
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhase relations, and mechanical and electronic properties of nickel borides, carbides, and nitrides from ab initio calculations†
Nursultan E. Sagatov *ab,
Aisulu U. Abuova*b,
Dinara N. Sagatovaac,
Pavel N. Gavryushkinac,
Fatima U. Abuovab and
Konstantin D. Litasovd
*ab,
Aisulu U. Abuova*b,
Dinara N. Sagatovaac,
Pavel N. Gavryushkinac,
Fatima U. Abuovab and
Konstantin D. Litasovd
aSobolev Institute of Geology and Mineralogy, Siberian Branch of the Russian Academy of Sciences, Novosibirsk 630090, Russian Federation. E-mail: sagatinho23@gmail.com
bL. N. Gumilyov Eurasian National University, Nur-Sultan 010008, Republic of Kazakhstan. E-mail: aisulu-us1980@yandex.ru
cNovosibirsk State University, Novosibirsk 630090, Russian Federation
dVereshchagin Institute for High Pressure Physics, Russian Academy of Sciences, Troitsk, Moscow 108840, Russian Federation
First published on 15th October 2021
Abstract
Based on density functional theory and the crystal structure prediction methods, USPEX and AIRSS, stable intermediate compounds in the Ni–X (X = B, C, and N) systems and their structures were determined in the pressure range of 0–400 GPa. It was found that in the Ni–B system, in addition to the known ambient-pressure phases, the new nickel boride, Ni2B3-Immm, stabilizes above 202 GPa. In the Ni–C system, Ni3C-Pnma was shown to be the only stable nickel carbide which stabilizes above 53 GPa. In the Ni–N system, four new phases, Ni6N-R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) , Ni3N-Cmcm, Ni7N3-Pbca, and NiN2-Pa, were predicted. For the new predicted phases enriched by a light-element, Ni2B3-Immm and NiN2-Pa, mechanical and electronic properties have been studied.
, Ni3N-Cmcm, Ni7N3-Pbca, and NiN2-Pa, were predicted. For the new predicted phases enriched by a light-element, Ni2B3-Immm and NiN2-Pa, mechanical and electronic properties have been studied.
Introduction
Transition metal borides, carbides, and nitrides are of interest in terms of materials science due to the relatively high bulk moduli, ultra-high melting points, and high chemical stabilities. This combination of properties makes these materials highly desirable for a number of industrial applications. Specifically, TMX, TMX2, and TMX4 (X = B, C, and N; TM-transition metal) possess comparable bulk moduli to that of superhard cubic-BN (400 GPa), such as ReB2, WB, OsC, RuC, WC, OsN2, PtN2, and WN,1–3 and some of them have been put to the test and proved to be superhard materials, such as MoN, WB4, and FeB4.4–6 High pressure enables not just the production of new polymorphic modifications of known borides, carbides, and nitrides but also the synthesis of new compounds having stoichiometries not accessible at ambient pressure.7 Therefore, recently, the number of theoretical and experimental studies devoted to the search for stable TM–X (X = B, C, and N) compounds has increased. The study of borides, carbides, and nitrides has been carried out practically for all IV group transition metals, with the exception of nickel and copper. For instance, there are many works devoted to the crystal structure predictions and high pressure syntheses of Fe–B,6,8,9 Fe–C,10–12 and Fe–N13–16 compounds.In the present paper, we investigate nickel borides, carbides, and nitrides under high pressure using modern ab initio crystal structure prediction methods. The Ni–B system comprises several stable (NiB, Ni4B3, Ni2B, and Ni3B) and metastable (Ni23B6 and Ni7B3) crystalline compounds.17–19 Recently, Wang et al.20 estimated the stability of ambient-pressure phases up to 100 GPa. They showed that Ni3B and Ni4B3 are unstable at pressures of 80–100 GPa, while Ni2B and NiB remains stable relative to the decomposition into isochemical mixture. In the Ni–C system, there is no stable intermediate compounds at ambient pressure. Recently, first stable nickel carbide, Ni3C with cementite-type structure, was synthesized in a diamond anvil cell (DAC) with laser heating through a direct reaction of a nickel powder with carbon at 184 GPa.21 In the Ni–N system, there is one known stable intermediate compound, Ni3N, at ambient pressure. Guillaume et al.22 show that the ambient-pressure structure of Ni3N remains stable at 20 GPa. Recently, nickel pernitride (NiN2) with marcasite-type structure was synthesized at 40 GPa using a laser-heated DAC.23 It should be noted that no crystal structure prediction calculations were performed for these systems.
In our work, we search for new stable structures and study the phase stability, electronic and mechanical properties of the structures in the Ni–B, Ni–C, and Ni–N systems within first-principles calculations.
Computational methods
The search for new stable crystal structures of nickel borides, carbides, and nitrides were performed using evolutionary algorithms implemented in the USPEX package24–26 and the random sampling method implemented in the AIRSS software27,28 at 50, 100, 200, 300, and 400 GPa.The search with USPEX was performed in variable composition mode with 6–36 atoms per unit cell. The size of the first generation in the calculations was equal to 65 structures. 60% of the structures with the lowest enthalpy were selected after the optimization and then used to produce the next generation. A new generation was produced as follow: 35% of all structures were generated by heredity, 20% – by atomic mutation, 10%—by lattice permutation, and 35%—randomly. In average, 36–40 generations have been produced and relaxed at each pressure. Using AIRSS, about 3500–4000 structures were randomly generated and optimized for compounds with fixed stoichiometries (NiXn; X = B, C, and N; n = 1–4) at each pressure, and those with the lowest enthalpy were selected.
The total energies and forces were calculated by solving the Schrödinger equation based on projector augmented plane-wave implementation of density functional theory (DFT) using the VASP package.29,30 Exchange correlation effects were treated in the generalized gradient approximation (GGA) with Perdew–Bürke–Ernzerhof scheme.31
In all crystal structure prediction calculations, medium-quality optimization was performed using the conjugate gradient. The medium quality settings were as follows: plane-wave cutoff energy—420 eV; Monkhorst–Pack k-point sampling grid of spacing—0.5 Å−1; Gaussian smearing with parameter σ = 0.15 eV. The most promising predicted structures were then optimized at various pressures with higher accuracy: the cutoff energy—700 eV, k-point sampling grid of spacing—0.2 Å−1, and σ = 0.05 eV.
To investigate mechanical properties, static elastic stiffness tensor (Cij) was calculated from the stress (σ) – strain (ε) relation σi = Cijεj.
To investigate electronic properties, in addition to the standard PBE, Dudarev's GGA + U (ref. 32) and hybrid functional Heyd–Scuseria–Ernzerhof (HSE06)33 methods were used.
Structures were visualized in the VESTA package,34 and symmetries of the predicted structures were analyzed using the FindSym program.35
Results and discussion
Crystal structure prediction
The thermodynamic stability of the most favorable structures among the predicted ones was estimated using the convex hull diagrams, which give the convenient graphical representation of the thermodynamic stability of the predicted structures.Crystal structure prediction calculations have been performed only for the intermediate compounds in the Ni–X (X = B, C, and N) systems. Crystal structure prediction calculations of pure Ni performed earlier by similar procedure showed its stability in Fmm (fcc) structure up to 400 GPa.36,37 For pure boron, carbon, and nitrogen the following data have been used: B-Rm, -Pnnm, -Cmca, and -P63/mcm;38 C-P63mc (graphite) and -Fdm (diamond); N-Pa, -I213,39, -Pba2,40 and -I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3m.41 The pressures of phase transition between listed polymorphic modifications are shown in Fig. 1b–3b.
3m.41 The pressures of phase transition between listed polymorphic modifications are shown in Fig. 1b–3b.
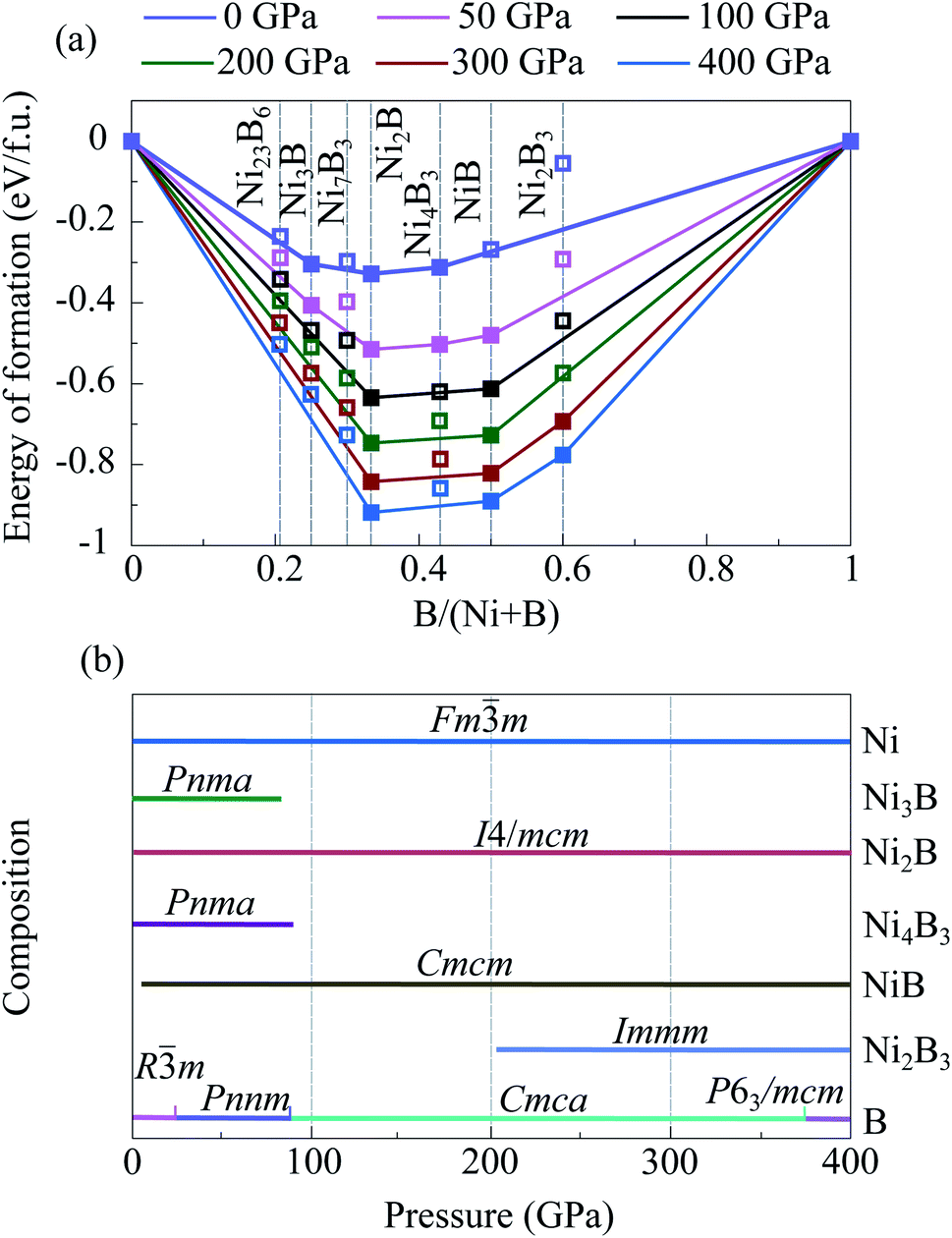 | ||
| Fig. 1 Thermodynamic convex hulls of Ni–B system at various pressures and zero temperature (a). Filled symbols denote stable structures, open symbols—metastable structures. Predicted pressure–composition phase diagram for the Ni–B system (b). | ||
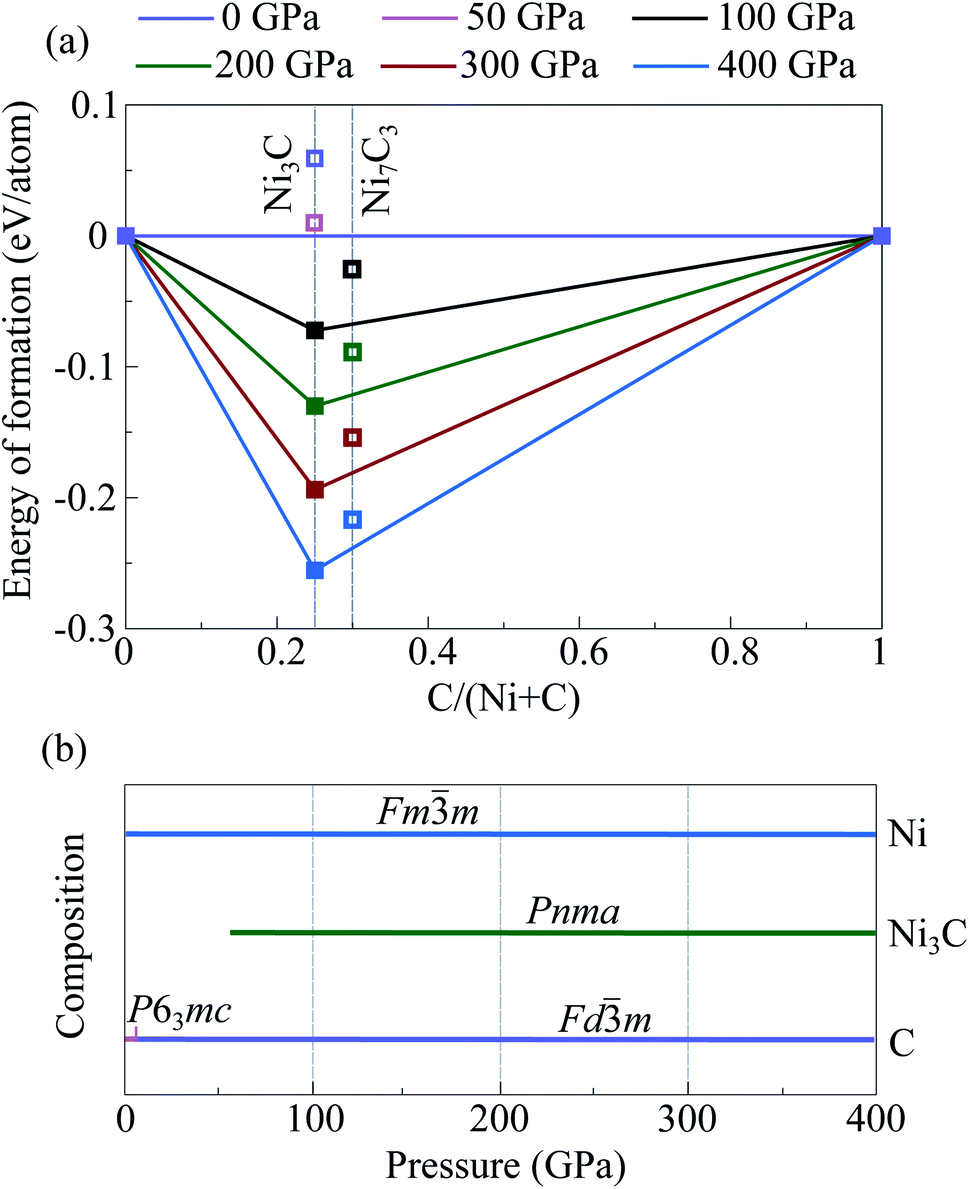 | ||
| Fig. 2 Stability convex hulls of the Ni–C system at various pressures and zero temperature (a). Filled symbols denote stable structures, open symbols—metastable structures. Predicted pressure–composition phase diagram for the Ni–C system (b). | ||
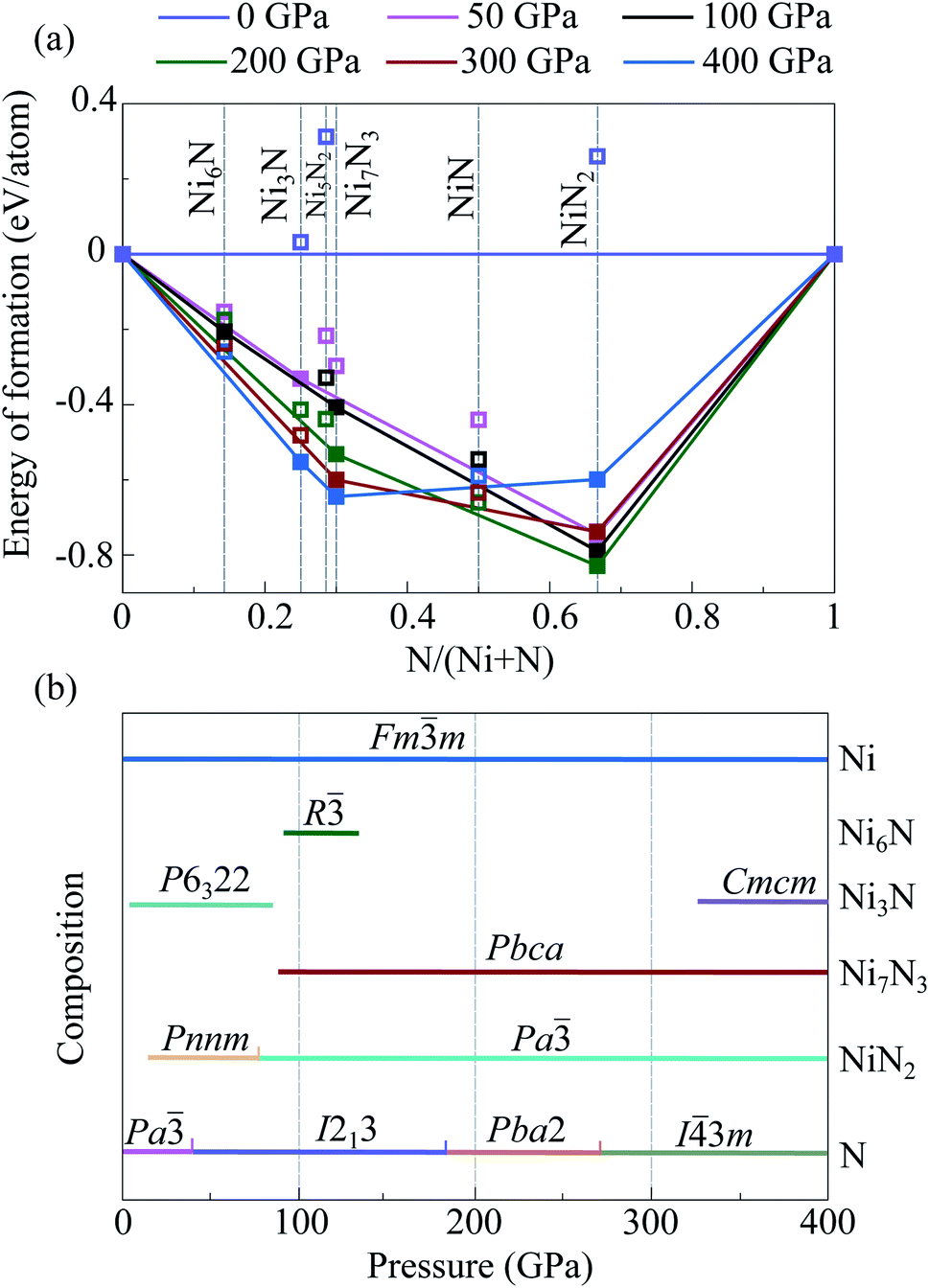 | ||
| Fig. 3 Stability convex hulls of the Ni–N system at various pressures and zero temperature (a). Filled symbols denote stable structures, open symbols—metastable structures. Predicted pressure–composition phase diagram for the Ni–N system (b). | ||
The Ni3B-Pnma phase was predicted at 50 GPa and it is stable from ambient pressure to 80 GPa. Above 80 GPa, Ni3B-Pnma decomposes into solid Ni and Ni2B (Fig. 1a and Table S1†). For Ni2B, the structure with the I4/mcm symmetry was predicted at 50, 100, 200, 300, and 400 GPa as the lowest-enthalpy one. This structure is stable in the entire considered pressure range without any phase transitions (Fig. 1b). The Ni4B3-Pnma was predicted at 50 GPa as the most energetically favorable. It is stable from 0 to 86 GPa, and above this pressure decomposes into the mixture of (Ni2B + NiB). The NiB was predicted in the Cmcm structure, which stabilizes at a pressure of 2 GPa and remains stable up to 400 GPa.
In the B-rich part of the Ni–B system, we predicted a new compound that was not previously known, namely Ni2B3 (Fig. 1 and Table S1†). This compound was found at 200, 300, and 400 GPa with Immm symmetry. Above 202 GPa, the enthalpy of Ni2B3-Immm becomes lower than that of mechanical mixture of (2NiB + B), indicating that the Ni2B3-Immm structure stabilizes relative to the decomposition reaction Ni2B3 ↔ 2NiB + B (Fig. S2a†).
All these Ni–B structures are dynamically stable, which were confirmed by the calculated phonon spectra (Fig. S3†).
Although the search of the structures stable at ambient pressure has not been performed, the comparison of the found structures with the known ones show, that all the structures stable at ambient pressure, which were observed in experiments, were revealed in our calculations at 50 GPa.42,43 In addition, we have considered Ni23B6,18 observed in experiments, and Ni7B3 (ref. 19) suggested theoretically. According to our calculations, Ni23B6 is metastable at 0 GPa. This is in agreement with experimental results.18 For Ni7B3, we considered two structures: Ni7B3-P63mc suggested in ref. 19 and Ni7B3-Pbca constructed from Fe7C3-Pbca,44 replacing Fe by Ni and C by B atoms. These structures are very similar and their distinguishing by X-ray powder diffraction technique is problematic. Our calculation showed that Ni7B3-Pbca has lower enthalpy than Ni7B3-P63mc in the pressure range of 0–155 GPa (Fig. S4a†). Based on this, we suggest that most likely the Pbca phase was formed in experiments of Hofmann et al.,19 than hexagonal phase suggested in that work. With pressure increase, Ni23B6 and Ni7B3 becomes even less favorable (Fig. 1).
In addition, we constructed Ni-analogs of stable iron carbide structures Fe7C3-P63mc,45 Fe7C3-Pbca,44 and Fe2C-Pnma,12 by replacing all iron atoms by nickel ones. According to the obtained results, constructed Ni7C3-Pbca is more favorable than constructed Ni7C3-P63mc (Fig. S4b†). Nevertheless, Ni7C3-Pbca is unstable relative to the mechanical mixtures of (Ni + C) or (Ni3C + C) in the entire considered pressure range (Fig. 2). Similar calculations for Ni2C-Pnma showed that this compound is not stable, and the energy difference reaches 0.4 eV f.u−1.
Crystal structure predictions in C-rich part of the Ni–C system were performed for fixed stoichiometries, namely NiC4, NiC3, NiC2, and NiC. For all these compounds the lowest-enthalpy structures are unstable and decompose into isochemical mixture in the entire considered pressure range.
Performed crystal structure prediction calculations show that there are two stable intermediate compounds in the Ni–N system at 50 GPa, Ni3N and NiN2 (Fig. 3 and Table S3†). Both predicted Ni3N-P6322 and NiN2-Pnnm were observed earlier in high-pressure experiments.22,23 According to obtained results, Ni3N-P6322 is stable in the pressure range of 4–96 GPa. Below 4 GPa, Ni3N-P6322 is unstable relative to mechanical mixture of (3Ni + N), and above 96 GPa it is unstable relative to mechanical mixture of (Ni7B3 + Ni). NiN2-Pnnm stabilizes above 14 GPa. At 100 GPa, there are three stable nickel nitrides, predicted Ni6N-R and NiN2-Pa, and constructed Ni7N3-Pbca. Ni6N-R stabilizes at 98 GPa, and above 114 GPa decomposes into solid Ni and Ni7N3. Constructed Ni7N3-Pbca is more favorable than constructed Ni7N3-P63mc in the entire pressure range (Fig. S2†). Ni7N3-Pbca become stable relative to Ni3N + NiN2 above 93 GPa and stable at least up to 400 GPa. NiN2 undergoes a phase transition from known NiN2-Pnnm to the new predicted NiN2-Pa at 96 GPa. NiN2-Pa is stable at least up to 400 GPa. At 400 GPa, in addition to Ni7N3-Pbca and NiN2-Pa, one new nitride Ni3N-Cmcm was predicted. It stabilizes relative to the mechanical mixture of (Ni7N3 + Ni) above 331 GPa. Phonon calculations show that the predicted nickel nitrides are dynamically stable; i.e., they have no imaginary modes in the phonon spectra (Fig. S6†).
Crystal chemistry of the predicted structures
Structural data of the new predicted compounds are given in Table 1.| Compound | P (GPa) | Space group | Lattice parameters (Å, deg) | Atom | Coordinates | ||||
|---|---|---|---|---|---|---|---|---|---|
| x | y | z | |||||||
| Ni2B3 | 250 | Immm (#71) | a = 2.695 | b = 3.357 | c = 5.622 | Ni1 | 0.000 | 0.000 | 0.804 |
| α = 90.00 | β = 90.00 | γ = 90.00 | B1 | 0.500 | 0.000 | 0.500 | |||
| B2 | 0.000 | 0.251 | 0.500 | ||||||
| Ni6N | 100 | R (#148) |
a = 4.058 | b = 4.058 | c = 11.653 | Ni1 | 0.316 | 0.331 | 0.246 |
| α = 90.00 | β = 90.00 | γ = 120.00 | N1 | 0.000 | 0.000 | 0.000 | |||
| Ni7N3 | 100 | Pbca (#61) | a = 4.131 | b = 12.671 | c = 10.978 | Ni1 | −0.080 | 0.534 | 0.686 |
| α = 90.00 | β = 90.00 | γ = 90.00 | Ni2 | 0.744 | 0.878 | 0.585 | |||
| Ni3 | 0.411 | 0.622 | 0.626 | ||||||
| Ni4 | 0.204 | 0.779 | 0.522 | ||||||
| Ni5 | 0.251 | 0.873 | 0.702 | ||||||
| Ni6 | −0.092 | 0.718 | 0.693 | ||||||
| Ni7 | 0.249 | −0.037 | 0.521 | ||||||
| N1 | −0.004 | −0.017 | 0.656 | ||||||
| N2 | −0.004 | 0.632 | 0.561 | ||||||
| N3 | 0.499 | 0.765 | 0.648 | ||||||
| Ni3N | 300 | Cmcm (#63) | a = 2.295 | b = 7.492 | c = 5.541 | Ni1 | 0.000 | 0.060 | 0.250 |
| α = 90.00 | β = 90.00 | γ = 90.00 | Ni2 | 0.000 | 0.637 | −0.059 | |||
| N1 | 0.000 | 0.741 | 0.250 | ||||||
| NiN2 | 200 | Pa (#205) |
a = 4.054 | b = 4.054 | c = 4.054 | Ni1 | 0.000 | 0.000 | 0.000 |
| α = 90.00 | β = 90.00 | γ = 90.00 | N1 | 0.592 | 0.592 | 0.592 | |||
The most crystallochemically interesting structures are the structures of compounds enriched by the light element. We have revealed two such compounds, NiN2 and Ni2B3. Two found structures of NiN2, NiN2-Pa, corresponds to the well-known structural type of pyrite with N–N molecular units.
The crystal structure of Ni2B3 is also characterised by the covalently bonded units. In this case, these are infinite (001) layers of boron atoms. In the layer each B atom is connected to four other B atoms forming the net with hexagonal and rhombic loops (Fig. 4a). The B–B bond distance within the layer is 1.67 Å at 250 GPa. The elemental boron is characterised by nearly the same B–B distance, 1.7 Å. The covalent nature of B–B bond in Ni2B3 structure is also confirmed by the electronic density distribution (Fig. S8†). Boron layers alternates with layers of Ni atoms, forming rectangular centered network (Fig. 4b). Each Ni is coordinated by 10 boron atoms, the six of which are from the top, and four are from the bottom layer. The performed analysis of the structures with the similar stoichiometry has revealed another structure, W2CoB2, belonging to the described structural type.47,48
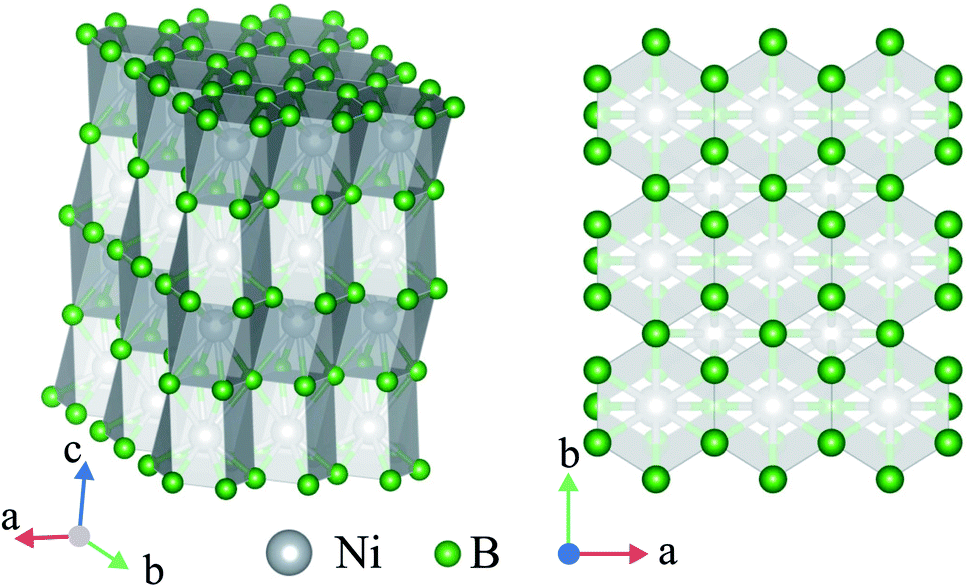 | ||
| Fig. 4 Crystal structure of Ni2B3-Immm in two projections. | ||
Graphical representation of the Ni6N-R, Ni3N-Cmcm, Ni7N3-Pbca, and NiN2-Pa crystal structures are shown in Fig. S7.†
Elastic properties
Transition metal borides, carbides, and nitrides often exhibit extraordinary mechanical properties. Elastic constants are helpful to understand the mechanical properties and provide useful information to estimate the hardness of a structure. To that end, we investigate the elastic properties of new predicted compounds enriched by a light-element, NiN2-Pa and Ni2B3-Immm, within the strain–stress method in combination with the DFT. Calculated elastic constants of NiN2-Pa and Ni2B3-Immm are listed in Table 2. From Table 2 and Fig. 5, it could be seen that the elastic constants Cij for these structures are all positive, increase monotonically with increasing pressure, and the whole set of the elastic constants of the structures satisfy the mechanical stability criteria.49 This means that NiN2-Pa and Ni2B3-Immm are mechanically stable in the considered pressure range.
and Ni2B3-Immm at different pressures
| Phase | P (GPa) | C11 | C12 | C13 | C22 | C23 | C33 | C44 | C55 | C66 |
|---|---|---|---|---|---|---|---|---|---|---|
| NiN2-Pa |
100 | 1311.6 | 326.4 | 326.2 | 1311.5 | 326.5 | 1311.6 | 204.8 | 204.8 | 204.8 |
| 200 | 1887.7 | 609.7 | 609.5 | 1887.7 | 609.7 | 1887.7 | 243.5 | 243.5 | 243.5 | |
| 300 | 2363.3 | 891.5 | 891.3 | 2363.3 | 891.5 | 2363.3 | 263.1 | 263.1 | 263.1 | |
| 400 | 2787.9 | 1175.5 | 1175.2 | 2787.9 | 1175.5 | 2787.9 | 272.8 | 272.8 | 272.8 | |
| Ni2B3-Immm | 200 | 1372.3 | 831.2 | 724.6 | 1354.5 | 774.6 | 1429.9 | 305.3 | 169.8 | 493.4 |
| 300 | 1773.5 | 1114.3 | 973.2 | 1750.0 | 1045.2 | 1895.3 | 400.4 | 239.6 | 643.6 | |
| 400 | 2153.0 | 1389.9 | 1218.3 | 2126.0 | 1307.1 | 2336.1 | 486.2 | 297.1 | 780.4 |
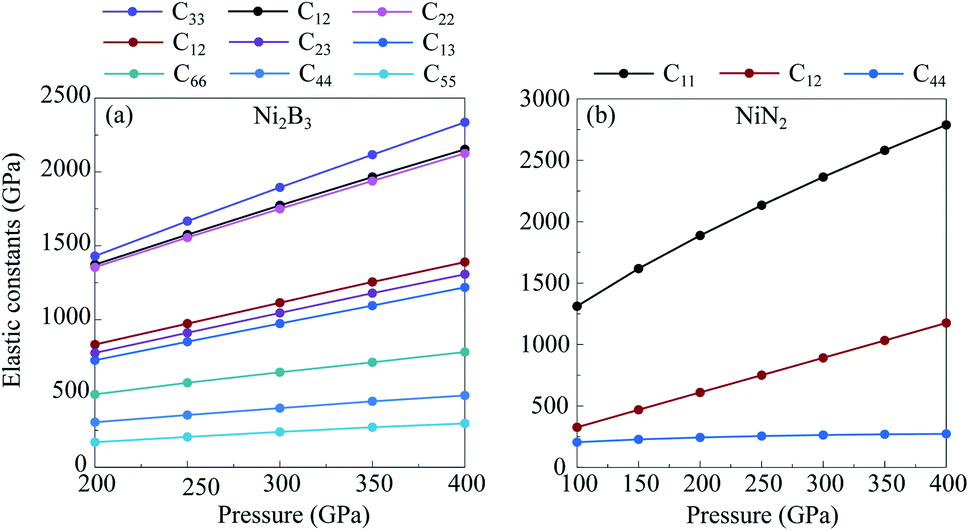 | ||
| Fig. 5 Calculated elastic constants of Ni2B3-Immm (a) and NiN2-Pa (b) as functions of pressure. | ||
Calculated bulk modulus B of NiN2-Pa and Ni2B3-Immm are ∼87% and ∼82% of that estimated for diamond, respectively, while shear modulus G of NiN2-Pa and Ni2B3-Immm are ∼35% and ∼38% of that estimated for diamond, respectively (Table 3). Predicted NiN2-Pa and Ni2B3-Immm are ductile materials since their B/G values (Table 3) are greater than 1.75.50
and Ni2B3-Immm at different pressures. For comparison, the data53 on diamond are presented
| Phase | P (GPa) | B | G | B/G | HChenV | HTianV |
|---|---|---|---|---|---|---|
| NiN2-Pa |
100 | 654.8 | 293.6 | 2.23 | 18.7 | 20.6 |
| 200 | 1035.7 | 362.7 | 2.86 | 15.4 | 18.1 | |
| 300 | 1382.1 | 403.2 | 3.43 | 12.8 | 15.8 | |
| 400 | 1712.9 | 428.6 | 4.00 | 10.7 | 13.9 | |
| Ni2B3-Immm | 200 | 979.7 | 297.5 | 3.29 | 10.9 | 13.4 |
| 300 | 1298.1 | 388.1 | 3.34 | 12.9 | 15.9 | |
| 400 | 1604.8 | 467.7 | 3.43 | 14.2 | 17.6 | |
| Diamond53 | 100 | 838 | 848 | — | — | — |
| 200 | 1188 | 1023 | — | — | — | |
| 300 | 1521 | 1168 | — | — | — | |
| 400 | 1839 | 1295 | — | — | — |
In order to investigate the hardness of NiN2-Pa and Ni2B3-Immm at various pressures we employ empirical Chen51 and Tian et al.52 models. The Vickers hardness of NiN2-Pa and Ni2B3-Immm are estimated to be 19 GPa and 12 GPa, respectively. The hardness of NiN2-Pa decreases with increasing pressure, while that of Ni2B3-Immm increases with increasing pressure. The estimated HV of both predicted structures are below minimal criteria of 20 GPa of hardness. Therefore, NiN2-Pa and Ni2B3-Immm are not classified as a hard materials.
Electronic structure
To investigate electronic characteristics of NiN2-Pa and Ni2B3-Immm, we calculate their total/partial electronic density of states (DOS) (Fig. 6) and band structures (Fig. 7).
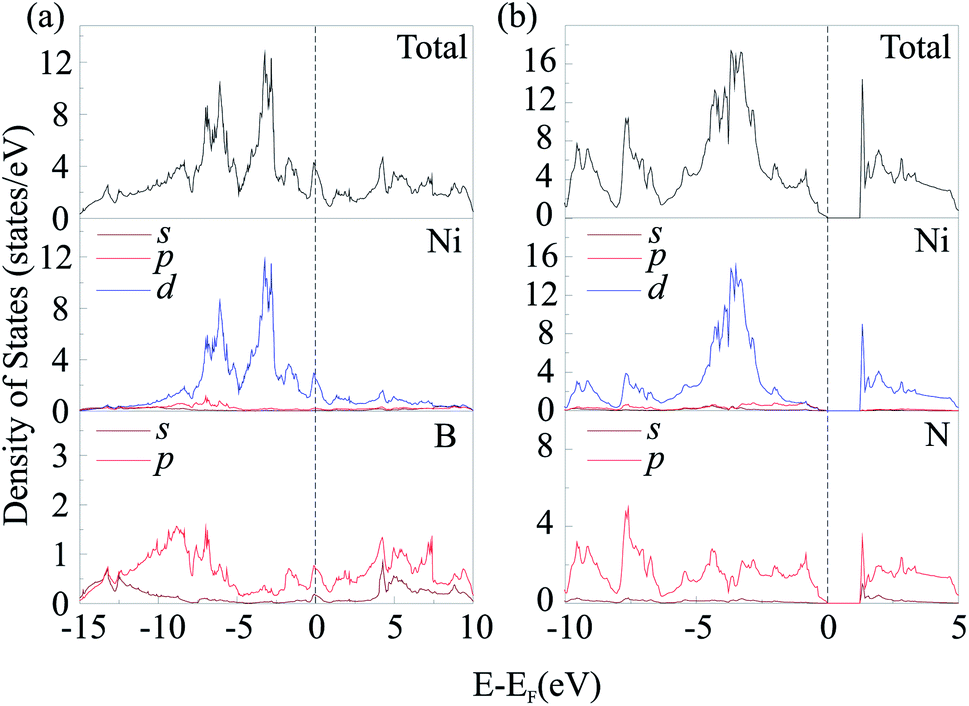 | ||
| Fig. 6 Total and partial DOS calculated for Ni2B3-Immm (a) and NiN2-Pa (b). | ||
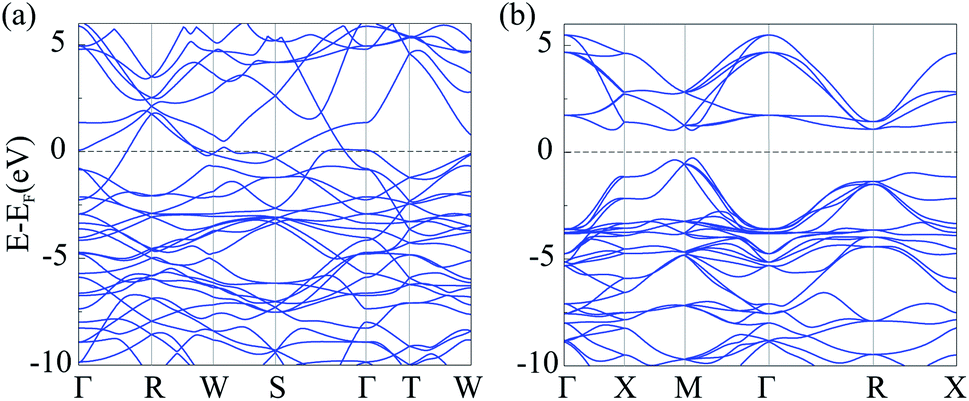 | ||
| Fig. 7 Calculated band structures of Ni2B3-Immm (a) and NiN2-Pa (b). | ||
The Fermi level of the considered phases is set to zero. The result shows that Ni2B3-Immm exhibit metallic features. From calculated partial DOS (Fig. 6a), we noticed that Ni d orbital has hybridization with B p orbital. The lower bands in the valence band region are mainly contributed by B 2p states, while near the top of valence bands main contributors are Fe 3d states. The Fermi level are mostly occupied by the Fe 3d electrons, with some contribution of B 2p electrons. On the other hand, NiN2-Pa is found to be a semiconductor with the indirect band gap of 1.297 eV. The electronic states at the lower energy range of the valence bands are mainly contributed by N 2p orbitals, while Ni 3d orbitals dominate at the top of valence bands. The main contributors in the conduction band region are unoccupied Fe 3d states, with some contribution of N 2p states.
It is well-known that GGA often underestimates the width of band gap. For this reason, and also to avoid misleading, we performed GGA + U (with U = 2, 4 eV) and hybrid (HSE06) calculations. The obtained results showed that PBE, GGA + U, and HSE06 give qualitatively the same DOS and band structures (Fig. S9 and S10†). This fact indicates that the results obtained by standard PBE calculations are valid. For NiN2-Pa, the band gap obtained by GGA + U with U = 2 eV was equal to 1.524 eV, by GGA + U with U = 4 – 1.001 eV, by HSE06 − 2.237 eV (Fig. S10b†).
To elucidate chemical bond properties of Ni2B3-Immm and NiN2-Pa, electron localization function (ELF) diagrams were constructed (Fig. 8). The ELFs of the structures are derived from various planes and different colours indicate diverse degrees of electron localization: the red means that the electron is totally localized at the realm and blue means the opposite situation, namely, the electron is not localized at all. According to the obtained result, in Ni2B3-Immm, we could observe that the boron atoms form a mesh structure. In NiN2-Pa, the nitrogen atoms form N2 dimers. In both structures, valence electrons are not localized around the Ni atoms. These data suggest that the chemical bonds among nickel mainly exhibit ionic characteristics and boron/nitrogen atoms form covalent chemical bonds, which seemingly do not strongly interact with nickel atoms.
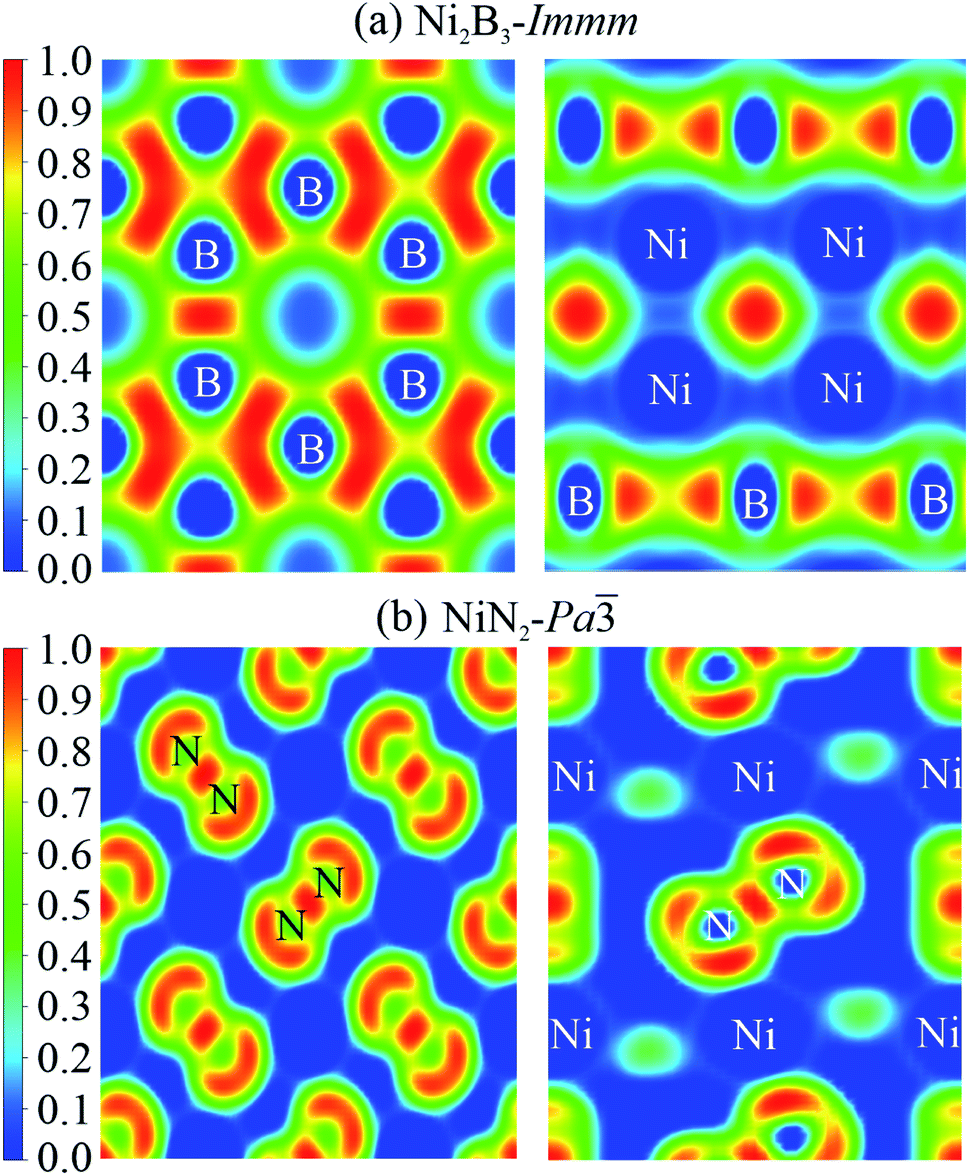 | ||
| Fig. 8 Electron localization functions of Ni2B3-Immm (a) and NiN2-Pa (b). | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was funded by the Ministry of Education and Science of the Republic of Kazakhstan under research project AP09562464 “Search for functional (superhard) materials in Ni–B, Ni–C, and Ni–N systems” and the state-assigned project of the IGM SB RAS. The computations were performed using resources provided by the Novosibirsk State University Supercomputer Center.Notes and references
- J. C. Crowhurst, A. F. Goncharov, B. Sadigh, C. L. Evans, P. G. Morrall, J. L. Ferreira and A. J. Nelson, Science, 2006, 311, 1275–1278 CrossRef CAS PubMed
.
- Z. Zhao, L. Xu, L.-M. Wang, B. Xu, M. Wang, Z. Liu and J. He, Comput. Mater. Sci., 2011, 50, 1592–1596 CrossRef CAS
- Y. Zhang, Z.-R. Liu, D.-W. Yuan, Q. Shao, J.-H. Chen, C.-L. Wu and Z.-L. Zhang, Acta Metall. Sin., 2019, 32, 1099–1110 CrossRef CAS
- M. Ürgen, O. Eryilmaz, A. Çakir, E. Kayali, B. Nilüfer and Y. Işik, Surf. Coat. Technol., 1997, 94–95, 501–506 CrossRef
- R. Mohammadi, A. T. Lech, M. Xie, B. E. Weaver, M. T. Yeung, S. H. Tolbert and R. B. Kaner, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10958–10962 CrossRef CAS PubMed
- H. Gou, N. Dubrovinskaia, E. Bykova, A. A. Tsirlin, D. Kasinathan, W. Schnelle, A. Richter, M. Merlini, M. Hanfland, A. M. Abakumov, D. Batuk, G. Van Tendeloo, Y. Nakajima, A. N. Kolmogorov and L. Dubrovinsky, Phys. Rev. Lett., 2013, 111, 157002 CrossRef PubMed
- A. Zerr, R. Riedel, T. Sekine, J. E. Lowther, W.-Y. Ching and I. Tanaka, Adv. Mater., 2006, 18, 2933–2948 CrossRef CAS
- I. Harran, H. Wang, Y. Chen, M. Jia and N. Wu, J. Alloys Compd., 2016, 678, 109–112 CrossRef CAS
- A. N. Kolmogorov, S. Shah, E. R. Margine, A. F. Bialon, T. Hammerschmidt and R. Drautz, Phys. Rev. Lett., 2010, 105, 217003 CrossRef CAS PubMed
- Z. G. Bazhanova, A. R. Oganov and O. Gianola, Phys.-Usp., 2012, 55, 489 CrossRef CAS
- G. L. Weerasinghe, R. J. Needs and C. J. Pickard, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 174110 CrossRef
- N. Sagatov, P. Gavryushkin, I. Medrish, T. Inerbaev and K. Litasov, Russ. Geol. Geophys., 2020, 61, 1345–1353 CrossRef
- L. Wu, R. Tian, B. Wan, H. Liu, N. Gong, P. Chen, T. Shen, Y. Yao, H. Gou and F. Gao, Chem. Mater., 2018, 30, 8476–8485 CrossRef CAS
- Y. Chen, X. Cai, H. Wang, H. Wang and H. Wang, Sci. Rep., 2018, 8, 10670 CrossRef PubMed
- Z. Wang, Y. Li, H. Li, I. Harran, M. Jia, H. Wang, Y. Chen, H. Wang and N. Wu, J. Alloys Compd., 2017, 702, 132–137 CrossRef CAS
- M. Bykov, E. Bykova, G. Aprilis, K. Glazyrin, E. Koemets, I. Chuvashova, I. Kupenko, C. McCammon, M. Mezouar and V. Prakapenka, et al., Nat. Commun., 2018, 9, 2756 CrossRef CAS PubMed
- V. Raghavan, J. Phase Equilib. Diffus., 2007, 28, 377–379 CrossRef CAS
- M. Baricco, E. Ferrari and L. Battezzati, MRS Proceedings, 1995, 398, 81 CrossRef
- K. Hofmann, N. Kalyon, C. Kapfenberger, L. Lamontagne, S. Zarrini, R. Berger, R. Seshadri and B. Albert, Inorg. Chem., 2015, 54, 10873–10877 CrossRef CAS PubMed
- M. Wang, J. Xie, K. Xue and L. Li, Comput. Mater. Sci., 2021, 194, 110465 CrossRef CAS
- T. Fedotenko, S. Khandarkhaeva, L. Dubrovinsky, K. Glazyrin, P. Sedmak and N. Dubrovinskaia, Minerals, 2021, 11, 516 CrossRef
- C. Guillaume, J. P. Morniroli, D. J. Frost and G. Serghiou, J. Phys.: Condens. Matter, 2006, 18, 8651–8660 CrossRef CAS PubMed
- K. Niwa, R. Fukui, T. Terabe, T. Kawada, D. Kato, T. Sasaki, K. Soda and M. Hasegawa, Eur. J. Inorg. Chem., 2019, 2019, 3753–3757 CrossRef CAS
- A. R. Oganov and C. W. Glass, J. Chem. Phys., 2006, 124, 244704 CrossRef PubMed
- A. R. Oganov, A. O. Lyakhov and M. Valle, Acc. Chem. Res., 2011, 44, 227–237 CrossRef CAS PubMed
- A. O. Lyakhov, A. R. Oganov, H. T. Stokes and Q. Zhu, Comput. Phys. Commun., 2013, 184, 1172–1182 CrossRef CAS
- C. J. Pickard and R. J. Needs, Phys. Rev. Lett., 2006, 97, 045504 CrossRef PubMed
- C. J. Pickard and R. J. Needs, J. Phys.: Condens. Matter, 2011, 23, 053201 CrossRef PubMed
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS
- H. T. Stokes and D. M. Hatch, J. Appl. Crystallogr., 2005, 38, 237–238 CrossRef CAS
- T. M. Inerbaev, N. Sagatov, D. Sagatova, P. N. Gavryushkin, A. T. Akilbekov and K. D. Litasov, ACS Earth Space Chem., 2020, 4, 1978–1984 CrossRef CAS
- N. E. Sagatov, A.-D. B. Bazarbek, T. M. Inerbaev, P. N. Gavryushkin, A. T. Akilbekov and K. D. Litasov, ACS Earth Space Chem., 2021, 5, 596–603 CrossRef CAS
- H. Dong, A. R. Oganov, V. V. Brazhkin, Q. Wang, J. Zhang, M. M. D. Esfahani, X.-F. Zhou, F. Wu and Q. Zhu, Phys. Rev. B, 2018, 98, 174109 CrossRef CAS
- M. I. Eremets, A. G. Gavriliuk, I. A. Trojan, D. A. Dzivenko and R. Boehler, Nat. Mater., 2004, 3, 558–563 CrossRef CAS PubMed
- Y. Ma, A. R. Oganov, Z. Li, Y. Xie and J. Kotakoski, Phys. Rev. Lett., 2009, 102, 065501 CrossRef PubMed
- C. J. Pickard and R. Needs, Phys. Rev. Lett., 2009, 102, 125702 CrossRef PubMed
- R. Gumeniuk, H. Borrmann and A. Leithe-Jasper, Z. Kristallogr. - New Cryst. Struct., 2006, 221, 425–426 CAS
- E. Havinga, H. Damsma and P. Hokkeling, J. Less-Common Met., 1972, 27, 169–186 CrossRef CAS
- C. Prescher, L. Dubrovinsky, E. Bykova, I. Kupenko, K. Glazyrin, A. Kantor, C. McCammon, M. Mookherjee, Y. Nakajima and N. Miyajima, et al., Nat. Geosci., 2015, 8, 220–223 CrossRef CAS
- N. Sagatov, P. N. Gavryushkin, T. M. Inerbaev and K. D. Litasov, RSC Adv., 2019, 9, 3577–3581 RSC
- P. N. Gavryushkin, N. Sagatov, D. Sagatova, M. V. Banaev, K. G. Donskikh and K. D. Litasov, J. Phys.: Conf. Ser., 2020, 1590, 012010 CrossRef CAS
- E. Partee and B. Chabot, Handb. Phys. Chem. Rare Earths, 1984, 6, 113–334 Search PubMed
- M. Pani, F. Merlo and M. L. Fornasini, Z. Kristallogr. - Cryst. Mater., 2002, 217, 415–419 CrossRef CAS
- Z. Wu, E. Zhao, H. Xiang, X. Hao, X. Liu and J. Meng, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 054115 CrossRef
- S. Pugh, London, Edinburgh Dublin Philos. Mag. J. Sci., 1954, 45, 823–843 CrossRef CAS
- X.-Q. Chen, H. Niu, D. Li and Y. Li, Intermetallics, 2011, 19, 1275–1281 CrossRef CAS
- Y. Tian, B. Xu and Z. Zhao, Int. J. Refract. Met. Hard Mater., 2012, 33, 93–106 CrossRef CAS
- E. Güler and M. Güler, Chin. J. Phys., 2015, 53, 195–205 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra06160g |
| This journal is © The Royal Society of Chemistry 2021 |