DOI:
10.1039/D1RA06691A
(Paper)
RSC Adv., 2021,
11, 38003-38015
Enhancing the activation of persulfate using nitrogen-doped carbon materials in the electric field for the effective removal of p-nitrophenol
Received
6th September 2021
, Accepted 5th November 2021
First published on 25th November 2021
Abstract
Degradation of nonbiodegradable organic compounds into harmless substances is one of the main challenges in environmental protection. Electrically-activated persulfate process has served as an efficient advanced oxidation process (AOP) to degrade organic compounds. In this study, we synthesized three nitrogen-doped carbon materials, namely, nitrogen-doped activated carbon plus graphene (NC), and nitrogen-doped activated carbon (NAC), nitrogen-doped graphene (NGE), and three nitrogen-doped carbon material-graphite felt (GF) cathodes. The three nitrogen-doped carbon materials (NC, NGE, NAC) were characterized using X-ray diffraction, Raman spectroscopy, X-ray electron spectroscopy, and nitrogen desorption–adsorption. The electron spin resonance technique was used to identify the presence of hydroxyl radicals (˙OH), sulfate radicals (SO4˙−) and singlet oxygen (1O2) species. The results showed that NC was more conducive for the production of free radicals. In addition, we applied NC-GF to an electro-activated persulfate system with the degradation of p-nitrophenol and investigated its performance for contaminant degradation under different conditions. In general, the nitrogen-doped carbon electrode electro-activated persulfate process is a promising way to treat organic pollutants in wastewater.
1. Introduction
With the increase of pollutants in industrial wastewater discharge and strict environmental laws and regulations, the efficient treatment of biodegradable substances in wastewater is gaining increasing attention. P-nitrophenol (PNP) is an important organic synthetic material commonly used in pharmaceutical industries such as phenacetin, manufacture of paracetamol, pesticide 1605 and dyes.1 Most nitrophenols, including PNP, enter the environment during production and processing, which are not easily decomposed in surface water. Because nitrate has strong electronic affinity, PNP has the characteristics of easy oxidation and toxicity in the environment.2 Therefore, if the wastewater containing PNP is improperly treated, the very high content will cause serious harm to the human body, water bodies, the environment and so on.
Currently, some traditional treatment techniques can be used for the treatment of PNP wastewater,3–6 but these techniques have some limitations. For example, the cost of physical treatment7 is high and the effect is poor. Although the efficiency of microbial degradation is guaranteed, the microbial domestication cycle is long and easy to impact, and the engineering application is difficult.7,8 The chemical oxidation method mainly uses advanced oxidation process (AOP) to complete the oxidation and degradation operations of organic matter, which can improve the oxidation efficiency, and has the characteristics of low oxidation cost, no secondary pollution, and easy promotion.9–11
The activated persulfate oxidation method is a new advanced oxidation technology developed in recent years. It is done with the aid of external energy or catalysis. The manner of the agent breaks the dioxygen bond O–O in the persulfate (S2O82−) to produce a sulfate radical (SO4˙−) with a solitary pair of electrons. The free radical has higher oxidation–reduction potential (E0 = 2.5–3.1 V) and stronger oxidation capacity than the persulfate itself, and theoretically can degrade most organic pollutants.12 Compared with the traditional advanced oxidation technology based on hydroxy free radical (˙OH), the activated persulfate oxidation method has higher reaction stability, loose requirements for pH, and is coupled with being environment-friendly. So, it has unique advantages and broad application prospects in organic pollution control.13 However, persulfate alone reacts slowly with most contaminants. So, it is necessary to activate the persulfate to produce free radicals. Persulfate can be activated to generate a sulfate radical (SO4˙−) by thermal,14 ultraviolet,15 transition metal ions,16–19 ultrasound,20 electrical,21 microwave,22 carbon,23 etc. Electrically-activated persulfate technology can effectively activate persulfate to mineralize organic pollutants in wastewater and introduce no metal ions throughout the whole process, reducing the risk of secondary contamination. At the same time, the electrically-activated persulfate process involves various reaction processes, such as free radical oxidation, non-free radical oxidation24 and electron direct transfer.25 Consequently, most hardly degradable organic pollutants can be oxidized. The mechanism of electrically-activated persulfate producing SO4˙− is shown in formulas (1-1)–(1-3):26
| | |
S2O82− + e− → SO42− + SO4˙−
| (1-1) |
| | |
HSO5− + e− → OH− + SO4˙−
| (1-2) |
| | |
HSO5− + e− → ˙OH + SO42−
| (1-3) |
The efficiency of the electro-activated persulfate depends largely on the selection of the electrodes, and the new electrodes may be more suitable for the practical application of the system.27 In the electro-activated persulfate system, the cathode material determines the pollutant degradation capacity of the system.
Recently, much attention has been attracted by carbon-based materials in environmental applications. The current commonly used carbon electrodes mainly include activated carbon,28 carbon felt, graphene,29 and carbon nanotubes.30 It has the advantages of low cost, huge specific surface area and strong adsorption capacity. The advantages of carbon material as a cathode are: (1) carbon material itself is enough to catalyze the generation of SO4˙−; (2) carbon materials have good conductivity and relatively low production costs; (3) carbon materials can increase the mass transfer efficiency of electrodes and persulfate ions, thus increasing the catalytic efficiency of persulfate and improving the system mineralization capacity. The ideal element atoms (such as nitrogen, sulfur, boron, phosphorus, etc.) or groups can produce heteroatom-doped carbon, which can change the material properties of the carbon material and increase the catalytic efficiency and electrical conductivity transfer rate. After modification, the material has strong stability and repeated reuse. Lee31 et al. studied that multi-walled carbon nanotubes (CNT) could effectively activate PS to oxidize phenols. Duan32 et al. used three-dimensional cubic mesoporous carbon to activate persulfate to remove phenols from an aqueous solution. The system was comparable to the zero-valent iron-activated persulfate system, without introducing any metal ions into the water. While Wang33 et al. doped nitrogen into graphene to induce a folded structure expansion. The material adsorption capacity is 1.75 times that before modification. The introduction of surface nitrogen-containing functional groups increased the charge density on the surface, fortified the medium of electron transfer, and improved the conductance transmission rate. Liu27 et al. used activated carbon fiber (ACF) with super-strong adsorption properties as cathodic-activated persulfate (E-ACF-PS) of electrochemical systems to remove nondegradable organic pollutants in water. Compared with the metal cathode, the ACF cathode enhanced the mass transfer effect of the solid–liquid interface, which improved the utilization efficiency of the oxidizer and reduced the energy consumption of the system. However, the electrochemically-activated persulfate technology has the unsatisfactory removal effect of low concentration of non-biodegradable organic pollutants in water, a limited mass transfer area of electrodes and persulfate, and an easy side reaction of returning H+ to H2. Although the activation of persulfate by carbon materials had some effects. Many studies showed that the carbon catalyst as a strong oxidizer would destroy surface catalytic groups by oxidation, with reduced catalytic activity and short service life. Therefore, we used carbon materials with good adsorptive effects as the cathode, and the activation of persulfate under the external electric field may achieve a complementary effect. The carbon cathode can increase the contact area of the electrode and persulfate, while the adsorption of low concentration of organic matter is beneficial to improve the oxidation efficiency of low concentration of organic matter. The external current can achieve both electric and carbon cathode activated persulfate to degrade organic pollutants.
In this study, we synthesized nitrogen-doped carbon material (NC) catalysts using a multistep approach to embellish the GF cathode and used NC-GF as a cathode during the electrically-activated persulfate process. We researched the degradation process of p-nitrophenol (PNP) in wastewater, and species and the yield of free radicals in the E-NC-PMS process. We compared the degradation of PNP contaminants using NAC-GF, NGE-GF, and NC-GF cathodes under an electric field. Furthermore, we investigated the effects of PMS concentration, pH, voltage and background ions on PNT degradation and electrode stability, and discussed the possible reaction mechanisms.
2. Experimental
2.1 Materials and reagents
The reagents used in this study were all of analytical grade reagents used without further purification. Activated carbon and graphene were purchased from Tangshan Jianhua Technology Development Co., Ltd. Conductive carbon black and GF were obtained from Suzhou Yilong Energy Technology Co., Ltd. Melyanamine, potassium hydrogen monosulfate and nitrophenol were purchased from Aladdin Shanghai Aladdin Biochemical Technology Co., Ltd. The experimental water was deionized water.
2.2 Preparation of nitrogen-doped carbon materials
First, we weighed accurately 0.1 g graphene and 0.4 g activated carbon, which were then dispersed in 50 mL of ethanol and treated under ultrasound for 30 min. Then, an appropriate amount of melamine powder and formaldehyde solution were added, and ultrasound treatment was continued for 2 h. After the ultrasound treatment, the resulting solution was placed in a 90 °C in a constant temperature water bath until the solution was completely dry. The resulting material was ground into powder and fit into a crucible and placed inside a tubular furnace. In the nitrogen environment, the dried mixed powder was first heated to 550 °C at a heating rate of 5 °C min−1 and heated at that temperature for 60 minutes. It was then heated to 850 °C at 5 °C min−1 followed by further heating at 850 °C for 60 minutes, then cooled to room temperature with a 5 °C min−1 procedure. Finally, we obtained nitrogen-doped carbon material (NC). Nitrogen-doped activated carbon (NAC) and nitrogen-doped graphene (NGE) were prepared by the same method.
2.3 Preparation of NC-GF
The carbon felt (GF) was cut of size 4 cm × 2 cm, which was immersed in acetone solution for 30 min, then washed repeatedly with deionized water and soaked overnight and finally in a drying box 24 h. A 0.02 g of nitrogen-doped carbon material with 0.08 g carbon black (XC-72R) were mixed and then added to 0.5 mL PTFE, 3 mL ethanol and 5 mL deionized water and mixed rapidly. After ultrasound treatment for 10 min, the slurry was coated with GF, dried in the drying box for 24 h. Finally, in heating furnace at 370 °C of annealing for 1 h, the NC-GF cathode material was obtained. The method for obtaining nitrogen-doped activated carbon and nitrogen-doped graphene-modified graphene felt was the same as above.
2.4 Physical characterizations
The prepared materials were characterized in accordance with the test criteria. Fourier transform infrared (FT-IR) spectroscopy (NicoletiS50) was used to determine the structure of the samples. The surface elements of the sample were analyzed by X-ray photoelectron spectroscopy (XPS, K-alpha). The Raman spectra were recorded using a Raman spectrometer (XploRA PLUS). The specific surface area and pore size distribution were determined from nitrogen adsorption–desorption isotherms collected on an Autosorb-iQ-C instrument using Brunauer–Emmett–Teller (BET) method. X-ray diffraction (XRD) patterns were recorded using a MiniFlex600 (Rigaku, Japan) with Cu Kαradiation (λ = 0.15406 nm) and scanning angle ranging from 10° to 80° of 2θ.
In addition, the electron spin resonance (ESR) was used to testify the existence of free radicals. 5,5-Dimethethyl-1-pyrroline-N-oxides (DMPO) and 2,2,6,6-tetramethylpiperidine (TEMP) were used as trapping agents. A 0.2 mL of sample was mixed with a 0.2 mL trapping agent for 30 s and then transferred to the capillary for detection.34
2.5 Degradation experiments
Experiments were conducted to decompose nitrophenol (PNP) in a 100 mL reactor with 0.05 M Na2SO4 as the supporting electrolyte. The prepared NC-GF (4.0 cm × 2.5 cm) was used as a cathode, with a stainless steel electrode (4.0 cm × 2.5 cm) as an anode, 1 cm apart between the electrodes. The reaction conditions were pH = 5, voltage 5 V, 0.05 M Na2SO4 and a constant temperature of 20 °C. The reaction was aerated at a rate of 0.3 L min−1. A 2 mL sample was taken every 20 minutes and then the PNP concentration was determined using ultraviolet-visible (UV-vis) spectroscopy at a maximum absorption wavelength of 317 nm. Eqn (2-1) was used to calculate the degradation rate. Fitted PNT degradation curve by first-order kinetic eqn (2-2):| |
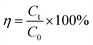 | (2-1) |
| |
 | (2-2) |
where C0 is the concentration of PNP (mg L−1) at the adsorption–desorption equilibrium state, Ct is the real-time concentration of PNP (mg L−1), η is the degradation rate of PNP, and k is the first-order rate constant (min−1).
3. Results and discussion
3.1 XRD analysis
The crystal structure of preprepared samples (NC, NGE, NAC, GE) was studied using powder XRD studies. As shown in Fig. 1, NGE, NC, NAC and GE showed two broad diffraction peaks centered at 23° and 44°, which may be attributed to (002) and (100) planes, indicating their microcrystalline structural carbon featuring weak graphitization. The presence of diffraction peaks at 2θ = 23° displayed parallel stacking and interconnection between the parts of the graphite layer in the carbon material. The peak at 2θ = 44° indicated that the sp2 hybridized carbon atoms interacted with the carbon material to form a hexagonal lattice structure. The (002) and (100) half-peak full widths of the diffraction peaks did not vary significantly, which indicated that all materials had a similar microcrystal size and a stable microcrystal structure.
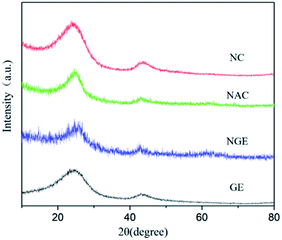 |
| | Fig. 1 XRD plots of GE, N-doped graphene (NGE), N-doped activated carbon (NAC) and N-doped carbon materials (NC). | |
3.2 XPS analysis
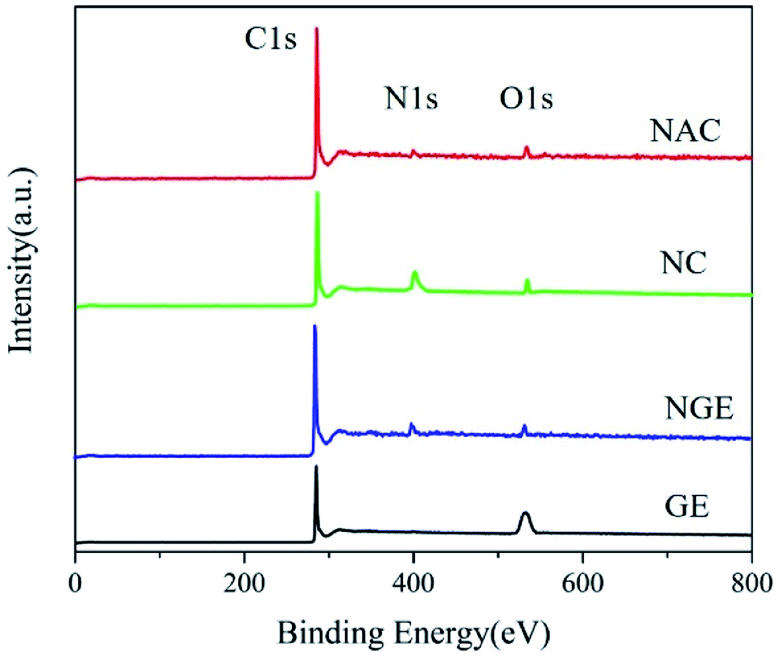
XPS was performed to analyze the elemental composition of GE, NGE, NAC and NC. As shown in Fig. 2, two peaks, C1 (285 eV) and O1s (533 eV) were seen in GE. The N1 peak was clearly visible near the binding energy of 400 eV after N doping, the nitrogen content was estimated to be 3.22%, 0.96%, and 5.96% in NGE, NAC and NC, respectively. Compared to the 13.87% oxygen content of graphene, the oxygen content of nitrogen–carbon material decreased slightly to 4.93%, 5.59%, and 4.69%, respectively, in the three materials. This suggested that the oxygen functional group had little influence on the catalytic performance, but the oxygen-containing functional group played an important role in the formation of C–N bonds with nitrogen precursors during doping.35,36
 |
| | Fig. 2 XPS spectra of GE, N-doped graphene (NGE), N-doped activated carbon (NAC) and N-doped carbon materials (NC). | |
N1s spectra of three nitrogen–carbon mixed materials could be divided into three peaks, 398.1 eV, 399.9 eV, and 402.0 eV,37 respectively, corresponding to pyridine, pyrrolic, and graphite nitrogen, as shown in Fig. 3a–c. The nitrogen-doped materials possess nitrogen-containing groups such as pyridine N, pyrrole N and graphite N with high charge density, spin density, and asymmetric electrons on the surface of the carbon materials, jointly promoting the redox reaction. Earlier, it was also pointed out that doping of N breaks the inertia of sp2 hybridized C and changed the chemical and electrochemical properties of graphene while producing new active sites for various reactions. The nitrogen-containing alkaline groups, such as pyridine N, pyrrole N, graphite N could act as Lewis alkaline sites to facilitate electron transfer.38 Doping of N changed the charge density of its adjacent C atoms, which gave the C atoms a positive charge to promote electron transfer with PMS; N promoted the adsorption of π-π bonds to organic molecules and collaborated the oxidation reaction. So, nitrogen doping of carbon materials was conducive to improving catalytic properties. In previous studies, we found that a higher graphite nitrogen ratio favored persulfate activation. Ren38 et al. also believed that PMS adsorption energy on graphite N(−1.418 eV) was much stronger than on pyridine N/pyrrole N(−0.892 eV) and graphite nitrogen was the main active site during the degradation process. We compared GE, NGE, NAC and NC in Table 1 and it was found that the NC had the highest proportion of graphite N at 49.77%, but the highest pyridine nitrogen was in NGE and NAC.
 |
| | Fig. 3 XPS high-resolution N1s spectra of NGE, NAC and NC. | |
Table 1 Types and content of nitrogen in GE and N doped carbon materials
| Sample |
N (%) |
| Pyridinic-N |
Pyrrolic-N |
Graphite-N |
| GE |
|
0.18 |
|
| NAC |
40.43 |
26.41 |
33.16 |
| NGE |
55.08 |
25.26 |
19.66 |
| NC |
32.43 |
17.80 |
49.77 |
 |
| | Fig. 4 FTIR spectra of GE, AC and NC. | |
3.3 FI-TR
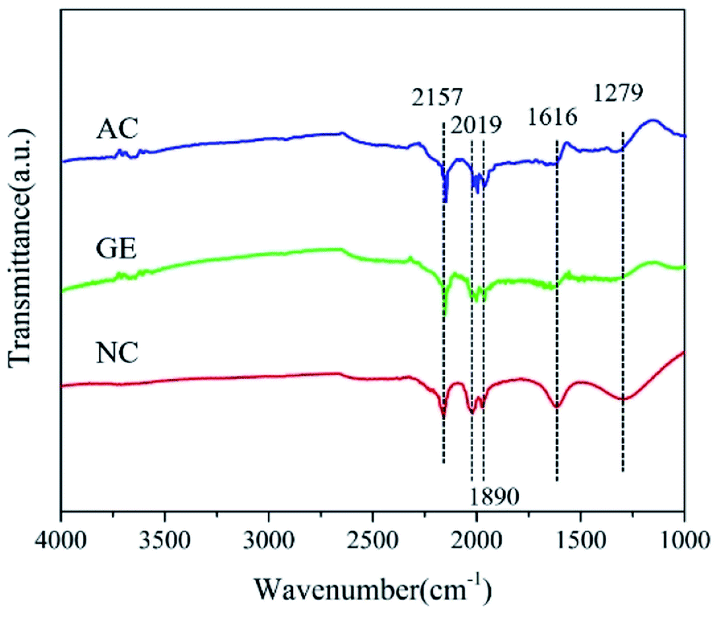
The functional group composition of graphene and activated carbon–nitrogen doping was further analyzed using FITR (Fig. 4). The peak of 2157 cm−1 was associated with C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C or CN. The peaks at 2019 cm−1 and 1890 cm−1 could possibly be linked to in-plane bending of the C–H bond and that associated with the COOH group at C
C or CN. The peaks at 2019 cm−1 and 1890 cm−1 could possibly be linked to in-plane bending of the C–H bond and that associated with the COOH group at C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, respectively. The peaks at 1616 cm−1 and 1279 cm−1 are linked to CN and C–N groups, respectively.39,40 As shown in the figure, the presence of CN and C–N in the nitrogen-doped carbon materials, which are not found in activated carbon and graphene, showed that doping of nitrogen atoms in the carbon skeleton of graphene was successful.
O, respectively. The peaks at 1616 cm−1 and 1279 cm−1 are linked to CN and C–N groups, respectively.39,40 As shown in the figure, the presence of CN and C–N in the nitrogen-doped carbon materials, which are not found in activated carbon and graphene, showed that doping of nitrogen atoms in the carbon skeleton of graphene was successful.
Table 2 Data obtained from N2 adsorption isotherms on different materials
| Sample |
Surface area cm2 g−1 |
Pore volume cm3 nm−1 g−1 |
Pore diameter N m |
| NAC |
130.61 |
0.373 |
3.841 |
| NC |
79.092 |
0.104 |
3.794 |
| NGE |
41.796 |
0.091 |
3.390 |
3.4 BET analysis
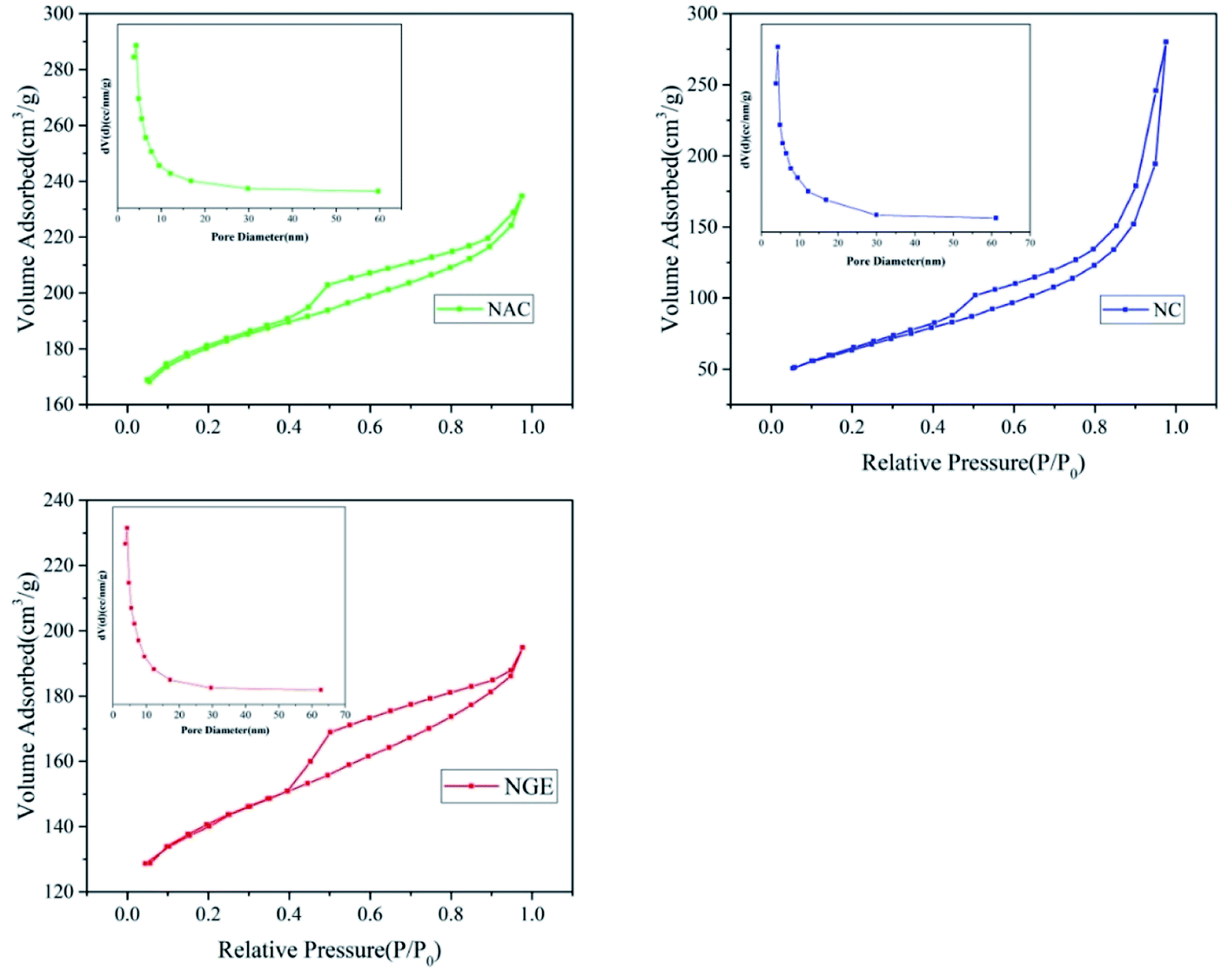
Fig. 5 shows the N2 adsorption–desorption isotherms of the three catalysts and the pore size distribution patterns calculated using the BJH method. According to the collected data, NGAC had a high specific surface area of 130.61 m2 g−1 then NC with a specific surface area of 79.092 m2 g−1 and NGE with 63.554 m2 g−1. In contrast, NAC had an absolutely high specific surface area, but the surface area of the NC was nearly 2 times that of the NGE, which indicated that the activated carbon made a contribution to the specific surface area of the modified catalyst. The narrow pore diameters of NC, NAC and NGE were observed at 3.794 nm, 3.841 nm and 3.390 nm, respectively (Table 2). The results of the N2 adsorption–desorption measurements indicated that the doping of nitrogen and carbon materials changed the morphology of the catalyst. Moreover, the analysis results manifested that there were part of the large pore and small pore aperture distribution, which constituted a large pore microporous multiporous structure. Free radicals were mainly produced from the surface pore structure of carbon materials, which was closely related to the surface materialization property and the composition of functional groups. The doped-nitrogen carbon materials had great potential in activating PMS. The multistage porous structure of the catalyst was conducive to the formation of a three-phase interface involving gas-electrode-liquid, and more active groups will be exposed because of the large specific surface area of the pore structure, thus greatly increasing the reaction rate for pollutant degradation on the electrode surface.
 |
| | Fig. 5 N2 adsorption–desorption isotherms and pore size distributions of NGE, NAC and NC. | |
3.5 Raman spectra analysis
The characterization of the cathode material by Raman spectroscopy was conducted to further determine the degree of surface defects in the three-carbon materials. In Fig. 6, Raman spectroscopy of the three materials showed that the band at 1345 cm−1 corresponds to a disorder-induced graphite carbon band (D band), while the band at 1580 cm−1 represented the bond extension of all sp2 carbon pairs (G bands). ID/IG is defined as the strength response ratio of D and G bands and is often used to characterize the degree of defect of the material.41 The larger the ID/IG value, the lower is the degree of the graphitization of the material and the higher the structural defects. The ID/IG values of NGE, NAC and NC are 1.04,1.15 and 1.25, respectively. It was seen that the ID/IG value of NC was the largest, which showed the crystal surface defects and a high degree of graphite.42 This data manifested an increased degree of surface defects in the material that could provide more active sites for the catalytic reaction.
 |
| | Fig. 6 Raman spectra of different materials. | |
 |
| | Fig. 7 The effect of oxidant on PNP degradation. | |
4. Degradation experiments
4.1 Oxidant
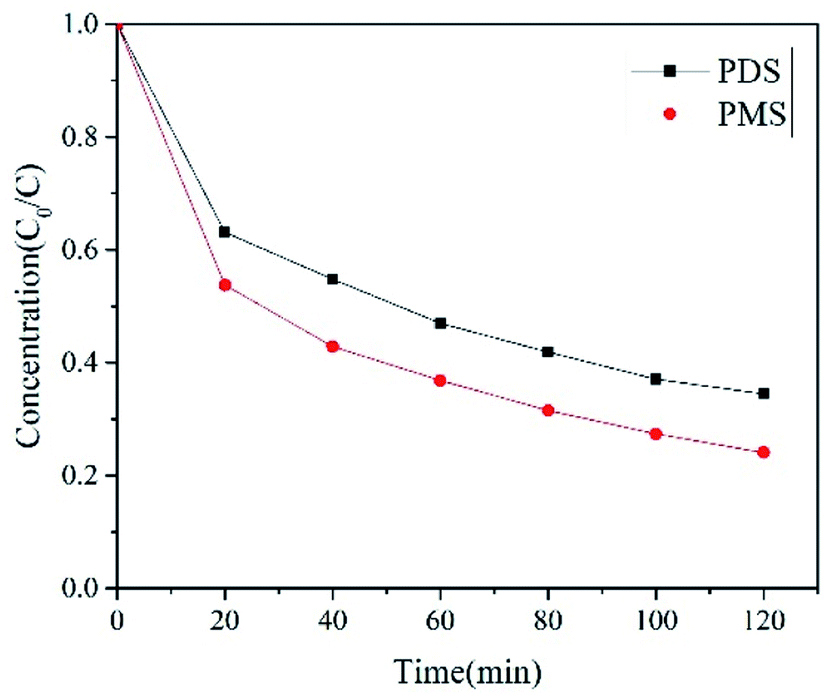
In electrochemical oxidation, the oxidant type is one of the important factors affecting the system's efficiency of removal of organic pollutants. Therefore, we had selected peroxymonosulfate (PMS) and persulfate (PDS) as oxidants for the E-NC system (Fig. 7). It was shown that both PDS and PMS as oxidizers could effectively remove PNP in water. Under equal oxide dosage and operating conditions, 75.91% of PNP in the aqueous solution was removed within the E-NC-PDS system in 120 min, while the removal efficiency of the E-NC-PMS system in 120 min reached 85.85%. The E-NC system was found to be more efficient in activating PMS, to obtain higher PNP removal and mineralization rates than E-NC-PDS. This was due to the stronger interaction between PMS and graphite N, and more PMS was adsorbed onto the nitrogen–carbon-doped materials to enhance the redox potential of the NC–PMS* complex. In contrast, the equilibrium of the PDS adsorption amount on carbon materials was much lower than that of PMS; nitrogen modification did not further improve the adsorption amount of PDS.38
4.2 Carbon material
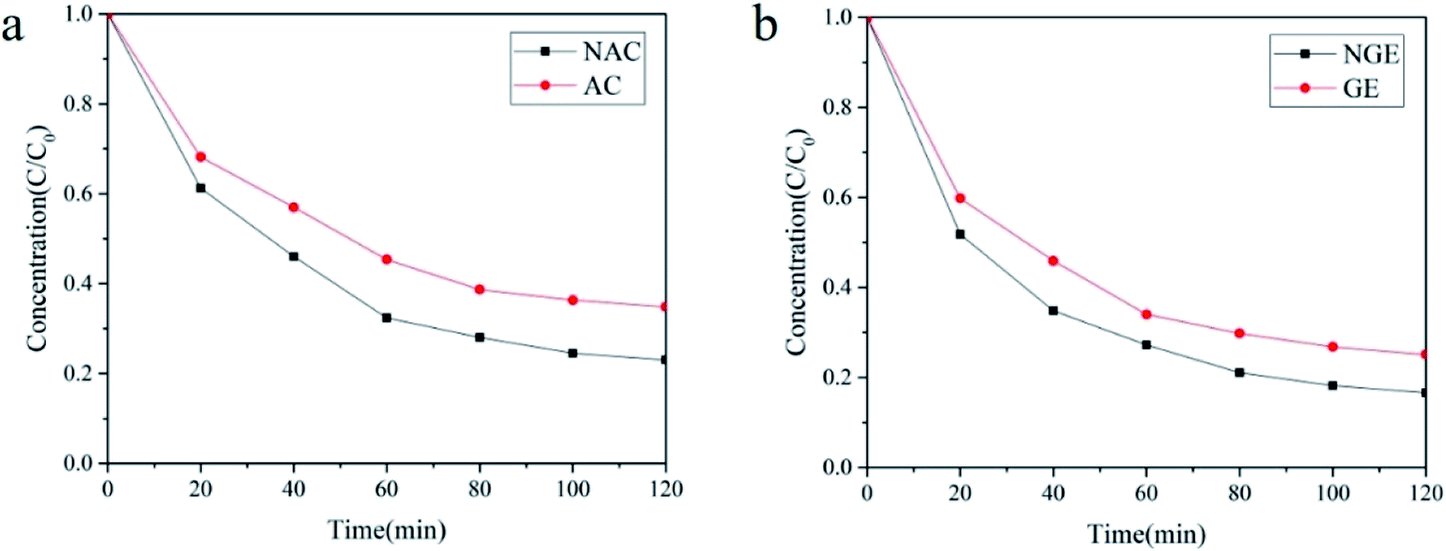
Fig. 8(a) shows the removal effect of activated carbon and nitrogen-mixed activated carbon as a cathode on PNP at pH = 5, voltage 5 V, 0.05 M Na2SO4, and a constant temperature of 20 °C. In the E-GAC-PMS system, the degradation rate of PNP in 120 min was 65.16%; in the E-NGAC-PMS system, the PNP degradation rate was 76.95%. Fig. 8(b) shows the removal effect of graphene and nitrogen-doped graphene on PNP at pH = 5 and a voltage of 5 V, 0.05 M Na2SO4, and a constant temperature of 20 °C. In the E-GNS-PMS system, the degradation rate of PNP in 120 min was 74.92% and the PNP degradation rate was 83.43% in the E-NGNS-PMS system. The introduction of atoms of ideal elements (such as nitrogen, sulfur, boron, and phosphorus) or groups can make mixed atomic carbon doping, which can change the material characteristics of the carbon material, increase the catalytic efficiency, electrical conductivity transmission rate and modify the material to have strong stability and reuse times. Nitrogen is adjacent to carbon in the periodic table, which is close to the atomic radius and has similar properties. Then, the atomic doping was stable, and the activation sites could be introduced. Relevant studies showed that nitrogen-doped carbon materials, such as nitrogen-doped graphene and nitrogen-doped carbon nanotubes have excellent activation properties for PMS, and graphite N played a key role in the catalytic process.29,41,43 Consequently, nitrogen-doped activated carbon and graphene have a better efficiency of activated persulfate to produce free radicals to degrade contaminants.
 |
| | Fig. 8 (a) The effect of NAC and AC on PNP degradation; (b) The effect of NGE and GE on PNP degradation. | |
Fig. 9(a) shows the degradation effect of nitrogen-doped carbon material, nitrogen-doped graphene and activated carbon on PNP as a cathode under the electric field. The experimental conditions are pH = 5, voltage 5 V, 0.05 M Na2SO4 and a constant temperature of 20 °C. In the E-NGE-PMS system, the degradation rate of PNP in 120 min was 83.43%, in the E-NAC-PMS system, it was 76.95%, while in the E-NC-PMS system, 91.96% of PNP was degraded after 120 min. The first 20 minutes of the treatment followed the first-order reaction kinetics. We obtained the disappearance rate constant of NC, NGE, NAC as 0.055 min−1, 0.034 min−1 and 0.024 min−1 (Fig. 9(c) and (d)), respectively. This suggested that the catalyst prepared by mixing graphene and activated carbon as the carbon skeleton was better effective in the activated persulfate process under an electric field than single graphene or activated carbon.
 |
| | Fig. 9 (a) The effect of material on PNP degradation; (b) the effect of different systems on PNP degradation; (c) K–L curve and (d) degradation rate constants. Conditions: pH = 5, voltage 5 V, 0.05 M Na2SO4 and a constant temperature of 20 °C; (e) E-NC-PMS systems for the UV-vis spectral analysis of PNP. Conditions: pH = 5, voltage 5 V, 0.05 M Na2SO4 and a constant temperature of 20 °C. | |
Fig. 9(b) shows the degradation effect of activated persulfate on PNP in different systems. The degradation rate of the PMS system, adsorption system, electrolysis system and E-NC-PMS system were 9.67%, 31.56%, 17.41% and 91.69%, respectively. As shown in Fig. 9(e), 50 mg L−1 of PNP can be essentially degraded in 120 min in the E-NC-PMS system. Furthermore, with the reaction time progressing, as shown in Fig. 9(e), it should be noted that the absorbance was rapidly reduced at 260 to 350 nm, when the reaction was initiated. The reaction rate was significantly reduced after 60 min and remained largely unchanged after 80 min. Compared with other systems, the E-NC-PMS system had greater PNP removal efficiency, and the efficiency was much higher than the sum of the removal efficiency in the other three systems (58.64%). The results revealed a synergistic effect of the three systems of individual electrolysis, adsorption and PMS oxidation so that the E-NC-PMS system, composed of three systems, could effectively remove PNP from water.
 |
| | Fig. 10 Effect of pH on PNP degradation. Conditions: pH = 5, voltage 5 V, 0.05 M Na2SO4 and a constant temperature of 20 °C. | |
4.3 pH
Under solution pH values kept constant at 3.0, 5.0, 7.0 and 9.0, we explored the effect of nitrogen-doped carbon electrodes under an electric field on the persulfate activation on nitrophenol (PNP) removal efficiency (Fig. 10). When the pH values of the solution are 3.0, 5.0, 7.0 and 9.0, the removal efficiencies of the PNP within 120 min of the system were 74.27%, 85.85%, 76.83%, and 72.9%, respectively. The results indicated that the system effectively removed PNP from water under both acidic, neutral and alkaline conditions. The system degraded PNP best under the condition of pH = 5. In the E-NC-PMS system, the removal of PNP was mainly attributed to five processes: carbon electrode adsorption, PMS oxidation alone, direct electron transfer, radical oxidation, and non-radical oxidation. Under acidic conditions, –OH binding H+ on nitrophenol was present in the form of a positive charge, which was more easily absorbed by the carbon electrode of the cathode and degraded on the carbon electrode surface.44 However, in the environment of very low pH, persulfate accelerated the decomposition to generate sulfate free radicals, resulting in a large number of sulfate-free genes in the solution that could not reach the organic surface in a timely manner and produced the quenching reaction of the radical itself, thus reducing the utilization rate of persulfate and sulfate-free radicals. Under neutral and alkaline conditions, the nitrophenol existed in the form of an anion, and was easier to move to the anode under the external electric field and not easy to be adsorbed and degraded by the cathode, which is the carbon electrode.
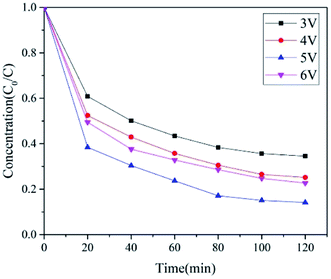 |
| | Fig. 11 The effect of voltage on PNP degradation. Conditions: pH = 5, voltage 5 V, 0.05 M Na2SO4 and a constant temperature of 20 °C. | |
4.4 Voltage
The voltage has always been used as a significant influence variable in electrochemistry. When the electrode potential is more than 6 V, the cathode is prone to hydrogen precipitation secondary reaction. We investigated the effect of voltage on nitrogen-doped carbon electrode activated PMS system for the removal of PNP below the hydrogen degradation potential (Fig. 11). As the voltage increased from 3 V to 6 V, the degradation rates of PNP were 65.47%, 74.82%, 85.85%, and 76.8%. On the one hand, as the voltage increased, the potential energy was greater at both ends of the electrode, so was the electron density, and PMS promoted the redox reaction of the cathode through electron transfer to produce the sulfate radical. On the other hand, the increase of voltage also favored direct anodic oxidation and non-radical oxidation of the transition state structure of PMS excited at the anode. However, the optimal voltage value was 5 V because the continued increase in voltage would cause more secondary reactions.45
4.5 Concentration of PMS
In electroactive PMS systems, PMS is the main source of ˙OH and SO4˙−, and the concentration of PMS in the solution directly affects the contact efficiency of the PMS and the electrode. Hence, the PMS concentration in the solution directly affected the efficiency of pollutant removal in the system. We examined the PNP removal efficiency in the E-NC-PMS system with PMS concentrations of 10 mM, 20 mM, 30 mM and 40 mM. As shown in Fig. 12, when the initial concentration of PMS increased from 10 mM to 40 mM, the removal rates for PNP from the E-AC-PMS system during 120 min were 72.46%, 76.95%, 85.85%, and 78.81%. The above results displayed that increasing the PMS concentration significantly improved the contaminant removal efficiency of the E-NC-PMS system. The degradation rate initially gradually increased because higher persulfate concentrations could produce more sulfate-free radicals per unit of time, facilitating the degradation of PNP. The PNP degradation rate appeared to decrease as the persulfate concentration continued to increase, possibly because the excess sulfate radicals could not reach the organic surface in time with their own quenching reaction.46
 |
| | Fig. 12 The effect of PMS concentration on PNP degradation. Conditions: pH = 5, voltage 5 V, 0.05 M Na2SO4 and a constant temperature of 20 °C. | |
4.6 Background ions
The ubiquitous anions in natural water bodies (e.g. HPO42−, HCO3−, Cl−, etc.) can affect the removal of organic contaminants during the AOPs process. The influence of various ions on the removal efficiency of PNP in the E-NC-PMS system is shown in Fig. 13. The presence of HPO42− inhibited the removal of PNP from the system. According to the literature report, HPO42−could catalyze the PMS production of ˙OH and SO4˙−. Simply considering it from this aspect, the presence of HPO42− may promote the activation of PMS, thus increasing the removal rate of PNP. However, the presence of HPO42− similarly lead to the occurrence of competitive side reactions, where HPO42− with ˙OH and SO4˙− produced in the system generated phosphate radicals, which had weaker oxidation capacity, thus reducing the concentration of ˙OH and SO4˙− and inhibiting the removal of PNP. Cl− had a significant role in the degradation of PNP. The promotion of chloride ions on PNP degradation may be due to a series of chain reactions producing new radicals Cl˙ and Cl2−˙. HCO3− reacted with the radicals in the system to produce a less active carbonate radical (CO3−˙˙), thus causing the suppression of the removal of the system PNP. Meanwhile, HCO3− in the solution reacted with PMS to generate HCO4−, which caused an invalid breakdown of PMS in the system and affected the removal efficiency of the system PNP. Furthermore, there was a competition between the HCO3− and PMS. The excess HCO3− shared the contact site of the electrode plate surface with PMS to reduce the contact efficiency between PMS and electrode plate, reducing the activation efficiency of the PMS.
 |
| | Fig. 13 The effect of background ions on PNP degradation. Conditions: pH = 5, voltage 5 V, 0.05 M Na2SO4 and a constant temperature of 20 °C. | |
4.7 TOC and COD
Total organic carbon (TOC) is an important water quality indicator and TOC reflected the degree of mineralization of pollutants in wastewater. Fig. 14(a) shows the removal rate of the TOC for four different cathode materials. The removal rates of TOC by SS, NC, NGE and NAC as cathode materials were 51.22%, 81.41%, 71.71%, and 63.48% within 2 h. The experiment showed that NC as a cathode material on the mineralization degree of pollutants and had significant improvement compared with stainless steel electrodes.
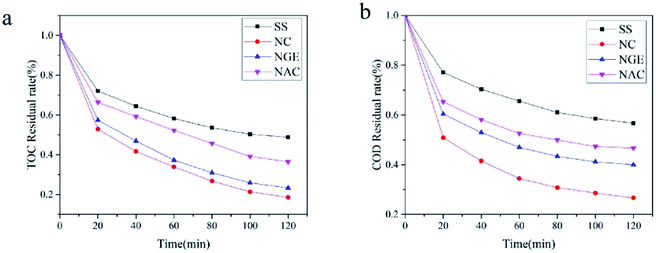 |
| | Fig. 14 TOC and COD of PNP degradation. Conditions: pH = 5, voltage 5 V, 0.05 M Na2SO4 and a constant temperature of 20 °C. | |
Chemical oxygen demand (COD) is also a vital indicator of water treatment. The removal rate of four materials is shown in Fig. 14(b), with NC as a cathode material reaching 73.33% in 2 h. The COD removal rates were increased by 13.33%, 20% and 30% compared to NGE, NAC and SS electrodes, respectively. This indicated that the NC electrode had the highest degradation rate as a cathode.
4.8 Identification of the major active substances
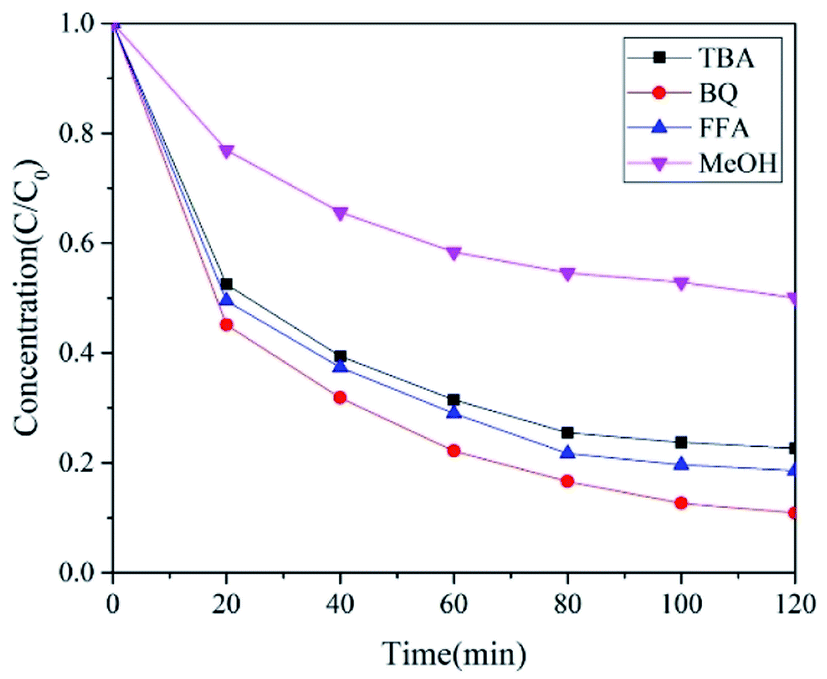
It has been reported that PMS and PDS under the electrochemical action can be activated to generate active substances such as ˙OH and SO4˙−. In the E-NC-PMS system, both ˙OH and SO4˙− were the main active substances for degradation. The capture agents currently commonly used to capture ˙OH and SO4˙− are mainly methanol (MeOH) and tertiary butanol (TBA). The reaction rate constants of methanol for ˙OH and SO4˙− are 9.7 × 108 M−1 S−1 and 2.5 × 107 M−1 S−1, respectively. The reaction rate of tertiary butanol for ˙OH is 6.0 × 108 M−1 S−1, and for SO4˙− reaction it is only 7.6 × 105 M−1 S−1, which differed in three orders of magnitude.12 Therefore, in systems with ˙OH and SO4˙−, the type of free radical produced in the system is often judged by adding methanol and tertiary butanol to the system. The 10 mM methanol and tertiary butanol were added to the E-NC-PMS system to capture ˙OH and SO4˙− produced during the reaction of the system.
When no free radical trap was present, the PNP removal rate in 120 min was 91.69%. When 10 miM tertiary butanol was added to the system, the removal rate of PNP in the system decreased from 91.69% to 81.44%, which showed that ˙OH played a role in the removal of PNP. After the addition of 10 mM methanol to the system, the removal rate of PNP in the system decreased from 91.69% to 49.94%, which indicated that SO4˙− also did some work in the removal of PNP. The above results proved that the E-NC-PMS system can produce ˙OH and SO4˙−, which effectively degrade PNP in the solution.
Many studies have shown that singlet oxygen (1O2) could be produced in PMS activation systems, while the self-decomposition of PMS may slowly produce 1O2. However, it is not clear whether there is a 1O2 generation in the E-NC-PMS system. Hence, furan methanol (FFA)12 is used as a trapping agent to clean the 1O2 produced in the system. The reaction rate constant of furan methanol for 1O2 is 1.2 × 108 M−1 S−1, which is five orders of magnitude higher than the reaction rate constants of methanol (3.0 × 103 M−1 S−1) and tertiary butanol (1.8 × 103 M−1 S−1), thus effectively cleaning the 1O2 generated in the system. The addition of 10 mM furan methanol to the E-NC-PMS system reduced the PNP degradation rate from 91.69% to 77.36%, which showed that 1O2 also plays a vital role in the degradation of PNP.
Finally, 10 mM of p-benzoquinone was added to the system to capture O2˙−. It can be seen from Fig. 15 that after the addition of p-benzoquinone, the degradation rate of PNP in the E-NC-PMS system decreased from 91.69% to 89.13%. The results discussed above indicated that no production or a small amount of O2˙˙− was not produced in this system. In the E-NC-PDS system, the ˙OH contribution rate was 20.24%, and that for SO4˙− was 34.36%, and 1O2 was 24.69%. It was seen that in the E-NC-PDS system, 1O2, SO4˙−, and ˙OH played dominant roles in PNP removal.
 |
| | Fig. 15 The degradation of PNP at different experimental conditions. Conditions: pH = 5, voltage 5 V, 0.05 M Na2SO4 and a constant temperature of 20 °C. | |
 |
| | Fig. 16 ESR spectrum in the E-NC-PMS, E-NAC-PMS and E-NGE-PMS systems. Conditions: pH = 5, voltage 5 V, 0.05 M Na2SO4 and a constant temperature of 20 °C. | |
To further verify the presence of ˙OH, SO4˙−, O2˙−, and 1O2 during the reaction, we used the ESR technique to detect the signals of the associated radicals (Fig. 16). DMPO and TEMP were added to the system to capture possible active species. DMPO was used to capture ˙OH, SO4˙− and O2˙− to generate the corresponding spin-additive products DMPO-OH, DMPO-SO4, and DMPO-OO. TEMP was used to capture 1O2 to generate TEMP-1O2. During the reaction, ESR spectra were used to detect the characteristic signal profile of the DMPO-OH additive compound that showed four split lines with a strength ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:1. The characteristic signal map of the DMPO-SO4 additive had six split lines.47 The ESR spectra of all electrodes showed the 1:1:1 signal, which is characteristic of theTEMP-1O2 adduct. Consequently, the ESR results showed that the peaks of DMPO-OH, DMPO-SO4, and TEMP-1O2 all existed, which indicated that OH, SO4˙−, and 1O2 all existed in the reaction system. However, the characteristic peaks of the DMPO-OO additive compound were not obvious. Combined with previous quenched experiments, it was further illustrated that although ˙OH, SO4˙−, O2˙−, and 1O2 were generated during the reaction, O2˙− is not the main active species for the PNP degradation.
:2:2:1. The characteristic signal map of the DMPO-SO4 additive had six split lines.47 The ESR spectra of all electrodes showed the 1:1:1 signal, which is characteristic of theTEMP-1O2 adduct. Consequently, the ESR results showed that the peaks of DMPO-OH, DMPO-SO4, and TEMP-1O2 all existed, which indicated that OH, SO4˙−, and 1O2 all existed in the reaction system. However, the characteristic peaks of the DMPO-OO additive compound were not obvious. Combined with previous quenched experiments, it was further illustrated that although ˙OH, SO4˙−, O2˙−, and 1O2 were generated during the reaction, O2˙− is not the main active species for the PNP degradation.
 |
| | Fig. 17 Electrode stability. Conditions: pH = 5, voltage 5 V, 0.05 M Na2SO4 and a constant temperature of 20 °C. | |
4.9 Study of electrode stability
We performed 10 consecutive cycles to remove organic wastewater with a running time of 120 min (Fig. 17). The experimental conditions were 0.05 M Na2SO4, the voltage of 5v, pH = 5, and a constant temperature of 20 °C. The removal rate of PNP with ten consecutive applications of the same electrode decreased by 20% and the catalytic properties were reduced. It may be because, during the oxidative degradation of the E-NC-PMS system, the micropores of the NC are covered with the adsorbed electrolyte and contaminant molecules, which decreased the NC surface area, reduced catalytic effects, and decreased the ability of reuse.
4.10 The degradation pathway of PNP
Many studies at home and abroad use PNP in oxidative and degradation environments, and with different treatments, the intermediates obtained by the methods are different. The most studied one is the use of free radicals to degrade PNP. The intermediates formed in its oxidative degradation are generally 4-nitrophthalphenol, 3,4,5-trihydroxynitrobenzene, 1,2,4-trihydroxybenzene, o-dihydroxybenzene, p-benzenediol, phenol, para-benzoquinone, phthalquinone, cis-butenedioic acid, allomaleic acid, dicarboxyl, acetic acid and formic acid and so on.48,49
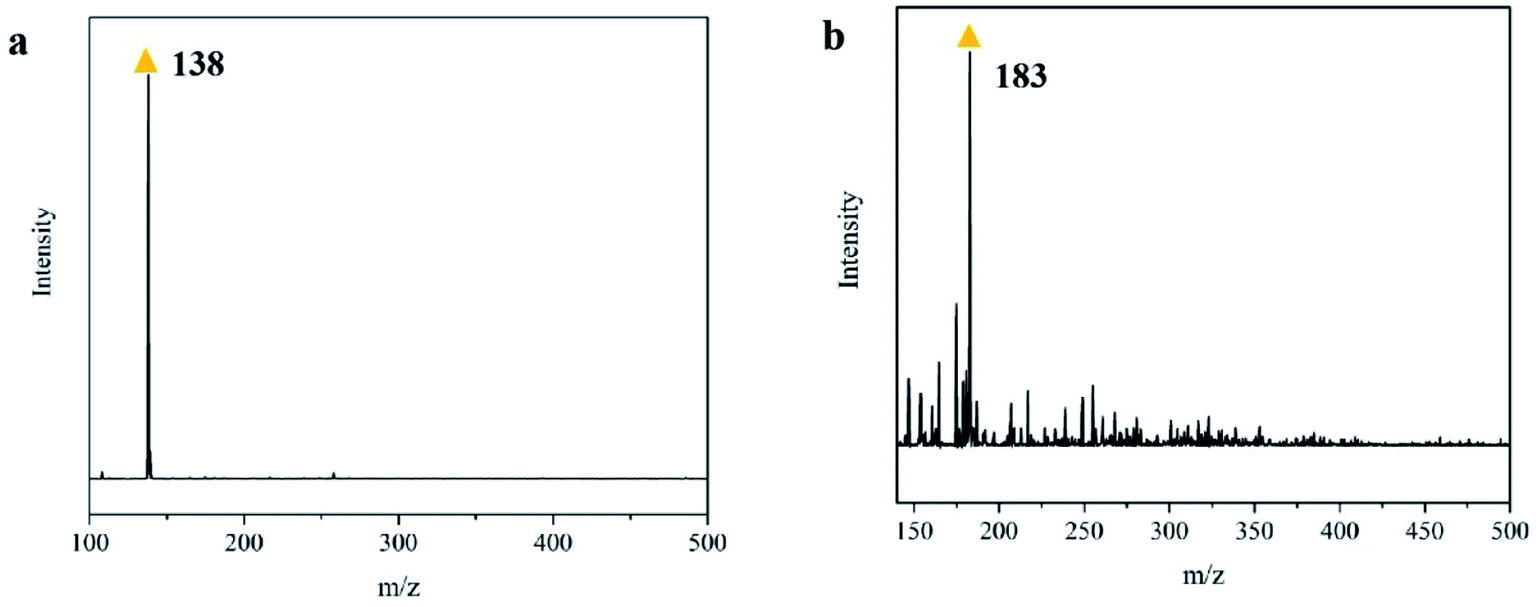
To explore the major intermediates of PNP in the E-NC-PMS system, samples at different degradation times in the system were taken. We analyzed the degradation intermediates in these samples using LC-MS, confirming them based on the peak-out time of the material and the mass-charge ratio (m/z) in mass spectrometry. The results of the intermediate analysis are shown in Table 3, where 2,4-dinitrophenol (2,4-DNP), phenol, p-benzoquinone, cis-butenedioic acid and 2,5-dihydroxyl-1,4-benzoquinone were detected to be the major intermediates in the PNP degradation system. As we can see from Table 3, through the analysis of LC-MS, the product with the peak time of 18.5 min is 2,4-DNP, the peak time at 12.3 min is phenol, the peak time of 7.6 min is p-benzoquinone, and the peak time of 4.5 min is cis-butenedioic acid. To further detect the degradation intermediates of PNP, the substances in the above samples were also analyzed by mass spectrometry. The results showed that 2,5-dihydroxy-1,4-phenylquinone is also an intermediate during PNP degradation, and additionally it was further verified that 2,4-DNP is an intermediate during PNP degradation. Among them, the test results of LC-MS were analyzed as described below. As seen from Fig. 18(b), the molecular mass-to-charge ratio of 2,4-DNP is 182.5, which indicates that this substance is 2,4-DNP. It has been found that, among other treatments,50 2,4-DNP was also one of the intermediates of PNP degradation. Also shown in Fig. 18(a), the molecular mass-to-charge ratio of this intermediate is 138, so we speculate that the intermediate with a mass-to-charge ratio of 138 was 2,5-dihydroxy-1,4-phenylquinone.
Table 3 The intermediates of PNP during the degradation process
| Number |
Reservation time (min) |
Test method |
Intermediate product |
| 1 |
4.5 |
LC-MS |
Cis-butenedioic acid |
| 2 |
7.6 |
LC-MS |
P-benzoquinone |
| 3 |
12.3 |
LC-MS |
Phenol |
| 5 |
6.1 |
LC-MS |
2,5-Dihydroxy-1,4-benzoquinone |
| 6 |
18.5 |
LC-MS |
2,4-Dinitrophenol |
 |
| | Fig. 18 Full-scan MS spectra of main oxidation products of 2,4-dinitrophenol (a) and 2,5-dihydroxy-1,4-phenylquinone (b). | |
 |
| | Fig. 19 The proposed degradation pathway of PNP. | |
Based on the previous analysis of the intermediates during PNP degradation, the PNP degradation pathway in the E-NC-PMS system is proposed (Fig. 19). First, with electric fields, SO4˙− in the system would attack the N–C bond on PNP molecules to form phenol-containing radicals. Subsequently, the intermediates phenol and 2,4-DNP are formed in the system. Phenol formed a intermediate p-benzoquinone under further attack from SO4˙−, then 2,5-dihydroxy-1,4-phenyl quinone was formed in the presence of hydroxyl radicals. The C–C bond was broken to form an open loop (cis-butenedioic acid was mainly detected in this experiment). It is eventually degraded to carbon dioxide and water. On the other hand, the formation of 2,4-DNP in the system is mainly the effect of the nitro group, which has the electron-pulling effect.50 Nitro groups removed from the benzene ring would further attack the PNP molecules, thus forming a 2,4-DNP. Open-ring material is then formed under attack by SO4˙− and is finally degraded into carbon dioxide and water.
5. Conclusions
This study proposed the degradation of persulfate using nitrogen-doped carbon electrodes. We discussed the relationship between persulfate concentration, voltage, pH, background ions and degradation efficiency. The PNP degradation rate by the NC-GF cathode in the electro-activated PMS system was 91.69%, and the COD removal rate was 73.33%. The results indicated that the proposed technique has advantages of low energy consumption and high efficiency over a wide pH range. The study is important for the application of activated persulfate oxidation processes under an electric field using modified carbon electrodes.
Conflicts of interest
There are no conflicts to declare.
References
- J. Li, Y. Ren, F. Z. Ji and B. Lai, Chem. Eng. J., 2017, 324, 63–73 CrossRef CAS.
- C. S. D. Rodrigues and L. M. Madeira, Environ. Technol. Innovation, 2021, 21, 101265 CrossRef CAS.
- Q. Wang, L. X. Zhang, Y. K. Guo, M. Shen, M. Wang, B. Li and J. L. Shi, Chem. Eng. J., 2020, 396, 125347 CrossRef CAS.
- W. J. Kong, Q. Y. Yue, Y. Gao, Q. Li, X. Xu, Y. Kong and B. Y. Gao, Chem. Eng. J., 2021, 413, 127456 CrossRef CAS.
- Q. Wang, H. M. Zhu and B. Li, Chem. Eng. J., 2019, 378, 122072 CrossRef CAS.
- Q. Wang, C. M. Fan, G. M. Li, J. S. Luo and B. Li, Catal. Sci. Technol., 2019, 9, 7023–7033 RSC.
- Z. Dhaouefi, A. Toledo-Cervantes, K. Ghedira, L. Chekir-Ghedira and R. Munoz, Chemosphere, 2019, 234, 356–364 CrossRef CAS PubMed.
- M. A. Musa and S. Idrus, Sustainability, 2021, 13, 4656 CrossRef CAS.
- E. Domingues, E. Fernandes, J. Gomes and R. C. Martins, Sci. Total Environ., 2021, 776, 145958 CrossRef CAS.
- R. Dewil, D. Mantzavinos, I. Poulios and M. A. Rodrigo, J. Environ. Manage., 2017, 195, 93–99 CrossRef CAS PubMed.
- A. Krzywicka and A. Kwarciak-Kozlowska, Water Sci. Technol., 2014, 69, 1875–1878 CrossRef CAS PubMed.
- F. Ghanbari and M. Moradi, Chem. Eng. J., 2017, 310, 41–62 CrossRef CAS.
- J. Liu, S. Zhong, Y. Song, B. Wang and F. Zhang, J. Electroanal. Chem., 2018, 809, 74–79 CrossRef CAS.
- Y. Q. Gao, N. Y. Gao, Y. Deng, D. Q. Yin, Y. S. Zhang, W. L. Rong and S. D. Zhou, Desalin. Water Treat., 2015, 56, 2225–2233 CrossRef CAS.
- Y. Q. Chen, S. X. Gao, Z. Z. Liu, S. L. Shao, W. Z. Yin, Z. Fang and L. Z. Huang, Environ. Chem. Lett., 2019, 17, 1017–1021 CrossRef CAS.
- T. V. Chung, T. Q. Anh, D. Q. Phung and T. D. Luong, Asian J. Chem., 2012, 24, 1371–1374 CAS.
- X. D. Du, Y. Q. Zhang, F. Si, C. H. Yao, M. M. Du, I. Hussain, H. Kim, S. B. Huang, Z. Lin and W. Hayat, Chem. Eng. J., 2019, 356, 178–189 CrossRef CAS.
- Q. Wang, B. B. Wang, Y. Ma and S. T. Xing, Chem. Eng. J., 2018, 354, 473–480 CrossRef CAS.
- M. A. Imran, Y. Z. Tong, Q. Hu, M. Z. Liu and H. H. Chen, Water, 2020, 12, 354 CrossRef CAS.
- S. Chakma, S. Praneeth and V. S. Moholkar, Ultrason. Sonochem., 2017, 38, 652–663 CrossRef CAS PubMed.
- H. R. Song, L. X. Yan, J. Jiang, J. Ma, Z. X. Zhang, J. M. Zhang, P. X. Liu and T. Yang, Water Res., 2018, 128, 393–401 CrossRef CAS PubMed.
- D. Miao, S. Zhao, K. C. Zhu, P. Zhang, T. C. Wang, H. Z. Jia and H. W. Sun, Chemosphere, 2020, 253, 126679 CrossRef CAS PubMed.
- W. Q. Huang, S. Xiao, H. Zhong, M. Yan and X. Yang, Chem. Eng. J., 2021, 418, 129297 CrossRef CAS.
- Y. B. Ding, X. R. Wang, L. B. Fu, X. Q. Peng, C. Pan, Q. H. Mao, C. J. Wang and J. C. Yan, Sci. Total Environ., 2021, 765, 142794 CrossRef CAS PubMed.
- H. Lee, H. I. Kim, S. Weon, W. Choi, Y. S. Hwang, J. Seo, C. Lee and J. H. Kim, Environ. Sci. Technol., 2016, 50, 10134–10142 CrossRef CAS PubMed.
- C. H. Zhao, B. B. Shao, M. Yan, Z. F. Liu, Q. H. Liang, Q. Y. He, T. Wu, Y. Liu, Y. Pan, J. Huang, J. J. Wang, J. Liang and L. Tang, Chem. Eng. J., 2021, 416, 128829 CrossRef CAS.
- Z. Liu, H. J. Ding, C. Zhao, T. Wang, P. Wang and D. D. Dionysiou, Water Res., 2019, 159, 111–121 CrossRef CAS PubMed.
- Y. M. Tan, C. F. Xu, G. X. Chen, Z. H. Liu, M. Ma, Q. J. Xie, N. F. Zheng and S. Z. Yao, ACS Appl. Mater. Interfaces, 2013, 5, 2241–2248 CrossRef CAS PubMed.
- Y. F. Song, J. Yang, K. Wang, S. Haller, Y. G. Wang, C. X. Wang and Y. Y. Xia, Carbon, 2016, 96, 955–964 CrossRef CAS.
- K. Adu, D. H. Ma, Y. X. Wang, M. Spencer, R. Rajagopalan, C. Y. Wang and C. Randall, Nanotechnology, 2018, 29, 035605 CrossRef PubMed.
- H. Lee, H. J. Lee, J. Jeong, J. Lee, N. B. Park and C. Lee, Chem. Eng. J., 2015, 266, 28–33 CrossRef CAS.
- X. Duan, H. Sun, M. Tade and S. Wang, Catal. Today, 2018, 307, 140–146 CrossRef CAS.
- X. B. Wang, Y. L. Qin, L. H. Zhu and H. Q. Tang, Environ. Sci. Technol., 2015, 49, 6855–6864 CrossRef CAS PubMed.
- T. Luo, Y. Peng, L. Chen, J. Li, F. Wu and D. Zhou, Environ. Sci. Technol., 2020, 54, 10261–10269 CrossRef PubMed.
- X. L. Li, H. L. Wang, J. T. Robinson, H. Sanchez, G. Diankov and H. J. Dai, J. Am. Chem. Soc., 2009, 131, 15939–15944 CrossRef CAS PubMed.
- Z. H. Sheng, L. Shao, J. J. Chen, W. J. Bao, F. B. Wang and X. H. Xia, ACS Nano, 2011, 5, 4350–4358 CrossRef CAS PubMed.
- C. Chu, J. Yang, D. Huang, J. Li, A. Wang, P. J. J. Alvarez and J.-H. Kim, Environ. Sci. Technol., 2019, 53, 10352–10360 CrossRef CAS PubMed.
- W. Ren, G. Nie, P. Zhou, H. Zhang, X. Duan and S. Wang, Environ. Sci. Technol., 2020, 54, 6438–6447 CrossRef CAS PubMed.
- X. J. Wei, S. G. Wan and S. Y. Gao, Nano Energy, 2016, 28, 206–215 CrossRef CAS.
- X. N. Lu, L. Y. Li, B. Song, K. S. Moon, N. N. Hu, G. L. Liao, T. L. Shi and C. P. Wong, Nano Energy, 2015, 17, 160–170 CrossRef CAS.
- J. L. Long, X. Q. Xie, J. Xu, Q. Gu, L. M. Chen and X. X. Wang, ACS Catal., 2012, 2, 622–631 CrossRef CAS.
- Y. Li, J. Yang, J. P. Huang, Y. Z. Zhou, K. Xu, N. Zhao and X. N. Cheng, Carbon, 2017, 122, 237–246 CrossRef CAS.
- F. Addison, N.-A. Offiong, Q. Han, R. Wang and N. Liu, Environ. Technol., 2020 DOI:10.1080/09593330.2020.1782994.
- N. Liu, L. Y. Zhang, Y. Xue, J. Lv, Q. M. Yu and X. L. Yuan, Sep. Purif. Technol., 2017, 184, 213–219 CrossRef CAS.
- D. Miao, G. Liu, Q. Wei, N. Hu, K. Zheng, C. Zhu, T. Liu, K. Zhou, Z. Yu and L. Ma, Water Sci. Technol., 2020, 81, 925–935 CrossRef CAS PubMed.
- A. R. Rahmani, H. Rezaeivahidian, M. Almasi, A. Shabanlo and H. Almasi, Res. Chem. Intermed., 2016, 42, 1441–1450 CrossRef CAS.
- W. Y. Dong, T. Cai, Y. T. Liu, L. L. Wang, H. Chen, W. G. Zeng, J. Li and W. L. Li, J. Colloid Interface Sci., 2021, 585, 400–407 CrossRef CAS PubMed.
- M. A. Oturan, J. Peiroten, P. Chartrin and A. J. Acher, Environ. Sci. Technol., 2000, 34, 3474–3479 CrossRef CAS.
- V. Kavitha and K. Palanivelu, J. Photochem. Photobiol., A, 2005, 170, 83–95 CrossRef CAS.
- M. Martin-Hernandez, J. Carrera, M. E. Suarez-Ojeda, M. Besson and C. Descorme, Appl. Catal., B, 2012, 123, 141–150 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab and
Yonggang Zhang
ab and
Yonggang Zhang