
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDoes electrophilic activation of nitroalkanes in polyphosphoric acid involve formation of nitrile oxides?†
Alexander V. Aksenov *a,
Nicolai A. Aksenova,
Nikita K. Kirilova,
Anton A. Skomorokhova,
Dmitrii A. Aksenova,
Igor A. Kurenkova,
Elena A. Sorokinab,
Mezvah A. Nobic and
Michael Rubin*ac
*a,
Nicolai A. Aksenova,
Nikita K. Kirilova,
Anton A. Skomorokhova,
Dmitrii A. Aksenova,
Igor A. Kurenkova,
Elena A. Sorokinab,
Mezvah A. Nobic and
Michael Rubin*ac
aDepartment of Chemistry, North Caucasus Federal University, 1a Pushkin St., Stavropol 355017, Russia. E-mail: aaksenov@ncfu.ru
bOrganic Chemistry Department, Peoples' Friendship University of Russia (RUDN University), 6, Miklukho-Maklaya St., Moscow, 117198, Russian Federation
cDepartment of Chemistry, University of Kansas, 1567 Irving Hill Road, Lawrence, KS 66045, USA. E-mail: mrubin@ku.edu
First published on 4th November 2021
Abstract
The mechanistic rationale involving activation of nitroalkanes towards interaction with nucleophilic reagents in the presence of polyphosphoric acid (PPA) was re-evaluated. Could nitrile oxide moieties be formed during this process? This experiment demonstrates that at least in some cases this could happen, as generated nitrile oxides were successfully intercepted as adducts of [3 + 2] cycloadditions.
Introduction
Nitroalkanes possessing acidic α-CH bonds are traditionally employed as versatile nucleophilic components in a variety of base-assisted C–C bond-forming reactions.1–8 Another mode of activation of nitroalkanes enables acid-promoted tautomerization into nitronic acid (aci form), which is electrophilic in nature and can interact with a number of nucleophiles. In aqueous media, facile acid-assisted reaction with water takes place, commonly known as the Nef reaction,9–12 resulting in the formation of aldehydes. In anhydrous media, alternative reactivity patterns are possible, involving interaction with various C- and N-based nucleophiles. Over the last decade, our group has been instrumental in the development of multistep cascade transformations triggered by initial electrophilic activation of nitroalkanes in the presence of polyphosphoric acid (PPA).13–17 Proposed mechanistic rationales for all these processes involve formation of a phosphorylated nitronate species as a key electrophilic intermediate. Herein, we wish to disclose our recent studies proving that formation of nitrile oxide species is also possible under the same conditions. This suggests that alternative mechanisms could be potentially considered, at least for some of the earlier described processes.Results and discussion
As discussed above, it was previously stated that interaction of nitroalkane 1 with PPA leads to the formation of electrophilic nitronate species 2, enabling subsequent reactions with various nucleophiles. The resulting phosphorylated N-hydroxy-N-alkyl hydroxylamine 4 may undergo further elimination of the ortho-phosphoric acid molecule to afford species 5 (oximes or amidines resulting from reaction with carbon or nitrogen-based nucleophiles, respectively, Scheme 1). Oximes 5 (Nu = Ar), as was previously demonstrated, may participate in Beckmann-type rearrangements, which was employed in acetamidation and carboxyamination reactions.18,19 Generation of amidines 5 (Nu = NHR) were utilized in design of cascade transformations leading to various heterocyclic scaffolds.13,20,21 We pondered if elimination of H3PO4 entity can take place in species 2 to form nitrile oxide 3 under the described reaction conditions.22 This species is also inherently electrophilic and reactions with nucleophiles should provide the same type of product 5 (Scheme 1).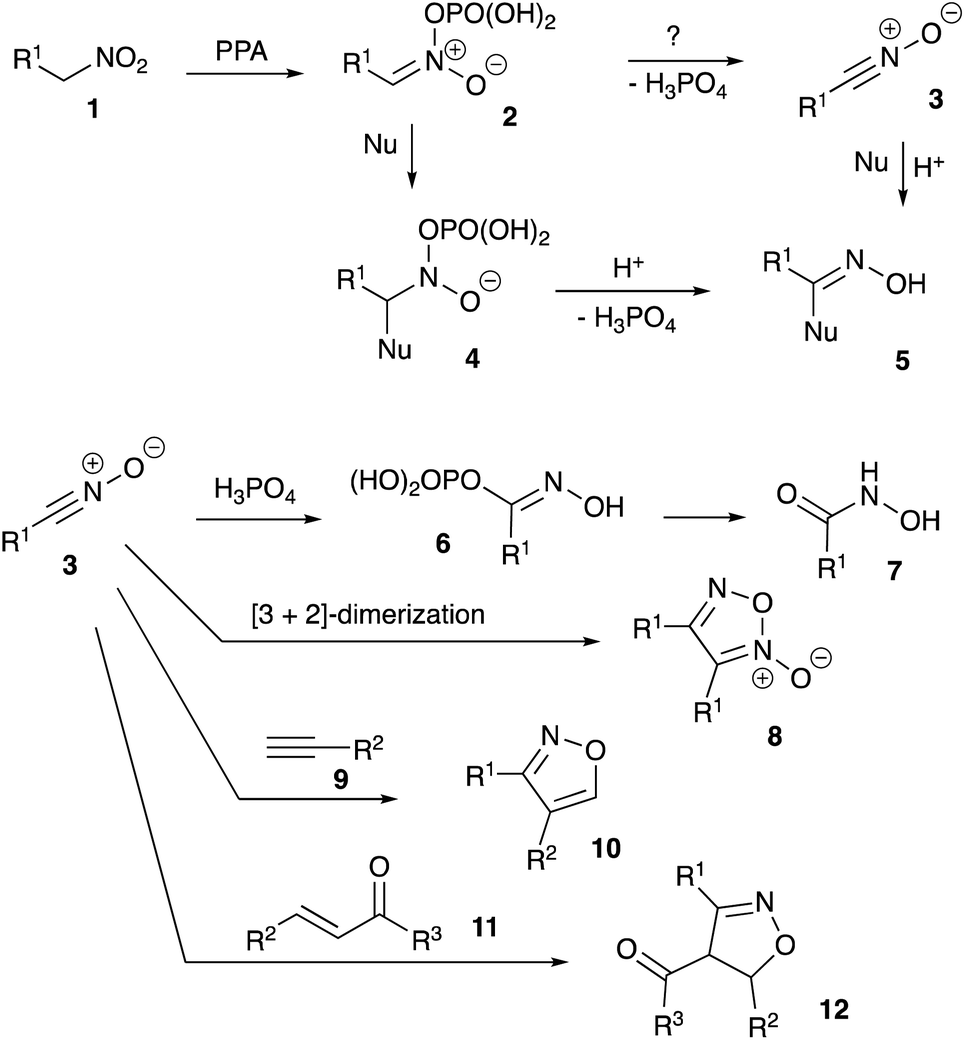 | ||
| Scheme 1 | ||
Furthermore, reaction of 3 with ortho-phosphoric acid in the absence of other nucleophiles could be envisioned, producing N-hydroxyalkanimidic phosphoric anhydride 6, which after hydrolytic cleavage, should provide hydroxamic acid 7. It should be noted that the isomerization of nitroalkanes into hydroxamic acids taking place in the presence of PPA is well documented.23 On the other hand, in most typical reactions nitrile oxides act as 1,3-dipoles. If nitrile oxides are indeed generated from nitroalkanes in the presence of PPA, then in the absence of external dipolarophiles, they should undergo [3 + 2]-dimerization to provide 1,2,5-oxadiazole 2-oxides (furoxans) 8 (Scheme 1). Alternatively, cross-cycloaddition reactions with alkynes 9 and chalcones 11 could be performed to generate isoxazoles 10 or 4,5-dihydroisoxazoles 12, respectively (Scheme 1).
First, we attempted to conduct the experiment involving interaction of 1-nitropropane (1, R1 = Et, Scheme 1) with PPA in the absence of any other reagents by targeting detection for furoxan 8 (R1 = Et) resulting from [3 + 2]-homodimerization. However, formation of such product was not detected, as almost the entire amount of starting nitroalkane was transformed into N-hydroxypropionamide 7 (R1 = Et).24 Evidently, even if formed under the described reaction conditions, propionitrile oxide (3, R1 = Et) is much too reactive and quickly interacts with the medium. To address this issue, we decided to test the same idea by employing 2-nitro-1-phenylethan-1-one (1a), producing greater concentration of aci-form, which should translate in higher concentration of benzoyl cyanide N-oxide (3a). Therefore, this starting material should be more prompt towards the desired dimerization reaction.
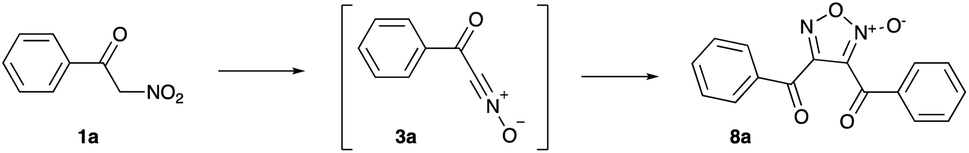
Initial tests were carried out in PPA with 80% content of P2O5 (corresponding to diphosphoric acid, H4P2O7). A mixture of 1a (1 mmol) with PPA (1.5 g) was stirred and heated at 50 °C for 2 hours, stopping only after TLC analysis proved complete consumption of starting material. Delightfully, some nitroalkane was indeed converted into furoxan 8a, albeit in marginal yield (Table 1, entry 1). Increasing the amount of PPA (2.0 g) improved the yield, though not significantly (entry 2). We thought that switching to 87% PPA might enhance the reaction performance, since this medium has less nominal content of water and thus less nucleophilic; however, the reaction proceeded sluggishly and at 50 °C, the yield of 8a was essentially the same as in 80% PPA (entry 3). Further improvement was achieved upon heating to a higher temperature, which also allowed to cut down reaction time. At 70 °C, a maximum yield of 51% was obtained (entry 4) and further increases of temperature proved detrimental (entry 5). The purified product 8a was isolated in a 46% yield (Scheme 2). With optimized reaction conditions in hand, we moved towards scope and limitation studies, the results of which are shown in Scheme 2. Generally, the reactions of substituted 2-nitro-1-phenylethan-1-ones 1b–h proceeded smoothly, well tolerating alkyl and aromatic halide substituents and affording the corresponding furoxans in moderate yields. We also managed to execute successful transformations of ethyl 2-nitroacetate (1i) into furoxan 8i, bearing two ester functions under the same reaction conditions (Scheme 2).
 | ||
| Scheme 2 | ||
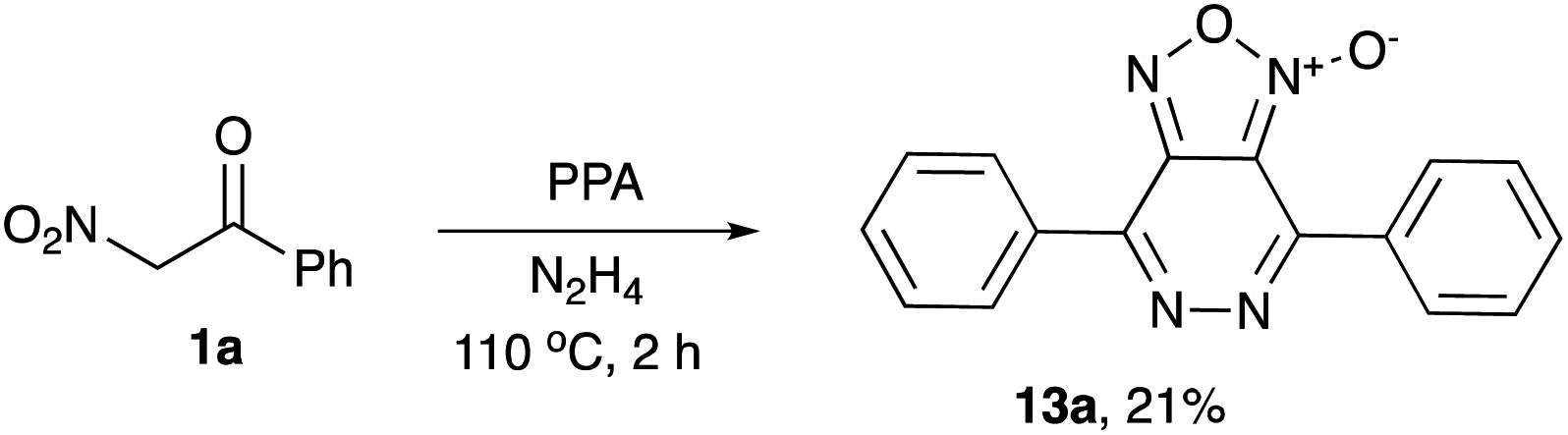
The possibility for a one-pot assembly of a bicyclic structural core, including a furoxan moiety fused to pyridazine ring, was also evaluated. To this end, nitroketone 1a was treated with PPA in the presence of hydrazine hydrate. The bicyclic structure 13a was formed as a sole isolable product, although in low quantities (Scheme 3).
 | ||
| Scheme 3 | ||
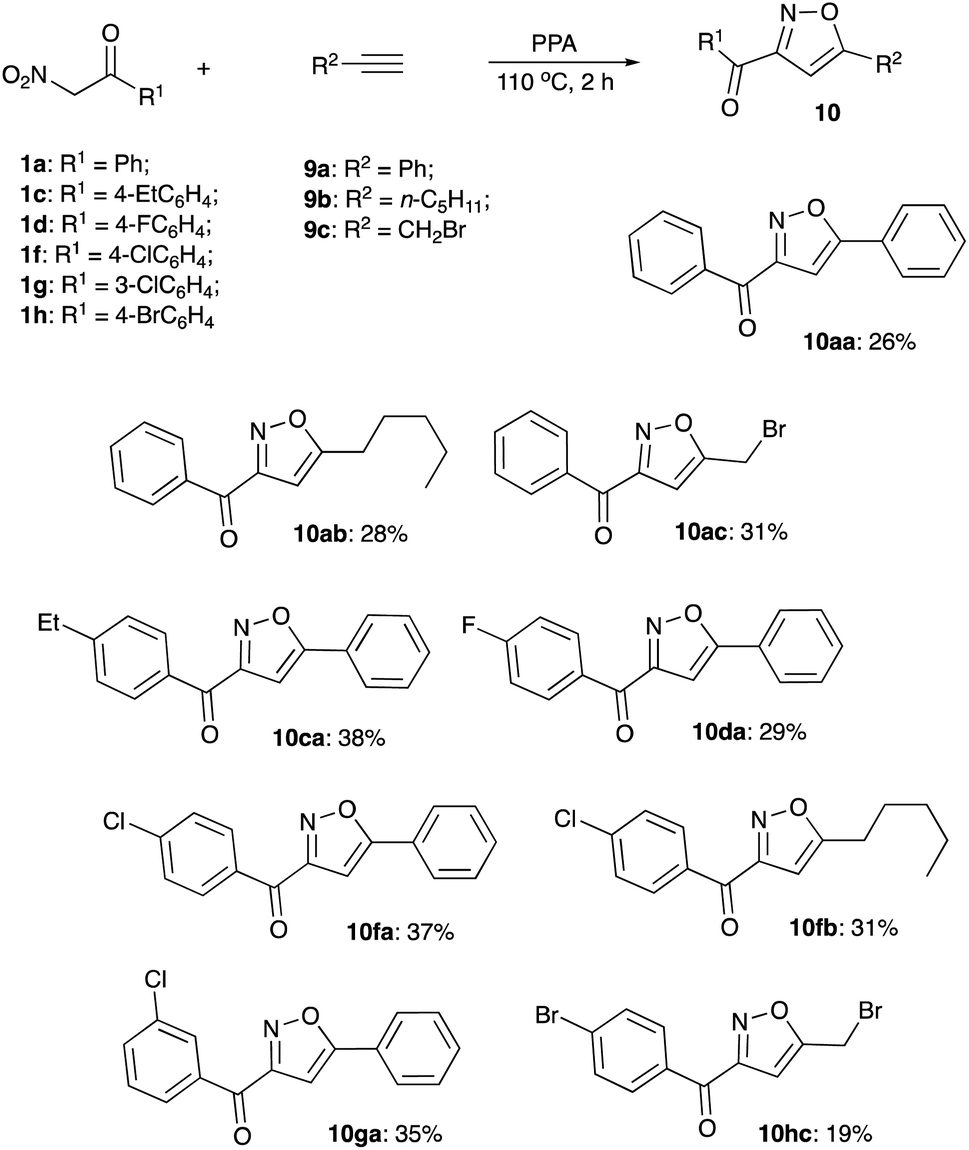
Next, we decided to explore the possibility to intercept the nitrile oxides generated from nitroalkanes with external dipolarophiles, such as terminal acetylenes. Treatment of various nitroalkanes with PPA was carried out in the presence of phenyl acetylene (9a), 1-heptene (9b), or propargyl bromide (9c) to yield the desired isoxazoles 10 (Scheme 4). Formation of isoxazole 10da was unambiguously confirmed by single crystal X-ray crystallography (Fig. 1).
 | ||
| Scheme 4 | ||
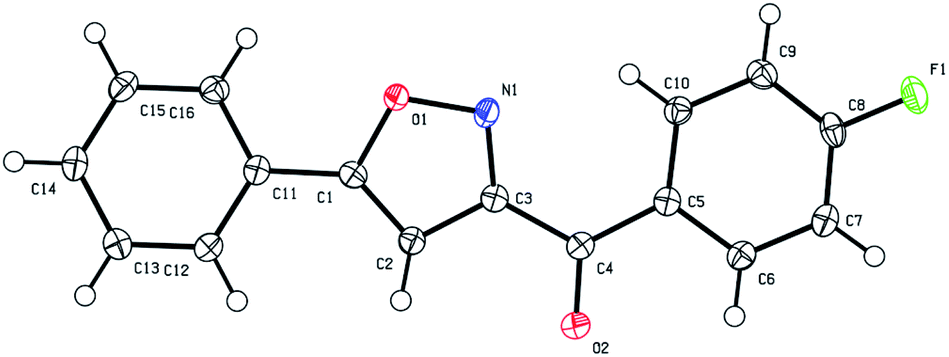 | ||
| Fig. 1 ORTEP drawing of X-ray structures of (4-fluorophenyl)(5-phenylisoxazol-3-yl)methanone (10da, CCDC #2099762†). The thermal ellipsoids are shown at 50% probability. | ||
This reaction cannot be recommended as a preparative protocol towards these materials, since most amount of nitroalkanes were converted into the corresponding hydroxamic acids. Despite that, it clearly confirms that nitrile oxides were indeed generated under the described reaction conditions.
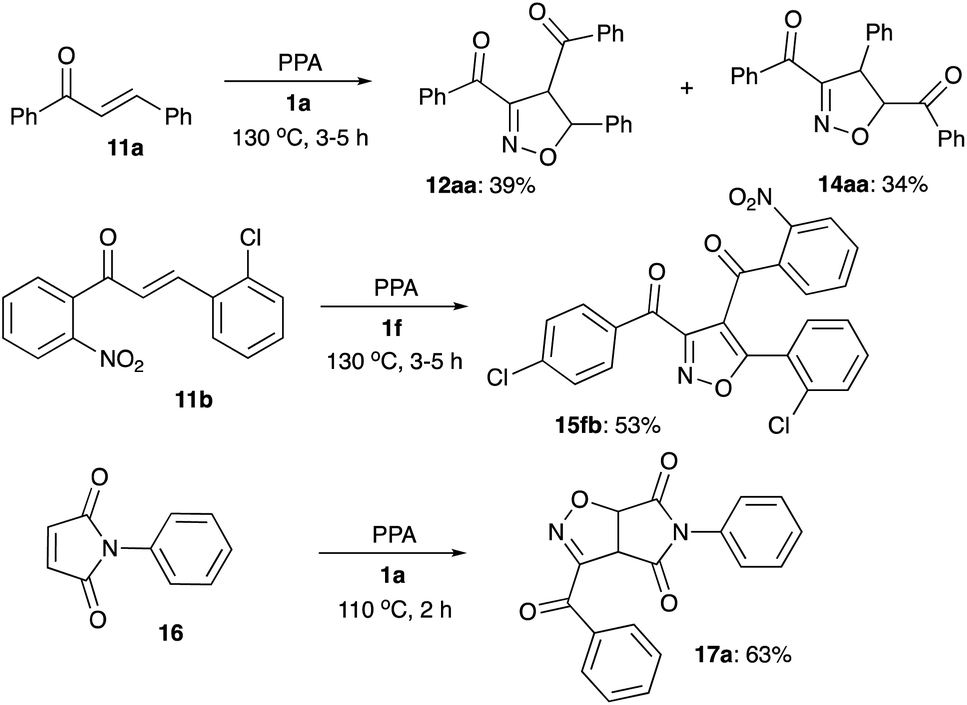
Interception of nitrile oxides with alkenes were tested as well. Reaction of 1a in the presence of chalcone 11a gave nearly an equimolar mixture of regioisomeric dihydroisoxazoles 12aa and 14aa, formation of which are most likely controlled by electronic and steric factors, respectively (Scheme 5). Reaction of 1f in the presence of chalcone 11b, with enhanced polarization of double bond, resulted in perfect electronically-controlled regiochemistry, but was accompanied by oxidative aromatization of the cycloaddition product 15fb (Scheme 5). Finally, reaction of 1a in the presence of malonic imide 16 proceeded smoothly, providing bicyclic imide 17a in good yield (Scheme 5).
 | ||
| Scheme 5 | ||
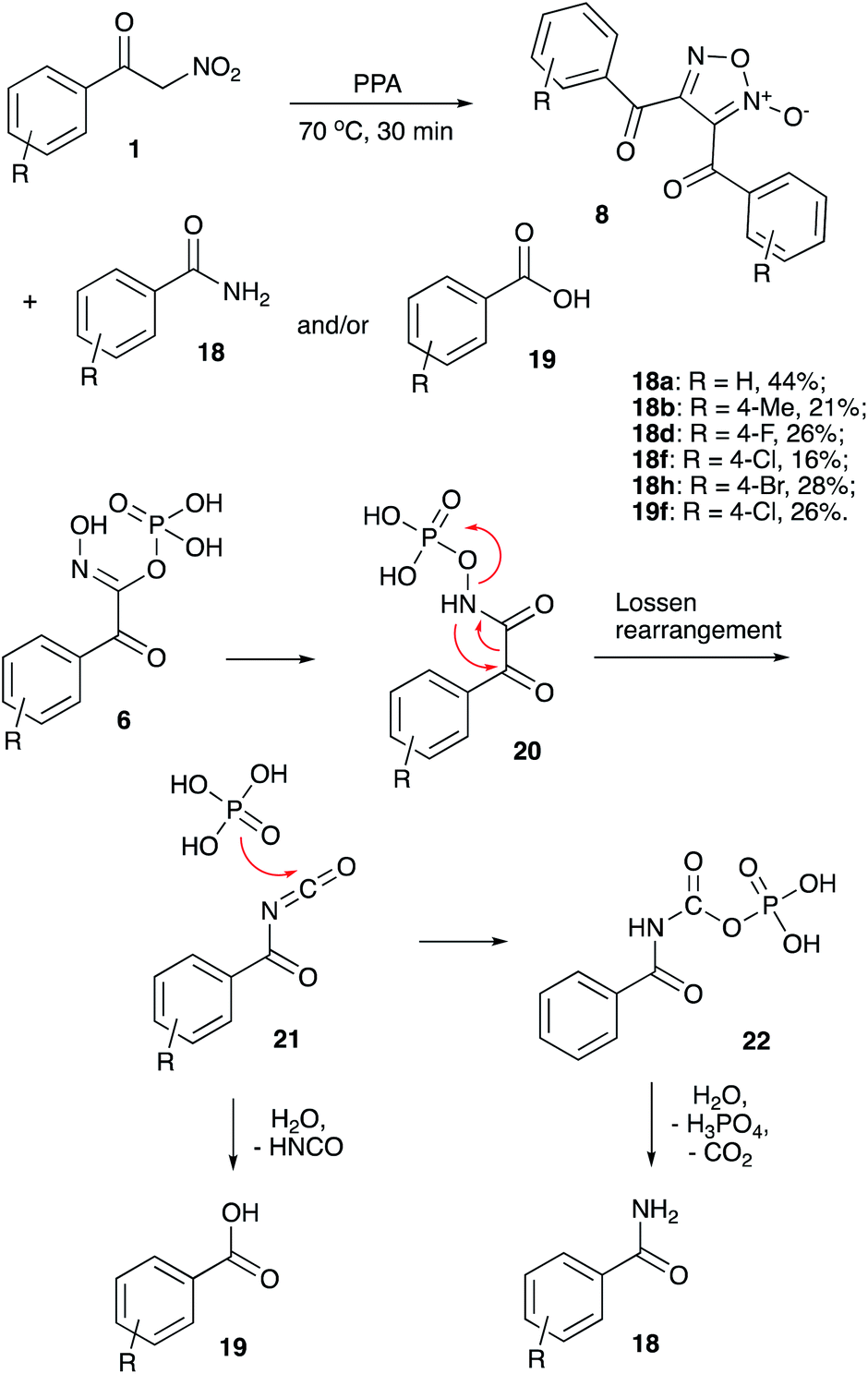
Since the yields obtained were quite moderate, we decided to investigate the material balance by tracing the byproducts formed in dimerization reactions towards furoxans 8 (Scheme 2). Initially we thought we will be dealing with hydroxamic acids 7, however, benzamides 18 (sometimes in a mixture with benzoic acids 19) were observed and isolated instead. Evidently, under the listed reaction conditions hydroxamic acids 7 undergo facile Lossen rearrangement or competing hydrolysis as shown in Scheme 6. It is believed, that O-phosphorylated hydroxamic acid 6 (initially formed after nucleophilic attack on nitrile oxide 2 with ortho-phosphoric acid as shown in Scheme 1, vide infra) could be re-esterified to afford NO-phosphorylated form 20. The later would undergo Lossen rearrangement into benzoyl isocyanate 21, which can be hydrolyzed with extrusion of isocyanic acid and formation of benzoic acid 19 (Scheme 6). More commonly, an alternative pathway was operating, involving reaction of isocyanate 21 with phosphoric acid to afford benzoylcarbamic phosphoric anhydride 22, which then underwent hydrolysis to afford benzamide species 18. These compounds were isolated in moderate yield from most of the reaction mixtures along with normal furoxan products 8.
 | ||
| Scheme 6 | ||
Conclusions
In conclusion, we demonstrated that electrophilic activation of nitroalkanes in the presence of polyphosphoric acid can, at least in some cases, provide nitrile oxide moieties, which likely exists in equilibrium with more ordinary nitronate species. In the presence of PPA, nitrile oxides tend to decompose to hydroxamic acids, but for most stable electron-deficient dipolar species, typical [3 + 2] cycloaddition reactions can be studied. Nevertheless, formation of hydroxamic acid is a dominant process in most cases, so this reaction cannot be recommended as a preparative method for generation of nitrile oxides.Experimental
General
NMR spectra, 1H and 13C was measured in solutions of CDCl3 or DMSO-d6 on Bruker AVANCE-III HD instrument (at 400.40 or 100.61 MHz, respectively). Residual solvent signals were used as internal standards, in DMSO-d6 (2.50 ppm for 1H, and 40.45 ppm for 13C nuclei) or in CDCl3 (7.26 ppm for 1H, and 77.16 ppm for 13C nuclei). HRMS spectra was measured on Bruker maXis impact (electrospray ionization, in MeCN solutions, employing HCO2Na–HCO2H for calibration). IR spectra was measured on FT-IR spectrometer Shimadzu IRAffinity-1S equipped with an ATR sampling module. Reaction progress, purity of isolated compounds, and Rf values were monitored with TLC on Silufol UV-254 plates. Column chromatography was performed on silica gel (32–63 μm, 60 Å pore size). Melting points were measured with Stuart SMP30 apparatus. Polyphosphoric acid samples were prepared by dissolving precisely measured amounts of P2O5 in 85% ortho-phosphoric acid. Nitroacetophenones and chalcones were prepared according to the previously published procedures.25–27 All other reagents and solvents were purchased from commercial venders and used as received.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5, v/v). 1H NMR (400 MHz, DMSO) δ 7.87 (d, J = 8.3 Hz, 2H), 7.44 (d, J = 8.2 Hz, 2H), 6.50 (s, 2H), 2.71 (q, J = 7.6 Hz, 2H), 1.20 (t, J = 7.6 Hz, 3H). 13C NMR (101 MHz, DMSO) δ 187.9, 151.6, 131.2, 128.7 (2C), 128.5 (2C), 82.8, 28.3, 15.1. IR, vmax/cm−1: 3027, 2974, 2935, 1961, 1933, 1694, 1607, 1549, 1328, 1229, 1185. HRMS (ES TOF) calcd for C10H11NNaO3 ([M + Na]+): 216.0631, found 216.0633 (−0.7 ppm).:4 v/v) to give pure furoxan. Colorless oil, Rf 0.5, EtOAc/hexane (1:4, v/v). Yield 72 mg (0.24 mmol, 49%). 1H NMR (400 MHz, CDCl3) δ 8.25–8.18 (m, 2H), 7.93–7.80 (m, 2H), 7.76–7.65 (m, 2H), 7.60–7.49 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 182.2, 180.9, 154.8, 135.9, 135.8, 134.3, 134.3, 131.0 (2C), 130.1 (2C), 129.7 (2C), 129.5 (2C), 112.1. IR, vmax/cm−1: 1735, 1684, 1602, 1559, 1455, 1419, 1325, 1246, 1178, 1101. HRMS (ES TOF) calcd for C16H10N2NaO4 ([M + Na]+): 317.0533, found 317.0532 (0.4 ppm).
:5, v/v). 1H NMR (400 MHz, DMSO) δ 7.87 (d, J = 8.3 Hz, 2H), 7.44 (d, J = 8.2 Hz, 2H), 6.50 (s, 2H), 2.71 (q, J = 7.6 Hz, 2H), 1.20 (t, J = 7.6 Hz, 3H). 13C NMR (101 MHz, DMSO) δ 187.9, 151.6, 131.2, 128.7 (2C), 128.5 (2C), 82.8, 28.3, 15.1. IR, vmax/cm−1: 3027, 2974, 2935, 1961, 1933, 1694, 1607, 1549, 1328, 1229, 1185. HRMS (ES TOF) calcd for C10H11NNaO3 ([M + Na]+): 216.0631, found 216.0633 (−0.7 ppm).:4 v/v) to give pure furoxan. Colorless oil, Rf 0.5, EtOAc/hexane (1:4, v/v). Yield 72 mg (0.24 mmol, 49%). 1H NMR (400 MHz, CDCl3) δ 8.25–8.18 (m, 2H), 7.93–7.80 (m, 2H), 7.76–7.65 (m, 2H), 7.60–7.49 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 182.2, 180.9, 154.8, 135.9, 135.8, 134.3, 134.3, 131.0 (2C), 130.1 (2C), 129.7 (2C), 129.5 (2C), 112.1. IR, vmax/cm−1: 1735, 1684, 1602, 1559, 1455, 1419, 1325, 1246, 1178, 1101. HRMS (ES TOF) calcd for C16H10N2NaO4 ([M + Na]+): 317.0533, found 317.0532 (0.4 ppm).Benzamide (18a)28 was also isolated from the same reaction mixture as colorless solid. Yield 53 mg (0.44 mmol, 44%). 1H NMR (400 MHz, CDCl3) δ 7.84 (dt, J = 7.1, 1.4 Hz, 2H), 7.59–7.50 (m, 1H), 7.46 (t, J = 7.5 Hz, 2H), 6.35 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 170.2, 133.8, 132.4, 129.1 (2C), 127.8 (2C).
:5). Yellow solid, mp 118–119.5 °C (EtOH), lit29 mp 124 °C (EtOH); yield 83 mg (0.26 mmol, 52%). Rf 0.46, EtOAc/hexane (1:5, v/v). 1H NMR (400 MHz, CDCl3) δ 8.11 (d, J = 8.3 Hz, 2H), 7.76 (d, J = 8.3 Hz, 2H), 7.33 (dd, J = 13.4, 8.3 Hz, 4H), 2.45 (d, J = 10.1 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 181.3, 180.0, 154.6, 147.0, 146.8, 131.5, 131.4, 130.7 (2C), 130.0 (2C), 129.9 (2C), 129.8 (2C), 111.9, 22.0 (2C). IR, vmax/cm−1: 1675, 1646, 1600, 1470, 1328, 1251, 1181, 1123, 1104. HRMS (ES TOF) calcd for C18H14N2NaO4 ([M + Na]+): 345.0846, found 345.0840 (1.7 ppm).4-Methylbenzamide (18b)28 was also isolated from the same reaction mixture as colorless solid. Yield 28 mg (0.21 mmol, 21%). 1H NMR (400 MHz, DMSO-d6) δ 7.90 (s, 1H), 7.77 (d, J = 8.2 Hz, 2H), 7.29 (s, 1H), 7.24 (d, J = 8.0 Hz, 2H), 2.34 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.8, 141.1, 131.5, 128.8 (2C), 127.6 (2C), 21.0.
:4). Colorless oil, Rf 0.60, EtOAc/hexane (1:5, v/v). Yield 86 mg (0.25 mmol, 49%). 1H NMR (400 MHz, CDCl3) δ 8.18–8.07 (m, 2H), 7.83–7.70 (m, 2H), 7.36 (dd, J = 13.6, 8.1 Hz, 4H), 2.75 (dq, J = 9.8, 7.6 Hz, 4H), 1.28 (q, J = 7.8 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 181.5, 180.1, 154.7, 153.2, 152.9, 131.8, 131.7, 131.0 (2C), 130.1 (2C), 128.9 (2C), 128.7 (2C), 112.1, 29.4, 29.3, 15.1, 15.1. IR, vmax/cm−1: 2964, 2877, 1733, 1595, 1460, 1427, 1383, 1337, 1272, 1147, 1096. HRMS (ES TOF) calcd for C20H18N2NaO4 ([M + Na]+): 373.1159, found 373.1151 (2.0 ppm).:4). Yellow oil, Rf 0.53, EtOAc/hexane (1:5, v/v). Yield 58 mg (0.18 mmol, 35%). 1H NMR (400 MHz, CDCl3) δ 8.40–8.34 (m, 2H), 8.03–7.97 (m, 2H), 7.32 (m, J = 11.1, 8.4 Hz, 4H). 13C NMR (101 MHz, CDCl3) δ 180.2, 178.9, 167.30 (d, J = 259.7 Hz), 167.28 (d, J = 259.3 Hz), 154.3, 133.7 (d, J = 9.9 Hz, 2C), 132.8 (d, J = 10.3 Hz, 2C), 130.3 (d, J = 2.9 Hz), 130.2 (d, J = 2.9 Hz), 116.9 (d, J = 22.4 Hz, 2C), 116.6 (d, J = 22.0 Hz, 2C), 111.7. IR, vmax/cm−1: 1769, 1684, 1663, 1593, 1508, 1472, 1414, 1325, 1236, 1157, 1101. HRMS (ES TOF) calcd for C16H8F2N2NaO4 ([M + Na]+): 353.0344, found 353.0338 (1.7 ppm).4-Fluorobenzamide (18b)28 was also isolated from the same reaction mixture as colorless solid. Yield 36 mg (0.26 mmol, 26%). 1H NMR (400 MHz, DMSO-d6) δ 8.01 (s, 1H), 7.97–7.87 (m, 2H), 7.41 (s, 1H), 7.32–7.19 (m, 2H). 13C NMR (101 MHz, DMSO) δ 166.0 (d, J = 164.9 Hz), 162.7, 130.8 (d, J = 3.0 Hz) 130.2 (d, J = 9.0 Hz, 2C), 115.2 (d, J = 21.6 Hz, 2C).
:4). Yellow oil, Rf 0.32, EtOAc/hexane (1:4, v/v). Yield 81 mg (0.25 mmol, 49%). 1H NMR (400 MHz, CDCl3) δ 7.92 (td, J = 7.8, 1.8 Hz, 1H), 7.82 (td, J = 7.4, 1.8 Hz, 1H), 7.70–7.61 (m, 2H), 7.36–7.29 (m, 2H), 7.17 (m, J = 25.1, 11.0, 8.4, 1.2 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 180.1, 177.1, 162.0 (d, J = 259.0 Hz), 161.6 (d, J = 256.4 Hz), 154.4, 136.8 (d, J = 7.6 Hz), 136.7 (d, J = 7.4 Hz), 131.5, 131.1, 125.2 (d, J = 3.3 Hz), 124.8 (d, J = 3.7 Hz), 123.4 (d, J = 10.3 Hz), 123.2 (d, J = 11.0 Hz), 117.0 (d, J = 21.6 Hz), 116.6 (d, J = 22.0 Hz), 112.7. IR, vmax/cm−1: 1655, 1602, 1458, 1439, 1330, 1238, 1181, 1159, 1099, 1041. HRMS (ES TOF) calcd for C16H8F2N2NaO4 ([M + Na]+): 353.0344, found 353.0334 (3.0 ppm).:4). Colorless solid, mp 120–121 °C (EtOH), lit30 mp 124 °C; Rf 0.57, EtOAc/hexane (1:6, v/v). Yield 92 mg (0.25 mmol, 51%). 1H NMR (400 MHz, CDCl3) δ 8.18 (d, J = 8.7 Hz, 2H), 7.80 (d, J = 8.6 Hz, 2H), 7.53 (m, J = 12.0, 8.7 Hz, 4H). 13C NMR (101 MHz, CDCl3) δ 180.5, 179.2, 154.0, 142.41, 142.39, 132.01, 132.00, 131.9 (2C), 131.0 (2C), 129.8 (2C), 129.5 (2C), 111.4. IR, vmax/cm−1: 1773, 1682, 1610, 1586, 1472, 1405, 1323, 1243, 1176, 1089. HRMS (ES TOF) calcd for C16H8Cl2N2NaO4 ([M + Na]+): 384.9753, found 384.9760 (−1.8 ppm).4-Chlorobenzamide (18f)28 and 4-chlorobenzoic acid (19f)31 were also isolated from the same reaction mixture as colorless solid. 18f: yield 25 mg (0.16 mmol, 16%). 1H NMR (400 MHz, DMSO-d6) δ 8.06 (br. s, 1H), 7.93–7.84 (m, 2H), 7.55–7.50 (m, 2H), 7.49 (br. s, 1H). 13C NMR (101 MHz, DMSO-d6) δ 166.9, 136.1, 133.1, 129.5 (2C), 128.4 (2C). 19f: yield 41 mg (0.26 mmol, 26%). 1H NMR (400 MHz, DMSO-d6) δ 13.19 (br. s, 1H), 7.93 (d, J = 8.6 Hz, 2H), 7.55 (d, J = 8.5 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 166.5, 137.8, 131.2 (2C), 129.7, 128.8 (2C).
:4). Yellow oil; Rf 0.49, EtOAc/hexane (1:4, v/v). Yield 102 mg (0.28 mmol, 56%). 1H NMR (400 MHz, CDCl3) δ 8.17 (m, J = 1.9 Hz, 1H), 8.12 (m, J = 7.8, 1.4 Hz, 1H), 7.84 (m, J = 1.9 Hz, 1H), 7.73–7.64 (m, 3H), 7.50 (m, J = 15.8, 7.9 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 180.5, 179.2, 153.8, 135.8, 135.50, 135.46, 135.3, 135.2, 135.11, 130.6, 130.4, 130.3, 129.4, 128.7, 127.8, 111.1. IR, vmax/cm−1: 1754, 1687, 1614, 1571, 1467, 1419, 1323, 1236, 1079. HRMS (ES TOF) calcd for C16H8Cl2N2NaO4 ([M + Na]+): 384.9753, found 384.9762 (−2.2 ppm).:4). Yellowish solid, mp 135.9–136.7 °C (EtOAc), Rf 0.57, EtOAc/hexane (1:4, v/v). Yield 90 mg (0.20 mmol, 40%). 1H NMR (400 MHz, CDCl3) δ 8.12–8.05 (m, 2H), 7.76–7.64 (m, 6H).13C NMR (101 MHz, CDCl3) δ 180.7, 179.4, 154.0, 132.7 (2C), 132.5 (2C), 132.4 (2C), 131.9 (2C), 131.3 (2C), 131.0 (2C), 111.3. IR, vmax/cm−1: 928, 1012, 1067, 1173, 1212, 1251, 1325, 1395, 1467, 1581, 1612, 1655, 1689, 3089. HRMS (ES TOF) calcd for C16H8Br2N2NaO4 ([M + Na]+): 472.8743, found 472.8741 (0.4 ppm).4-Bromobenzamide (18h)28 was also isolated from the same reaction mixture as colorless solid. Yield 56 mg (0.28 mmol, 28%). 1H NMR (400 MHz, DMSO-d6) δ 8.06 (s, 1H), 7.84–7.78 (m, 2H), 7.70–7.63 (m, 2H), 7.48 (s, 1H). 13C NMR (101 MHz, DMSO) δ 167.4, 133.8, 131.7 (2C), 130.1 (2C), 125.5.
:4). Colorless oil, Rf 0.46, EtOAc/hexane (1:4, v/v). Yield 49 mg (0.21 mmol, 43%). 1H NMR (400 MHz, DMSO-d6) δ 4.45 (qd, J = 7.1, 1.0 Hz, 2H), 4.36 (qd, J = 7.1, 1.1 Hz, 2H), 1.33 (td, J = 7.1, 1.0 Hz, 3H), 1.27 (td, J = 7.1, 1.1 Hz, 3H). 13C NMR (101 MHz, DMSO) δ 156.5, 154.8, 148.8, 107.4, 63.5, 63.1, 13.72, 13.7. IR, vmax/cm−1: 2988, 1742, 1619, 1472, 1373, 1335, 1246, 1195, 1063, 1022. HRMS (ES TOF) calcd for C8H10N2NaO6 ([M + Na]+): 253.0431, found 253.0426 (2.1 ppm).:PE, 1:4 v/v) affording the titled product as orange solid, mp 199–201 °C (EtOAc), lit30 mp 210 °C; Rf 0.44, EtOAc/hexane (1:4, v/v). Yield 31 mg (0.11 mmol, 21%). 1H NMR (400 MHz, CDCl3) δ 8.68–8.60 (m, 2H), 8.00–7.90 (m, 2H), 7.68–7.56 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 150.4, 148.3, 145.1, 132.6, 131.9, 131.8, 130.3, 129.6 (2C), 129.3 (2C), 129.2 (2C), 128.7 (2C), 106.9. IR, vmax/cm−1: 2974, 2882, 1600, 1508, 1460, 1429, 1388, 1263, 1147, 1089, 1048. HRMS (ES TOF) calcd for C16H10N4NaO2 ([M + Na]+): 313.0696, found 313.0691 (1.7 ppm).:2 v/v) to afford the titled compound as yellowish solid, mp 85.4–86 °C (EtOH), lit32 mp 88–90 °C, Rf 0.46, benzene/hexane (1:2, v/v). Yield 65 mg (0.26 mmol, 26%). 1H NMR (400 MHz, CDCl3) δ 8.38–8.32 (m, 2H), 7.86 (m, J = 7.8, 1.9 Hz, 2H), 7.67 (m, J = 7.4 Hz, 1H), 7.53 (m, J = 17.5, 7.9 Hz, 5H), 7.06 (s, 1H). 13C NMR (101 MHz, CDCl3) δ 185.9, 170.8, 162.4, 135.8, 134.1, 130.8, 130.7 (2C), 129.2 (2C), 128.6 (2C), 126.7, 126.0 (2C), 100.3. IR, vmax/cm−1: 1655, 1598, 1573, 1451, 1426, 1236, 1181, 1147, 1101, 1075. HRMS (ES TOF) calcd for C16H11NNaO2 ([M + Na]+): 272.0682, found 272.0678 (1.3 ppm).:4). Colorless oil; Rf 0.72, EtOAc/hexane (1:4, v/v). Yield 68 mg (0.28 mmol, 28%). 1H NMR (400 MHz, CDCl3) δ 8.33–8.26 (m, 2H), 7.67–7.61 (m, 1H), 7.54–7.48 (m, 2H), 6.52 (s, 1H), 2.86–2.80 (m, 2H), 1.80–1.71 (m, 2H), 1.42–1.34 (m, 4H), 0.95–0.89 (m, 3H). 13C NMR (101 MHz, CDCl3) δ 185.2, 173.8, 160.8, 134.9, 132.9, 129.6 (2C), 127.5 (2C), 100.6, 30.1, 26.1, 25.6, 21.3, 12.9. IR, vmax/cm−1: 2930, 1774, 1660, 1602, 1455, 1434, 1258, 1239, 1181, 1104. HRMS (ES TOF) calcd for C15H17NNaO2 ([M + Na]+): 266.1151, found 266.1145 (2.6 ppm).:4). Yellow solid, mp 62–65 °C; Rf 0.73, EtOAc/hexane (1:4, v/v). Yield 82 mg (0.31 mmol, 31%). 1H NMR (400 MHz, CDCl3) δ 8.31–8.28 (m, 2H), 7.68–7.63 (m, 1H), 7.55–7.50 (m, 2H), 6.85 (d, J = 0.6 Hz, 1H), 4.54 (d, J = 0.6 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 185.3, 168.5, 162.3, 135.6, 134.4, 130.8 (2C), 128.8 (2C), 104.9, 18.1. IR, vmax/cm−1: 3152, 2959, 1766, 1645, 1599, 1578, 1452, 1291, 1224, 1187, 999. HRMS (ES TOF) calcd for C11H8BrNaNO2 ([M + Na]+): 287.9631, found 287.9624 (2.2 ppm).:PE 1:4). White solid, mp 84–85 °C (EtOH), Rf 0.49, benzene/hexane (1:2, v/v). Yield 105 mg (0.38 mmol, 38%). 1H NMR (400 MHz, CDCl3) δ 8.28 (d, J = 8.3 Hz, 2H), 7.85 (dd, J = 7.8, 1.8 Hz, 2H), 7.55–7.47 (m, 3H), 7.36 (d, J = 8.3 Hz, 2H), 7.04 (s, 1H), 2.75 (q, J = 7.6 Hz, 2H), 1.29 (t, J = 7.6 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 184.4, 169.6, 161.6, 150.3, 132.4, 129.9 (2C), 129.7, 128.1 (2C), 127.2 (2C), 125.8, 125.0 (2C), 99.3, 28.1, 14.1. IR, vmax/cm−1: 2964, 1653, 1602, 1566, 1439, 1296, 1255, 1183, 1151, 1046. HRMS (ES TOF) calcd for C18H15NNaO2 ([M + Na]+): 300.0995, found 300.0997 (−0.6 ppm).:2 v/v). White solid, mp 111–113 °C (EtOH), lit32 mp 112–114 °C; yield 77 mg (0.29 mmol, 29%). Rf 0.49, benzene/hexane (1:2, v/v). 1H NMR (400 MHz, CDCl3) δ 8.48–8.39 (m, 2H), 7.89–7.82 (m, 2H), 7.55–7.48 (m, 3H), 7.25–7.18 (m, 2H), 7.05 (s, 1H). 13C NMR (101 MHz, CDCl3) δ 183.0, 169.9, 165.4 (d, J = 256.8 Hz), 161.4, 132.5 (d, J = 9.5 Hz, 2C), 131.1 (d, J = 2.8 Hz), 129.8, 128.2, 125.6, 125.0, 114.8 (d, J = 21.6 Hz, 2C), 99.2. IR, vmax/cm−1: 3133, 1655, 1600, 1506, 1441, 1296, 1251, 1231, 1147, 1104. HRMS (ES TOF) calcd for C16H10FNNaO2 ([M + Na]+): 290.0588, found 290.0582 (1.9 ppm).:2 v/v). Yellowish solid, mp 139–141 °C (EtOH), lit32 mp 143–145 °C; Rf 0.49, benzene/hexane (1:2, v/v). Yield 105 mg (0.37 mmol, 37%). 1H NMR (400 MHz, CDCl3) δ 8.38–8.30 (d, 2H), 7.85 (dd, J = 7.6, 2.0 Hz, 2H), 7.51 (m, J = 8.4, 2.0 Hz, 5H), 7.05 (s, 1H). 13C NMR (101 MHz, CDCl3) δ 183.4, 169.9, 161.3, 139.7, 133.0, 131.1 (2C), 129.8, 128.2 (2C), 127.9 (2C), 125.6, 125.0 (2C), 99.2. IR, vmax/cm−1: 3133, 3060, 1930, 1884, 1648, 1588, 1443, 1395, 1251, 1181, 1089. HRMS (ES TOF) calcd for C16H10ClNNaO2 ([M + Na]+): 306.0292, found 306.0295 (−0.9 ppm).:4). Colorless oil; Rf 0.74, EtOAc/hexane (1:4, v/v). Yield 86 mg (0.31 mmol, 31%). 1H NMR (400 MHz, CDCl3) δ 8.33–8.28 (m, 2H), 7.53–7.47 (m, 2H), 6.55 (s, 1H), 2.86 (t, J = 7.6 Hz, 2H), 1.83–1.75 (m, 2H), 1.44–1.35 (m, 4H), 0.95–0.92 (m, 3H). 13C NMR (101 MHz, CDCl3) δ 184.9, 175.1, 161.9, 140.7, 134.3, 132.2 (2C), 129.0 (2C), 101.8, 31.3, 27.3, 26.7, 22.4, 14.0. IR, vmax/cm−1: 2935, 2853, 1923, 1665, 1590, 1455, 1400, 1255, 1210, 1178, 1096. HRMS (ES TOF) calcd for C15H16ClNNaO2 ([M + Na]+): 300.0762, found 300.0760 (0.7 ppm).:4). Colorless solid, mp 117–117.5 °C (EtOH); Rf 0.46, benzene/hexane (1:2, v/v). Yield 99 mg (0.35 mmol, 35%). 1H NMR (400 MHz, CDCl3) δ 8.34 (t, J = 2.0 Hz, 1H), 8.27 (d, J = 7.8 Hz, 1H), 7.85 (m, J = 7.8, 2.0 Hz, 2H), 7.63 (m, J = 8.1, 2.2, 1.1 Hz, 1H), 7.54–7.48 (m, 4H), 7.06 (s, 1H). 13C NMR (101 MHz, CDCl3) δ 184.5, 171.1, 162.2, 137.2, 134.9, 134.0, 130.9, 130.6, 129.9, 129.2 (2C), 128.9, 126.6, 126.0 (2C), 100.2. IR, vmax/cm−1: 1959, 1896, 1761, 1655, 1571, 1436, 1236, 1159, 1082, 1046. HRMS (ES TOF) calcd for C16H10ClNNaO2 ([M + Na]+): 306.0292, found 306.0285 (2.2 ppm).:4). Brown solid, mp 83–85 °C; Rf 0.40, EtOAc/hexane (1:4, v/v). Yield 66 mg (0.19 mmol, 19%). 1H NMR (400 MHz, CDCl3) δ 8.22–8.18 (m, 2H), 7.69–7.66 (m, 2H), 6.85 (d, J = 0.6 Hz, 1H), 4.54 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 184.0, 168.6, 162.1, 134.1, 132.2 (2C), 132.0 (2C), 129.8, 104.7, 17.9. IR, vmax/cm−1: 3147, 2920, 2853, 1934, 1652, 1585, 1448, 1397, 1295, 1224, 1178, 932. HRMS (ES TOF) calcd for C11H7Br2NaNO2 ([M + Na]+): 365.8736, found 365.8727 (2.4 ppm).:4 v/v) to obtain two regioisomeric products 12aa and 14aa.
Product 12aa. Brown solid, mp 105–107 °C; Rf 0.30, EtOAc/hexane (1
:4, v/v). Yield 138 mg (0.39 mmol, 39%). 1H NMR (400 MHz, CDCl3) δ 8.27 (dt, J = 8.3, 1.3 Hz, 2H), 7.90–7.85 (m, 2H), 7.62 (dddd, J = 7.5, 5.2, 3.9, 1.9 Hz, 2H), 7.52–7.41 (m, 7H), 7.33 (dd, J = 6.3, 2.6 Hz, 2H), 5.78 (d, J = 8.2 Hz, 1H), 5.58 (dd, J = 8.3, 1.1 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 195.7, 185.8, 157.1, 138.3, 135.6, 135.5, 134.4, 134.1, 130.7 (2C), 129.5, 129.4 (2C), 129.3 (2C), 129.1 (2C), 128.6 (2C), 126.4 (2C), 89.5, 63.1. IR, vmax/cm−1: 3060, 2925, 1684, 1643, 1578, 1448, 1366, 1277, 1195, 1016, 932. HRMS (ES TOF) calcd for C23H17NaNO3 ([M + Na]+): 378.1101, found 378.1091 (2.6 ppm).
Product 14aa. Yellow viscous oil; Rf 0.29, EtOAc/hexane (1
:4, v/v). Yield 121 mg (0.34 mmol, 34%). 1H NMR (400 MHz, CDCl3) δ 8.17 (dt, J = 8.5, 1.2 Hz, 2H), 8.01 (dt, J = 8.5, 1.2 Hz, 2H), 7.66–7.56 (m, 2H), 7.53–7.32 (m, 9H), 5.86 (dd, J = 5.4, 1.0 Hz, 1H), 5.37 (dd, J = 5.4, 1.0 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 192.1, 184.9, 159.1, 137.6, 135.9, 134.4, 134.0, 133.8, 130.5 (2C), 129.6 (2C), 129.6 (2C), 129.1 (2C), 128.5 (2C), 128.47, 127.8 (2C), 90.6, 55.4. IR, vmax/cm−1: 3062, 2927, 1686, 1645, 1579, 1448, 1369, 1279, 1196, 1026, 932. HRMS (ES TOF) calcd for C23H17NaNO3 ([M + Na]+): 378.1101, found 378.1092 (2.3 ppm).
:4). Brown viscous oil; Rf = 0.41, benzene. Yield 248 mg (0.53 mmol, 53%). 1H NMR (400 MHz, CDCl3) δ 8.05–7.92 (m, 2H), 7.77 (dd, J = 8.5, 1.2 Hz, 1H), 7.56–7.49 (m, 3H), 7.41–7.16 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 185.0, 184.3, 171.0, 160.2, 145.8, 141.6, 134.3, 134.1, 133.9, 133.9, 132.7, 131.5 (2C), 131.4, 131.3, 129.9, 129.4 (2C), 129.1, 126.8, 125.0, 123.9, 118.6. IR, vmax/cm−1: 3089, 2925, 1732, 1674, 1587, 1522, 1346, 1224, 1084, 1014, 930. HRMS (ES TOF) calcd for C23H12Cl2N2NaO5 ([M + Na]+): 489.0015, found 489.0013 (0.6 ppm).:4). Colorless solid, mp 147–150 °C, lit33 mp 100–102 °C (EtOAc); Rf 0.16, EtOAc/hexane (1:4, v/v). Yield 201 mg (0.63 mmol, 63%). 1H NMR (400 MHz, CDCl3) δ 8.27–8.17 (m, 2H), 7.68–7.62 (m, 1H), 7.54–7.42 (m, 5H), 7.31–7.26 (m, 2H), 5.70 (d, J = 9.8 Hz, 1H), 5.24 (d, J = 9.8 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 183.4, 170.0, 168.8, 153.2, 134.9, 134.7, 130.8, 130.6 (2C), 129.5 (2C), 129.4, 128.8 (2C), 126.2 (2C), 81.2, 54.9. IR, vmax/cm−1: 3065, 1901, 1713, 1651, 1598, 1501, 1448, 1381, 1279, 1200, 1070, 1024. HRMS (ES TOF) calcd for C18H12N2NaO4 ([M + Na]+): 343.0689, found 343.0679 (2.9 ppm).Author contributions
A. V. Aksenov – conceptualization, supervision, funding acquisition; N. A. Aksenov – methodology, formal analysis, funding acquisition; N. K. Kirilov – investigation; A. A. Skomorokhov – investigation; D. A. Aksenov – investigation; I. A. Kurenkov – investigation; E. A. Sorokina – investigation, formal analysis; M. A. Nobi – formal analysis, writing (review and editing); M. Rubin – conceptualization, supervision, writing (original draft, review, and editing).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the grant of the President of the Russian Federation (grant #MD-3505.2021.1.3) and the Ministry of Education and Science of the Russian Federation (grant #0795-2020-0031). Support for the NMR instruments used in this project was provided by the Center of Shared Instrumentation, NCFU (Grant #075-15-2021-672).Notes and references
- R. Ballini, L. Barboni, L. Castrica, F. Fringuelli, D. Lanari, F. Pizzo and L. Vaccaro, Adv. Synth. Catal., 2008, 350, 1218–1224 CrossRef CAS.
- E. M. Budynina, K. L. Ivanov, A. O. Chagarovskiy, V. B. Rybakov, I. V. Trushkov and M. Y. Melnikov, Chem.–Eur. J., 2016, 22, 3692–3696 CrossRef CAS PubMed.
- M. N. Grayson, J. Org. Chem., 2017, 82, 4396–4401 CrossRef CAS PubMed.
- Y. Liu, W. Zhen, W. Dai, F. Wang and X. Li, Org. Lett., 2013, 15, 874–877 CrossRef CAS PubMed.
- G. R. Reddy, D. Mukherjee, A. K. Chittoory and S. Rajaram, Org. Lett., 2014, 16, 5874–5877 CrossRef CAS PubMed.
- D. K. Romney, N. S. Sarai and F. H. Arnold, ACS Catal., 2019, 9, 8726–8730 CrossRef CAS PubMed.
- H. Ueda, K. Yoshida and H. Tokuyama, Org. Lett., 2014, 16, 4194–4197 CrossRef CAS PubMed.
- L. Wen, L. Yin, Q. Shen and L. Lu, ACS Catal., 2013, 3, 502–506 CrossRef CAS.
- R. Ballini and M. Petrini, Tetrahedron, 2004, 60, 1017–1047 CrossRef CAS.
- R. Ballini and M. Petrini, Adv. Synth. Catal., 2015, 357, 2371–2402 CrossRef CAS.
- R. Ballini, M. Petrini and G. Rosini, Molecules, 2008, 13, 319–330 CrossRef CAS PubMed.
- R. Ballini, M. Petrini and G. Rosini, Molecules, 2008, 13, 319–330 CrossRef CAS PubMed.
- A. V. Aksenov, V. Khamraev, N. A. Aksenov, N. K. Kirilov, D. A. Domenyuk, V. A. Zelensky and M. Rubin, RSC Adv., 2019, 9, 6636–6642 RSC.
- D. A. Aksenov, N. A. Arutiunov, V. V. Maliuga, A. V. Aksenov and M. Rubin, Beilstein J. Org. Chem., 2020, 16, 2903–2910 CrossRef CAS PubMed.
- N. A. Aksenov, A. V. Aksenov, N. K. Kirilov, N. A. Arutiunov, D. A. Aksenov, V. Maslivetc, Z. Zhao, L. Du, M. Rubin and A. Kornienko, Org. Biomol. Chem., 2020, 18, 6651–6664 RSC.
- N. A. Aksenov, A. V. Aksenov, S. N. Ovcharov, D. A. Aksenov and M. Rubin, Front. Chem., 2020, 8, 77 CrossRef CAS PubMed.
- N. A. Aksenov, N. A. Arutiunov, N. K. Kirillov, D. A. Aksenov, A. V. Aksenov and M. Rubin, Chem. Heterocycl. Compd., 2020, 56, 1067–1072 CrossRef CAS.
- A. V. Aksenov, N. A. Aksenov, O. N. Nadein and I. V. Aksenova, Synlett, 2010, 2628–2630, DOI:10.1055/s-0030-1258767.
- A. V. Aksenov, N. A. Aksenov, O. N. Nadein and I. V. Aksenova, Synth. Commun., 2012, 42, 541–547 CrossRef CAS.
- A. V. Aksenov, A. N. Smirnov, N. A. Aksenov, A. S. Bijieva, I. V. Aksenova and M. Rubin, Org. Biomol. Chem., 2015, 13, 4289–4295 RSC.
- N. A. Aksenov, A. V. Aksenov, O. N. Nadein, D. A. Aksenov, A. N. Smirnov and M. Rubin, RSC Adv., 2015, 5, 71620–71626 RSC.
- K.-i. Itoh, T. Aoyama, H. Satoh, Y. Fujii, H. Sakamaki, T. Takido and M. Kodomari, Tetrahedron Lett., 2011, 52, 6892–6895 CrossRef CAS.
- A. V. Aksenov, A. N. Smirnov, I. V. Magedov, M. R. Reisenauer, N. A. Aksenov, I. V. Aksenova, A. L. Pendleton, G. Nguyen, R. K. Johnston, M. Rubin, A. De Carvalho, R. Kiss, V. Mathieu, F. Lefranc, J. Correa, D. A. Cavazos, A. J. Brenner, B. A. Bryan, S. Rogelj, A. Kornienko and L. V. Frolova, J. Med. Chem., 2015, 58, 2206–2220 CrossRef CAS PubMed.
- M. S. Maurice and S. L. Bearne, Biochemistry, 2004, 43, 2524–2532 CrossRef PubMed.
- N. A. Aksenov, D. A. Aksenov, A. A. Skomorokhov, L. A. Prityko, A. V. Aksenov, G. D. Griaznov and M. Rubin, J. Org. Chem., 2020, 85, 12128–12146 CrossRef CAS PubMed.
- Z. Wang, X. Wu, Z. Li, Z. Huang and F. Chen, Org. Biomol. Chem., 2019, 17, 3575–3580 RSC.
- Z. Lian, S. D. Friis and T. Skrydstrup, Chem. Commun., 2015, 51, 3600–3603 RSC.
- Q. Song, Q. Feng and K. Yang, Org. Lett., 2014, 16, 624–627 CrossRef CAS PubMed.
- A. R. Daniewski, M. Witanowski and T. Urbanski, J. Org. Chem., 1967, 32, 4050–4052 CrossRef CAS.
- H. R. Snyder and N. E. Boyer, J. Am. Chem. Soc., 1955, 77, 4233–4238 CrossRef CAS.
- L. Tang, X. Guo, Y. Li, S. Zhang, Z. Zha and Z. Wang, Chem. Commun., 2013, 49, 5213–5215 RSC.
- P. Gao, H.-X. Li, X.-H. Hao, D.-P. Jin, D.-Q. Chen, X.-B. Yan, X.-X. Wu, X.-R. Song, X.-Y. Liu and Y.-M. Liang, Org. Lett., 2014, 16, 6298–6301 CrossRef CAS PubMed.
- H. Wang, R. Cheng, G. Wang, Y. Shi, J. Wang, H. Guo, L. Trigoura, Y. Xing and S. Sun, Tetrahedron Lett., 2020, 61, 151652 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral charts. CCDC 2099762. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra06503c |
| This journal is © The Royal Society of Chemistry 2021 |