
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConvenient one-pot synthesis of 1,2,4-oxadiazoles and 2,4,6-triarylpyridines using graphene oxide (GO) as a metal-free catalyst: importance of dual catalytic activity†
Puja Basak,
Sourav Dey and
Pranab Ghosh *
*
Department of Chemistry, University of North Bengal, Darjeeling, West Bengal, India. E-mail: pizy12@yahoo.com; Fax: +91-353-2699001; Tel: +91-353-2776381
First published on 28th September 2021
Abstract
A convenient and efficient process for the synthesis of 3,5-disubstituted 1,2,4-oxadiazoles and 2,4,6-triarylpyridines has been described using an inexpensive, environmentally benign, metal-free heterogeneous carbocatalyst, graphene oxide (GO). GO plays a dual role of an oxidizing agent and solid acid catalyst for synthesizing 1,2,4-oxadiazoles and triarylpyridines. This dual catalytic activity of GO is due to the presence of oxygenated functional groups which are distributed on the nanosheets of graphene oxide. A broad scope of substrate applicability and good sustainability is offered in this developed protocol. The results of a few control experiments reveal a plausible mechanism and the role of GO as a catalyst was confirmed by FTIR, XRD, SEM, and HR-TEM analysis.
Introduction
Nitrogen-containing heterocyclic compounds are valuable due to their potential application as a key intermediate in the synthesis of numerous drugs.1 3,5-Disubstituted 1,2,4-oxadiazoles are a remarkably important class of nitrogen-containing heterocyclic scaffold as they are widely used as pharmacophores, bioactive molecules, and functional materials.1,2 Among the oxadiazole derivatives, the 1,2,4-oxadiazole motif has received interest due to its application as a stable bioisostere in place of an amide, ester, or urea functionality.3 These compounds when selectively functionalized, have performed as various muscarinic agonists,4 benzodiazepine receptor partial agonists,5 serotonergic (5-HT3) antagonists,6 dopamine transporters,7 antischistosomal drugs,8 G-quadruplex ligands for probing DNA superstructure in antitumor research.9,10 Another nitrogen-containing heterocycle, pyridines are ubiquitous and have attracted much attention due to their unique biological, medicinal, and pharmaceutical properties.11–13 2,4,6-Triarylpyridines are frequently used as a synthon in supramolecular chemistry owing to their π-stacking ability.14 In addition, pyridines have received a growing interest as monomeric building blocks in thin films and organometallic polymers.15It is noteworthy that, in the last decade many efficient protocols have been developed to synthesize these significant heterocyclic moieties. Among the known synthetic strategies of 1,2,4-oxadiazoles, the most conventional approach involves the use of amidoximes as starting materials or intermediates. Other common approaches involve O-acylation of amidoximes by an activated carboxylic acid derivative, followed by cyclodehydration,16 the 1,3-dipolar cycloaddition of nitrile oxide to nitriles, and intermolecular cyclodehydration reaction of amidoximes with aldehydes followed by oxidative dehydrogenation.17,18 Besides this, base-mediated one-pot synthesis, MnO2/GO based synthesis, microwave-assisted efficient synthesis of oxadiazoles using PTSA and ZnCl2 have also been reported.17,19–21 On the other hand, efficient protocols for the synthesis of another important heterocycle 2,4,6-triarylpyridines involve condensation reaction between benzaldehydes, acetophenones, and ammonium acetate in presence of different acid catalysts22–24 e.g. pentafluorophenylammonium triflate,23 heteropolyacid,25 HClO4–SiO2,26 Brønsted-acidic ionic liquid,27 and nano-metal catalyst.23,28,29 Nevertheless, most of the traditional synthetic method requires harsh reaction condition, prolonged heating, and use of toxic transition metal catalyst. However, only a few protocols have shown greener context and high atom economy. Multicomponent reaction (MCR) is considered to be an effective and straightforward approach for the synthesis of heterocycles in an atom economical way. Considering the efficiency of MCRs and the aspects of green chemistry,30–33 there is a need for new methods which involve metal-free, environmentally friendly catalytic protocol to synthesize 1,2,4-oxadiazoles and 2,4,6-triarylpyridines.
Recently, carbonaceous nanomaterials have gained considerable attention in green chemistry, especially in the development of metal-free sustainable heterogeneous catalysts.34–36 Among the carbonaceous nanomaterials, graphene oxide (GO) has been reported to accelerate several organic transformation reactions replacing different hazardous chemical reagents. GO, a thin two-dimensional unique nanomaterial contains different oxygen functionalities like carbonyl (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), carboxyl (–COOH), epoxy (–O–), and hydroxyl (–OH) on its edges and basal plane.37–39 On account of the presence of large surface area, and diverse oxygen functionalities, GO has been identified as a heterogeneous solid acid catalyst (pH 4.5 at 0.1 mg mL−1) as well as a benign oxidizing agent.40–42 Its abundance from low-cost natural carbon sources, low toxicity, reusability, and metal-free catalytic activity makes this heterogeneous carbon material (GO) as a promising carbocatalyst. Due to the inherent acidic and oxidation property of GO, it is explored as a catalyst in different organic transformations like C–H oxidations,43 oxidative coupling of amines44 to the imines, oxidation of thioanisole,45 glutaraldehyde to glutaric acid,46 5-hydroxymethylfurfural,47 benzylpyrazolyl coumarins,48 Fisher esterification,49 and transamidation.50–54 The versatility and sustainability of GO as a catalyst leads us to employ GO as a metal-free catalyst for the synthesis of substituted 1,2,4-oxadiazoles and 2,4,6-triarylpyridines to overcome the drawbacks of the reported protocols and reduce environmental hazards. Our present study explores the role of GO as an acid catalyst as well as an oxidizing agent using the surface-bound oxygen-containing functional groups. To unleash the dual catalytic activity of GO, a plausible oxidative cyclization pathway to the synthesis of oxadiazoles and triarylpyridines under benign conditions has also been established.
O), carboxyl (–COOH), epoxy (–O–), and hydroxyl (–OH) on its edges and basal plane.37–39 On account of the presence of large surface area, and diverse oxygen functionalities, GO has been identified as a heterogeneous solid acid catalyst (pH 4.5 at 0.1 mg mL−1) as well as a benign oxidizing agent.40–42 Its abundance from low-cost natural carbon sources, low toxicity, reusability, and metal-free catalytic activity makes this heterogeneous carbon material (GO) as a promising carbocatalyst. Due to the inherent acidic and oxidation property of GO, it is explored as a catalyst in different organic transformations like C–H oxidations,43 oxidative coupling of amines44 to the imines, oxidation of thioanisole,45 glutaraldehyde to glutaric acid,46 5-hydroxymethylfurfural,47 benzylpyrazolyl coumarins,48 Fisher esterification,49 and transamidation.50–54 The versatility and sustainability of GO as a catalyst leads us to employ GO as a metal-free catalyst for the synthesis of substituted 1,2,4-oxadiazoles and 2,4,6-triarylpyridines to overcome the drawbacks of the reported protocols and reduce environmental hazards. Our present study explores the role of GO as an acid catalyst as well as an oxidizing agent using the surface-bound oxygen-containing functional groups. To unleash the dual catalytic activity of GO, a plausible oxidative cyclization pathway to the synthesis of oxadiazoles and triarylpyridines under benign conditions has also been established.
Results and discussion
For screening the reaction parameter benzonitrile (1.5 mmol), hydroxylamine hydrochloride (1.5 mmol), and base (1.5 mmol) were taken as model substrates to find out suitable conditions for the synthesis of amidoxime (intermediate). To satisfy our curiosity, the reaction was performed in different solvents e.g. polar protic, polar aprotic, and nonpolar. However, in absence of a base, a low yield was obtained (Table 1, entry 6). Gratifyingly, the reaction results showed (Table 1) the formation of amidoxime is highly favored in mixed solvent ethanol–water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) using K2CO3 as a base. To control the reaction conditions, after completion of the reaction, the solvent was removed by a rotary evaporator to separate the intermediate. While monitoring the TLC, only one spot was observed other than the reactant. After workup and purification by column chromatography, 91% yield of the intermediate (amidoxime) was obtained (Table 1, entry 7). Although other bases were also employed (Table 1, entries 2, 5 and 9), K2CO3 exerted the best result in an ethanol–water solvent. The synthesized amidoxime was characterized by NMR (300 MHz) and the spectral data was shown in ESI.†
:3) using K2CO3 as a base. To control the reaction conditions, after completion of the reaction, the solvent was removed by a rotary evaporator to separate the intermediate. While monitoring the TLC, only one spot was observed other than the reactant. After workup and purification by column chromatography, 91% yield of the intermediate (amidoxime) was obtained (Table 1, entry 7). Although other bases were also employed (Table 1, entries 2, 5 and 9), K2CO3 exerted the best result in an ethanol–water solvent. The synthesized amidoxime was characterized by NMR (300 MHz) and the spectral data was shown in ESI.†
|
||||
|---|---|---|---|---|
| Entry | Solvent | Temp (°C) | Base | Yieldb (%) |
| a Reaction condition: benzonitrile (1.5 mmol), hydroxylamine hydrochloride (1.5 mmol), base (1.5 mmol) and solvent (5 mL).b Isolated yield.c No base was added.d The reaction was carried out for 24 h. | ||||
| 1 | Water | 100 | K2CO3 | 68 |
| 2 | Water | 100 | Cs2CO3 | 72 |
| 3 | Ethanol | 80 | K2CO3 | 66 |
| 4 | Ethanol | 80 | TEA | 70 |
| 5 | Ethanol–water | 80 | TEA | 80 |
| 6 | Ethanol–water | 80 | — | <50c |
| 7 | Ethanol–water | 80 | K2CO3 | 91 |
| 8 | Ethanol–water | 80 | K2CO3 | 94d |
| 9 | Ethanol–water | 80 | Cs2CO3 | 93 |
| 10 | THF | 120 | K2CO3 | 54 |
| 11 | Toluene | 110 | K2CO3 | <50 |
| 12 | CH3CN | 82 | K2CO3 | 68 |
| 13 | DMF | 120 | K2CO3 | 76 |
In the second step of the reaction, benzaldehyde (1 mmol) and the catalyst were added to the reaction mixture to prioritize the synthesis of 3,5-disubstituted 1,2,4-oxadiazole. In presence of a small amount of GO, 73% yield of the product was obtained at 80 °C temperature (entry 2). Further increase in the amount of GO, proved to be favorable in the formation of 1,2,4-oxadiazole. No product was obtained when the reaction was carried out in absence of GO (Table 2, entry 1). High yield of the product was observed in aqueous ethanolic solution with a ratio ethanol–water (1:3). The outstanding catalytic activity of GO in ethanol–water (1:3) is revealed due to its better dispersibility. To establish the catalytic activity of GO, few controlled experiments were carried out using various catalysts. Other carbonaceous nanomaterials e.g. powdered graphite, reduced graphene oxide (rGO) showed less catalytic activity than GO because they do not contain as many hydroxyl and carboxylic groups, indicating oxygen-containing functional groups in graphene oxide have a profound effect in catalyzing the synthesis of 3,5-disubstituted 1,2,4-oxadiazole. The reaction was also carried out in presence of GO and an oxidant H2O2, the reason for the low yield may be due to the oxidation of benzaldehyde to benzoic acid in presence of H2O2 (Table 2, entry 12). The yield was not improved when only an H2O2 oxidant was used (entry 13). These control experiments infer the significant catalytic role of GO in the reaction.
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (mg) | Solvent | Temperature | Time (h) | Yield% |
| a Reaction condition: benzaldehyde (1 mmol), amidoxime (1 mmol) and ethanol–water(5 mL), pristine GO (25 mg).b Graphite powder was used.c Reduced graphene oxide (rGO).d GO and extra oxidant 30% H2O2 (1 mmol) were used.e Only H2O2 was used.f Under inert atmospheric condition. | |||||
| 1 | — | Ethanol | 80 | 12 | Trace |
| 2 | 15 (GO) | Ethanol | 80 | 12 | 73 |
| 3 | 15 (GO) | Water | 100 | 12 | 77 |
| 4 | 15 (GO) | DMF | 100 | 12 | 60 |
| 5 | 15 (GO) | Ethanol–water | 80 | 12 | 79 |
| 6 | 15 (GO) | Ethanol–water | 80 | 24 | 83 |
| 7 | 25 (GO) | Ethanol–water | 80 | 12 | 89 |
| 8 | 25 (GO) | Ethanol–water | 80 | 8 | 88 |
| 9 | 25 (GO) | Ethanol–water | RT | 12 | 52 |
| 10 | 25 (graphite) | Ethanol–water | 80 | 8 | 40b |
| 11 | 25 (rGO) | Ethanol–water | 80 | 8 | 45c |
| 12 | 25 (GO)/oxidant | Ethanol–water | 80 | 8 | 67d |
| 13 | Oxidant | Ethanol–water | 80 | 8 | <40e |
| 14 | 25 (GO) | Neat | 80 | 8 | 69 |
| 15 | 25 (GO) | Ethanol–water | 80 | 8 | 85f |
| 16 | — | Ethanol–water | 80 | 8 | Nilf |
The scope and the substrate applicability of the reaction were also examined and results were summarized in Table 3.
|
||||
|---|---|---|---|---|
| Entry | R | R1 | Product | Yieldb (%) |
| a In the first step, benzonitrile (1 mmol), hydroxylamine hydrochloride (1.5 mmol), K2CO3 (1.5 mmol), and ethanol–water (5 mL) were stirred for 8 h and in the 2nd step benzaldehyde (1 mmol) and GO (x mg) were added and stirred for another 8 h.b Isolated yield after purification through column chromatography.c 4-(Dimethylamino)benzaldehyde (1 mmol) was used.d Heptaldehyde was used.e Acetonitrile (1 mmol) was used. | ||||
| 1 | 4-H | 4-H | 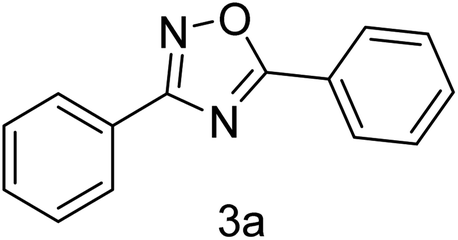 |
83 |
| 2 | 4-H | 4-CH3 | 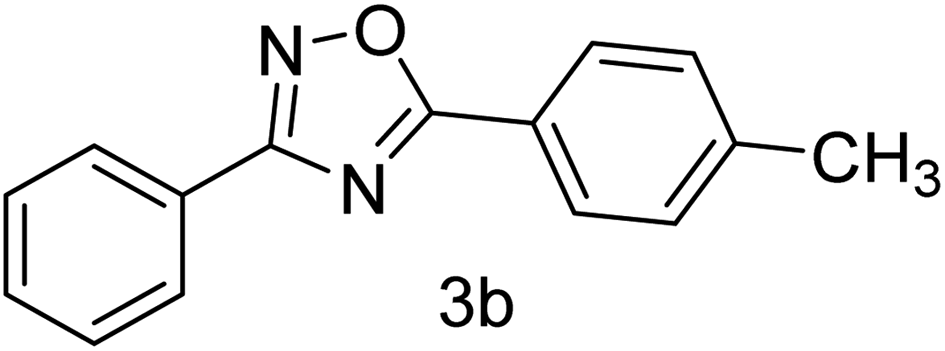 |
81 |
| 3 | 4-H | 4-OCH3 | 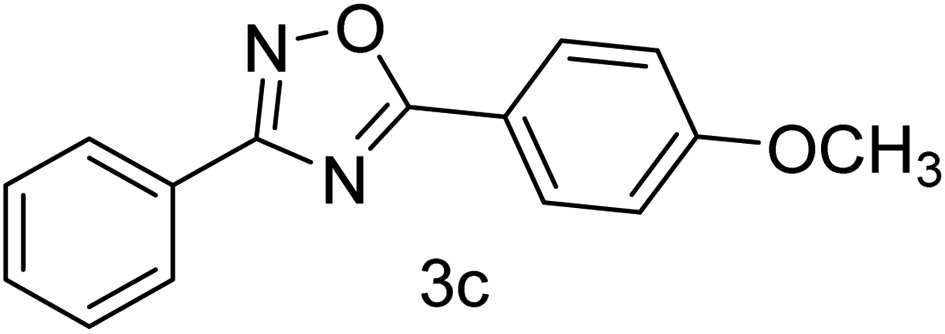 |
80 |
| 4 | 4-H | 4-F | 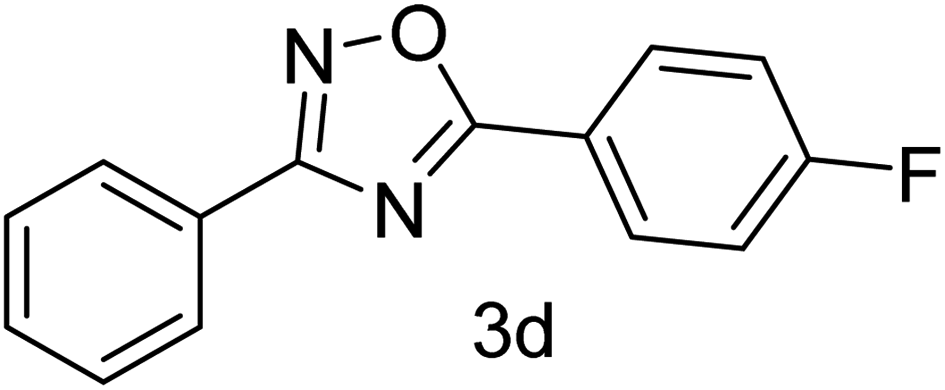 |
78 |
| 5 | 4-H | 3-NO2 | 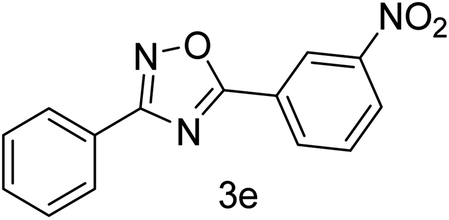 |
75 |
| 6c | 4-H | 4-N(CH3)2 | No 1,2,4-oxadiazole, only imine formation | — |
| 7 | 4-H | 1-Napthaldehyde | 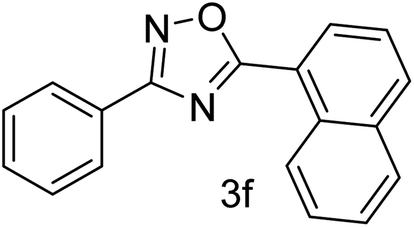 |
62 |
| 8 | 4-H | Furan-2-carbaldehyde | 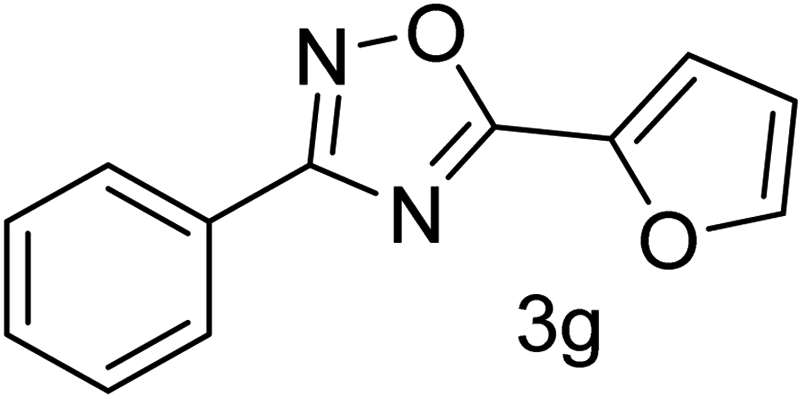 |
72 |
| 9 | 4-H | Thiophene-2-carbaldehyde | 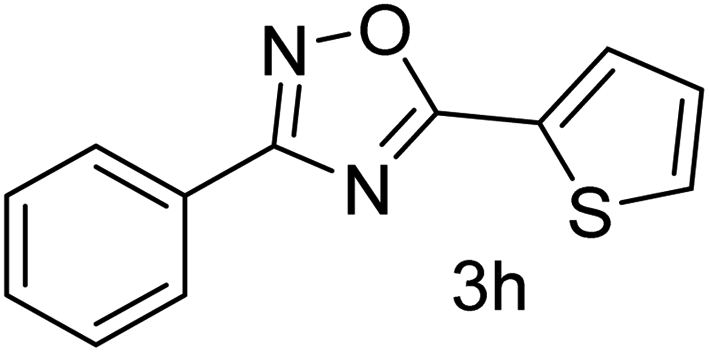 |
70 |
| 10 | 4-CH3 | 4-H | 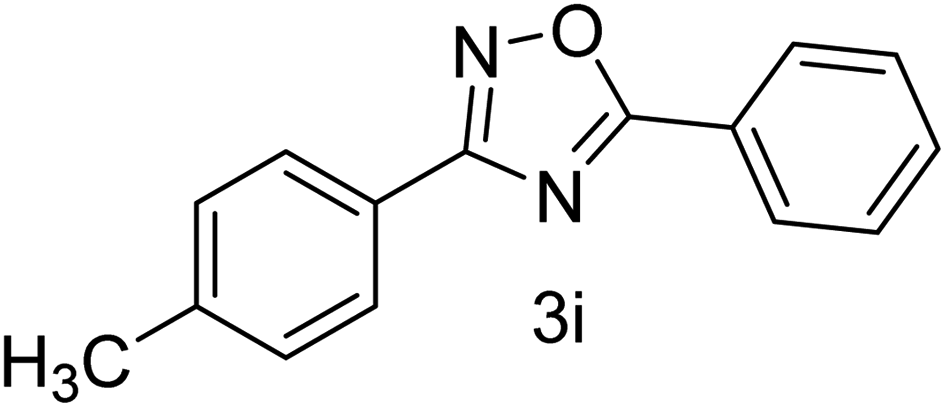 |
80 |
| 11 | 4-OCH3 | 4-H | 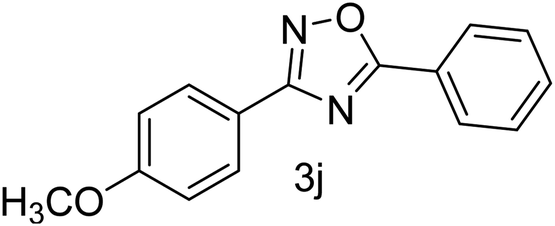 |
78 |
| 12 | 4-OCH3 | 4Cl | 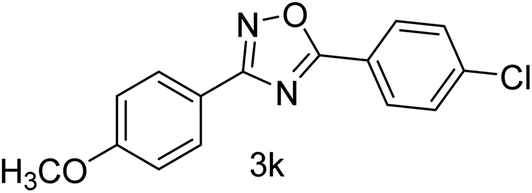 |
82 |
| 13 | 4-Pyridinecarbonitrile | 4-H | 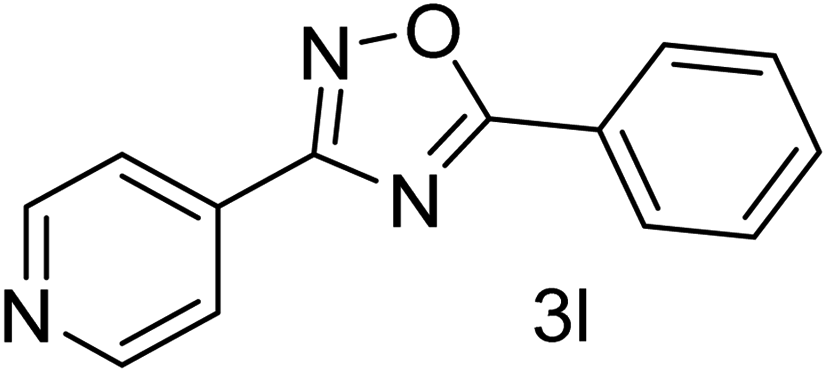 |
68 |
| 14 | 4-H | CH3CHO | 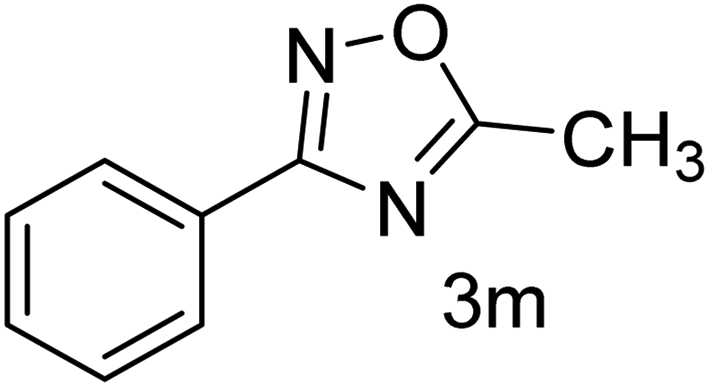 |
75 |
| 15d | 4-H | Heptaldehyde | NR | — |
| 16e | CH3CN | 4-H | NR | — |
With the optimized condition in hand, we have extended the substrate scope in organic transformations and a series of diversely substituted aldehydes and benzonitriles are subjected to the synthesis of 3,5-disubstituted 1,2,4-oxadiazole (Table 3). Both the electron-donating (Table 3, entries 2, 3, 10 and 11) and electron-withdrawing groups (entries 4 and 5) in the substituents afforded the corresponding product in good to excellent yield which indicates that the electronic nature of the substituents is not much influential to determine the yield of the reaction. 1-Napthaldehyde offered the product with low yield and the reason may be due to steric hindrance (Table 3, entry 7). In the case of 4-N,N-(dimethylamino) benzaldehyde, the reaction was stopped at amidoxime, no desired oxadiazole is obtained (Table 3, entry 6). The present catalytic condition showed a wide tolerance to heterocyclic aldehydes (Table 3 entries 8, 9) and they were found to be highly effective to afford the corresponding product. The generality of the reaction was examined in the case of aliphatic aldehydes also. Interestingly, acetaldehyde was equally effective to yield the product with excellent quantity (entry 14). However, no product was found with increasing the side chain of aliphatic aldehydes (entry 15). It was disappointing that acetonitrile did not exert the corresponding product (entry 16). Due to the heterogeneous nature of GO, it can be easily isolated from the reaction mixture and reused. The catalytic activity of GO was examined for five consecutive cycles for the synthesis of 3,5-disubstituted 1,2,4-oxadiazole from benzaldehyde and amidoxime under reflux conditions for 8 h to ascertain the recyclability potential of graphene oxide. The catalyst was separated after each recycles and washed thoroughly with ethanol and reused. A marginal decrease in the yield of oxadiazole is observed after each cycle which indicates a slight loss of catalytic activity of GO with recycling (Fig. 1).
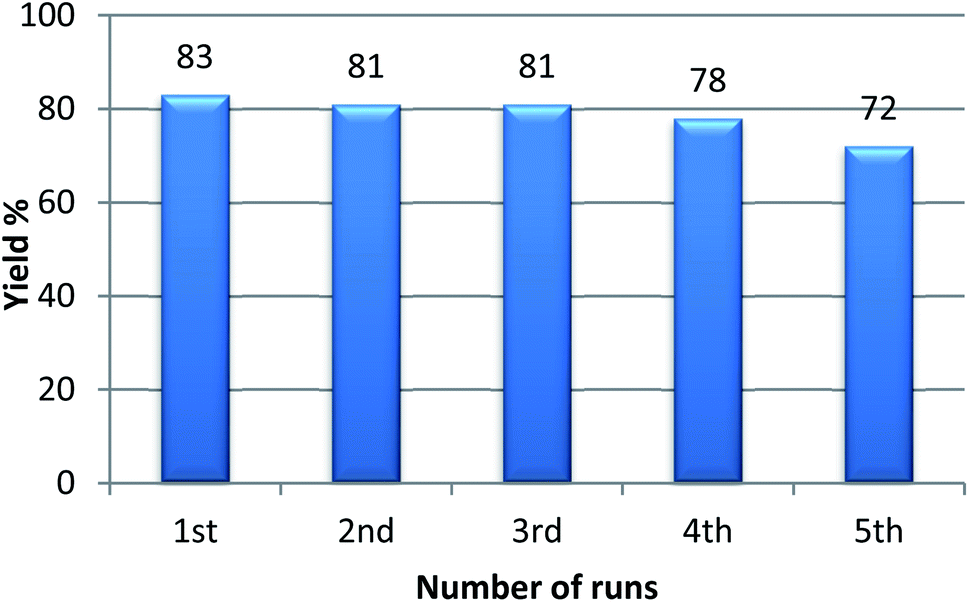 | ||
| Fig. 1 Recyclability study of GO for the synthesis of 3,5-disubstituted 1,2,4-oxadiazole. | ||
The catalytic activity arises some structural changes in GO which were analyzed by FTIR, XRD, SEM, HR-TEM, and EDX analysis. The XRD spectra of fresh GO and recycled catalyst (GO after 3rd run and 5th run) are shown in Fig. 2. A comparison of spectra indicates the reduction in the intensity of the first characteristic peak of GO (2θ = 10.01) and the appearance of a new peak at (2θ = 24.62) due to the formation of partially reduced GO/reduced graphene oxide upon reuse. These results confirm the reduction of the functional groups of GO during the reaction.
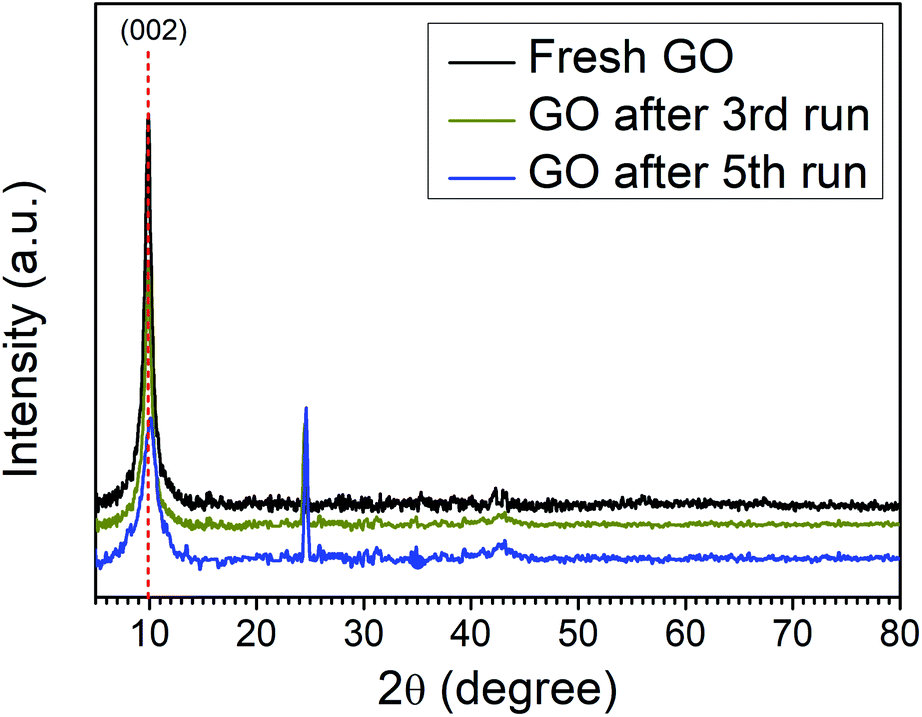 | ||
| Fig. 2 XRD spectra of fresh GO, after 3rd run and 5th run. | ||
The comparison of the FTIR spectra revealed that the peak at 1720 cm−1 in fresh GO has completely disappeared after reuse. In addition to this, the peak intensity of the hydroxyl group at 3400 cm−1 decreases after reuse. FTIR data strongly support the reduction of GO to rGO in this oxidative cyclization reaction (Fig. 3).
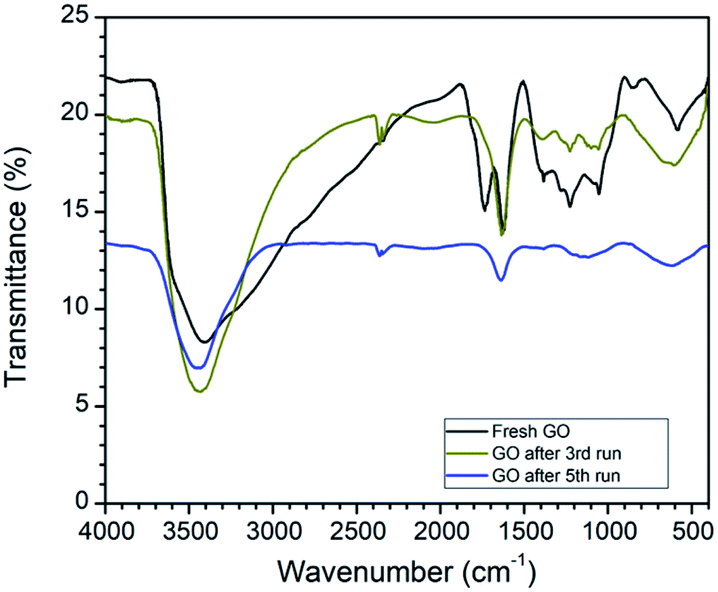 | ||
| Fig. 3 Comparative FTIR of fresh GO, after 3rd run and 5th run. | ||
A morphological study of GO and GO after the 5th run was carried out using SEM and HR-TEM to investigate the disintegration of graphene oxide sheets after the reaction. In HR-TEM, the graphene oxide sheets are disintegrated into smaller sheets with slight aggregation after recycle (Fig. 4).
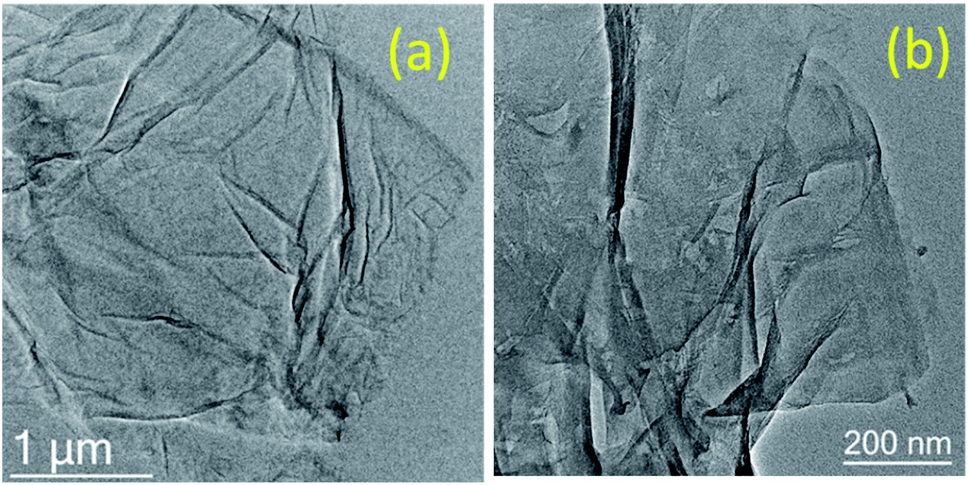 | ||
| Fig. 4 HR-TEM images of (a) GO and (b) GO after the 5th run. | ||
Moreover, the SEM images (Fig. 5) also reveal the formation of multiple small GO sheets after reuse. As GO catalyzes the reaction, its reduction to reduced graphene oxide possibly leads to its disintegration into smaller sheets.
 | ||
| Fig. 5 SEM images of (a) GO and (b) GO after the 5th run. | ||
The contribution of oxygen-containing functionalities during the reaction was further confirmed by the EDX analysis (Fig. 6). The carbon content was increased from 52.65% (fresh GO) to 71.79% (GO after 5th run) and the oxygen content was decreased from 47.35% (fresh GO) to 28.21% (GO after 5th run). The decrease in the oxygen content, therefore, indicates the role of GO in this cyclization reaction as an oxidizing agent. The universality and the dual catalytic activity of GO were established by a plausible mechanism (Scheme 1).
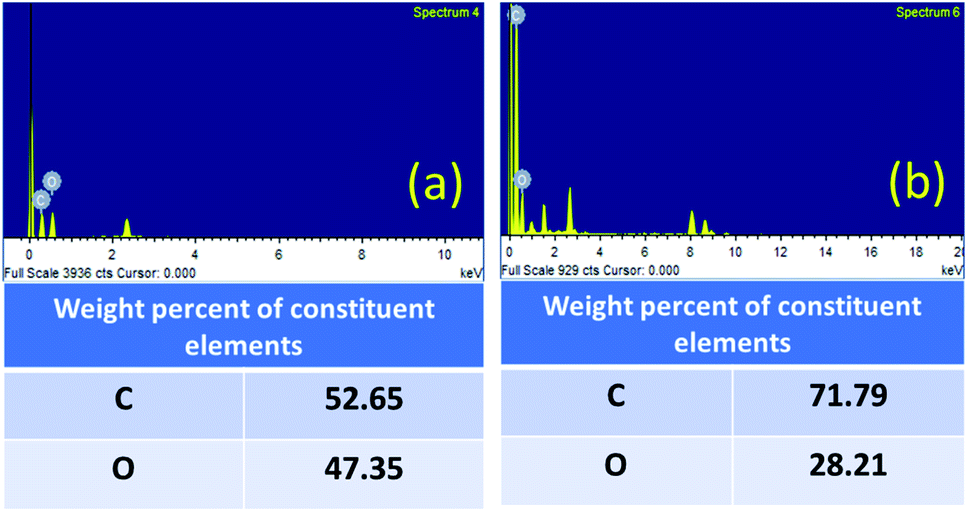 | ||
| Fig. 6 EDX spectra of (a) GO and (b) GO after the 5th run. | ||
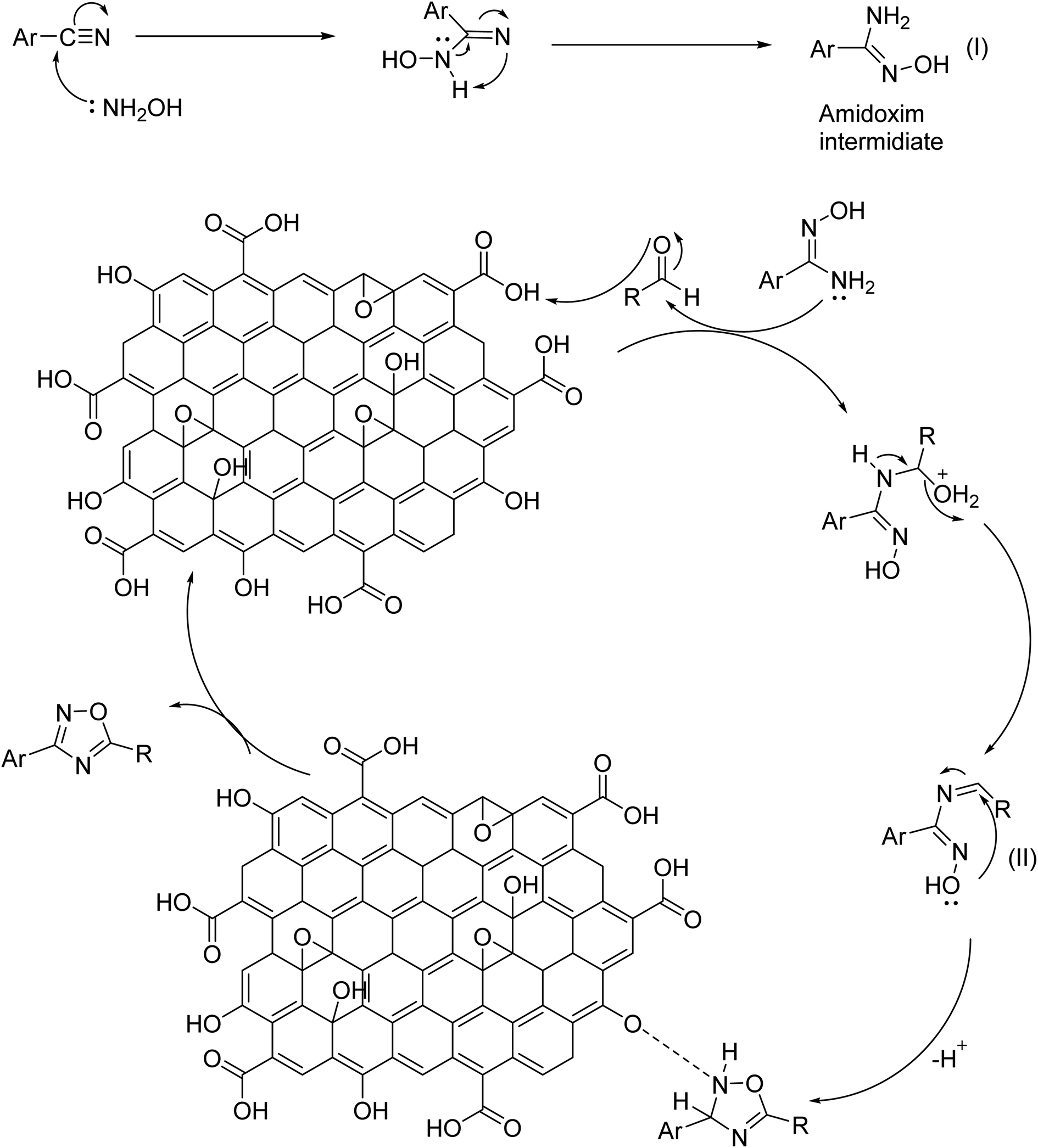 | ||
| Scheme 1 A plausible route to the synthesis of 3,5-disubstituted 1,2,4-oxadiazole. | ||
Mechanism
A plausible mechanism of GO catalyzed synthesis of 3,5-disubstituted 1,2,4-oxadiazole has been proposed (Scheme 1) based on literature reports55 and our controlled experiments (Table 2). Now, we propose the formation of amidoxime intermediate (I) from benzonitrile and hydroxylamine hydrochloride. However, in the first step, a base is required to neutralize hydroxylamine hydrochloride. In the 2nd step protonation of aldehyde, oxygen occurs and subsequently, a nucleophilic attack by amidoxime occurs at the electrophilic center of aldehyde. After that, the intermediate (II) undergoes an oxidative cyclization in presence of GO to produce 1,2,4-oxadiazoles. This mechanism is in good agreement with the control experiments as described in Table 2. However, in presence of only H2O2 oxidant the yield of the reaction was diminished (Table 2, entry 11). The role of GO as an acid catalyst and an oxidant was confirmed as its absence did not lead to the oxadiazole product. The oxygen containing functional groups of GO are consumed during the reaction and the activity of GO gradually decreases. The activity of recycled GO is lower than that of the pristine GO. Good yield of the product was obtained even under an inert atmosphere which strongly establish (Table 2, entry 15), the prime role of GO in absence of atmospheric oxygen.In connection to our previous work, the catalytic activity of synthesized GO was investigated in the case of 2,4,6-triarylpyridine synthesis. To find out the optimized condition of the reaction, acetophenone (2 mmol), benzaldehyde (1 mmol), and ammonium acetate (2 mmol) were selected as model substrates and the results were summarized in Table 4. As can be seen from Table 4 that neither polar nor non-polar solvents were found suitable for the reaction. The best result was obtained under neat or solvent-free conditions (Table 4, entry 11). The effect of temperature and the amount of catalyst was also examined to find out the optimized condition. Studies reveal that the yield increases with increasing temperature. Room-temperature reaction afforded only 20% of the product which strongly indicates the vital role of temperature in governing the reaction (entry 16). However, after 120 °C the yield decreases with a further increase in temperature (Table 4, entry 10). To ascertain the catalytic function of GO, the reaction was performed in absence of catalyst and only a trace amount of product was obtained. The amount of the catalyst was also altered and optimum condition offered a neat reaction with 30 mg of GO at 100 °C temperature. Ammonia sources other than ammonium acetate produced the corresponding product with a low yield (Table 4, entries 14 and 15).
| Entry | Temp (°C) | Solvent | Catalyst GO (mg) | Ammonia source | Yieldb (%) |
|---|---|---|---|---|---|
| a Reaction condition: acetophenone (2 mmol), benzaldehyde (1 mmol), ammonium acetate (2 mmol), reaction time: 2 hb Isolated yields. | |||||
| 1 | 100 | H2O | 15 | NH4OAc | 65 |
| 2 | 80 | Ethanol | 15 | NH4OAc | 55 |
| 3 | 100 | DMF | 15 | NH4OAc | 53 |
| 4 | 100 | DMSO | 15 | NH4OAc | 45 |
| 5 | 100 | Toluene | 15 | NH4OAc | 50 |
| 6 | 80 | CH3CN | 15 | NH4OAc | 30 |
| 7 | 100 | Ethylene glycol | 15 | NH4OAc | 60 |
| 8 | 100 | Neat | 15 | NH4OAc | 83 |
| 9 | 120 | Neat | 30 | NH4OAc | 90 |
| 10 | 150 | Neat | 30 | NH4OAc | 86 |
| 11 | 100 | Neat | 30 | NH4OAc | 92 |
| 12 | 80 | Neat | 30 | NH4OAc | 80 |
| 13 | 100 | Neat | — | NH4OAc | Trace |
| 14 | 100 | Neat | 30 | (NH4)2CO3 | 48 |
| 15 | 100 | Neat | 30 | (NH4)2SO4 | Trace |
| 16 | RT | Neat | 30 | NH4OAc | <20 |
To explore the catalytic activity of GO, a wide variety of aromatic aldehydes and substituted acetophenones were subjected to synthesize 2,4,6-triarylpyridines. Based on the above-optimized results, GO catalyzed reaction was carried out at 100 °C temperature under solvent-free condition and the results are summarized in Table 5. First, the compatibility of the substituents in the phenyl ring of acetophenone and benzaldehyde was examined. All the electron-donating and electron-withdrawing substituents on the aromatic ring are equally capable of producing the corresponding product with a good yield. However, aldehydes with electron-withdrawing groups (Table 5, entries 3, 4 and 9) exerted excellent yield and reacted faster than the aromatic aldehydes with electron-donating groups (Table 5, entries 2, 5, 7). In the case of heterocyclic aldehydes, the reaction has smoothly proceeded as can be seen from entry 6.
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | Product | Time (h) | Yieldb (%) |
| a Reaction condition: acetophenone (2 mmol), benzaldehyde (1 mmol), ammonium acetate (2 mmol) and GO (30 mg).b Isolated yields after purification through column chromatography on silica gel. | |||||
| 1 | 4-H | 4-H | 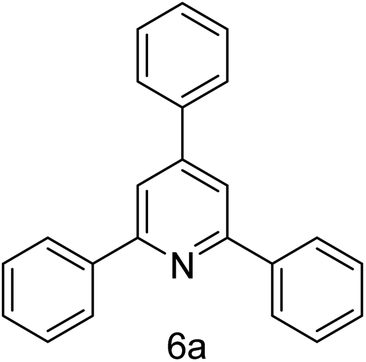 |
2 h | 92 |
| 2 | 4-H | 4-Me | 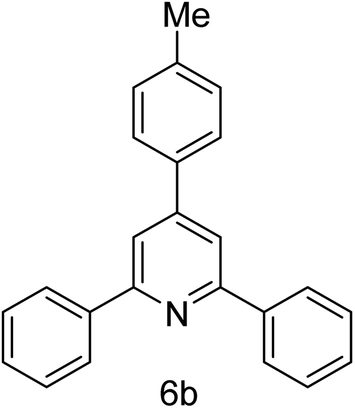 |
2 h | 86 |
| 3 | 4-H | 4-Cl | 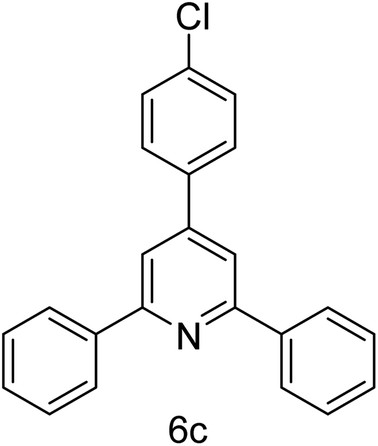 |
1 h | 93 |
| 4 | 4-H | 4-NO2 | 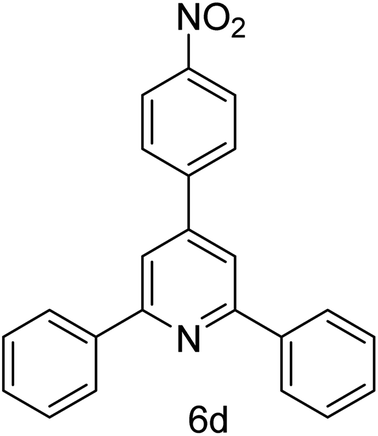 |
1 h | 88 |
| 5 | 4-H | 4-OMe | 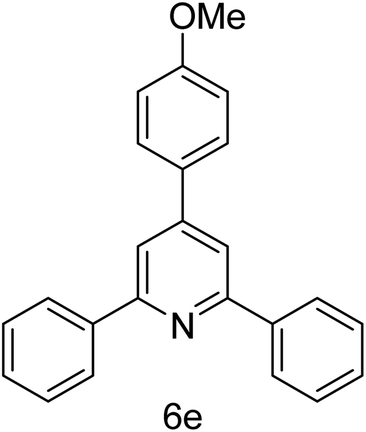 |
2 h | 83 |
| 6 | 4-H | Furan-2-carbaldehyde | 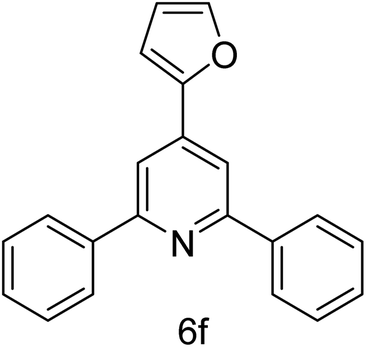 |
2 h | 78 |
| 7 | 4-Me | 4-H | 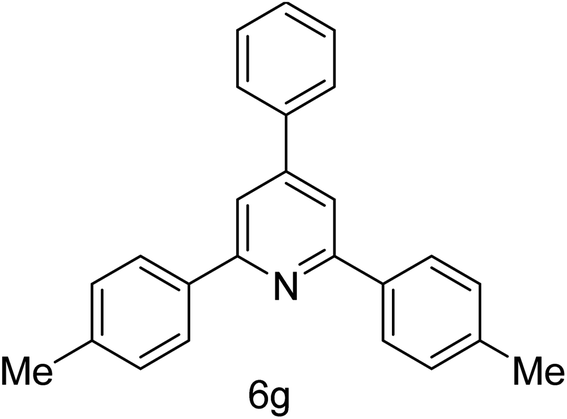 |
2 h | 87 |
| 8 | 4-Br | 4-H | 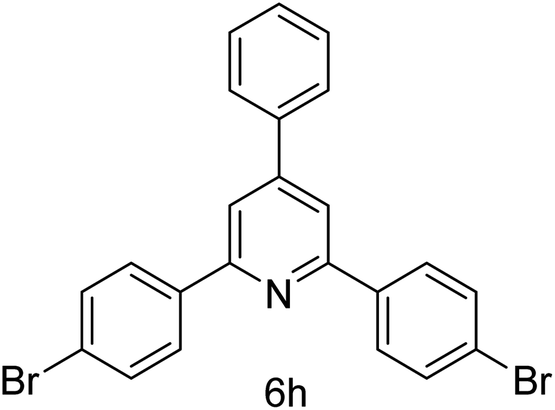 |
1 h | 90 |
| 9 | 4-Br | 4-Cl | 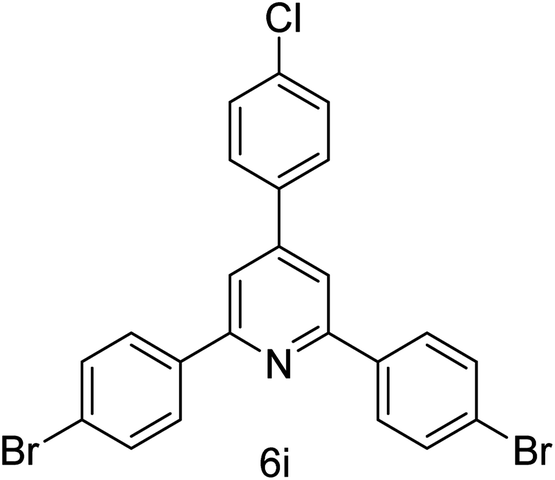 |
1 h | 94 |
The probable mechanism for the synthesis of 2,4,6-triarypyridines using GO is described in Scheme 2. At the very first step, aldol condensation occurs between acetophenone and aromatic aldehyde. Acetophenone is activated by the acidic group of GO and the nucleophilic attack occurs at the carbonyl carbon of aromatic aldehyde. After that, an acetophenone molecule is reacted with an ammonia source to form enamine (II). In the third stage, Michael's addition between enamine (II) and the aldol condensation product (I) occurs. GO protonates the condensation product (I), thereby facilitating the Michael addition by enamine (II). The intermediate (III) is formed by Michael's addition and undergoes cyclization to form dihydropyridine (V). At the last step, oxidation to dihydropyridine occurs and gives the ultimate product 2,4,6-triarylpyridine (VI).
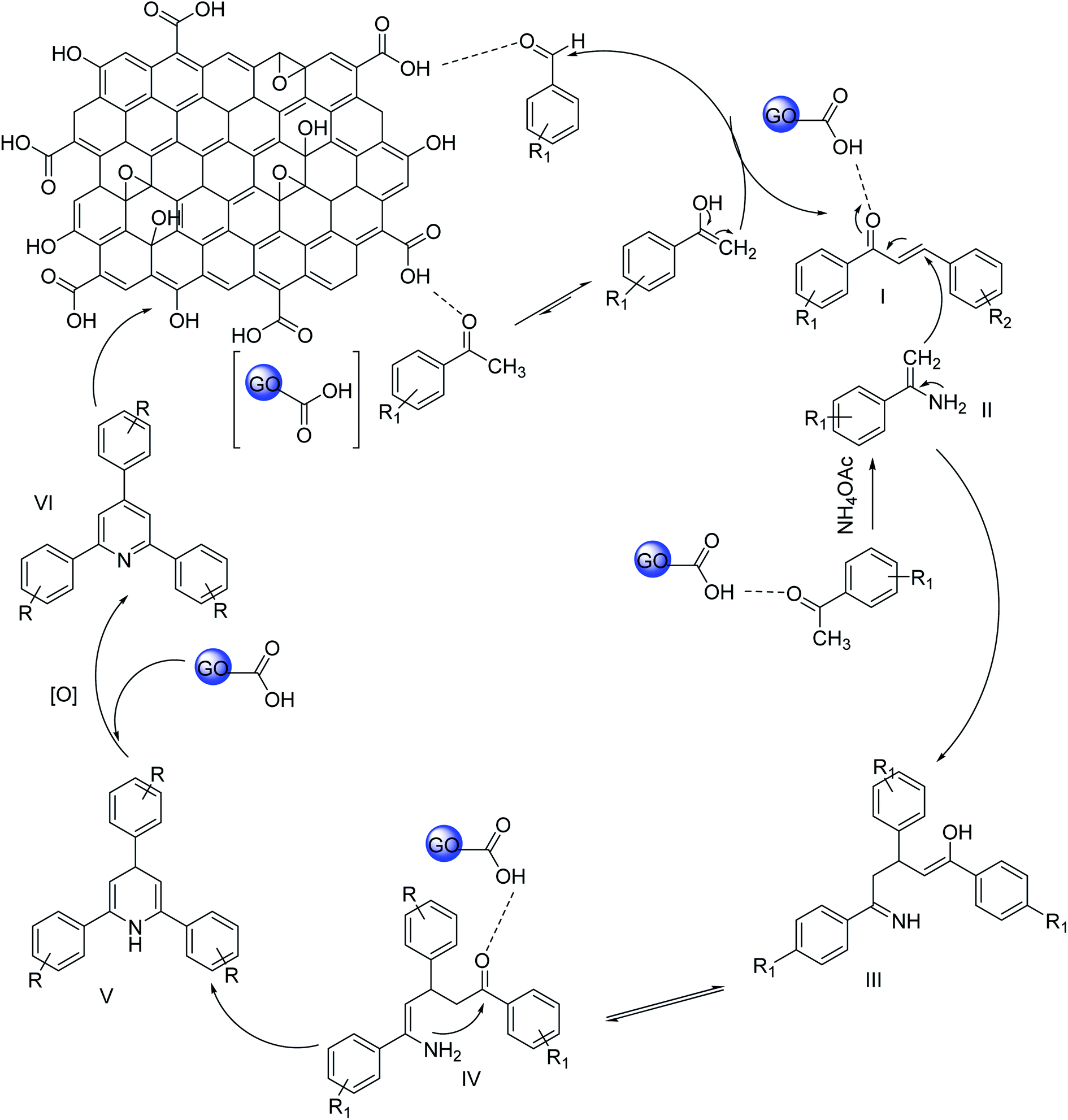 | ||
| Scheme 2 A possible route of GO catalyzed synthesis of 2,4,6-triarylpyridine. | ||
The main advantage of heterogeneous catalysts is their reusability in organic transformation. For this purpose, acetophenone, benzaldehyde, and ammonium acetate were taken in a reaction vial in presence of 120 mg of GO. The model reaction was carried out for an adequate time and after completion of the reaction, ethyl acetate (30 mL) was added into the reaction vial and centrifuged for four times. The supernatant liquid after centrifugation was decanted off and the residual catalyst was washed repeatedly with water and acetone. The dry GO was then collected and reused for the 2nd run. It was observed that GO could easily retain its acidic property without significant loss in its catalytic activity even after 5 successive runs (Fig. 7). Although there may be loss of some oxygenated groups due to subsequent runs, the recovered catalyst shows almost equal efficiency with the fresh GO.
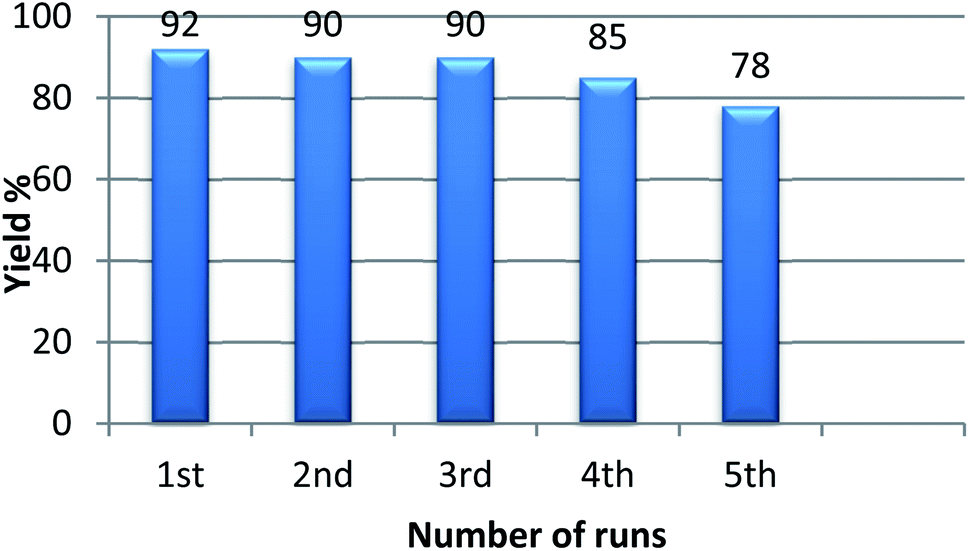 | ||
| Fig. 7 Recyclability experiment of catalyst GO for the synthesis of 2,4,6-triarylpyridines. | ||
Conclusion
In conclusion, carbocatalyst based metal-free catalytic pathway for the synthesis of 3,5-disubstituted 1,2,4-oxadiazoles and 2,4,6-triarylpyridines has been established. The solid acid catalyst, GO facilitates the synthesis of oxadiazoles and triarylpyridines with good yield, easy recovery, and under mild reaction conditions. The dual catalytic activity of GO has been demonstrated without any undesired by-product under benign conditions. The present protocol gives a clean strategy to provide a wide variety of substituted oxadiazoles and pyridines.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
PB is thankful to CSIR-New Delhi, India for the CSIR-Senior Research Fellowship [File no: 09/285(0072)/2016-EMR-I].References
- M. Carbone, Y. Li, C. Irace, E. Mollo, F. Castelluccio, D. A. Pascale, G. Cimino, R. Santamaria, W. Y. Guo and M. Gavagnin, Org. Lett., 2011, 13, 2516–2519 CrossRef CAS PubMed.
- A. V. Gulevich, A. S. Dudnik, N. Chernyak and V. Gevorgyan, Chem. Rev., 2013, 113, 3084–3213 CrossRef CAS PubMed.
- R. J. Mathvink, A. M. Barritta, M. R. Candelore, M. A. Cascieri, L. Deng, L. Tota, C. D. Strader, M. J. Wyvratt, M. H. Fisher and A. E. Weber, Bioorg. Med. Chem. Lett., 1999, 9, 1869–1874 CrossRef CAS PubMed.
- F. I. Carroll, J. L. Gray, P. Abrahm, M. A. Kuzemko, A. H. Lewin, J. W. Boja and M. J. Kuhar, J. Med. Chem., 1993, 36, 2886–2890 CrossRef CAS PubMed.
- J. W. Clitherow, P. Beswick, W. J. Irving, D. I. C. Scopes, J. C. Barnes, J. Clapham, J. D. Brown, D. J. Evans and A. G. Hayes, Bioorg. Med. Chem. Lett., 1996, 6, 833–838 CrossRef CAS.
- C. B. Vu, E. G. Corpuz, T. J. Merry, S. G. Pradeepan, C. Bartlett, R. S. Bohacek, M. C. Botfield, C. J. Eyermann, B. A. Lynch, I. A. MacNeil and M. K. Ram, J. Med. Chem., 1999, 42, 4088–4098 CrossRef CAS PubMed.
- J. Matsumoto, T. Takahashi, M. Agata, H. Toyofuku and N. Sasada, Jpn. J. Pharmacol., 1994, 65, 51–57 CrossRef CAS PubMed.
- B. L. Mylari, T. A. Beyer, P. J. Scott, C. E. Aldinger, M. F. Dee, T. W. Siegel and W. J. Zembrowski, J. Med. Chem., 1992, 35, 457–465 CrossRef CAS PubMed.
- B. S. Orlek, F. E. Blaney, F. Brown, M. S. G. Clark, M. S. Hadley, J. Hatcher, G. J. Riley, H. E. Rosenberg, H. J. Wadsworth and P. Wyman, J. Med. Chem., 1991, 34, 2726–2735 CrossRef CAS PubMed.
- T. Nakamura, M. Asano, Y. Sekiguchi, Y. Mizuno, K. Tamaki, F. Nara, Y. Kawase, Y. Yabe, D. Nakai, E. Kamiyama, Y. Urasaki-Kaneno, T. Shimozato, H. Doi-Komuro, T. Kagari, W. Tomisato, R. Inoue, M. Nagasaki, H. Yuita, K. Oguchi-Oshima, R. Kaneko and T. Nishi, Eur. J. Med. Chem., 2012, 51, 92–98 CrossRef CAS PubMed.
- B. Y. Kim, J. BokAhn, H. W. Lee, S. K. Kang, J. H. Lee, J. S. Shin, S. K. Ahn, C. Hong and S. S. Yoon, Eur. J. Med. Chem., 2004, 39, 433–447 CrossRef CAS PubMed.
- L. Tian, J. Song, J. Wang and B. Liu, Chin. Chem. Lett., 2009, 20, 288–291 CrossRef CAS.
- R. D. Allen and G. A. R Johnston, Med. Res. Rev., 1983, 3, 91–118 CrossRef PubMed.
- E. C. Constable, C. E. Housecroft, M. Neuburger, D. Phillips, P. R. Raithby, E. Schofield, E. Sparr, D. A. Tocher, M. Zehnder and Y. Zimmermann, Dalton Trans., 2000, 13, 2219–2228 RSC.
- E. Figgemeier, E. C. Constable, C. E. Housecroft and Y. C. Zimmermann, Langmuir, 2004, 20, 9242–9248 CrossRef CAS PubMed.
- (a) Y. Wang, R. L. Miller, D. R. Sauer and S. W. Djuric, Org. Lett., 2005, 7, 925–928 CrossRef CAS PubMed; (b) S. Kandre, P. R. Bhagat, R. Sharma and A. Gupte, Tetrahedron Lett., 2013, 54, 3526–3529 CrossRef CAS; (c) B. Kaboudin and L. Malekzadeh, Tetrahedron Lett., 2011, 52, 6424–6426 CrossRef CAS; (d) B. Kaboudin and F. Saadati, Tetrahedron Lett., 2007, 48, 2829–2832 CrossRef CAS.
- J. K. Augustine, V. Akabote, S. G. Hegde and P. Alagarsamy, J. Org. Chem., 2009, 74, 5640–5643 CrossRef CAS PubMed.
- D. S. Bolotin, K. I. Kulish, N. A. Bokach, G. L. Starova, V. V. Gurzhiy and V. Y. Kukushkin, Inorg. Chem., 2014, 53, 10312–10324 CrossRef CAS PubMed.
- (a) M. Okimoto and Y. Takahashi, Bull. Chem. Soc. Jpn., 2003, 76, 427–428 CrossRef CAS; (b) D. B. Repke, H. P. Albrecht and J. G. Moffat, J. Org. Chem., 1975, 40, 2481–2487 CrossRef CAS PubMed.
- W. Wang, H. Xu, Y. Xu, T. Ding, W. Zhang, Y. Ren and H. Chang, Org. Biomol. Chem., 2016, 14, 9814–9822 RSC.
- F. Saadati, B. Kaboudin, R. Hasanloei, Z. Namazifar, X. Marset and G. Guillena, Appl. Organomet. Chem., 2020, 34, 5838–5849 CrossRef.
- M. Adib, A. H. Jahromi, N. Tavoosi, M. Mahdavi and H. R. Bijanzadeh, Tetrahedron Lett., 2006, 47, 2965–2967 CrossRef CAS.
- N. Montazeri and S. Mahjoob, Chin. Chem. Lett., 2012, 23, 419–422 CrossRef CAS.
- J. Safari, S. Gandomi-Ravandi and M. Borujeni, Chem. Sci., 2013, 125, 1063–1070 CrossRef CAS.
- M. M. Heravi, K. Bakhtiari, Z. Daroogheha and F. F. Bamoharram, Catal. Commun., 2007, 8, 1991–1994 CrossRef CAS.
- L. Nagarapu, A. R. Peddiraju and S. Apuri, Catal. Commun., 2007, 8, 1973–1976 CrossRef CAS.
- A. Davoodnia, M. Bakavoli, R. Moloudi, N. Tavakoli-Houseini and M. Khashi, Monatsh. Chem., 2010, 141, 867–870 CrossRef CAS.
- A. Maleki and R. Firouzi-Haji, Sci. Rep., 2018, 8, 17303–17311 CrossRef PubMed.
- E. Tabrizian, A. Amoozadeh, S. Rahmani, E. Imanifar, S. Azhari and M. Malmir, Chin. Chem. Lett., 2015, 26, 1278–1282 CrossRef CAS.
- G.-L. Wu and Q.-P. Wu, Adv. Synth. Catal., 2018, 360, 1949–1953 CrossRef CAS.
- R. Mishra, A. Jana, A. K. Panday and L. H. Choudhury, Org. Biomol. Chem., 2018, 16, 3289–3302 RSC.
- C.-Y. Chen, W.-P. Hu, P.-C. Yan, G. C. Senadi and J.-J. Wang, Org. Lett., 2013, 15, 6116–6119 CrossRef CAS PubMed.
- C. C. Răzvan, R. Eelco and V. A. O. Romano, Green Chem., 2014, 16, 2958–2975 RSC.
- D. S. Su, S. Perathoner and G. Centi, Chem. Rev., 2013, 113, 5782–5816 CrossRef CAS PubMed.
- O. Mohammadi, M. Golestanzadeh and M. Abdouss, New J. Chem., 2017, 41, 11471–11497 RSC.
- N. Oger, Y. F. Lin, E. L. Grognec, F. Rataboul and F. X. Felpin, Green Chem., 2016, 18, 1531–1537 RSC.
- D. R. Dreyer and C. W. Bielawski, Chem. Sci., 2011, 2, 1233–1240 RSC.
- S. Navalon, A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Chem. Rev., 2014, 114, 6179–6212 CrossRef CAS PubMed.
- S. Zhu, J. Wang and W. Fan, Catal.: Sci. Technol., 2015, 5, 3845–3858 RSC.
- C. Su and K. P. Loh, Acc. Chem. Res., 2013, 46, 2275–2285 CrossRef CAS PubMed.
- (a) D. R. Dreyer, H. P. Jia and C. W. Bielawski, Angew. Chem., Int. Ed., 2010, 49, 6813–6816 CAS; (b) D. R. Dreyer, H. P. Jia, A. D. Todd, G. Jeng and C. W. Bielawski, Org. Biomol. Chem., 2011, 9, 7292–7295 RSC.
- T. Szabo, E. Tombacz, E. Illes and I. Dekany, Carbon, 2006, 44, 537–545 CrossRef CAS.
- H. P. Jia, D. R. Dreyer and C. W. Bielawski, Tetrahedron, 2011, 67, 4431–4434 CrossRef CAS.
- H. Huang, J. Huang, Y. M. Liu, H. Y. He, Y. Cao and K. N. Fan, Green Chem, 2012, 14, 930–934 RSC.
- G. A. B. Gonçalves, S. M. G. Pires, M. M. Q. Simoes, M. G. P. M. S. Neves and P. A. A. P. Marques, Chem. Commun., 2014, 50, 7673–7676 RSC.
- X. Chu, Q. Zhu, W.-L. Dai and K. Fan, RSC Adv., 2012, 2, 7135–7139 RSC.
- G. Lv, H. Wang, Y. Yang, T. Deng, C. Chen, Y. Zhu and X. Hou, ACS Catal., 2015, 5, 5636–5646 CrossRef CAS.
- T. A. J. Siddiqui, B. G. Ghule, S. Shaikh, P. V. Shinde, K. C. Gunturu, P. K. Zubaidha, J. M. Yun, C. O. Dwyer, R. S. Mane and K. H. Kim, RSC Adv., 2018, 8, 17373–17379 RSC.
- B. Roy, D. Sengupta and B. Basu, Tetrahedron Lett., 2014, 55, 6596–6600 CrossRef CAS.
- R. Wang, Z. Wu, Z. Qin, C. Chen, H. Zhu, J. Wu, G. Chen, W. Fan and J. Wang, Catal.: Sci. Technol., 2016, 6, 993–997 RSC.
- S. Bhattacharya, P. Ghosh and B. Basu, Tetrahedron Lett., 2018, 59, 899–903 CrossRef CAS.
- Z. Chen, Y. Wen, Y. Fu, H. Chen, M. Ye and G. Luo, Synlett, 2017, 28, 981–985 CrossRef CAS.
- J. Porwal, N. Karanwal, S. Kaul and S. L. Jain, New J. Chem., 2016, 40, 1547–1553 RSC.
- H. P. Mungse, N. Bhakuni, D. Tripathi, O. P. Sharma, B. Sain and O. P. Khatri, J. Phys. Org. Chem., 2014, 27, 944–951 CrossRef CAS.
- K. B. Dhopte, R. S. Zambare, A. V. Patwardhan and P. R. Nemade, RSC Adv., 2016, 6, 8164–8172 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: The scanned copies of 1H and 13C NMR are included in this section. See DOI: 10.1039/d1ra06331f |
| This journal is © The Royal Society of Chemistry 2021 |