
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigation of the exciton relaxation processes in poly(9,9-dioctylfluorene-co-benzothiadiazole):CsPbI1.5Br1.5 nanocrystal hybrid polymer–perovskite nanocrystal blend†
Antonio Balena‡
a,
Arianna Cretíb,
Mauro Lomascolob and
Marco Anni *a
*a
aDipartimento di Matematica e Fisica “Ennio De Giorgi”,Università del Salento, Via per Arnesano, 73100 Lecce, Italy. E-mail: marco.anni@unisalento.it
bIMM-CNR Institute for Microelectronic and Microsystems, Via per Monteroni, 73100 Lecce, Italy
First published on 14th October 2021
Abstract
The combination of lead halide perovskite nanocrystals and conjugated polymer in a blend film opens the way to the realization of hybrid active layers with widely tunable optical and electrical properties. However, the interaction between the polymeric and the perovskite component of the blends is mainly unexplored to date. In this work we perform temperature-dependent photoluminescence and time resolved photoluminescence measurements in order to deeply investigate the photophysics of a poly(9,9-dioctylfluorene-co-benzothiadiazole) (F8BT):CsPbI1.5Br1.5 nanocrystal hybrid film. Our results suggest that the primary interaction channel is charge transfer, both from F8BT to the NCs and from the NCs to F8BT, while Förster resonant energy transfer has no visible effects. Moreover, we show that the charge transfer is assisted by energy migration within the F8BT excited state distribution and that it is dependent on the local micromorphology of the film. Our work improves the current understanding of the polymer:perovskite NC interactions in hybrid films, and it is expected to be relevant for the development of hybrid organic–perovskite optoelectronic devices.
1. Introduction
After their first demonstration by Protesescu et al.1 fully inorganic lead halide perovskite nanocrystals (NCs) emerged as extremely fascinating systems for basic science investigations and as possible active materials for photonic and optoelectronic devices.2 Perovskite NCs show very high photoluminescence quantum yield,3,4 fully tunable emission from the blue to the NIR either acting on the chemical composition or on the size,1,5 narrow emission, and high optical gain at room temperature both under femtosecond and nanosecond excitation.6–10 These properties allowed the demonstration of light-emitting diodes (LEDs) with external quantum efficiency exceeding 20%11–13 and low threshold optically pumped lasers exploiting thin films of NCs as active layers.14In particular, the combination of broad emission wavelength tunability and narrow emission linewidth allows obtaining an ultra-wide color gamut, exceeding the National Television System Committee (NTSC) TV colour standard, and almost entirely covering the CIE 1931 color space.1,15
Despite these interesting features, the lack of long term operational stability to date still prevents the use of lead halide perovskites in general, and of their NCs in particular, in commercially available devices. In particular, the two main degradation channels of perovskites NCs are related to the interaction with the environmental moisture and oxygen7 and, in electrically driven devices, to ion migration under applied bias.16,17
A particularly promising strategy to reduce these effects is the NCs inclusion within inert or semiconducting polymeric matrices.18,19 This approach has been exploited to date to realize polymer–perovskites composites by mixing an inert polymer to the perovskite precursors, thus obtaining films made by perovskites nano and micro-crystals embedded in a polymer matrix.20–25 Ultrastable hybrid organic–perovskites composites containing perovskites nanocrystals can also be simply realized by directly mixing in solution the NCs and a hydrophobic polymer in order to obtain thin films that can be exploited as down converter layer for white LEDs26–28 or as active systems in highly efficient LEDs.29
Even more interesting is the possibility of blending perovskites NCs with semiconducting polymers, thus allowing the generation of hybrid composites to combine the electrical and optical properties of both components, thus obtaining active properties not available for the individual materials. This approach has been widely used in order to improve the film-forming and charge injection properties of II–VI colloidal nanocrystals, thanks to their inclusion in proper conjugated polymer matrices, thus obtaining functionalized hybrid active layers for LEDs (both monochromatic and white), lasers30 and solar cells.31
On the contrary, despite the conceptual similarity between polymer:II–VI NCs blends and polymer:perovskite NCs ones, that suggests their potential exploitation in the same broad range of devices, the work to realize, understand and optimize hybrid blends of conjugated polymers and perovskites NCs with properly engineered active properties is still in an initial stage, even if some very interesting preliminary results have been published. For example blends between blue emitting CsPbBrxCl3−x NCs and orange emitting poly[2-methoxy-5-(2-ethylhexyloxy)-1,4-phenylenevinylene] (MEH:PPV) have been used as active films in LEDs,32 showing an emission spectrum made by the superposition of the component materials ones and an emission color controllable by acting on the blend relative content. In particular the blend content optimization allowed the demonstration of a white emitting LED. The emission color tunability of hybrid polymer–NCs blends have been later investigated in details33 demonstrating full photoluminescence (PL) color tunability from the blue to the red, including white light generation, by combining a blue emitting poly(9,9-dioctylfluorene) (PFO) with green emitting CsPbBr3 or red emitting CsPbBr1.5I1.5 NCs and a yellow emitting poly(9,9-dioctylfluorene-co-benzothiadiazole) (F8BT) and the red NCs, in different proportions. Finally, improved lasing properties of conjugated polymer particles ascribed to Förster resonant energy transfer (FRET) from CH3NH3PbX3 NCs34 has been also reported.
A fundamental aspect of the development of hybrid active films with optimized optical and optoelectronic properties is a deep understanding of their photophysics and of the processes related to the interaction between the two component materials. This point has been deeply investigated in hybrid systems containing an organic molecule and II–VI colloidal NCs evidencing that the two main processes are FRET from host to the guest or charge transfer and that the dominant process is determined by the energy level alignment, by the type of ligand of the NC and on the donor–acceptor distance.30,35–40
The photophysics of active polymer:perovskite NCs blends is instead to date still largely unexplored, with only few preliminary experiments claiming the presence of FRET either from the NCs to the organic or from the organic to the NCs,32,34,41,42 based only on partial experimental evidences that do not allow to evaluate the possible presence and importance of charge transfer processes. In some experiments41,42 claims of energy transfer from the organic to the perovskite, or vice versa, are simply based on the evidence of good overlap between the candidate donor PL and the candidate acceptor absorption. Unfortunately the spectra overlap is only a necessary, but not sufficient, condition to have FRET thus preventing to conclude about the presence of FRET only by considering it. A deeper investigation of the photophysics of a CsPbBrxCl3−x:MEH:PPV blend32 evidenced a lifetime shortening of the NCs in presence of the MeH–PPV, thus demonstrating that an interaction between the two components takes place. Also in this case the made attribution of this effect to FRET is only tentative, as also a NC → MEH:PPV charge transfer would result in a NCs lifetime shortening. In a similar way the investigation of the relaxation dynamics variation of the NCs and the organic34 lead to the claim of FRET from the NCs to the polymer despite a NCs lifetime reduction dependence on the spectral overlap not fully consistent with the behavior expected for FRET.
The lack of a clear understanding of the kind of interaction between the polymer and the NCs in the blend is currently a strong limit in the understanding of the kind of devices that could efficiently exploit this kind of active systems. For example, the presence of efficient FRET from the polymer to the NCs would lead to a blend emission spectrum dominated by the NCs emission, and could be useful for the realization of monochromatic LEDs43,44 or low threshold optically pumped lasers.45,46 On the contrary the lack of FRET would allow to combine the emission of both components, thus generating widely tunable emission color, including white light,33 that could be interesting for white LEDs.32 Finally, the presence of efficient charge transfer between the polymer and the NCs could be exploited in photoelectric devices, like solar cells and photodetectors.47
In this paper, in order to improve the understanding of the basic photophysics of hybrid polymer:perovskite NCs blends, we exploit the temperature dependence of the photoluminescence spectra and relaxation dynamics in order to deeply investigate the interaction between a green-emitting conjugated polymer poly(9,9-dioctylfluorene-co-benzothiadiazole) (F8BT) and red-emitting CsPbBr1.5I1.5 fully inorganic perovskite NCs in a hybrid blend. We demonstrate that, despite the excellent spectral overlap between the donor (F8BT) photoluminescence and the acceptor (CsPbBr1.5I1.5 NCs) absorption, the F8BT → NCs FRET has no observable role in the exciton relaxation. On the contrary we observe the presence of an exciton quenching process both for the F8BT and for the NCs ascribed to charge transfer from F8BT to NCs and from NCs to F8BT, assisted by exciton energy migration within the organic matrix. We also demonstrate that the polymer–NC interaction is affected by the local micromorphology of the film.
Our results provide the first complete experimental investigation of the interaction between the two component materials of active polymer–perovskite NCs blends and are expected to be relevant for the development of optoelectronic devices based on hybrid blends.
2. Results and discussion
The investigated sample is a blend between F8BT and CsPbBr1.5I1.5 NCs with a relative NCs content of 2.1%.As a first step we investigated the temperature dependence of the PL spectra of the component materials and of the blend film. The PL spectrum of the F8BT film at low temperature (T = 10 K) shows (see Fig. S1†) a clear peak at about 547 nm, followed by a shoulder at about 586 nm that can be ascribed to a vibronic replica of the S1 → S0 transition. As the temperature increases we observe a progressive intensity reduction, a slight blue-shift and a broadening of the emission peaks, overall leading to a progressively reduced visibility of the vibronic band.
Concerning the NCs pure films (see Fig. S2†) we observe, at T = 10 K, a single emission peak at about 587 nm with a Full Width at Half Maximum (FWHM) of only 9.0 nm. As the temperature increases the emission peak progressively blue-shifts and broadens, as typically observed in this class of materials. In particular the FWHM increase it typically related to the progressive increase of exciton–phonon coupling.48 Concerning the NCs PL intensity we observe a progressive intensity decrease up to 230 K, followed by a PL intensity increase at higher temperatures. This anomalous behavior is qualitatively similar to previous results on formamidinium lead triiodide NCs,10,49 ascribed to the thermal activation of exciton detrapping from surface states. We also observe that a similar temperature induced PL intensity has been recently obtained in CsPbBr3 NCs films,48 only in positions of the film showing strong NCs aggregation, suggesting that the exciton detrapping involves surface states of NCs aggregates. Finally, the absence of the temperature induced PL intensity increase in other films of nominally comparable NCs allows us to exclude the attribution of this effect to eventual crystalline phase transitions in the investigated temperature range.50
Moving to the PL spectra of the F8BT:NC blend we observe, at low temperature (T = 10 K), two clear peaks at about 547 nm and 592 nm (see Fig. 1a) that can be ascribed, by comparison with the PL spectra of the blend components, to F8BT and to the NCs, respectively (see Fig. 1b). As the temperature increases both the F8BT and the NC peaks show a progressive blue-shift up to about 5 nm at 310 K, a broadening, and a gradual intensity reduction. The disappearance of the NCs PL intensity increase above 230 K is consistent with the aggregation reduction, due to NCs dispersion in the polymer matrix, and with the passivation of surface states from the polymer.51
 | ||
| Fig. 1 (a) PL spectra of the F8BT:NC blend as a function of the temperature. The spectra are vertically translated for clarity. (b) Comparison between the blend spectra at 10 K and 310 K and the F8BT and NC ones. | ||
In order to have a direct insight in the interaction between the F8BT and the NCs in the blend we also performed time resolved PL measurements at the peak wavelength of the corresponding PL bands.
The PL relaxation dynamics of the pure F8BT film (see Fig. S3a†) shows a monoexponential decay at all the temperatures, becoming progressively faster as the temperature increases. The decay time has been determined from a monoexponential fit of the experimental data, and it is about 2.1 ns at 10 K, and decreases down to about 1.8 ns at room temperature (see Fig. 2b).
 | ||
| Fig. 2 (a) Comparison between the relaxation dynamics of the F8BT in the blend at 10 K and 310 K with the pure F8BT ones. The curves are normalized to the peak value and vertically scaled for clarity. (b) Temperature dependence of the fast and slow decay time of the F8BT in the blend (full symbols) and of the decay time of the pure F8BT film (empty symbols). Inset: temperature dependence of the relative amplitude of the fast process. | ||
The relaxation dynamics of the F8BT in the blend (see Fig. S3b†) is, at all the temperatures, faster than the pure F8BT one, evidencing a variation of the F8BT excitons relaxation due to the presence of the NCs. A simple comparison between the PL relaxation dynamics at 10 K and at 310 K of the pure F8BT and of the F8BT in the blend (see Fig. 2a) allows to observe that the faster relaxation in the blend is due to the appearance of a fast relaxation process, superimposed to a slower relaxation similar to the pure F8BT one. This result evidences the presence of a further F8BT relaxation process in the blend, clearly demonstrating the presence of a F8BT–NCs interaction channel. The decay time related to this process (τfast) has been obtained from a bi-exponential fit of the experimental traces (see Fig. 2b), and it is about 1.2 ns up to 130 K, an then progressively decreases down to 0.90 ns at room temperature, while the second decay time (τslow) is identical to the one of the pure F8BT in all the investigated temperature range. As the temperature increases the relative importance of the faster process progressively increases from about 55% at 10 K up to about 95% at room temperature (see inset of Fig. 2b).
Concerning the NCs we observe, at low temperature, a non exponential relaxation (see Fig. S4a†), that can be reproduced by the superposition of three exponential decays with a short lifetime τ1 ≈ 0.7 ns, an intermediate one τ2 ≈ 2.7 ns and a long one τ3 ≈ 30 ns. All the decay times show a weak temperature dependence up to 230 K (see Fig. 3b and S5†), followed by the disappearance of the faster process, by an increase of the relative contribution of the slowest process, and by a progressive increase of τ2 and τ3 up to room temperature.
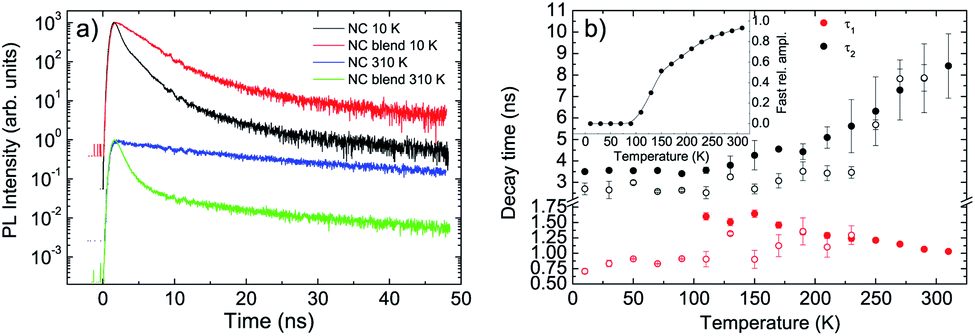 | ||
| Fig. 3 (a) Comparison between the relaxation dynamics of the NCs in the blend at 10 K and 310 K with the pure NCs ones. (b) Temperature dependence of the fast and slow decay time of the NCs in the blend (full symbols) and in the pure NC film (empty symbols). Inset: temperature dependence of the relative amplitude of the fast process. | ||
When the NCs are mixed to the F8BT in the blend we observe a PL relaxation dynamics different from the pure NCs film one (see Fig. 3a and S4b†). In particular, for temperatures between 10 K and 90 K, we observe the suppression of the faster relaxation process while τ2 and τ3 are basically unchanged (see Fig. 3b and S5†), overall leading to a PL relaxation slower than the pure NCs one. When the temperature reaches 110 K a further fast relaxation process appears, not present in the pure NCs, with a lifetime τ1 ≈ 1.6 ns becoming progressively faster as the temperature increases. The relative contribution of this additional fast relaxation process progressively increases with the temperature, up to about 93% at room temperature (see inset of Fig. 3b). The presence of this additional decay process demonstrates that the NCs in the blend interacts with the F8BT through a process causing NCs excitons relaxation.
Beyond the clear evidence of the presence of additional decay channels for both F8BT and NCs in the blends, related to their interaction, it is also interesting to evidence that, for both the blend components, the PL relaxation dynamics show the superposition of the decay related to the polymer–NCs interaction and the one due to exciton relaxation of the pure materials. This result strongly suggests that the F8BT–NCs interaction does not uniformly take place across the blend film otherwise the interaction process and the intrinsic relaxation processes would take place in parallel, leading to a general shortening of the intrinsic decay dynamics. On the contrary the coexistence in the blend of PL signal from F8BT molecules (NCs) interacting with the NCs (F8BT) and the pure material ones evidences that the F8BT–NCs (NCs–F8BT) interaction takes place only in some regions of the sample, leaving the PL relaxation unchanged in the regions in which the interaction is not present.
In order to quantitatively investigate the previous results we started from the analysis of the F8BT data, observing that the PL quenching and PL decay time decrease with the increase of the temperature are the typical signatures of the presence of thermally activated non-radiative processes. Assuming a temperature independent radiative decay time (τr) and a thermally activated non-radiative decay time (τnr (T)) the PL decay time τ is given by:
 | (1) |
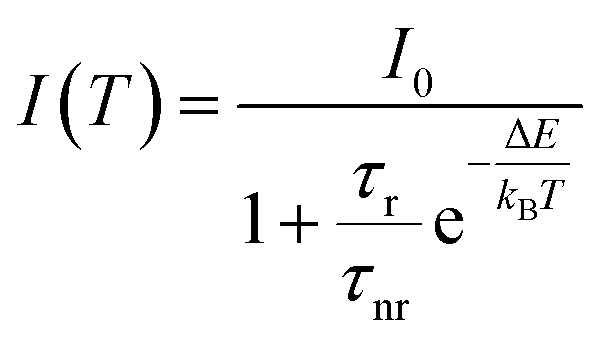 | (2) |
The best fit curve of the temperature dependence of the pure F8BT PL intensity with eqn (2) nicely reproduces the experimental data for ΔE = 30 ± 2 meV and τr/τnr = 0.60 ± 0.05 (see Fig. 4a). The obtained value of ΔE is of the same order of magnitude of the values reported in other polymers52,53 and allows to ascribe the observed PL quenching to thermal activation of up-hill energy migration within the disordered distribution of the polymer excited states, allowing exciton migration towards non-radiative defects.
 | ||
| Fig. 4 (a) Temperature dependence of the integrated PL intensity (dots) of pure F8BT, F8BT in the blend and NCs in the blend PL. The red lines are the best fit curves with eqn (2). (b) Temperature dependence of the slow decay time of pure F8BT (dots). The red line is the best fit curve with eqn (1). (c) Temperature dependence of the decay time due to the F8BT–NCs interaction (dots). The red line is the best fit curve with the thermally activated term of eqn (1). | ||
The temperature dependence of the pure F8BT PL decay time can be instead well reproduced by the best fit with eqn (1) (see Fig. 4b), with best fit ΔE = 40 ± 5 meV and τr/τnr = 0.55 ± 0.05, evidencing that the observed progressive lifetime shortening with the temperature is due to the thermal activation of a non radiative process. The similarity between the best fit values of ΔE and τr/τnr obtained from the best fit of the PL intensity and of the PL decay time temperature dependence allows to ascribe both effects to thermally activated energy migration toward defects in the F8BT film.
Concerning the PL intensity temperature dependence of the F8BT in the blend (see Fig. 4a, and ESI† for the procedure used to separate the F8BT and the NCs contribution to the blend PL intensity), we obtained a good fit of the experimental data with eqn (2) for ΔE = 33 ± 2 meV and τr/τnr = 1.41 ± 0.15. The activation energy of the thermal quenching of F8BT in the blend is thus the same of the pure F8BT one, evidencing that the quenching is still related to exciton migration in the F8BT film. On the other side, the ratio τr/τnr is strongly increased in the blend (about 2.4 times), evidencing a higher non-radiative rate in the blend, at any given temperature, assuming an unchanged F8BT radiative decay time. This clearly indicates the presence of a further F8BT PL quenching channel when NCs are present, which is assisted by intermolecular energy migration, as suggested by the unchanged activation energy.
Finally, the best fit of the PL intensity temperature dependence of the NCs in the blend evidences that the activation energy of the NCs PL quenching (ΔE = 33 ± 2 meV) is the same of the F8BT, and τr/τnr = 6.2 ± 0.6 (see Fig. 4a). The coincidence of the NCs quenching activation energy with the F8BT ones suggests that the same thermally activated process (energy migration) allows PL quenching of both F8BT and NCs.
Overall, these results allow us to conclude that the F8BT and the NCs interact in the blend, that the F8BT–NCs interaction determines a PL intensity quenching of both species, and that it is assisted by intermolecular energy migration within the F8BT molecules that allows F8BT excitons to reach the interface with NCs.
In order to have further insight into the polymer–NC interaction we also investigated the temperature dependence of the decay time of the additional relaxation channel observed from the time-resolved PL measurements (see Fig. 4c). In this case, considering that the additional fast relaxation process is observed in the time-resolved PL measurements of the NCs only from 130 K, we limited the analysis of the data to the temperature range 130–310 K both for F8BT and NCs and we fitted the data with the thermally activated term in eqn (1).
Both decay times show a thermally activated decrease with the temperature (see Fig. 4c), with a comparable activation energy of ΔE = 6.0 ± 0.3 meV and ΔE = 7.0 ± 0.9 meV for F8BT and NCs, respectively.
Concerning the nature of the F8BT–NCs interaction we preliminary observe that our results clearly allow to exclude any important role of F8BT → NCs FRET, despite the excellent spectral overlap between the F8BT PL and the NCs absorption spectra (see Fig. S7†). Actually FRET from F8BT to NCs should result in a F8BT PL quenching and lifetime shortening, as observed, but also to a NCs PL enhancement, that instead it is not present. Moreover the F8BT → NCs should not modify the PL decay dynamics of the acceptor (NCs), while a further decay process is observed. This conclusion, that is in clear contrast with the general assumptions done in literature in similar systems,32,34,41,42 is instead perfectly consistent with our previous data evidencing that the observed value of the fast lifetime in the F8BT relaxation dynamics is about 2.4 times shorter than the FRET lifetime lower limit,33 and thus that the F8BT–NCs interaction is much faster than the expected FRET lifetime.
On the contrary, the simultaneous presence of PL quenching and lifetime shortening that demonstrates the presence of an exciton quenching process for both F8BT and NCs, is consistent with the presence of F8BT → NCs and NCs → F8BT charge transfer.
The possibility to have charge transfer both from F8BT to the NCs and from the NCs to the F8BT can be understood by looking at the energy level scheme of our system (see Fig. 5), evidencing that the F8BT Lowest Unoccupied Molecular Orbital (LUMO) level54 is slightly above the NCs conduction band,55 while the F8BT Highest Occupied Molecular Orbital (HOMO) is below the NCs valence band edge. This makes energetically favored both an electron and a hole transfer from a photoexcited F8BT molecule to a close NC. Concerning the possibility of observing a charge transfer between a photoexcited NC to an unexcited F8BT molecule we observe that in both the blend components the energy levels are not at a unique position along the sample due to inhomogeneous broadening. At room temperature the PL linewidth is about 230 meV and about 100 meV for the F8BT and the NCs, respectively. Assuming that both the ground state and the excited state contribute in the same way to the linewidth we estimated a standard deviation of about 165 meV and 70 meV for F8BT and NCs, respectively.56 This makes statistically possible to have F8BT molecules with LUMO below the NCs conduction band, thus allowing also electron transfer from a photoexcited NC to a F8BT molecule. On the contrary the estimated level broadening makes unlikely to have hole transfer from NCs to F8BT energetically convenient.
 | ||
| Fig. 5 Energy level scheme and schematic chemical structure of F8BT (left) and NCs (right). The dotted lines are Gaussians curves with 165 meV and 70 meV standard deviation for F8BT and NCs, respectively, evidencing the role of the line broadening on the relative level position. The continuous black lines represent absorption of the pump laser, the colored continuous lines the possible charge transfer processes and the dotted line the unlikely hole transfer from NCs to F8BT, respectively. | ||
Our conclusion, that could be confirmed by photoinduced absorption measurements, is fully consistent with previous results on blends between polymers and II–VI NCs demonstrating a thermally activated interaction39 and a switching of the main interaction process from FRET to charge transfer when the ligand is changed from tri-octylphosphine oxide (TOPO) to oleic acid (OA).40
The evidence of a further common activation energy for the charge transfer (beyond the energy migration one) observed in the temperature dependence of the interaction times, demonstrates that after the thermal activation of energy migration a second process has to be thermally activated in order to allow the charge transfer. Considering that the activation energy of the interaction time is the same both for F8BT and for the NCs this further process is common both to F8BT → NCs and NCs → F8BT charge transfers, and it is thus likely related to charge motion along the ligand molecules separating the F8BT from the NCs.
In addition, we investigated the spatial uniformity of the blend emission properties by performing PL mapping measurements by fluorescence microscopy. The obtained results (see Fig. 6) clearly evidence that the sample emission properties and morphology are not uniform across the film surface. In particular we observe several elongated bright aggregates with a typical length of about 10 μm and a width of about 2 μm, that are likely related to NCs aggregation during the drop-casting.7,48 The regions surrounding the aggregates are bright in the F8BT emission channel and dark in the NCs one. The regions with a uniform morphology show instead rather uniform PL intensity both for the F8BT and for the NCs. Considering the spontaneous aggregation tendency of the NCs during the solvent evaporation it is likely that the aggregates are due to NCs aggregation and that the surrounding regions have a locally lower density of NCs. This would explain both the weak NCs PL, due to the low NCs density, and the bright F8BT PL, due to the absence of non-radiative interaction with the NCs. In these regions we thus expect to observe the PL properties of the pure NCs, in the clusters, and of F8BT, in the surrounding regions, that are responsible for the presence of the PL relaxation dynamics of the pure materials in the PL decay dynamic of the blend. The regions with uniform morphology and PL intensity suggest that F8BT and NCs are finely and uniformly mixed, thus allowing their interaction, resulting in the decay time shortening and PL quenching for both materials.
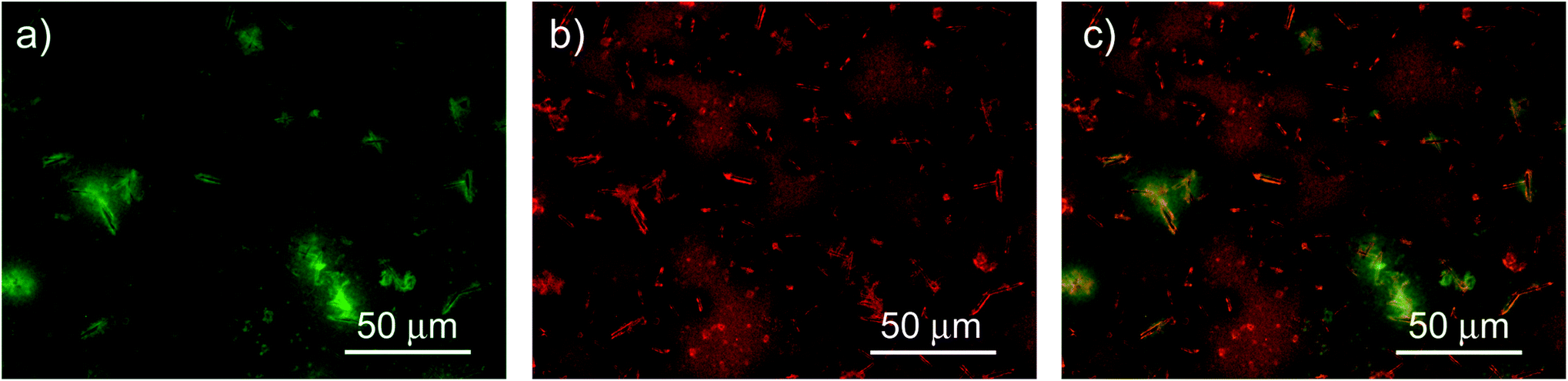 | ||
| Fig. 6 200 μm × 150 μm fluorescence map of F8BT (a) and NCs (b) in the blend and composite image (c). | ||
Finally, in order to probe the generality of our conclusions about the polymer–NCs interaction type and the effects of the dominance of charge transfer over FRET on the blend emission properties we also realized a different blend, between blue emitting poly(9,9-dioctylfluorene) (PFO) and green emitting CsPbBr3 NCs (GNC) with a relative NCs content of 5.5%, and we investigated its Amplified Spontaneous Emission (ASE) properties under nanosecond pulsed excitation.
Also in this film the two used materials allow to obtain a good spectral overlap between the PFO (host) PL and the GNCs (guest) absorption (see Fig. S8a†), thus fulfilling the necessary condition to allow FRET from the host to the guest.
Moreover, the GNCs show efficient ASE properties in neat films7,9 under nanosecond pumping that, in case of PFO → GNC FRET should be further enhanced, as widely demonstrated in host:guest organic blends interacting by FRET.45,46,57,58
The PL spectrum of the blend at low excitation density shows (see Fig. S8b†) the superposition of the PFO59 and of the GNC PL7,33 but, as the excitation density increases, only a general intensity increase is observed, without any evidence of ASE from the GNCs. This behavior clearly demonstrates that the GNC ASE properties are not only not improved by the dispersion in PFO, as expected in case of PFO → GNC FRET, but are even suppressed in the blend. This result is fully consistent with the presence of a non radiative interaction of the GNCs with the PFO due to charge transfer, that suppressed the ASE due to both the NCs exciton quenching and to the spectral overlap between the GNCs gain and the PFO polaron photoinduced absorption band.60
3. Conclusions
In conclusion, we investigated the interaction between a green-emitting conjugated polymer poly(9,9-dioctylfluorene-co-benzothiadiazole) (F8BT) and fully inorganic red-emitting CsPbBr1.5I1.5 perovskite NCs in a hybrid blend. The obtained results demonstrate that, contrary to what typically believed in literature, F8BT → NCs FRET has a negligible role in the exciton relaxation of the film, and strongly suggest that charge transfer both from F8BT to NCs and from NCs to F8BT takes place, resulting in a PL quenching and PL lifetime decrease. These results, beyond improving the understanding of the photophysics of hybrid polymer–perovskite films, also clarify the potential device applications of these systems. In particular, the lack of efficient FRET explains the previous evidence of coexistence of polymer and NCs emission in the blend emission spectra32,33 and suggests hybrid polymer:perovskite NCS blends as possible active material for white LEDs but not for low threshold laser exploiting host → guest FRET. Moreover, the dominance of charge transfer processes suggests the possibility of exploiting these blends as active materials of photoelectric devices, like solar cells and photodetectors, whose working principle requires efficient exciton dissociation is free charges.4. Experimental section
4.1. Nanocrystal synthesis and thin film deposition
The oleic acid/oleylamine coated CsPbI1.5Br1.5 (NC) and CsPbBr3 (GNC) nanocrystals are identical to the ones used in A. Perulli et al.33 (please refer to this paper for details on the synthesis and basic properties like shape, size and PL quantum yield). The CsPbI1.5Br1.5 active film has been deposited by drop-casting on glass substrates, at room temperature and in air. In order to improve the film morphology the film was left to dry under a Petri dish, thus slowing the solvent evaporation. The room temperature PL spectrum is peaked at 578 nm (525 nm), with a FWHM of 26 nm (19 nm) for NC (GNC). The FWHM values are in line with the ones of state of the art nanocrystals,1,50 thus evidencing a narrow size distribution. The F8BT and PFO have been obtained from ADSdyes and used as received. The pure F8BT film has been deposited by spin coating at 2500 rpm for 2 minutes from a 10 mg ml−1 solution in toluene.The blend was realized by mixing the F8BT solution to the CsPbI1.5Br1.5 solution and the PFO to the CsPbBr3 one, all in toluene and with a concentration of 10 mg ml−1. The relative concentration (30% of polymer solution and 70% of NCs solution) has been converted into the percentage of emitters by considering the different molecular weights of the polymers and the NCs. The realized blend has thus a relative content of NCs of 2.1% for the F8BT:CsPbI1.5Br1.5 and 5.5% for the PFO:CsPbBr3 one.
The used polymer:NCs relative content was determined taking into account that, as the NCs content in the blend increases, the effects of their interaction with the polymer become stronger,33 but the film uniformity decreases, due to the NCs aggregation. The lack of uniformity could make the spectroscopic investigation, that probes the optical properties of a region with a size of about 100 μm, not necessarily representative of the average macroscopic emission properties. The films realized by mixing 30% of polymer solution and 70% of NCs solution thus provide the best compromise between high NC content and uniform macroscopic morphology.
The films were obtained by drop-casting on glass substrates pre-cleaned with toluene, thus obtaining films with a typical thickness of about 300 nm ± 30 nm.
4.2. PL and time-resolved PL
The time-resolved PL (TR-PL) measurements were performed by exciting the samples by a solid-state pulsed laser (mod. PLP-10, Hamamatsu), which provides pulse at a wavelength of 400 nm of about 58 ps at a repetition rate of 1 MHz. The PL has been dispersed using an iHR320 (focal length of 0.32 m) Horiba monochromator equipped with a Peltier cooled Hamamatsu photomultiplier (head-on mod R943-02), operating in single-photon counting. The time-resolved PL measurements have been performed by Time Correlated Single Photon Counting (TCSPC) technique using an Edinburg Instruments TCC900 TSCPC electronics card. The temporal resolution of the system is about 0.46 ns. The sample temperature has been changed by a closed circle He cryostat, allowing to change the temperature in the range 10–310 K. The fluorescence microscopy measurements have been performed with a Nikon C1 microscope, exciting the sample with a Hg lamp in the spectral range 340–380 nm, and collecting the F8BT and the NCs PL with band pass filter in the 515–555 nm and 580–630 nm range, respectively.4.3. Amplified spontaneous emission measurements
The excitation density dependence of the photoluminescence under pulsed nanosecond excitation has been investigated by pumping the films with a LBT MNL 100 nitrogen laser, delivering 3 ns pulses at a wavelength of 337 nm, with a repetition rate of 10 Hz. The pump beam was focused onto the sample surface by a cylindrical lens in a rectangular stripe with a length of about 4 mm and width of 80 μm, collecting the PL from the sample edge after waveguiding in the active film. The emission was dispersed with an Acton SpectraPro-750 spectrometer equipped with a Peltier cooled CCD (Andor). The spectral resolution was about 0.5 nm.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. V. Kovalenko and G. Nedelcu from ETH Zürich (Switzerland) and Laboratory for Thin Films and Photovoltaics, Empa – Swiss Federal Laboratories for Materials Science and Technology, Dübendorf (Switzerland) are deeply acknowledged for providing us the perovskite nanocrystals used for the realization of the samples investigated in our experiments.Notes and references
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed
.
- A. Dey, J. Ye, A. De, E. Debroye, S. K. Ha, E. Bladt, A. S. Kshirsagar, Z. Wang, J. Yin, Y. Wang, L. N. Quan, F. Yan, M. Gao, X. Li, J. Shamsi, T. Debnath, M. Cao, M. A. Scheel, S. Kumar, J. A. Steele, M. Gerhard, L. Chouhan, K. Xu, X.-g. Wu, Y. Li, Y. Zhang, A. Dutta, C. Han, I. Vincon, A. L. Rogach, A. Nag, A. Samanta, B. A. Korgel, C.-J. Shih, D. R. Gamelin, D. H. Son, H. Zeng, H. Zhong, H. Sun, H. V. Demir, I. G. Scheblykin, I. Mora-Seró, J. K. Stolarczyk, J. Z. Zhang, J. Feldmann, J. Hofkens, J. M. Luther, J. Pérez-Prieto, L. Li, L. Manna, M. I. Bodnarchuk, M. V. Kovalenko, M. B. J. Roeffaers, N. Pradhan, O. F. Mohammed, O. M. Bakr, P. Yang, P. Müller-Buschbaum, P. V. Kamat, Q. Bao, Q. Zhang, R. Krahne, R. E. Galian, S. D. Stranks, S. Bals, V. Biju, W. A. Tisdale, Y. Yan, R. L. Z. Hoye and L. Polavarapu, ACS Nano, 2021, 15, 10775–10981 CrossRef CAS PubMed
- F. Liu, Y. Zhang, C. Ding, S. Kobayashi, T. Izuishi, N. Nakazawa, T. Toyoda, T. Ohta, S. Hayase, T. Minemoto, K. Yoshino, S. Dai and Q. Shen, ACS Nano, 2017, 11, 10373–10383 CrossRef CAS PubMed
- B. A. Koscher, J. K. Swabeck, N. D. Bronstein and A. P. Alivisatos, J. Am. Chem. Soc., 2017, 139, 6566–6569 CrossRef CAS PubMed
- L. Protesescu, S. Yakunin, S. Kumar, J. Bär, F. Bertolotti, N. Masciocchi, A. Guagliardi, M. Grotevent, I. Shorubalko, M. I. Bodnarchuk, C.-J. Shih and M. V. Kovalenko, ACS Nano, 2017, 11, 3119–3134 CrossRef CAS PubMed
- S. Yakunin, L. Protesescu, F. Krieg, I. B. Maryna, G. Nedelcu, M. Humer, G. De Luca, M. Fiebig, W. Heiss and M. V. Kovalenko, Nat. Commun., 2015, 6, 8056 CrossRef CAS PubMed
- M. L. De Giorgi, F. Krieg, M. V. Kovalenko and M. Anni, Sci. Rep., 2019, 9, 17964 CrossRef PubMed
- M. L. De Giorgi and M. Anni, Appl. Sci., 2019, 9, 4591 CrossRef CAS
- A. Balena, A. Perulli, M. Fernandez, M. L. De Giorgi, G. Nedelcu, M. V. Kovalenko and M. Anni, J. Phys. Chem. C, 2018, 122, 5813–5819 CrossRef CAS
- P. Papagiorgis, A. Manoli, L. Protesescu, C. Achilleos, M. Violaris, K. Nicolaides, T. Trypiniotis, M. I. Bodnarchuk, M. V. Kovalenko, A. Othonos and G. Itskos, ACS Photonics, 2018, 5, 907–917 CrossRef CAS
- T. Chiba, Y. Hayashi, H. Ebe, K. Hoshi, J. Sato, S. Sato, Y.-J. Pu, S. Ohisa and J. Kido, Nat. Photonics, 2018, 12, 681–687 CrossRef CAS
- K. Lin, J. Xing, L. N. Quan, F. P. G. de Arquer, X. Gong, J. Lu, L. Xie, W. Zhao, C. Zhang, D. Yan, W. Li, X. Liu, Y. Lu, J. Kirman, E. H. Sargent, Q. Xiong and Z. Wei, Nature, 2018, 562, 245–248 CrossRef CAS PubMed
- Y. Cao, N. Wang, H. Tian, J. Guo, Y. Wei, H. Chen, Y. Miao, W. Zou, K. Pan, Y. He, H. Cao, Y. Ke, M. Xu, Y. Wang, M. Yang, K. Du, Z. Fu, D. Kong, D. Dai, Y. Jin, G. Li, H. Li, Q. Peng, J. Wang and W. Huang, Nature, 2018, 562, 249–253 CrossRef CAS PubMed
- C.-Y. Huang, C. Zou, C. Mao, K. L. Corp, Y.-C. Yao, Y.-J. Lee, C. W. Schlenker, A. K. Y. Jen and L. Y. Lin, ACS Photonics, 2017, 4, 2281–2289 CrossRef CAS
- G. Lozano, J. Phys. Chem. Lett., 2018, 9, 3987–3997 CrossRef CAS PubMed
- W. Tress, J. Phys. Chem. Lett., 2017, 8, 3106–3114 CrossRef CAS PubMed
- X. Shan, J. Li, M. Chen, T. Geske, S. G. R. Bade and Z. Yu, J. Phys. Chem. Lett., 2017, 8, 2412–2419 CrossRef CAS PubMed
- S. N. Raja, Y. Bekenstein, M. A. Koc, S. Fischer, D. Zhang, L. Lin, R. O. Ritchie, P. Yang and A. P. Alivisatos, ACS Appl. Mater. Interfaces, 2016, 8, 35523–35533 CrossRef CAS PubMed
- T. Xuan, J. Huang, H. Liu, S. Lou, L. Cao, W. Gan, R.-S. Liu and J. Wang, Chem. Mater., 2019, 31, 1042–1047 CrossRef CAS
- G. Li, Z.-K. Tan, D. Di, M. L. Lai, L. Jiang, J. H.-W. Lim, R. H. Friend and N. C. Greenham, Nano Lett., 2015, 15, 2640–2644 CrossRef CAS PubMed
- J. Li, S. G. R. Bade, X. Shan and Z. Yu, Adv. Mater., 2015, 27, 5196–5202 CrossRef CAS PubMed
- X. Ji, X. Peng, Y. Lei, Z. Liu and X. Yang, Org. Electron., 2017, 43, 167–174 CrossRef CAS
- Y. Xin, H. Zhao and J. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 4971–4980 CrossRef CAS PubMed
- S. Masi, A. Rizzo, F. Aiello, F. Balzano, G. Uccello-Barretta, A. Listorti, G. Gigli and S. Colella, Nanoscale, 2015, 7, 18956–18963 RSC
- X. Ji, X. Peng, Q. Wang, J. Ren, Z. Xiong and X. Yang, Org. Electron., 2018, 52, 350–355 CrossRef CAS
- T. Xuan, J. Huang, H. Liu, S. Lou, L. Cao, W. Gan, R.-S. Liu and J. Wang, Chem. Mater., 2019, 31, 1042–1047 CrossRef CAS
- Y. Wang, J. He, H. Chen, J. Chen, R. Zhu, P. Ma, A. Towers, Y. Lin, A. J. Gesquiere, S.-T. Wu and Y. Dong, Adv. Mater., 2016, 28, 10710–10717 CrossRef CAS PubMed
- J. Zhu, Z. Xie, X. Sun, S. Zhang, G. Pan, Y. Zhu, B. Dong, X. Bai, H. Zhang and H. Song, ChemNanoMat, 2019, 5, 346–351 CrossRef CAS
- B. Zhao, S. Bai, V. Kim, R. Lamboll, R. Shivanna, F. Auras, J. M. Richter, L. Yang, L. Dai, M. Alsari, X.-J. She, L. Liang, J. Zhang, S. Lilliu, P. Gao, H. J. Snaith, J. Wang, N. C. Greenham, R. H. Friend and D. Di, Nat. Photonics, 2018, 12, 783–789 CrossRef CAS
- M. Anni, Nanomaterials, 2019, 9, 1036 CrossRef CAS PubMed
- M. J. Greaney and R. L. Brutchey, Mater. Today, 2015, 18, 31–38 CrossRef CAS
- E.-P. Yao, Z. Yang, L. Meng, P. Sun, S. Dong, Y. Yang and Y. Yang, Adv. Mater., 2017, 29, 1606859 CrossRef PubMed
- A. Perulli, A. Balena, M. Fernandez, G. Nedelcu, A. Cretí, M. V. Kovalenko, M. Lomascolo and M. Anni, Appl. Phys. Lett., 2018, 112, 171904 CrossRef
- A. Mikosch, S. Ciftci, G. Tainter, R. Shivanna, B. Haehnle, F. Deschler and A. J. C. Kuehne, Chem. Mater., 2019, 31, 2590–2596 CrossRef CAS
- N. C. Greenham, X. Peng and A. P. Alivisatos, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 17628–17637 CrossRef CAS PubMed
- D. S. Ginger and N. C. Greenham, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 10622–10629 CrossRef CAS
- M. Anni, L. Manna, R. Cingolani, D. Valerini, A. Cretí and M. Lomascolo, Appl. Phys. Lett., 2004, 85, 4169–4171 CrossRef CAS
- S. Kaufmann, T. Stöferle, N. Moll, R. F. Mahrt, U. Scherf, A. Tsami, D. V. Talapin and C. B. Murray, Appl. Phys. Lett., 2007, 90, 071108 CrossRef
- T. Stöferle, U. Scherf and R. F. Mahrt, Nano Lett., 2009, 9, 453–456 CrossRef PubMed
- S. N. Sharma, T. Vats, N. Dhenadhayalan, P. Ramamurthy and A. Narula, Sol. Energy Mater. Sol. Cells, 2012, 100, 6–15 CrossRef CAS
- B. A. Al-Asbahi, S. M. H. Qaid and A. S. Aldwayyan, Polymers, 2020, 12, 444 CrossRef CAS PubMed
- S. M. Qaid, B. Al-Asbahi, H. M. Ghaithan, M. AlSalhi and A. S. Al dwayyan, J. Colloid Interface
Sci., 2020, 563, 426–434 CrossRef CAS PubMed
- P. T. K. Chin, R. A. M. Hikmet and R. A. J. Janssen, J. Appl. Phys., 2008, 104, 013108 CrossRef
- L. z. Borg, D. Lee, J. Lim, W. K. Bae, M. Park, S. Lee, C. Lee, K. Char and R. Zentel, J. Mater. Chem. C, 2013, 1, 1722–1726 RSC
- M. Anni and S. Lattante, J. Phys. Chem. C, 2015, 119, 21620–21625 CrossRef CAS
- M. Anni, J. Lumin., 2019, 215, 116680 CrossRef CAS
- R. Soltani, B. M. D. Puscher, A. A. Katbab, I. Levchuk, N. Kazerouni, N. Gasparini, N. Camaioni, A. Osvet, M. Batentschuk, R. H. Fink, D. M. Guldi and T. Ameri, Phys. Chem. Chem. Phys., 2018, 20, 23674–23683 RSC
- M. Anni, A. Cretí, M. L. De Giorgi and M. Lomascolo, Nanomaterials, 2021, 11, 1470 CrossRef CAS PubMed
- H.-H. Fang, L. Protesescu, D. M. Balazs, S. Adjokatse, M. V. Kovalenko and M. A. Loi, Small, 2017, 1700673 CrossRef PubMed
- B. T. Diroll, G. Nedelcu, M. V. Kovalenko and R. D. Schaller, Adv. Funct. Mater., 2017, 27, 1606750 CrossRef
- E. Ochoa-Martinez, M. Ochoa, R. D. Ortuso, P. Ferdowsi, R. Carron, A. N. Tiwari, U. Steiner and M. Saliba, ACS Energy Lett., 2021, 6, 2626–2634 CrossRef CAS
- J. M. Hodgkiss, G. Tu, S. Albert-Seifried, W. T. S. Huck and R. H. Friend, J. Am. Chem. Soc., 2009, 131, 8913–8921 CrossRef CAS PubMed
- P. L. Santos, B. B. A. Costa, K. S. Araujo, L. A. Cury, E. W. Snedden, K. N. Bourdakos, F. B. Dias and A. P. Monkman, J. Phys.: Condens. Matter, 2011, 24, 015801 CrossRef PubMed
- J. S. Park, B. R. Lee, E. Jeong, H.-J. Lee, J. M. Lee, J.-S. Kim, J. Y. Kim and H. Young, Appl. Phys. Lett., 2011, 99, 163305 CrossRef
- V. K. Ravi, G. B. Markad and A. Nag, ACS Energy Lett., 2016, 1, 665 CrossRef CAS
- The PL spectrum energy distribution depends on the convolution between the ground and the excited states distribution. Assuming Gaussian density of states both for the HOMO (valence band) and LUMO (conduction band) of F8BT (NCs) with the same variance σ2, and remembering that the convolution of two Gaussians is a Gaussian, with a variance given by the sum of the variances of the two convoluted Gaussians we have σPL2 = 2σ2.
- V. G. Kozlov, V. Bulovic, P. E. Burrows, M. Baldo, V. B. Khalfin, G. Parthasarathy, S. R. Forrest, Y. You and M. E. Thompson, J. Appl. Phys., 1998, 84, 4096–4108 CrossRef CAS
- R. Munoz-Marmol, N. Zink-Lorre, J. M. Villalvilla, P. G. Boj, J. A. Quintana, C. Vazquez, A. Anderson, M. J. Gordon, A. Sastre-Santos, F. Fernandez-Lazaro and M. A. Diaz-Garcia, J. Phys. Chem. C, 2018, 122, 24896–24906 CrossRef CAS
- M. Anni, A. Perulli and G. Monti, J. Appl. Phys., 2012, 111, 093109 CrossRef
- T. Virgili, G. Grancini, E. Molotokaite, I. Suarez-Lopez, S. K. Rajendran, A. Liscio, V. Palermo, G. Lanzani, D. Polli and G. Cerullo, Nanoscale, 2012, 4, 2219–2226 RSC
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra06821k |
| ‡ Present address: Istituto Italiano di Tecnologia (IIT), Center for Biomolecular Nanotechnologies, Via Barsanti 14, 73010 Arnesano (Lecce), Italy. |
| This journal is © The Royal Society of Chemistry 2021 |