CrossRef CAS.
- J. Lee, X. Cheng, J. M. Swails, M. S. Yeom, P. K. Eastman, J. A. Lemkul, S. Wei, J. Buckner, J. C. Jeong, Y. Qi, S. Jo, V. S. Pande, D. A. Case, C. L. Brooks III, A. D. MacKerell Jr, J. B. Klauda and W. Im, J. Chem. Theory Comput., 2016, 12, 405–413 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- T. E. Cheatham, J. L. Miller, T. Fox, T. A. Darden and P. A. Kollman, J. Am. Chem. Soc., 1995, 117, 4193–4194 CrossRef CAS.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- S. Nose, Prog. Theor. Phys. Suppl., 1991, 103, 1–46 CrossRef CAS.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- E. A. Berry and B. L. Trumpower, Anal. Biochem., 1987, 161, 1–15 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra06888a |
| ‡ Present address: Graduate School of Science, University of Hyogo, 3-2-1 Koto, Kamigori-cho, Ako-gun, Hyogo 678-1297, Japan. |
| This journal is © The Royal Society of Chemistry 2021 |