DOI:
10.1039/D1RA06151H
(Paper)
RSC Adv., 2021,
11, 32269-32274
X-ray absorption spectroscopy of exemplary platinum porphyrin and corrole derivatives: metal- versus ligand-centered oxidation†
Received
14th August 2021
, Accepted 23rd September 2021
First published on 30th September 2021
Introduction
Platinum, a renowned transition metal,1,2 has long been a cornerstone of the field of catalysis. In the same vein, over the last half-century, cisplatin and related Pt(II) complexes have emerged as a mainstay of cancer chemotherapy, accounting for some 40% of all such treatment.3–5 To avert off-target reactions and side effects of Pt(II) complexes, Pt(IV) complexes are also being actively studied as prodrugs, since they can generate the active cytotoxic Pt(II) drugs via intracellular reduction at their target sites.6 In yet a third domain, Pt(II) porphyrins played an important role in Martin Gouterman's optical taxonomy of porphyrin derivatives7 and, as strongly phosphorescent, oxygen-sensing materials, they famously found applications as pressure-sensitive paints on airplane wings.8,9 Platinum(IV) porphyrins have also been known for many years.10,11 Much more recently, Pt(IV) corroles have been synthesized,12 albeit in poor yields, and found to exhibit near-IR phosphorescence under ambient conditions.13,14 Given the importance of multiple oxidation states in Pt chemistry, X-ray absorption spectroscopy (XAS)15,16 plays a major role in studies of Pt speciation in complex catalytic systems17,18 and increasingly also in biological systems.19 To assist such analyses, we carried out a Pt L3 XAS study of a set of well-characterized Pt porphyrin and corrole derivatives. (Given their broad importance, there have been surprisingly few XAS and related measurements on porphyrin and corrole derivatives.20–30) The results, interpreted with density functional theory (DFT) calculations, provide detailed insights into the factors influencing the pre-edge shifts of Pt complexes.
Results and discussion
X-ray absorption spectroscopy (XAS)
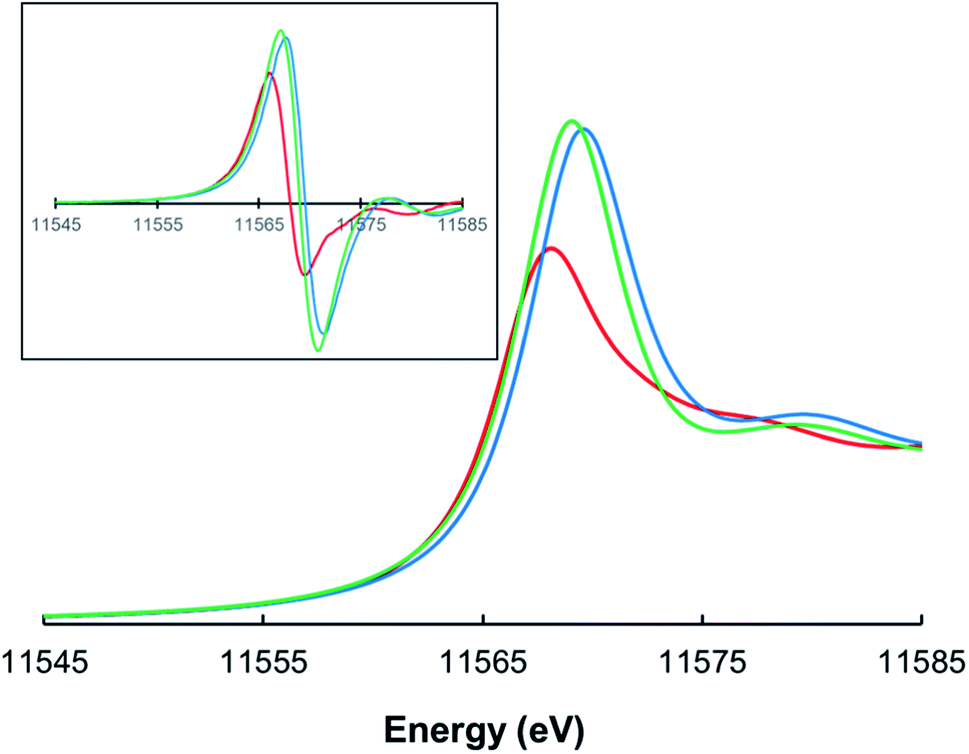
Pt L3-edge X-ray absorption spectroscopy (XAS), including X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) analysis, was performed on three complexes – PtII[TpCF3PP], PtIV[TpCF3PP]Cl2, and PtIV[TpCF3PC](Ar)(py) [TpCF3PP2− = meso-tetrakis(p-trifluoromethylphenyl)porphyrinato, TpCF3PC3− = meso-tris(p-trifluoromethylphenyl)corrolato, Ar = m-cyanophenyl, and py = pyridine; Fig. 1] – to interrogate their electronic structure and bonding parameters. (The choice of CF3-substituted porphyrinoid ligands was prompted by the general expectation that the resulting complexes should be more resistant toward aerial oxidation and conducive to long-term storage.) The analyses were carried out on powdered samples. The three complexes yielded Pt L3-edge energies of 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 566.0 eV, 11567.2 eV, and 11567.6 eV, respectively (Fig. 2). The Pt L3-edge energies and intensities are sensitive to the charge (or electrostatic potential) and the number of empty 5d states, respectively, at the Pt center.31,32 The increase in the L3-edge energy and intensity of PtIV[TpCF3PP]Cl2 when compared to PtII[TpCF3PP] is thus consistent with oxidation of the Pt center from, formally, Pt(II) to Pt(IV). The slight increase in the edge position of PtIV[TpCF3PC](Ar)(py), compared to PtIV[TpCF3PP]Cl2, is consistent with an oxidized species with an increase in the strength of the ligand field due to the TpCF3PC, Ar and/or pyridine ligands. Support for this interpretation also comes from the approximately equal pre-edge intensities of PtIV[TpCF3PP]Cl2 and PtIV[TpCF3PC](Ar)(py), as expected for two low spin d6 Pt(IV) species.
566.0 eV, 11567.2 eV, and 11567.6 eV, respectively (Fig. 2). The Pt L3-edge energies and intensities are sensitive to the charge (or electrostatic potential) and the number of empty 5d states, respectively, at the Pt center.31,32 The increase in the L3-edge energy and intensity of PtIV[TpCF3PP]Cl2 when compared to PtII[TpCF3PP] is thus consistent with oxidation of the Pt center from, formally, Pt(II) to Pt(IV). The slight increase in the edge position of PtIV[TpCF3PC](Ar)(py), compared to PtIV[TpCF3PP]Cl2, is consistent with an oxidized species with an increase in the strength of the ligand field due to the TpCF3PC, Ar and/or pyridine ligands. Support for this interpretation also comes from the approximately equal pre-edge intensities of PtIV[TpCF3PP]Cl2 and PtIV[TpCF3PC](Ar)(py), as expected for two low spin d6 Pt(IV) species.
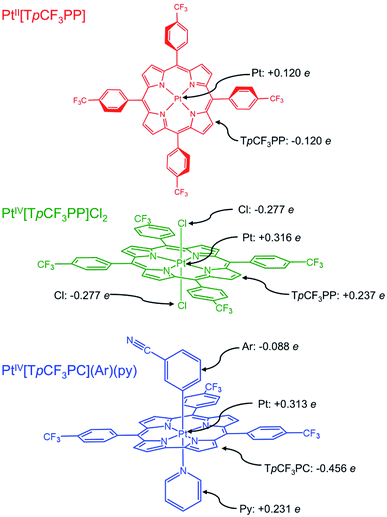 |
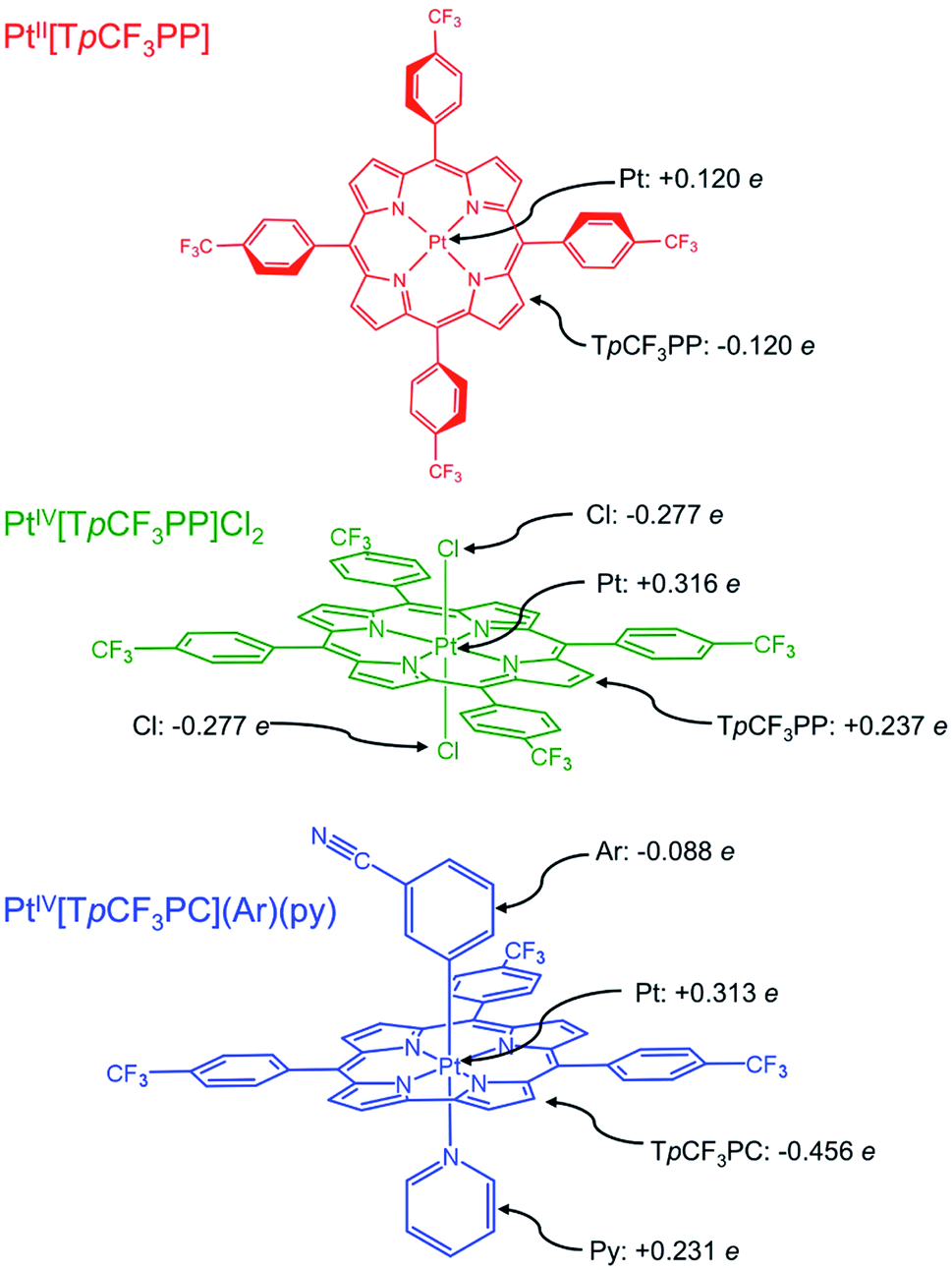
| | Fig. 1 Platinum porphyrin and corrole derivatives studied in this work, along with selected TPSS Hirshfeld charges. | |
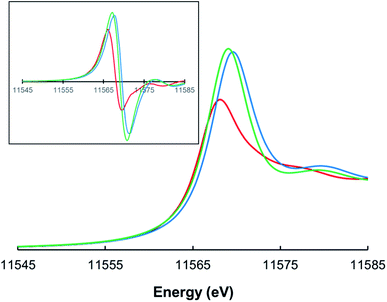 |
| | Fig. 2 Normalized Pt L3-edge XAS spectra for PtII[TpCF3PP] (red), PtIV[TpCF3PP]Cl2 (green), and PtIV[TpCF3PC](Ar)(py) (blue). The inset depicts first derivative spectra. | |
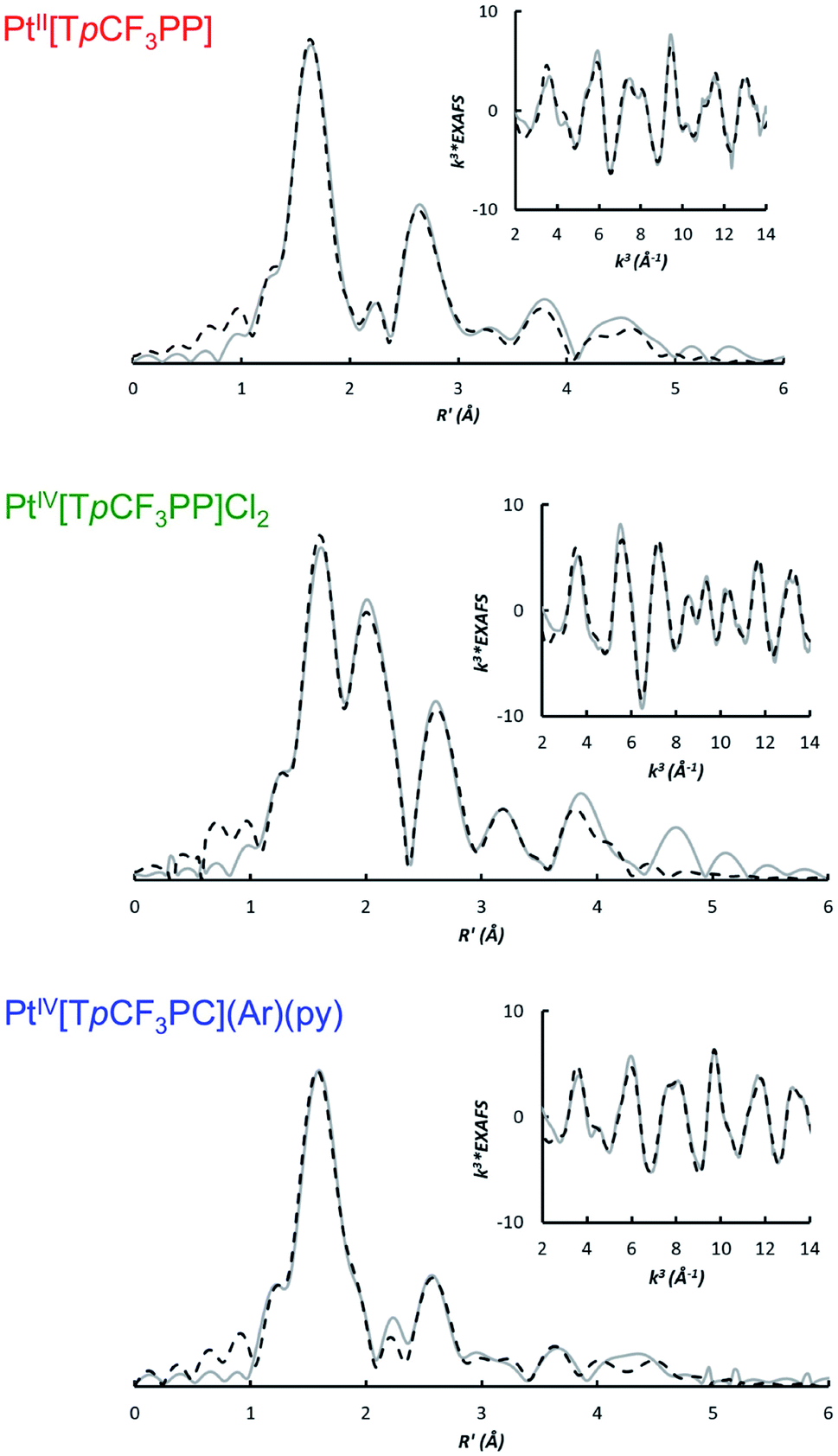
The bonding parameters of all three species were determined via EXAFS analysis (Fig. 3 and Table 1) and found to agree well with DFT optimized structures. Thus, the two Pt porphyrins PtIV[TpCF3PP]Cl2 and PtII[TpCF3PP] were found to exhibit similar Pt–N distances of 2.03 Å and 2.01 Å, respectively. The Pt–Cl distance in PtIV[TpCF3PP]Cl2 was determined to be 2.31 Å. EXAFS analysis of PtIV[TpCF3PC](Ar)(py) revealed 4 shortened Pt–N distances of 1.96 Å, as well as distances of 1.98 Å and 2.26 Å for the axial Pt–CPh and Pt–Npy bonds, respectively. These distances also served as proof of the chemical integrity of the samples under the conditions of the XAS experiments.
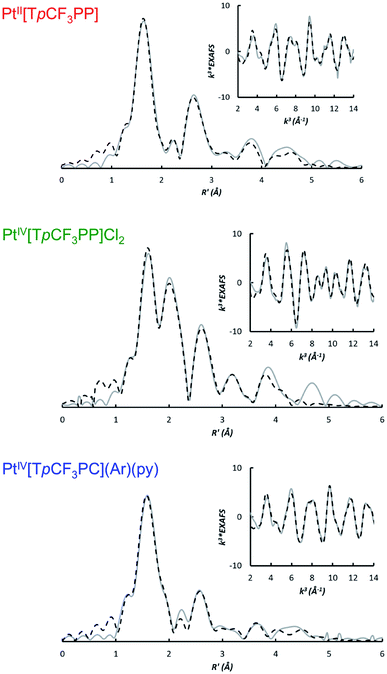 |
| | Fig. 3 Non-phase shift corrected Fourier transforms of the Pt L-edge EXAFS data (gray, solid) and corresponding fits (black, dashed) for PtII[TpCF3PP] (top), PtIV[TpCF3PP]Cl2 (middle), and PtIV[TpCF3PC](Ar)(py) (bottom). Insets show the EXAFS regions and their fits. | |
Table 1 Pt L-edge EXAFS curve-fitting results
| Complex |
Path |
Ra (Å) |
σ2b (Å2) |
ΔE0 |
| The estimated standard deviations for the distances are in order of ±0.02 Å. The σ2 values are multiplied by 105. A * indicates that the σ2 value was linked to that of the Pt–N path. |
| PtII[TpCF3PP] |
4 Pt–N |
2.01 |
388 |
10.56 |
| 8 Pt–Cpyr |
3.04 |
227 |
| 16 Pt–N–Cpyr |
3.20 |
1688 |
| 4 Pt–Cmeso |
3.42 |
364 |
| 16 Pt–Cmeso–Cpyr |
3.91 |
385 |
| 16 Pt–Cpyr–Cpyr′ |
4.32 |
62 |
| 8 Pt–Cmeso–Cph |
4.92 |
202 |
| PtIV[TpCF3PP]Cl2 |
4 Pt–N |
2.03 |
214 |
9.25 |
| 2 Pt–Cl |
2.31 |
182 |
| 8 Pt–Cpyr |
3.05 |
258 |
| 4 Pt–Cmeso |
3.39 |
389 |
| 16 Pt–N–Cl |
3.78 |
214* |
| 4 Pt–N–N′ |
4.02 |
846 |
| 16 Pt–N–Cpyr |
4.31 |
214* |
| PtIV[TpCF3PC](Ar)(py) |
4 Pt–N |
1.96 |
211 |
7.46 |
| 1 Pt–Cph |
1.98 |
211* |
| 1 Pt–Npy |
2.26 |
211* |
| 6 Pt–Cpyr |
2.96 |
211* |
| 3 Pt–Cmeso |
3.31 |
211* |
| 8 Pt–N–Npy |
3.90 |
211* |
| 8 Pt–N–C |
4.18 |
184 |
| 6 Pt–Cmeso–C |
4.82 |
157 |
Density functional theory
To shed additional light on the electronic structure of the three Pt species, DFT (TPSS33) calculations were performed with SARC-ZORA (Pt)34 and def2-TZVP35–37 basis sets. As noted above, the Pt L3-edge blueshifts by 1.2 eV upon oxidation of PtII[TpCF3PP] to PtIV[TpCF3PP]Cl2. While this shift to higher energy is expected upon oxidation, the magnitude of the energy shift is significantly lower than expected for 2-e− oxidation, i.e., Pt(II) to Pt(IV). Hirshfeld charges obtained from the calculations showed that both the Pt centers and TpCF3PP ligands are oxidized by an appreciable amount, when going from PtII[TpCF3PP] to PtIV[TpCF3PP]Cl2. In PtII[TpCF3PP], the charge on the Pt center is calculated to be +0.120e (e being the absolute value of the electronic charge) and, accordingly, the charge on the TpCF3PP ligand is −0.120e in the neutral species. By comparison, PtIV[TpCF3PP]Cl2 shows an increased charge on Pt (+0.316e) and TpCF3PP (+0.237e), with the two chlorido groups (−0.277e each) balancing out yield the neutral species. Taken together, these Hirshfeld charges suggest that the relatively small Pt L3-edge blueshift upon oxidation of PtII[TpCF3PP] to PtIV[TpCF3PP]Cl2 reflects substantial oxidation of the TpCF3PP ligand (Δq = +0.357e), as well as, of course, the Pt center (Δq = +0.196e).
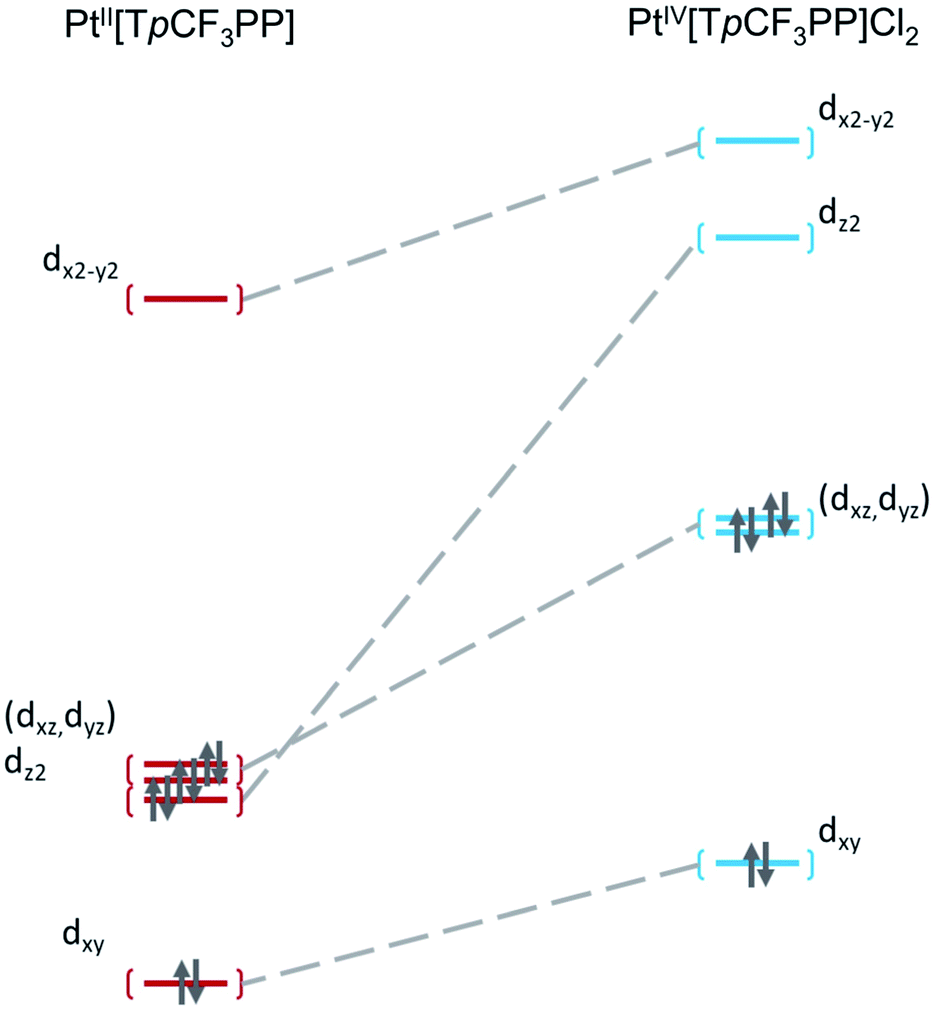
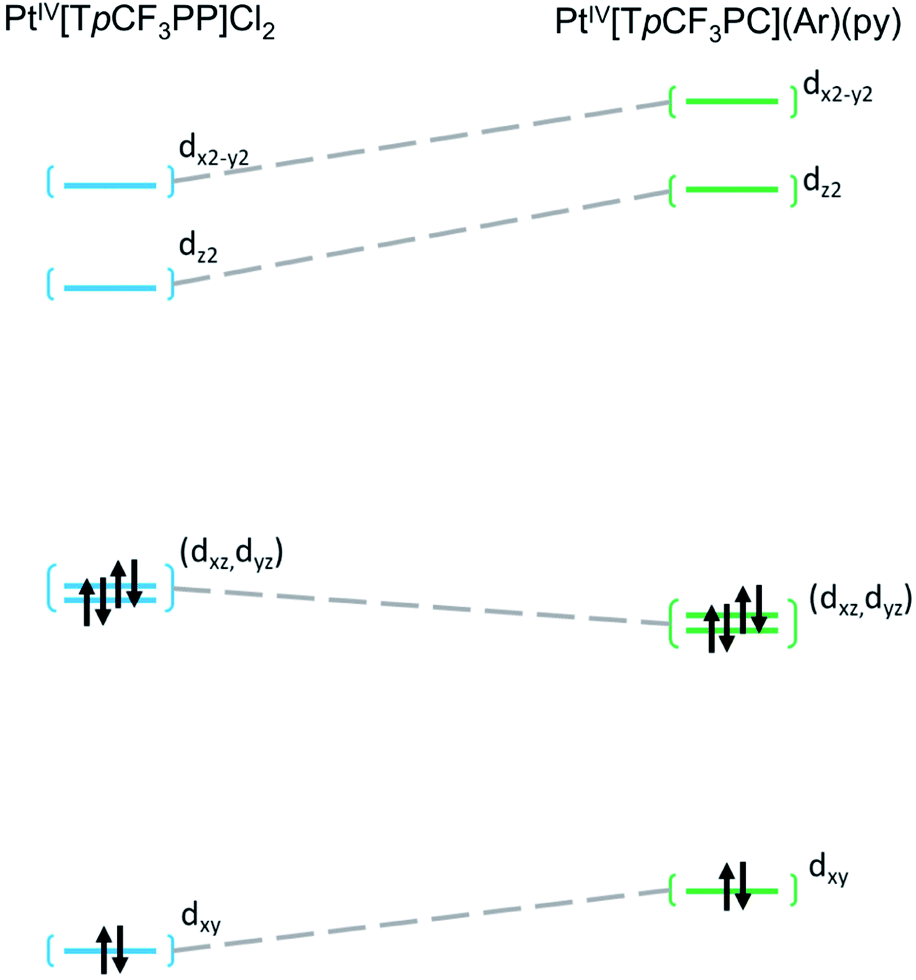
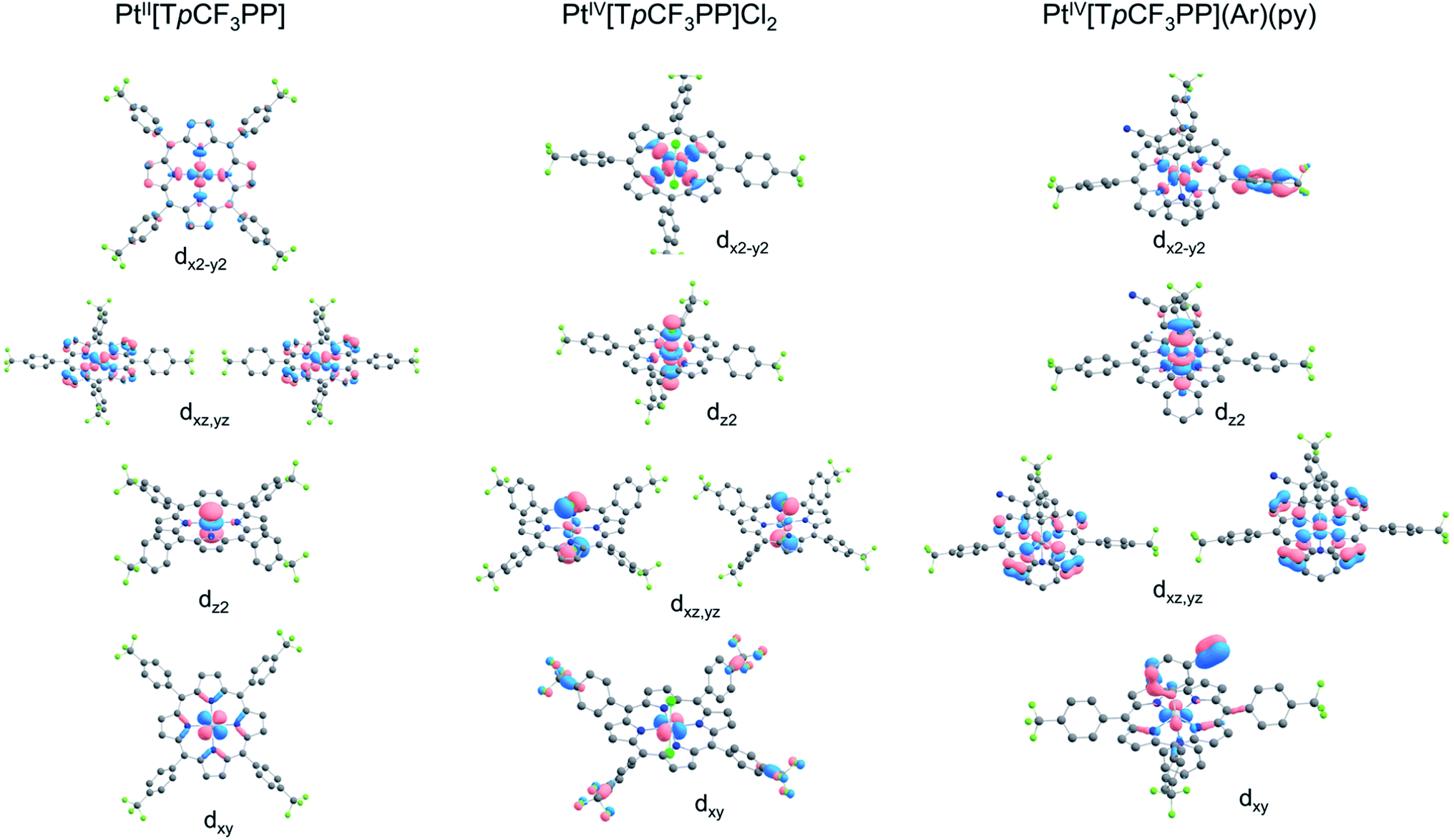
Further insight into the electronic structures came from visualization of the d-based frontier molecular orbitals (FMOs, Fig. 4–6). The 6-coordinate, d6 species PtIV[TpCF3PP]Cl2 (Fig. 5) shows the expected d-manifold splitting in which the fully occupied, non-bonding dxy orbital lies lowest in energy and a fully occupied, degenerate set of (dxz, dyz)-derived π-nonbonding orbitals at a relative energy of +2.9 eV. Higher still in energy are the unoccupied σ* orbitals derived from dz2 (+5.3 eV relative to dxy) and dx2−y2 (+6.1 eV relative to dxy). Comparison of these FMOs to those of the structurally similar PtIV[TpCF3PC](Ar)(py) reveals some crucial differences in the bonding and ligand field strength of the associated ligands. Although the d-based FMOs appear in the same order in both species, they occur at slightly different orbital energies. Thus, the dxy orbital has a slightly higher orbital energy (by ∼1 eV) in PtIV[TpCF3PC](Ar)(py) relative to PtIV[TpCF3PP]Cl2. The difference appears to be related to the decrease in symmetry in PtIV[TpCF3PC](Ar)(py), which allows the orbital, which is strictly non-bonding in PtIV[TpCF3PP]Cl2, to engage in weak σ/σ* interactions. The (dxz, dyz) pair shown in Fig. 5 exhibits the opposite behavior, with these orbitals lying slightly lower in energy (∼0.3 eV) in PtIV[TpCF3PC](Ar)(py) compared to PtIV[TpCF3PP]Cl2, indicating the combined effects of a number of interactions, especially the π-antibonding interactions with the chlorido ligands. Finally, the dz2 and dx2−y2 orbitals appear at higher energies (∼1.3 and 1.2 eV, respectively) in PtIV[TpCF3PC](Ar)(py), as expected for the higher σ-donating abilities of both the equatorial corrole and axial aryl and pyridine ligands and consistent with the higher Pt L3-edge energy in PtIV[TpCF3PC](Ar)(py).
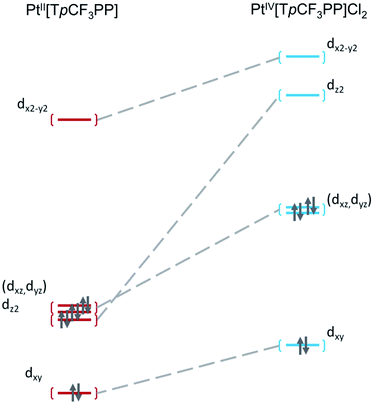 |
| | Fig. 4 Comparative Kohn–Sham (TPSS) d-orbital energy level diagrams for PtII[TpCF3PP] and PtIV[TpCF3PP]Cl2. | |
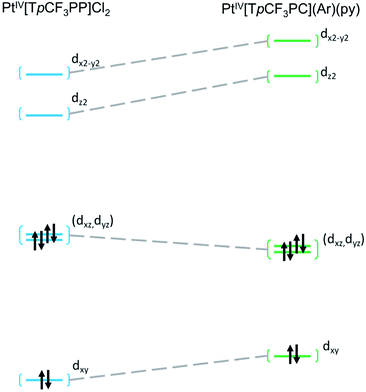 |
| | Fig. 5 Comparative Kohn–Sham (TPSS) d-orbital energy level diagrams for PtIV[TpCF3PP]Cl2 and PtIV[TpCF3PC](Ar)(py). | |
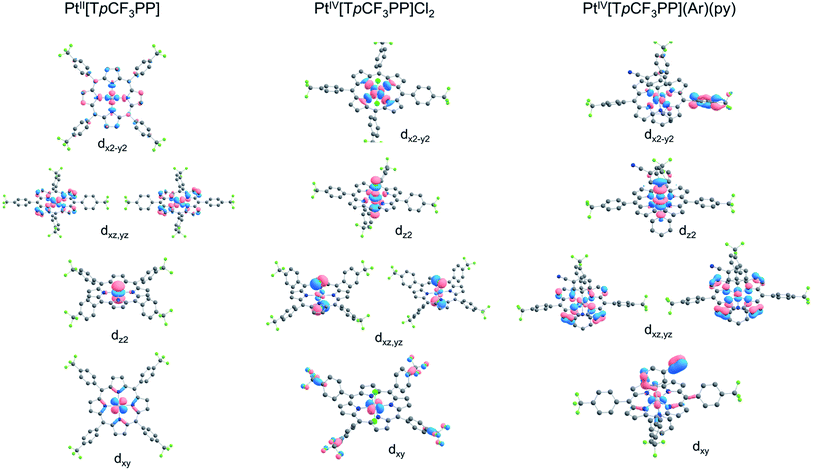 |
| | Fig. 6 Platinum 5d-based FMOs of the complexes studied. | |
Conclusion
A combined XAS and DFT study of a set of three well-characterized Pt porphyroind complexes has shed light on the factors affecting Pt L3 pre-edge energies. From PtII[TpCF3PP] to PtIV[TpCF3PP]Cl2, the Pt L3 pre-edge energy upshifts by 1.2 V, which is unexpectedly small for a two-electron oxidation of the metal. An explanation comes from DFT calculations, which showed that the oxidation is far from purely metal-centered; instead, the porphyrin ligand in PtIV[TpCF3PP]Cl2 is substantially oxidized. From PtIV[TpCF3PP]Cl2 to PtIV[TpCF3PC](Ar)(py), the Pt L3 pre-edge energy upshifts by a further 0.4 eV. This upshift is largely attributable to an increase in overall ligand field strength in PtIV[TpCF3PC](Ar)(py).
Experimental section
Materials and instruments
Benzonitrile was pre-dried over and distilled from P4O10 and stored over activated 4 Å molecular sieves. Ultraviolet-visible (UV-vis) spectra were recorded on an HP 8454 spectrophotometer in CH2Cl2. 1H (400 MHz) and 19F (376 MHz) NMR spectra were acquired on a 400 MHz Bruker Avance III HD spectrometer equipped with a 5 mm SmartProbe BB/1H (BB = 19F, 31P-15N) in CDCl3 and referenced to CHCl3 (δ = 7.26 ppm) for 1H and to 2,2,2-trifluoroethanol-d3 (δ = −77.8 ppm) for 19F. High resolution electrospray ionization mass spectra (HRMS) were recorded on an LTQ Orbitrap XL spectrometer.
Syntheses
PtII[TpCF3PP] was synthesized according to Buchler et al.10 by refluxing free-base meso-tetra(4-trifluoromethylphenyl)porphyrin with PtCl2 (3 equiv.) in dry benzonitrile for 2.5 h.38 Purification of the crude product was performed via column chromatography on silica gel with n-hexane/CH2Cl2 as eluent. Subsequent crystallization from 1:2 CHCl3/MeOH yielded the desired product as an orange-red solid. Spectroscopic data obtained for PtII[TpCF3PP] matched those reported earlier.39 PtIV[TpCF3PC](Ar)(py) was also synthesized as reported recently.13
PtIV[TpCF3PP]Cl2 was prepared according to Mink et al.,40 with a few modifications, as follows: a saturated solution Cl2(g) in CHCl3 (1 mL) solution was added dropwise over 2 min to a stirred, chilled (0 °C) solution of PtII[TpCF3PP] (40 mg, 0.037 mmol) in CHCl3 (5 mL). The orange solution thereupon turned red and was stirred for an additional 20 min at room temperature. UV-vis spectroscopy at this point showed that PtII[TpCF3PP] was still not fully consumed. An additional 1 mL of saturated Cl2/CHCl3 solution was accordingly added dropwise and the reaction mixture was stirred for a further 40 min, whereupon the 399 nm soret band of Pt[TpCF3PP] disappeared completely. The dark red solution was evaporated under vacuum and the distillate, essentially Cl2/CHCl3, was dechlorinated with a saturated aqueous solution of sodium thiosulfate (Na2S2O3) prior to disposal. The residue was redissolved in a minimum volume of CHCl3 and the resulting solution was layered with three times its volume of cold MeOH. After 3 days, PtIV[TpCF3PP]Cl2 was obtained as shiny, purple crystals (31 mg, 0.021 mmol, 73%). UV-vis λmax [nm, ε × 104 (M−1 cm−1)]: 324 (2.22), 420 (27.82), 536 (2.21), 600 (0.44). 1H NMR δ (CDCl3, δ = 7.26 ppm): 9.05 (s, 8H, β-H); 8.42 (d, 8H, J = 8.0 Hz, 5,10,15,20-o or -m, pCF3Ph); 8.11 (d, 8H, J = 8.0 Hz, m or o,pCF3Ph) 19F NMR δ: −62.63 (s, 12F, 5,10,15,20-pCF3Ph). HRMS (ESI, major isotopomer) [M]+: 1150.0842 (expt), 1150.0825 (calcd); [M − 2Cl]+: 1079.1475 (expt), 1079.1455 (calcd).
X-ray absorption spectroscopy
The Pt L3-edge X-ray absorption spectra of the three complexes of interest were measured at the Stanford Synchrotron Radiation Lightsource (SSRL) on the unfocused 20-pole 2 T wiggler sidestation beamline 7–3 under nonstandard ring conditions of 3 GeV and ∼100 mA (low-alpha operations mode at SSRL). An Si(220) double crystal monochromator was used for energy selection. A Rh-coated harmonic rejection mirror was used on beamline 7–3 to reject components of higher harmonics. The monochromator was further detuned by 50% to eliminate components of higher harmonics. All species were ground with boron nitride (BN) as a dilutant and then placed in 1 mm aluminum spacer for solid-state analysis. During data collection, the samples were maintained at a constant temperature of ∼10–15 K using an Oxford Instruments CF 1208 liquid helium cryostat. Data were collected to k = 14 Å−1 using an ionization chamber detector placed directly downstream of the sample. Internal energy calibration was accomplished via simultaneous measurement of the absorption of a Pt foil placed between two ionization chambers situated after the sample. The first inflection point of the foil spectrum was fixed at 11563.7 eV. No visual change in the rising-edge energy was observed over successive scans, indicating that the samples survived photoreduction/damage under experimental conditions. The data presented here are averages over 2 to 4 scans. The data were processed in the Athena utility of the Demeter 0.9.26 package41 by fitting a second order polynomial to the pre-edge region and subtracting this from the entire spectrum as background. A three-region spline of orders 2, 3, and 3 was used to model the smoothly decaying post-edge region.
Theoretical EXAFS signals χ(k) were calculated by using FEFF42–44 (Macintosh version 8.4). Structural models were obtained from crystal structures. The input structures were successful in generating reasonable phase and amplitude parameters required to obtain a good fit. Data fitting was performed in Artemis utility in the Demeter version 0.9.26 code.41 The structural parameters varied during the fitting process were the bond distance (R) and the bond variance σ2, which is related to the Debye–Waller factor resulting from thermal motion, and static disorder of the absorbing and scattering atoms. The nonstructural parameter ΔE0 (E0 is the energy at which k = 0) was also allowed to vary but was restricted to a common value for every component within a given fit. Coordination numbers were systematically varied in the course of the fits but were fixed within a given fit.
Computational details
All DFT calculations were performed using ORCA 3.0.3,45 the TPSS meta-GGA functional, Pantazi and Neese's SARC-ZORA basis set on Pt34 and Ahlrich's all-electron def2-TZVP basis set on all other atoms.35–37
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported in part by grants 262229 and 324139 to AG from the Research Council of Norway. The Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH.
References
- D. McDonald and L. B. Hunt, A History of Platinum and its Allied Metals, Johnson Matthey, London, 1982, p. 450 Search PubMed.
- G. Pinto, J. Chem. Educ., 2017, 94, 970–975 CrossRef CAS.
- B. Rosenberg, L. van Camp and T. Krigas, Nature, 1965, 205, 698–699 CrossRef CAS PubMed.
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed.
- S. Rottenberg, C. Disler and P. Perego, Nat. Rev. Cancer, 2021, 21, 37–50 CrossRef CAS PubMed.
- Z. Wang, Z. Deng and G. Zhu, Dalton Trans., 2019, 48, 2536–2544 RSC.
- M. Gouterman, in The Porphyrins, ed. D. Dolphin, Academic Press, New York, 1978, pp. 1–165 Search PubMed.
- J. Kavandi, J. Callis, M. Gouterman, G. Khalil, D. Wright, E. Green, D. Burns and B. McLachlan, Rev. Sci. Instrum., 1990, 61, 3340–3347 CrossRef.
- For a historical account, see: A. Ghosh, Angew. Chem., Int. Ed., 2021, 60, 9760–9770 CrossRef CAS PubMed.
- J. W. Buchler, K.-L. Lay and H. Stoppa, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1980, 35, 433–438 CrossRef.
- D. Kim, D. Holten, M. Gouterman and J. W. Buchler, J. Am. Chem. Soc., 1984, 106, 4015–4017 CrossRef CAS.
- A. B. Alemayehu, H. Vazquez-Lima, C. M. Beavers, K. J. Gagnon, J. Bendix and A. Ghosh, Chem. Commun., 2014, 50, 11093–11096 RSC.
- A. B. Alemayehu, L. J. McCormick, K. J. Gagnon, S. M. Borisov and A. Ghosh, ACS Omega, 2018, 3, 9360–9368 CrossRef CAS PubMed.
- A. B. Alemayehu, K. E. Thomas, R. F. Einrem and A. Ghosh, Acc. Chem. Res., 2021, 54, 3095–3107 CrossRef CAS PubMed.
- R. Sarangi, Coord. Chem. Rev., 2013, 257, 459–472 CrossRef CAS PubMed.
- X-Ray Absorption and X-Ray Emission Spectroscopy: Theory and Applications, ed. J. A. van Bokhoven and C. Lamberti, Wiley, New York, 2016, p. 895 Search PubMed.
- A. M. Gänzler, M. Casapu, P. Vernoux, S. Loridant, F. J. Cadete Santos Aires, T. Epicier, B. Betz, R. Hoyer and J.-D. Grunwaldt, Angew. Chem., Int. Ed., 2017, 56, 13078–13082 CrossRef PubMed.
- T. J. P. Hersbach, A. C. Garcia, T. Kroll, D. Sokaras, M. T. M. Koper and A. T. Garcia-Esparza, ACS Catal., 2021, 11, 9904–9915 CrossRef CAS.
- A. A. Hummer and A. Rompel, Metallomics, 2013, 5, 597–614 CrossRef CAS PubMed.
- R. Cao, K. E. Thomas, A. Ghosh and R. Sarangi, RSC Adv., 2020, 10, 20572–20578 RSC.
- S. Ganguly, L. J. McCormick, J. Conradie, K. J. Gagnon, R. Sarangi and A. Ghosh, Inorg. Chem., 2018, 57, 9656–9669 CrossRef CAS PubMed.
- R. K. Hocking, S. DeBeer George, Z. Gross, F. A. Walker, K. O. Hodgson, B. Hedman and E. I. Solomon, Inorg. Chem., 2009, 48, 1678–1688 CrossRef CAS PubMed.
- J. J. Yan, T. Kroll, M. L. Baker, S. A. Wilson, R. Decréau, M. Lundberg, D. Sokaras, P. Glatzel, B. Hedman, K. O. Hodgson and E. I. Solomon, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 2854–2859 CrossRef CAS PubMed.
- S. Ganguly, J. Conradie, J. Bendix, K. J. Gagnon, L. J. McCormick and A. Ghosh, J. Phys. Chem. A, 2017, 121, 9589–9598 CrossRef CAS PubMed.
- S. Ganguly, L. J. Giles, K. E. Thomas, R. Sarangi and A. Ghosh, Chem.–Eur. J., 2017, 23, 15098–15106 CrossRef CAS PubMed.
- J. H. Palmer and K. M. Lancaster, Inorg. Chem., 2012, 51, 12473–12482 CrossRef CAS PubMed.
- H. Lim, K. E. Thomas, B. Hedman, K. O. Hodgson, A. Ghosh and E. I. Solomon, Inorg. Chem., 2019, 58, 6722–6730 CrossRef CAS PubMed.
- R. Sarangi, L. J. Giles, K. E. Thomas and A. Ghosh, Eur. J. Inorg. Chem., 2016, 3225–3227 CrossRef CAS.
- S. Ganguly and A. Ghosh, Acc. Chem. Res., 2019, 52, 2003–2014 CrossRef CAS PubMed.
- J. Herritsch, J.-N. Luy, S. Rohlf, M. Gruber, B. P. Klein, M. Kalläne, P. Schweyen, M. Bröring, K. Rossnagel, R. Tonner and J. M. Gottfried, ECS J. Solid State Sci. Technol., 2020, 9, 061005 CrossRef CAS.
- J. A. Horsley, J. Chem. Phys., 1982, 76, 1451–1458 CrossRef CAS.
- A. N. Mansour, J. W. Cook Jr and D. E. Sayers, J. Phys. Chem., 1984, 88, 2330–2334 CrossRef CAS.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- D. A. Pantazis, X.-Y. Chen, C. R. Landis and F. Neese, J. Chem. Theory Comput., 2008, 4, 908–919 CrossRef CAS PubMed.
- A. Schaefer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef CAS.
- A. Schaefer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- R. C. Kwong, S. Sibley, T. Dubovoy, M. Baldo, S. R. Forrest and M. E. Thompson, Chem. Mater., 1999, 11, 3709–3713 CrossRef CAS.
- C. Arunkumar, F. R. Kooriyaden, X. Zhang, S. Sujatha and J. Zhao, New J. Chem., 2017, 41, 4908–4917 RSC.
- L. M. Mink, M. L. Neitzel, L. M. Bellomy, R. E. Falvo, R. K. Boggess, B. T. Trainum and P. Yeaman, Polyhedron, 1997, 16(16), 2809–2817 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- J. Mustre de Leon, J. J. Rehr, S. I. Zabinsky and R. C. Albers, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 4146–4156 CrossRef CAS PubMed.
- J. J. Rehr, J. Mustre de Leon, S. I. Zabinsky and R. C. Albers, J. Am. Chem. Soc., 1991, 113, 5135–5140 CrossRef CAS.
- S. I. Zabinsky, J. J. Rehr, A. Ankudinov, R. C. Albers and M. J. Eller, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, 2995–3009 CrossRef CAS PubMed.
- F. Neese, ORCA: An ab initio, DFT and semiempirical SCF-MO package, Version 3.0.1, University of Bonn, Germany, 2013 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra06151h |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a and
Ritimukta Sarangi
*a and
Ritimukta Sarangi