
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical synthesis of the organoarsenical antibiotic arsinothricin†
A. Hasan Howlader‡
 a,
Sazzad H. Suzol§
a,
Venkadesh Sarkarai Nadarb,
Adriana Emilce Galvánb,
Aleksandra Nedovicc,
Predrag Cudicc,
Barry P. Rosenb,
Masafumi Yoshinaga*b and
Stanislaw F. Wnuk*a
a,
Sazzad H. Suzol§
a,
Venkadesh Sarkarai Nadarb,
Adriana Emilce Galvánb,
Aleksandra Nedovicc,
Predrag Cudicc,
Barry P. Rosenb,
Masafumi Yoshinaga*b and
Stanislaw F. Wnuk*a
aDepartment of Chemistry and Biochemistry, Florida International University, Miami, Florida 33199, USA. E-mail: wnuk@fiu.edu
bDepartment of Cellular Biology and Pharmacology, Herbert Wertheim College of Medicine, Florida International University, Miami, Florida 33199, USA. E-mail: myoshina@fiu.edu
cDepartment of Chemistry and Biochemistry, Charles E. Schmidt College of Science, Florida Atlantic University, Boca Raton, Florida 33431, USA
First published on 3rd November 2021
Abstract
We report two routes of chemical synthesis of arsinothricin (AST), the novel organoarsenical antibiotic. One is by condensation of the 2-chloroethyl(methyl)arsinic acid with acetamidomalonate, and the second involves reduction of the N-acetyl protected derivative of hydroxyarsinothricin (AST-OH) and subsequent methylation of a trivalent arsenic intermediate with methyl iodide. The enzyme AST N-acetyltransferase (ArsN1) was utilized to purify L-AST from racemic AST. This chemical synthesis provides a source of this novel antibiotic for future drug development.
Introduction
Arsenic has been utilized therapeutically since the eras of Ancient Greece and China. Numerous arsenic species present in the environment1 are used in the treatment of a number of human diseases2 and are biotransformed in microbes and animals, including humans.3 Synthesis of organoarsenic compounds is crucial to characterize arsenic species produced by arsenic biotransformations. A variety of synthetic protocols are being developed for novel organoarsenicals.4,5 The structures of the new aromatic arsenical metabolites discovered in chicken liver were confirmed by chemical synthesis.6Recently a new arsenic-containing compound, arsinothricin [2-amino-4-(hydroxymethylarsinoyl)butanoic acid, AST (1); Scheme 1], was shown to be synthesized by the rice rhizosphere bacterium Burkholderia gladioli GSRB05.7 A three-gene cluster, arsQML, responsible for the AST biosynthesis has been identified recently.8 AST, a nonproteogenic amino acid analog of glutamate, is a broad-spectrum antibiotic effective against both Gram-positive and Gram-negative bacteria.9 Importantly, AST effectively inhibits growth of major pathogens such as carbapenem-resistant Enterobacter cloacae, one of the World Health Organization's global priority pathogens. It can control growth of Mycobacterium bovis, a causative agent of both human and animal tuberculosis. The mechanism of action of AST is by inhibition of bacterial glutamine synthetase (GS), an essential enzyme for production of glutamine and for control of ammonia toxicity. AST is chemically similar to the glutamate acylphosphate intermediate in the GS catalytic cycle and binds to the active site of the enzyme, producing inhibition.
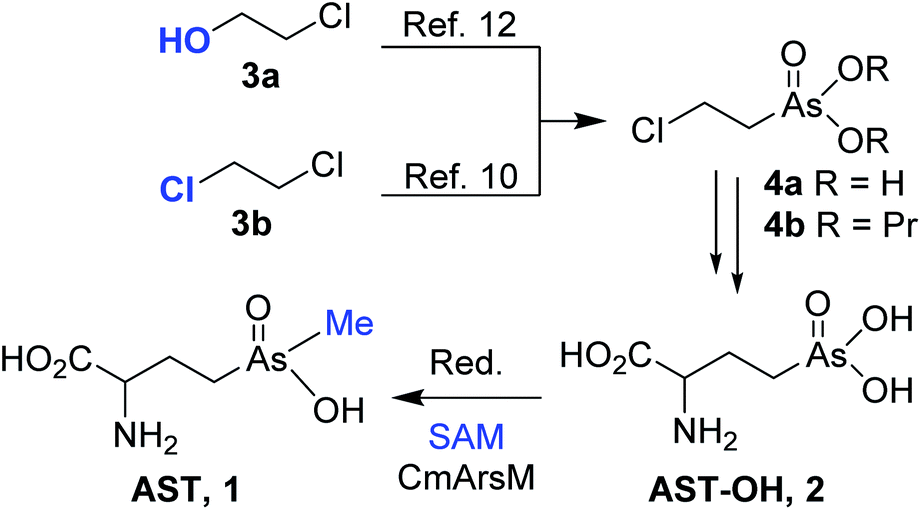
We predict that 1 and related arsenic-containing compounds may be the progenitors of a new class of antibiotics. They may prove to be more effective as drugs than chemically related phosphonates, including some of the most effective commercially available herbicides, pesticides and human drugs. While modest amounts of 1 can be generated using the source organism, drug development requires a reliable source of 1. For that reason, we recently developed a semi-synthesis of racemic 1 that involves chemical synthesis of the 2-amino-4-(dihydroxyarsonoyl)butanoic acid [hydroxyarsinothricin, AST-OH; (2); Scheme 1] precursor; and its enzymatic methylation to AST10 using the robust thermostable enzyme CmArsM, the As(III) S-adenosylmethionine (SAM) methyltransferase from the acidothermophilic eukaryotic alga Cyanidioschyzon sp. 5508.11
Synthesis of AST-OH from (2-chloroethyl)arsenate ester 4b that was prepared from 2-chloroethanol 3a in 5 steps that included elaboration via trivalent arsenic species has been reported (Scheme 1).12 Coupling of 4b with sodium salt of diethyl acetamidomalonate followed by deprotection and decarboxylation gave 2. We reported that condensation of 1,2-dichloroethane 3b with sodium arsenite provided (2-chloroethyl)arsonic acid 4a in a single step; which eliminates the necessity of conversion of the pentavalent (2-hydroxyethyl)arsonic acid to trivalent dichloro(2-hydroxyethyl)arsine and challenging displacement of hydroxy group with chloride. We also found that esterification of 4a to 4b was not necessary, as subjection of 4a directly to coupling with acetamidomalonate and deprotection/decarboxylation sequence provided 2 in higher yield.10 Reduction of 2 and enzymatic methylation of the resulting trivalent arsenicals by CmArsM provided 1.10 To reduce effort and complexities associated with synthesis of pure 1 from either bacterial culture medium7 or enzymatic buffer10 and to provide reliable source of larger quantities of 1 for future drug development, we report here two straightforward methods for the synthesis of racemic AST: one by condensation of the 2-chloroethyl(methyl)arsinic acid with acetamidomalonate and the second by methylation of an AST-OH derivative with methyl iodide. Enzymatic separation of racemic AST is also described.
Results and discussion
In our first route, pentavalent 2-hydroxyethyl(methyl)arsinic acid 5 was designed as crucial precursor for the synthesis of AST, which in turn could be converted to 2-chloroethyl(methyl)arsinic acid 7 via direct chlorination or a path involving trivalent arsines of type 6b. Thus, nucleophilic displacement of chloride in 2-chloroethanol 3a with sodium methylarsonite [MeAs(ONa)2] provided 5 in 86% yield (Scheme 2). The sodium methylarsonite13 was prepared in 97% yield by in situ reduction of the sodium salt of methyl arsonate [MeAs(O)(OH)ONa] with SO2 gas in the presence of HCl and catalytic amount of KI14 followed by hydrolysis of the resulting diiodo(methyl)arsine (MeAsI2) with aqueous NaOH. Reduction of 5 with SO2/HCl/KI yielded less polar trivalent chloro(2-hydroxyethyl)(methyl)arsine 6a, which appears to be susceptible to hydrolysis as it was observed for dichloro(2-hydroxyethyl)arsine.10 Treatment of crude 6a with SOCl2 resulted in vigorous reaction and failed to give 6b, instead producing dichloro(2-hydroxyethyl)arsine with loss of the methyl group. Since other approaches for the synthesis of 6b by halogenation of 6a were also unsuccessful,15 we turned our attention to prepare 7 by direct chlorination of 5.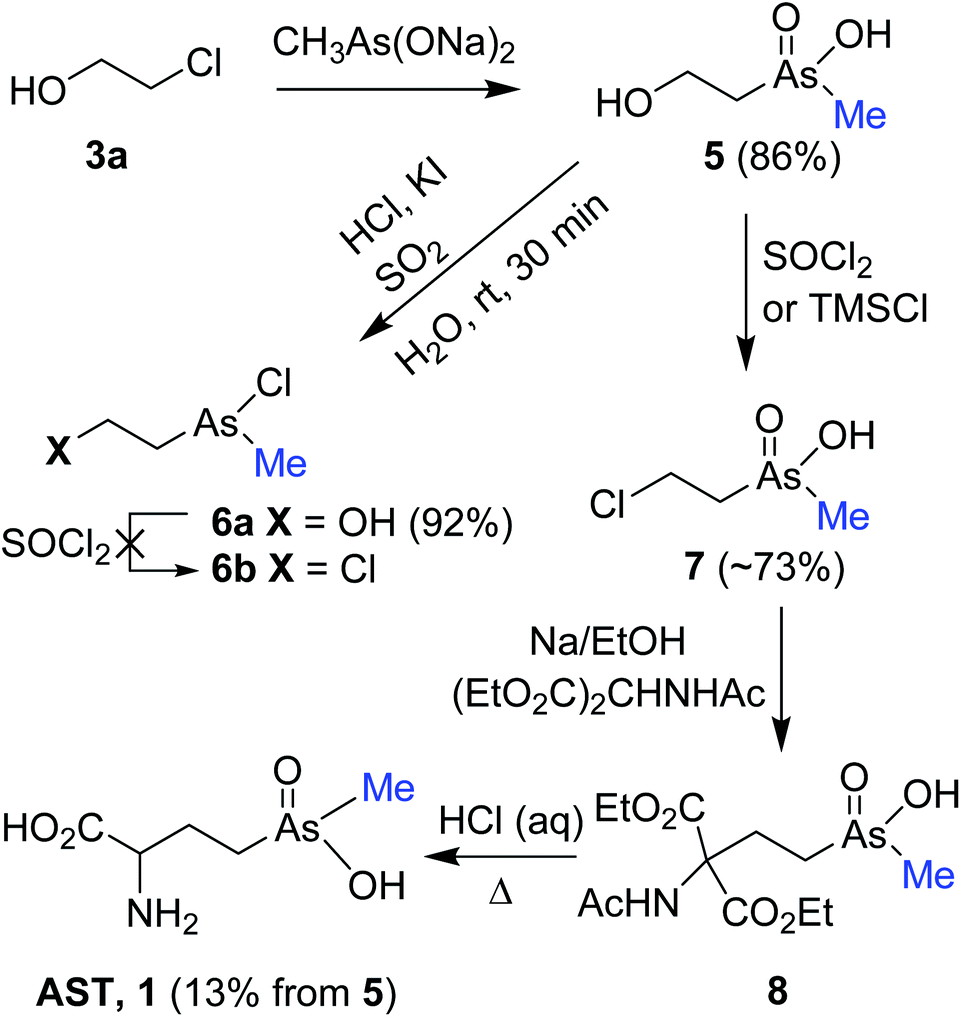 | ||
| Scheme 2 Synthesis of 2-chloroethyl(methyl)arsinic acid 7 and its conversion to AST 1. | ||
Treatment of the purified and iodide-free sodium salt of 5 with TMSCl in DMSO afforded minor quantities of 7 in addition to unchanged 5 [8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :92 mixture based on 1H NMR and HPLC coupled with ICP-MS (inductively coupled plasma mass spectrometry)]. Subsequent reaction of this mixture with acetamidomalonate in the presence of sodium ethoxide at 70 °C yielded malonate 8. Reflux of crude 8 in 6 M HCl effected global deprotection and decarboxylation providing a mixture of products containing 5% of AST as estimated based on ICP-MS and 1H NMR. However, we were fortunate to find that treatment of sodium salt of 5 with SOCl2 effected efficient chlorination to provide 7 contaminated by the acidic form of substrate 5 as 80:20 mixture (based on 1H NMR).16 Additional purification yielded pure 7 (4%) and a mixture of 7 and 5 (85:15, ∼69%). Treatment of the latter mixture with acetamidomalonate followed by deprotection and decarboxylation of the resulting 8 yielded AST17 (13% from 5) after purification by Dowex and Sephadex column chromatography.
:92 mixture based on 1H NMR and HPLC coupled with ICP-MS (inductively coupled plasma mass spectrometry)]. Subsequent reaction of this mixture with acetamidomalonate in the presence of sodium ethoxide at 70 °C yielded malonate 8. Reflux of crude 8 in 6 M HCl effected global deprotection and decarboxylation providing a mixture of products containing 5% of AST as estimated based on ICP-MS and 1H NMR. However, we were fortunate to find that treatment of sodium salt of 5 with SOCl2 effected efficient chlorination to provide 7 contaminated by the acidic form of substrate 5 as 80:20 mixture (based on 1H NMR).16 Additional purification yielded pure 7 (4%) and a mixture of 7 and 5 (85:15, ∼69%). Treatment of the latter mixture with acetamidomalonate followed by deprotection and decarboxylation of the resulting 8 yielded AST17 (13% from 5) after purification by Dowex and Sephadex column chromatography.
Building on our enzymatic methylation of AST-OH to AST,10 we chemically methylated reduced As(III)T-OH 9 with MeI as a source of an electrophilic methyl group. Reduction of 2 with SO2/HCl/KI (rt/15 min) followed by treatment with 6 M NaOH gave the reduced arsenic salt 9 (Scheme 3). Treatment of the alkaline solution of crude 9 with excess MeI effected methylation at arsenic atom. However, the reaction also resulted in methylation of the amino group yielding, after purification on cation exchange resin (Dowex® H+ form) with NH4OH, the trimethylammonium salt 10 (70%, from 2).
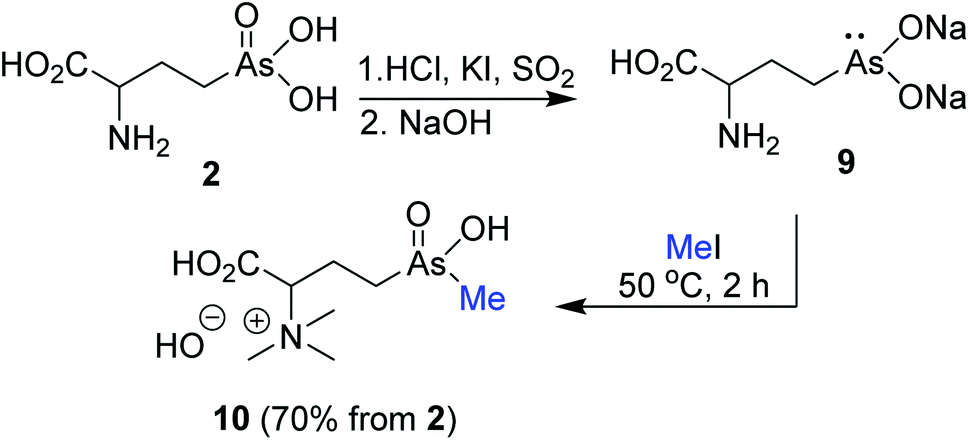 | ||
| Scheme 3 Attempted synthesis of AST via methylation of AST-OH with methyl iodide. | ||
Following encouraging methylation of 9 with MeI, we envisioned that reduction/methylation sequence of the AST-OH derivative bearing the protected amino group would result in an alternative straightforward synthetic route to AST. We selected N-acyl protection for the amino group in AST-OH, especially since the original synthesis of AST-OH12 and our improved protocol10 required synthesis of the N-acetyl protected derivative of type 11 (ref. 10) (Scheme 4). Thus, treatment of 4a with acetamidomalonate as described10 following purification from the excess of malonate afforded 11. Reduction of pure 11 with SO2/HCl and catalytic KI followed by pH adjustment to ∼11 with 6 M NaOH gave sodium salt of the trivalent arsenic compound 12. Subsequent treatment of 12 with MeI (50 °C/4 h) resulted in the methylation at the arsenic atom, providing protected pentavalent AST derivative 8. Excess MeI and elevated temperature were critical for the optimal yield. The progress of the methylation reaction was monitored by HPLC-ICP-MS. Formation of RAs(O)Me(OMe) byproduct was not observed in agreement with the comparable methylation of trivalent phenylarsenicals6 with MeI. Reflux of sodium salt of 8 in 6 M HCl effected global deprotection and decarboxylation providing crude AST. Purification on Dowex (H+ form) column with 0.25 M NH4OH followed by size-exclusion chromatography on Sephadex LH-20 with 70% (v/v) of EtOH/H2O afforded AST 1 (65%, from 11) as a racemic mixture. The formation of AST and reduced As(III)T-OH byproduct(s) were also observed, whereas formation of dimethylated product was not detected.
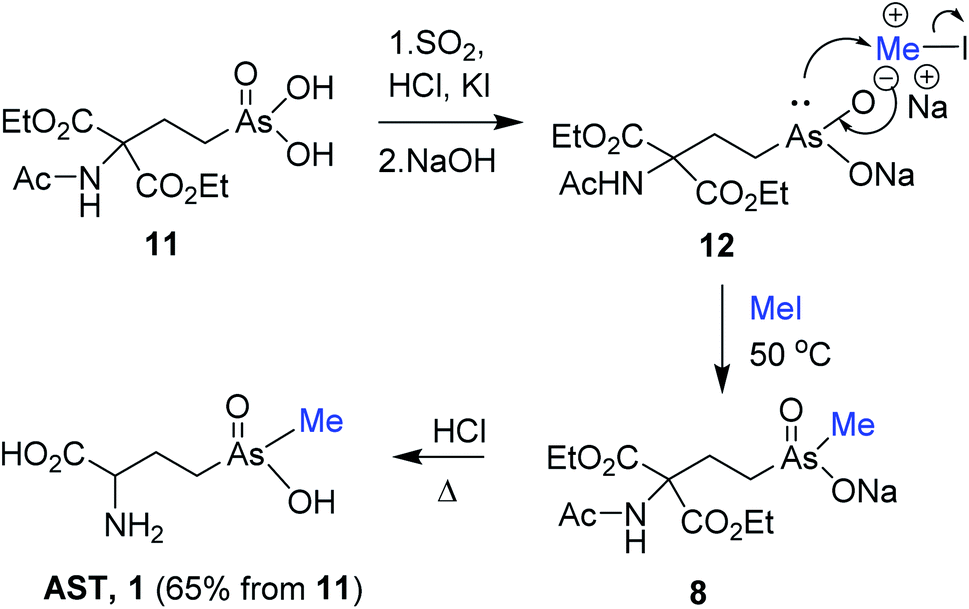 | ||
| Scheme 4 Synthesis of AST from N-acetyl protected AST-OH derivative 11 via reduction and methylation. | ||
We propose that the methylation of 12 with MeI in basic solution involves SN2 attack of the nucleophilic arsenic species on the electrophilic methyl iodide with concurrent formation of the arsenic–oxygen double bond, which also oxidized trivalent arsenic to the pentavalent species 8. The reaction resembles a Michaelis–Arbuzov reaction of trivalent phosphorus esters with alkyl halides to form pentavalent phosphonate esters. Analogous conversion of trivalent to pentavalent organoarsenicals with alkyl halides has been noted.18
The antibiotic properties of the chemically synthesized AST 1 (cAST, a mixture of the D/L-enantiomers) were characterized and compared with those of biogenic AST7,9 (bAST, the L-enantiomer) and semisynthesized AST10 (sAST, a mixture of the D/L-enantiomers). Approximately twice as much cAST or sAST was required to inhibit growth (Fig. 1) and GS activity (Table 1) of Escherichia coli as bAST, consistent with the L-enantiomer of b-AST as the active species.9 ArsN1, the bacterial enzyme that confers AST resistance, catalyzes transfer of the acetyl group of acetyl coenzyme A (AcCoA) to the amine group of 1, generating acetyl-AST (AcAST, 13; Scheme 5). Purified PpArsN1 (ArsN1 from Pseudomonas putida KT2440) nearly completely converted bAST to an arsenic species predicted to be AcAST, while only 50% of racemic cAST or sAST were converted to the putative species and the other half was unmodified (Fig. 2), consistent with only the L-enantiomer being the substrate of ArsN1, as predicted from L-AST-bound ArsN1 crystal structures.9
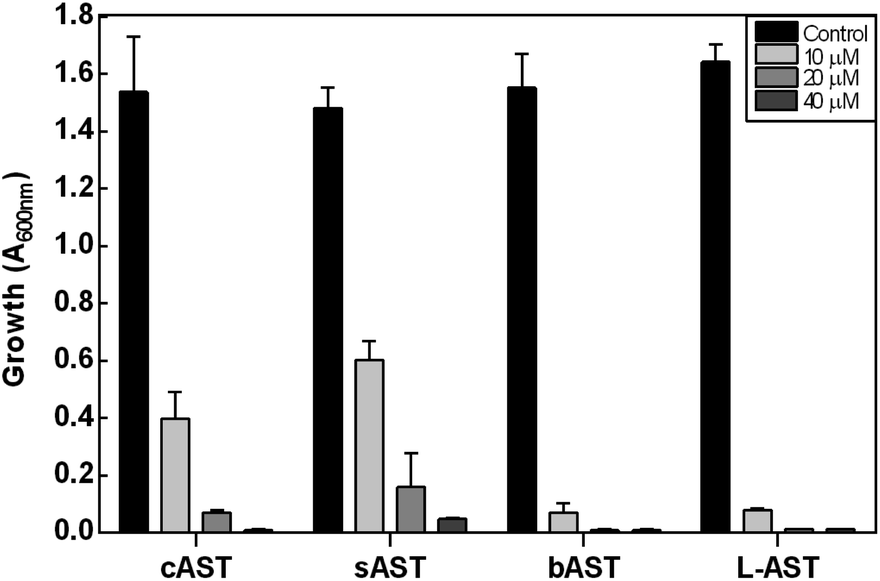 | ||
| Fig. 1 AST inhibits growth of E. coli. Cells were cultured in M9 medium in the absence or presence of the indicated concentrations of D/L-cAST, D/L-sAST, L-bAST, L-AST. Growth was estimated from the A600nm after 16 h. Data are the mean ± SE (n = 3). | ||
| AST | Ki (μM) |
|---|---|
| Chemically synthesized (D/L-cAST) | 0.75 ± 0.20 |
| Semisynthetic (D/L-sAST) | 0.65 ± 0.20 |
| Biogenic (L-bAST) | 0.30 ± 0.10 |
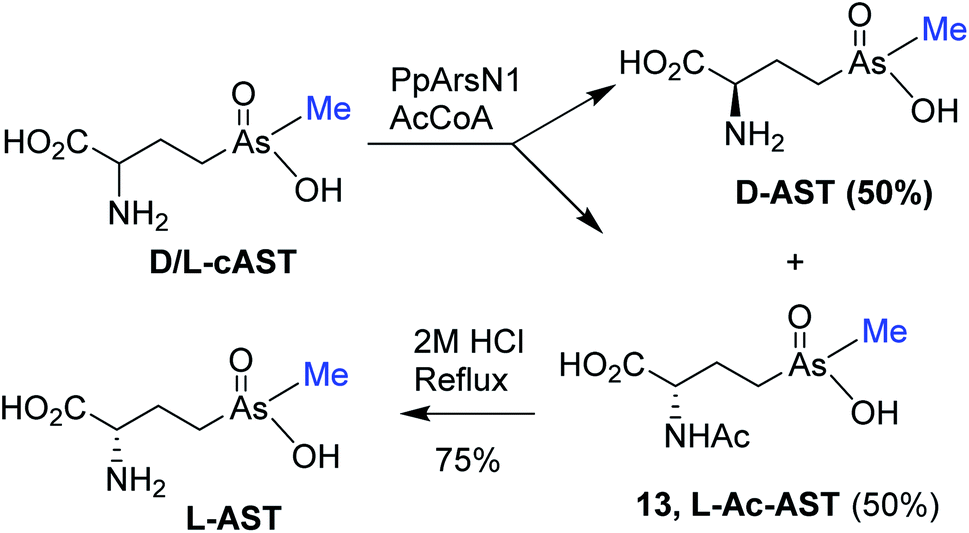 | ||
| Scheme 5 Enzymatic acetylation of cAST to L-AcAST 13 and chemical deacetylation of 13 to L-AST. | ||
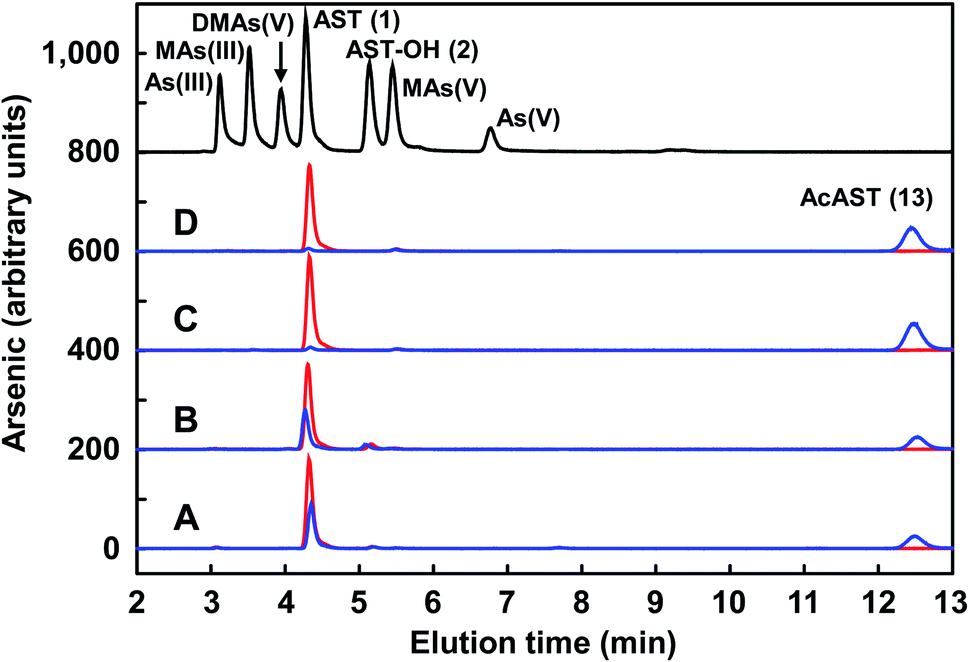 | ||
| Fig. 2 Enzymatic acetylation of 1 to produce 13. Chemically synthesized D/L-cAST (A), semisynthetic D/L-sAST (B), biogenic L-bAST (C) or L-enantiomeric (D) 1 was incubated in the absence (red lines) or presence (blue lines) of PpArsN1. The arsenic species in the reaction solutions were analyzed by HPLC-ICP-MS. Abbreviations: As(III), arsenite; MAs(III), methylarsenite; DMAs(V), dimethylarsenate; AST (1), arsinothricin; AST-OH (2), hydroxyarsinothricin; MAs(V), methylarsenate; As(V), arsenate; AcAST (13), N-acetylarsinothricin. | ||
We utilized PpArsN1 to purify L-AST from D/L-cAST. 7 mg of cAST was incubated with purified PpArsN1 and AcCoA overnight, resulting in a mixture of D-AST and L-AcAST 13 (Scheme 5). Purification by size-exclusion chromatography on Sephadex LH-20 afforded 13 (3.0 mg, 36%) and D-AST (2.1 mg, 30%). Reflux of 13 in 2 M HCl effected acetyl deprotection providing L-AST after purification on Sephadex LH-20 column with 70% (v/v) of EtOH/H2O. Chiral purity of L-AST (ee 87%) has been established by derivatization with Marfey's reagent (see Fig. S1 in ESI†).19 The L-AST when treated with PpArsN1/AcCoA was acetylated to 13 proving further its enantiomeric purity (Fig. 2). L-AST inhibited growth of E. coli as effectively as bAST (Fig. 1), additionally supporting the enantiomeric purity.
Conclusions
In summary, we developed two chemical methods for synthesis of the novel antibiotic arsinothricin. One is by reduction of the N-acetyl protected analogue of AST-OH and subsequent methylation of the resulting trivalent arsenic intermediate with methyl iodide, while the second involves condensation of the 2-chloroethyl(methyl)arsinic acid with acetamidomalonate. Enzymatic separation by enantioselective acetylation with PpArsN1 was utilized to purify L-AST from racemic AST. This convenient chemical synthesis can be scaled up to gram quantities to produce AST in sufficient amounts for further drug development.Experimental section
General Information
1H NMR spectra at 400 MHz and 13C NMR at 100.6 MHz were recorded in D2O unless otherwise noted. All chemical shift values are reported in parts per million (ppm) and referenced to the residual solvent peaks of CDCl3 (7.26) and D2O (4.79 ppm) for 1H NMR and the CDCl3 (77.16) peaks for 13C NMR spectra, with coupling constant (J) values reported in Hz. HRMS were obtained in TOF (ESI) negative or positive mode. TLC was performed on Merck Kieselgel 60-F254, and products were detected by staining with 1% ninhydrin solution. Merck Kieselgel 60 (230–400 mesh) was used for column chromatography. All reagents and solvents were purchased from commercial suppliers and used without further purification.(a) Preparation of diiodo(methyl)arsine (MeAsI2). A solution of KI (41.5 g, 0.252 mol) in H2O (40 mL) was added into the solution of monosodium salt of the commercially available methylarsonate (MeAs(O)(OH)ONa; 0.126 mol, 3.15 M/H2O, 40 mL). Conc. HCl (30 mL) was slowly added into the mixtures with continuous stirring. Then SO2 gas was passed into the mixtures for 30 min. The resulting mixture was extracted with CH2Cl2 (3 × 70 mL) and dried over anhydrous Na2SO4. The volatiles were evaporated to afford MeAsI2 (46 g, 97%) as orange liquid.14
(b) Condensation with 2-chloroethanol. 12 M aqueous NaOH (40 mL, 19.2 g, 0.48 mol) was slowly added into MeAsI2 (46 g, 0.122 mol) placed in a round bottom flask (0 °C, ice bath) over a 20 min with vigorous stirring. During the addition of NaOH, the yellow color was disappeared resulting in colorless solution. The resulting MeAs(ONa)2 solution was stirred for 15 min and then 2-chloroethanol (3a; 8.2 mL, 9.8 g, 0.122 mol) was slowly added over 10 min. The mixture was allowed to warm to ambient temperature (approximately 1 h) and stirring was continued for 12 h. The mixture was acidified with 6 M HCl to pH ∼4 and white precipitate was filtered out. The filtrate was evaporated at reduced pressure yielding a white solid, which was suspended in MeOH. The white precipitate was removed by vacuum filtration and the mother liquor was evaporated at reduced pressure to give 5 (19.9 g, 86%) as a white solid: 1H NMR δ 1.92 (s, 3H), 2.55 (t, J = 6.4 Hz, 2H), 3.97 (t, J = 6.4 Hz, 2H); 13C NMR δ 18.06, 37.30, 55.63; HRMS m/z calcd for C3H8AsO3 [M − H]− 1669694, found 166.9693.
Note: removal of volatiles from the reaction mixture prior neutralization with HCl and purification of the residue on silica column with 10–30% MeOH/CH2Cl2 provided iodide-free sodium salt of 5.
Note: treatment (0 °C to rt, 3 h) of the stirred solution of 6a (12 g, 70.4 mmol) in anhydrous CH2Cl2 (60 mL) with SOCl2 (12.3 mL, 20.1 g, 0.17 mol) followed by removal of volatiles under reduced pressure provided dichloro-(2-hydroxyethyl)arsine (8.7 g, 65%) as brown liquid with the 1H and 13C NMR spectra as reported10 (HRMS m/z calcd for C2H4AsOCl2 [M − H]− 188.8860, found 188.8862) instead of desired chloro(2-chloroethyl)(methyl)arsine 6b.
Method A. SOCl2 (10 mL) was added to an iodide-free sodium salt of 5 (500 mg, 2.63 mmol) in a dry flask at 0 °C (ice-bath) and was stirred at rt for 15 h. The reaction was quenched by adding 20 mL water and extracted with CH2Cl2. The aqueous phase was separated and volatiles were evaporated at reduced pressure. The residue was purified by column chromatography (20 → 30% MeOH/CH2Cl2) to give mixture of 7 and 5 as a sticky solid (450 mg; 80
:20). Second silica column purification afforded pure 7 (20 mg, 4%) as colorless gummy solid in addition to mixture of 7 and acidic form of 5 as a gummy solid (85:15; 400 mg, ∼69% of 7) to give overall yield for the conversion of 5 to 7 of approximately 73%. Compound 7 had: 1H NMR δ 2.08 (s, 2H), 2.94 (t, J = 6.6 Hz, 1H), 3.98 (t, J = 6.8 Hz, 1H); 13C NMR δ 17.47, 36.86, 36.97; HRMS m/z calcd for C3H9AsClO2 [M + H]+ 186.9501, found 186.9500.
Method B. Trimethylsilyl chloride (755 μL, 647 mg, 5.96 mmol) was added to a stirring solution of sodium salt of 5 (500 mg, 2.98 mmol) in DMSO (1 mL) at rt. The resulting mixture was stirred at rt for 14 h. The reaction was quenched by adding 10 mL water and extracted with EtOAc (5 × 10 mL) to remove DMSO. The aqueous phase was separated, and volatiles were evaporated at reduced pressure to give a gummy solid (∼400 mg) containing 7 (8%, based on 1H NMR and HPLC-ICP-MS) and unchanged 5 (92%); HRMS m/z calcd for C3H9AsClO2 [M + H]+ 186.9501, found 186.9500 for 7 and m/z calcd for C3H10AsO3 [M + H]+ 168.9840, found 168.9840 for 5.
(a) Reduction. AST-OH10 (2; 50 mg, 0.22 mmol) was dissolved in the mixture of concentrated HCl and water (1
:1, 5 mL). Then catalytic amount of KI (2.2 mg, 0.013 mmol) was added and SO2 gas was passed into this solution for 15 min at rt. The pH was then adjusted around ∼11 with 6 M NaOH (aq) solution under N2.
(b) Methylation. To the solution from step a, MeI (1.1 mL) was added and the mixture was stirred at 50 °C for 2 h. After 2 h, the pH of the reaction mixture showed around 6.5. The volatiles were evaporated under reduced pressure and the residue was suspended in methanol. The off-white precipitate was removed by vacuum filtration. Evaporation of volatiles from the filtrate at reduced pressure gave brown solid. The residue was dissolved in 2 mL H2O and applied to a Dowex 50WX8 (H+ form) column (30 × 1 cm, 10 g) which was washed with 50 mL of H2O. The product was eluted with a solution of NH4OH (0.5 M, 50 mL). The appropriate fractions (TLC, Rf 0.70, i-PrOH/H2O/NH4OH, 5
:2:3; identified by staining with 1% ninhydrin solution) from the ammonium elution (∼20 mL) were evaporated under reduced pressure to afford 10 (41 mg, 70%) as an off-white solid: 1H NMR δ 1.69 (s, 3H), 1.90–2.06 (m, 2H), 2.09–2.19 (m, 1H), 2.23–2.32 (m, 1H), 3.19 (s, 9H), 3.68 (dd, J = 11.6, 3.6 Hz, 1H); 13C NMR δ 15.84, 19.61, 28.99, 51.95, 78.62, 170.65; HRMS m/z calcd for C8H19AsNNaO4 [M + H]+ 290.0344, found 290.0346.
Procedure A (from 11).
(a) Reduction. Compound 11 (500 mg, 1.35 mmol) was dissolved in the mixture of concentrated hydrochloric acid and water (1
:1, 14 mL). Then catalytic amount of KI (13.5 mg, 0.08 mmol) was added and SO2 gas was passed into this solution for 15 min at ambient temperature. The pH was then adjusted to around 11 with 6 M NaOH (aq) solution under N2 to give crude 12 which was directly used in next step.
(b) Methylation. To the product from step a, MeI (7 mL) was added and the mixture was stirred at 50 °C for 4 h. The reaction progress was monitored by HPLC-ICP-MSA. After 4 h, the pH of the reaction mixture showed around 6.7. The volatiles were evaporated under reduced pressure and the residue was suspended in methanol. The off-white precipitate was removed by vacuum filtration. Evaporation of volatiles from the filtrate at reduced pressure gave brown solid crude 8.
(c) Deprotection and decarboxylation. 6 M HCl (20 mL) was added into the crude 8, and the resulting mixture was refluxed at 120 °C in an oil bath for 3 h. The mixture was neutralized with 6 M HCl around pH ∼7 and white precipitate was filtered out. The volatiles were evaporated under reduced pressure and the residue was suspended in methanol. The off-white precipitate was removed by vacuum filtration. Evaporation of volatiles from the filtrate at reduced pressure gave brown solid crude 1. The residue was dissolved in 15 mL H2O and applied to a Dowex 50WX8 (H+ form) column (30 × 1 cm, 10 g) which was washed with 100 mL of H2O. The product was eluted with a solution of NH4OH (0.5 M, 100 mL). The appropriate fractions (TLC, Rf 0.70, i-PrOH/H2O/NH4OH, 5
:2:3; identified by staining with 1% ninhydrin solution) from the ammonium elution (∼40 mL) were evaporated under reduced pressure to afford 1 as an off-white solid with some impurities. The residue was dissolved in 5 mL H2O and was applied onto a Sephadex LH-20 (GE Healthcare) column with a mobile phase 70% (v/v) EtOH at a flow rate of 1.0 mL min−1. Arsenic species in each fraction was analyzed by HPLC-ICP-MS. Fractions containing AST with high purity (>95%) were combined and concentrated by a rotary evaporator to afford 1 (ref. 7 and 10) (200 mg, 65% from 11) as an off-white solid: 1H NMR δ 2.03 (s, 3H), 2.27–2.34 (m, 2H), 2.42–2.48 (m, 1H), 2.52–2.58 (m, 1H), 3.97 (t, J = 6.4 Hz, 1H); 13C NMR δ 16.39, 23.34, 29.39, 54.39, 172.93; HRMS m/z calcd for C5H13AsNO4 [M + H]+ 226.0055, found 226.0055.
Note: use of pure 11free of diethyl acetamidomalonate impurities is critical since during deprotection and decarboxylation steps these impurities are converted to glycine that is difficult to separate from the AST product during purification on Dowex.
Procedure B (from 7).
(a) Condensation. Sodium (198 mg, 8.60 mmol) was added into a dry flask containing anhydrous EtOH (5 mL) and the mixture was stirred at rt until the sodium dissolved. Then diethylacetamidomalonate (1.40 g, 6.45 mmol) was added, and the resulting mixture was stirred for 30 min, followed by addition of 7 as a mixture of 7 and 5 (85
:15, 400 mg from Method A) dissolved in 2 mL EtOH. The resulting mixture was stirred at 70 °C for 4 h. Volatiles were evaporated under reduced pressure, yielding crude 8.
(b) Deprotection and decarboxylation. Subjection of crude 8 to the same protocol as described for Procedure A (step c) gave 1 (72 mg, ∼17% from 7) as an off-white solid: 1H NMR δ 1.97 (s, 3H), 2.21–2.27 (m, 2H), 2.33–2.41 (m, 1H), 2.43–2.51 (m, 1H), 3.83 (t, J = 6.2 Hz, 1H); 13C NMR δ 16.17, 23.53, 29.26, 55.01, 173.73; HRMS m/z calcd for C5H13AsNO4 [M + H]+ 226.0055, found 226.0055.
Note: treatment of crude 7(350 mg, from Method B) with EtONa and diethylacetamidomalonate as described for Procedure B afforded 1(5% from 5, based on 1H NMR and HPLC-ICP-MS). The presence of 1was confirmed by HPLC-ICP-MS and HRMS: calcd for C5H11AsNO4 [M − H]− 223.9909, found 223.9909.
(a) Enzymatic N-acetylation of AST (1). PpArsN1, the AST-selective N-acetyltransferase from Pseudomonas putida KT2440, was purified as described previously.9 10 μM of 1 (cAST, sAST bAST, or L-AST) was incubated in a buffer consisting of 20 mM Tris–HCl (pH 7.4), 1 mM ethylenediaminetetraacetic acid, 0.2 mM acetyl coenzyme A (AcCoA) at 37 °C for 30 min, with or without 0.2 μM PpArsN1. The reaction solution was filtered using an Amicon Ultra centrifugal filter with a 3K cutoff membrane (MilliporeSigma), and arsenic species in the filtrate were analyzed by HPLC-ICP-MS.
(b) Purification of L-AcAST. A larger amount of D/L-cAST 1 (0.9 mM, 35 mL, 7 mg) was incubated overnight with 1 mM AcCoA and 20 μM PpArsN1 in a buffer consisting of 20 mM Tris–HCl, pH 7.4 at 37 °C. The reaction solution was filtered using an Amicon Ultra centrifugal filter with a 3K cutoff membrane to remove protein. The filtrate was concentrated to 5 mL by rotary evaporation at reduced pressure and separated by Sephadex LH-20 size-exclusion chromatography gave L-AcAST 13 and D-AST with little impurities. Arsenic species in each fraction was analyzed by HPLC-ICP-MS. Fractions containing putative L-AcAST with high purity (>90%) were combined and concentrated by a rotary evaporation. The concentrated L-AcAST solution was applied again to Sephadex LH-20 size-exclusion chromatography for further purification. Fractions containing purified L-AcAST (>95%) were combined and concentrated to give 13 (3.0 mg, 36%): 1H NMR δ 1.92 (s, 3H), 19.8–2.07 (m, 1H), 2.04 (s, 3H), 2.24–2.15 (m, 1H), 2.37–2.26 (m, 2H), 4.24 (dd, J = 8.2, 4.6 Hz, 1H); 13C NMR δ 15.22, 21.89, 24.07, 29.42, 55.01, 173.80, 177.32; HRMS m/z calcd for C7H15AsNO5 [M + H]+ 268.0161, found 268.0162.
Note: fractions containing D-AST (>95%) were combined and concentrated to give D-AST (2.1 mg, 30%); HRMS m/z calcd for: C5H13AsNO4 [M + H]+ 226.0055, found 226.0059.
Derivatization of AST with Marfey's reagent
Sample of racemic AST or L-AST (5 μmol) is dissolved in 100 μL of water. 2 mg of 1-fluoro-2,4-dinitrophenyl-5-L-alanine amide (Marfey's reagent) is dissolved in 200 μL of acetone (1% solution). Solution of Marfey's reagent is added to the dissolved AST sample followed by 40 μL of 1 M NaHCO3. Reaction is set up at 40 °C for 1 h after which is quenched by addition of 20 μL of 2 M HCl. Small aliquot is taken, diluted with DMSO and injected on RP HPLC (column: Vydac Denali C-18, 5 μM, 120 Å). Elution method 100% A for 5 min followed by linear gradient of 0–100% B over 40 min where A is 0.1% TFA in H2O and B is 0.1% TFA in ACN. Eluting products were detected by UV at 340 nm. Results are shown in Fig. S1 in ESI.†Arsenic speciation by HPLC-ICP-MS
Arsenic species were determined by HPLC-ICP-MS using the HPLC retention time of known standards as described previously.10Author contributions
AHH and SHS conducted the synthetic experiments. VSN, AEG and MY conducted the HPLC-ICP-MS analysis and biological studies. AN and PC established chiral purity of L-AST. AHH, BPR, MY and SFW designed the experiments and wrote the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by NSF BIO/MCB grant 1817962 to MY and NIH grants GM55425 and GM136211 to BPR. AHH is recipient of the FIU Presidential Fellowship and Dissertation Year Fellowship.Notes and references
- S. Foster and W. Maher, J. Environ. Sci., 2016, 49, 131 CrossRef CAS PubMed.
- B. Chen, Q. Liu, A. Popowich, S. Shen, X. Yan, Q. Zhang, X.-F. Li, M. Weinfeld, W. R. Cullen and X. C. Le, Metallomics, 2015, 7, 39 CrossRef CAS PubMed.
- Q. Q. Wang, D. J. Thomas and H. Naranmandura, Chem. Res. Toxicol., 2015, 28, 281 Search PubMed.
- W. S. Tay and S. A. Pullarkat, Chem.–Asian J., 2020, 15, 2428 CrossRef CAS PubMed.
- W. R. Cullen, Q. Liu, X. Lu, A. McKnight-Whitford, H. Peng, A. Popowich, X. Yan, Q. Zhang, M. Fricke, H. Sun and X. C. Le, J. Environ. Sci., 2016, 49, 7 CrossRef CAS PubMed.
- H. Peng, B. Hu, Q. Liu, J. Li, X.-F. Li, H. Zhang and X. C. Le, Angew. Chem., Int. Ed., 2017, 56, 6773 CrossRef CAS PubMed.
- M. Kuramata, F. Sakakibara, R. Kataoka, K. Yamazaki, K. Baba, M. Ishizaka, S. Hiradate, T. Kamo and S. Ishikawa, Environ. Chem., 2016, 13, 723 CrossRef CAS.
- A. E. Galván, N. P. Paul, J. Chen, K. Yoshinaga-Sakurai, S. M. Utturkar, B. P. Rosen, M. Yoshinaga and X. Tang, Microbiol. Spectrum, 2021, 9, e00502 Search PubMed.
- V. S. Nadar, J. Chen, D. S. Dheeman, A. E. Galván, K. Yoshinaga-Sakurai, P. Kandavelu, B. Sankaran, M. Kuramata, S. Ishikawa, B. P. Rosen and M. Yoshinaga, Commun. Biol., 2019, 2, 131 CrossRef PubMed.
- S. H. Suzol, A. Hasan Howlader, A. E. Galván, M. Radhakrishnan, S. F. Wnuk, B. P. Rosen and M. Yoshinaga, J. Nat. Prod., 2020, 83, 2809 CrossRef CAS PubMed.
- J. Qin, C. R. Lehr, C. Yuan, X. C. Le, T. R. McDermott and B. P. Rosen, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 5213 CrossRef CAS PubMed.
- S. R. Adams, M. J. Sparkes and H. B. F. Dixon, Biochem. J., 1983, 213, 211 CrossRef CAS PubMed.
- V. K. Kuskov and V. N. Vasil'ev, Zh. Obshch. Khim., 1951, 21, 90 CAS.
- A. M. Spuches, H. G. Kruszyna, A. M. Rich and D. E. Wilcox, Inorg. Chem., 2005, 44, 2964 CrossRef CAS PubMed.
- Attempted displacement of OH group in trivalent arsine 6a with NBS or X2/PPh3 (X = Br or I) also failed to produce the halogenated products of type 6b (X = Br or I).
- Direct halogenenation of 5 with HX (X = Br or I) failed to give halogenated products of type 7.
- Numerous attempts to displace activated hydroxyl group in the protected homoserine with diiodo(methyl)arsine (MeAsI2) in the presence of aqueous NaOH failed to produce AST 1, instead yielding dihydro-3-amino-2-(3H)-furanone lactone.
- B. E. Abalonin, Russ. Chem. Rev., 1991, 60, 1346 CrossRef.
- N. Bionda, M. Cudic, L. Barisic, M. Stawikowski, R. Stawikowska, D. Binetti and P. Cudic, Amino Acids, 2012, 42, 285 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: RP-HPLC traces after derivatization of AST with Marfey's reagent and copies of 1H, 13C NMR and HRMS spectra for all new compounds. See DOI: 10.1039/d1ra06770b |
| ‡ Present address: Department of Chemistry, Johns Hopkins University, Baltimore, Maryland, USA. |
| § Present address: Department of Chemistry & Biochemistry, Rowan University, Glassboro, New Jersey, USA. |
| This journal is © The Royal Society of Chemistry 2021 |