
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBlock copolymer synthesis using free-radical polymerization and thiol–maleimide ‘click’ conjugation†
Talena Rambarran* and
Heather D. Sheardown *
*
Department of Chemical Engineering, McMaster University, 1280 Main Street West, Hamilton, Ontario L8S 4L8, Canada. E-mail: sheardow@mcmaster.ca; rambart@mcmaster.ca
First published on 26th October 2021
Abstract
A method of making block copolymers utilizing free-radical polymerization and subsequent polymer conjugation is described. A disulphide functional radical initiator was used to polymerize methacrylic acid and 3-acrylamidophenylboronic acid. After purification, the disulphide bond of the end group was cleaved, revealing a thiol group which was used for subsequent conjugation to a polylactide containing the complementary maleimide functional group. The method is versatile and can be applied to the synthesis of various block copolymers without requiring the use of controlled/living radical polymerization methods.
The ability of block copolymers to self-organize in both the solid state and solutions have led to their use in a variety of applications ranging from polymer solar cells,1 electrodes,2 and as drug delivery vehicles in medicine.3 The synthesis of polymer blocks using radical chain-growth polymerization has been dominated by controlled, living radical polymerization techniques such as nitroxide-mediated polymerization (NMP), and to a greater extent, atom-transfer radical polymerization (ATRP) and reversible addition–fragmentation chain-transfer polymerization (RAFT).4 Such methods have many attractive features such as well-defined, narrow molecular weight distributions and tolerance to a range of functional groups.5 These controlled methods also enable facile synthesis of block copolymers. There are copious examples in the literature where ATRP or RAFT are used to polymerize the two blocks from the sequential addition of monomers, building the second block directly from the first,6,7 or where the incorporated end groups are modified to join polymers together through chemical ligation.8,9 Another attractive feature of these methods for block copolymer synthesis is the ability to polymerize acrylic and olefin monomers from a variety of polymeric macro-initiators. There are many mono- or di-carbonothioylthio functional polymers available as macro-RAFT agents, and similarly, alkyl halide functional polymers as macro-ATRP initiators. This simplifies block copolymer synthesis and enables the growth of one polymer chain directly off the other to generate a variety of polymer structures.
Previously, the Sheardown group created a novel block copolymer using RAFT, polymerizing methacrylic acid (MAA) and 3-acrylamidophenylboronic acid (PBA) from a commercially available macro-RAFT initiator, poly(D,L-lactide) (PLA) terminated with sulphanylthiocarbonylsulphanyl.10 The block copolymer self assembles in water to form micelles that can trap hydrophobic cargo in their core. The PBA imparts mucoadhesive properties to the micelles and these have been used to improve retention on mucosal surfaces. The Sheardown Lab leverages this property mainly for anterior ocular drug delivery, which suffers from many limitations such as fast clearance from the ocular surface. In an effort to move the ocular drug delivery technology forward towards commercial scale production, alternate methods for synthesizing the block copolymer that do not rely on controlled radical polymerization methods such as RAFT were required. Conventional free-radical polymerization was selected as the alternative since it does not require stringent process conditions and is easier to transfer to large-scale production.
Considerable amounts of the chemical literature focus on the use of controlled radical polymerization methods to make block copolymers where one component requires a radical chain growth mechanism. There are far fewer examples employing conventional free-radical chemistry to polymerize acrylic/olefin monomers. Wu et al. made a block copolymer of PLA and poly(N-vinylcaprolactam) (PNVCL) by end functionalizing PNVCL with an alkyne after polymerization and performing a Cu-catalyzed azide–alkyne cycloaddition with azido-PLA.11 Xu et al. used an alcohol functional initiator to polymerize N-isopropylacrylamide (NIPAAm) and subsequently ring opened D,L-lactide to make PNIPAAm-b-PLA.12 Finally, free radical polymerization of styrene was performed in the presence of 2-mercaptoethanol, which was used as a functional chain transfer agent. The polystyrene block obtained was used as a macroinitiator in the subsequent ring opening polymerization of L-lactide.13
Herein, a method to make a block copolymer using conventional free-radical polymerization for the acrylic block followed by subsequent ‘click’ ligation to the other polymer component (PLA) is presented. The thiol–maleimide ‘click’ reaction was chosen since it is highly efficient and it does not generate by-products. A review of the literature suggested the use of thiol–ene ‘click’ reactions are best suited for modification where one component is a small molecule and may not be efficient or practical when bringing polymers together.14 There are, however, examples of different ‘click’ reactions being successfully employed to join polymers together9 and with optimization of the reaction conditions, it was hypothesized the thiol–maleimide reaction could be used as a tool for the formation of block copolymers of comparable molecular weight to the previous RAFT-based examples. Making a di-block required both polymer components to be mono-functional, one containing a thiol and the other a maleimide. Maleimide terminated PLA is commercially available or can be made using the appropriate maleimide functional initiator to ring-open dilactide.15 The introduction of a functional group to one end of the free-radically polymerized block could be achieved by using a functional radical initiator. The functional group present on the initiator should not react during polymerization (i.e. will not react with the radical present during polymer growth), nor should it react with the functional groups present on the monomers being polymerized. Since thiols can react with both acrylic monomers and radicals, an initiator directly decorated with a thiol would not work. Additionally, thiols can oxidize to disulphides which would lead to an oligomeric initiator. The thiol group on the initiator must therefore be ‘protected’ during polymerization. For this reason, a disulphide seemed to be the ideal functionality to incorporate on the initiator because it is unreactive to radicals and it is already in the oxidized form. A disulphide functional radical initiator was therefore synthesized and used to polymerize MAA and PBA. After purification of the hydrophilic polymer block, the S–S can be cleaved to reveal the free thiol that can be used for subsequent conjugation to the maleimide–PLA to create the desired block copolymer without requiring controlled polymerization methods such as RAFT to make such architectures.
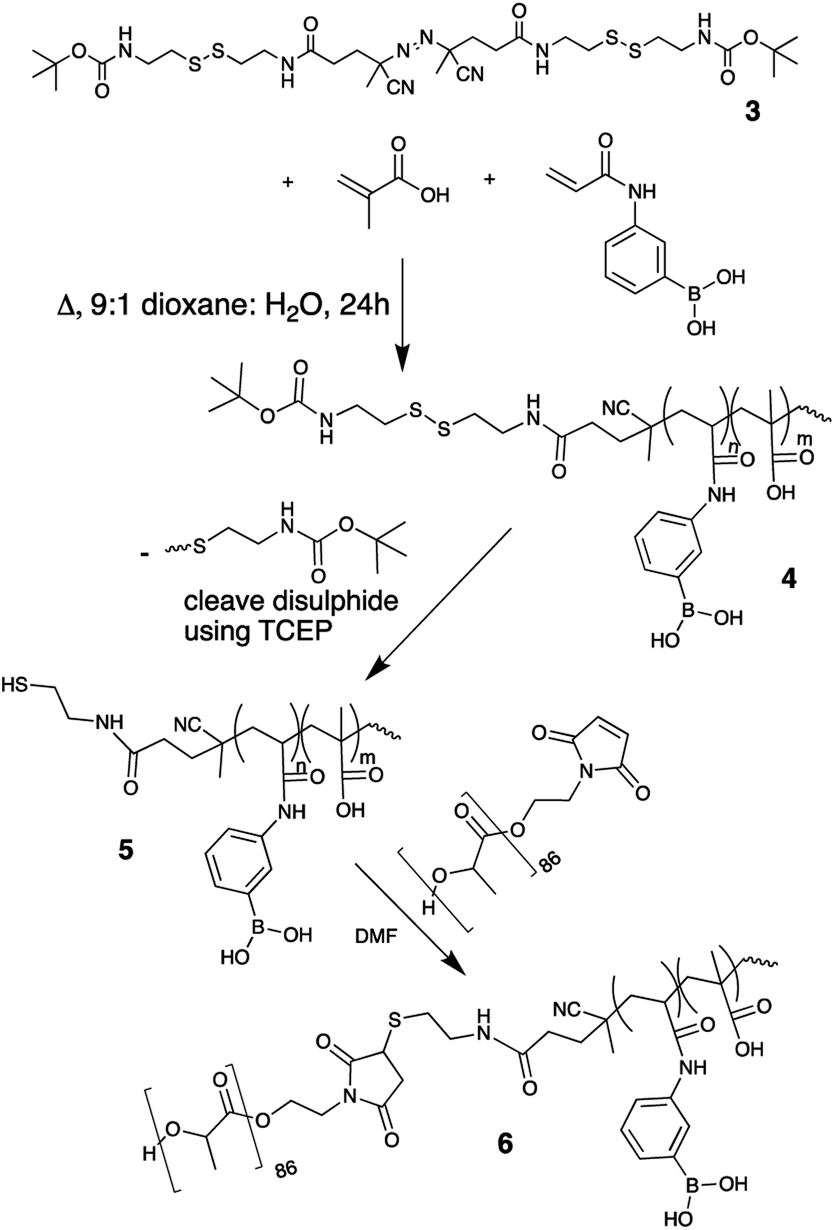
An azo-based initiator was chosen since it possesses the appropriate decomposition temperature for thermal initiation and functional azo starting materials are readily available to modify. 4,4′-Azobis(4-cyanovaleric acid) (ACVA) was first activated with N-hydroxysuccinimide (NHS) using an EDC coupling reaction to create NHS–ACVA, 1 (see ESI† for full synthetic details and reaction schemes).16 N-tert-Butoxycarbonyl cystamine, 2, was made from a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of cystamine and di-tert-butyl dicarbonate.17 Modifying the reaction from Fan et al.,18 1 and 2 were reacted to create 3, a tBoc disulphide functional thermal radical initiator depicted in Scheme 1. The structure of this initiator satisfies the requirements laid out above. The azo center will thermally degrade yielding a radical capable of initiating polymerization of acrylic groups and the disulphide group of the initiator will not react with the propagating radicals or monomers. 3 (95.6 mg, 0.13 mmol) was used to initiate the copolymerization of MAA (746 mg, 8.66 mmol) and PBA (405 mg, 2.24 mmol) in a 10% weight/volume solution in 9:1 dioxane:water at 80 °C, forming tBoc/disulphide terminated p-MAA-co-PBA, 4 (seen in Scheme 1). As desired, the hydrophilic block was now terminated with the disulphide which can be cleaved to reveal a thiol group. Polymer 4 was first purified by multiple precipitations into cold diethyl ether to remove any unreacted monomer, which would interfere with the next step of the reaction (cleaving the disulphide) since thiol groups can react with residual acrylate monomers and the thiol groups must be intact for ligation to the hydrophobic maleimide terminated PLA. 4 was characterized by 1H NMR in DMSO-d6. The absence of unreacted acrylic monomers was observed while the tBoc end group of the polymer was visible at 1.37 ppm (Fig. 1). Polymer 4 was then dissolved in a 1:1 acetone:water mixture and the disulphide bond was cleaved using (tris(2-carboxyethyl)phosphine), purified by dialysis to remove the tBoc cysteamine by-product and lyophilized to yield thiol terminated p-MAA-co-PBA, 5 (Scheme 1). 1H NMR suggests both the cleavage and purification were successful as the signal corresponding to the tBoc end group was absent (Fig. 1). 13C NMR also confirmed this (see the ESI†). GPC were run in a variety of solvents (THF, DMF, PBS) to characterize the molecular weight of the hydrophilic block, polymer 4. It was, however, unsuccessful in each case. For this reason, end group analysis using an Ellman's test for free thiols, was used to determine the average molecular weight of the of the hydrophilic polymer block, 5, instead. Ellman's reagent, 5,5′-dithio-bis-(2-nitrobenzoic acid), reacts with sulphhydryl groups in solution producing a measurable yellow colour and is a popular assay for –SH quantification. The test was performed immediately following lyophilization of 5 to limit the ability of the thiol groups to oxidize to disulphides. The published protocol from the Ellman's reagent manufacturer website (ThermoFisher) was followed (see ESI†). Based on the feed ratio of monomer to initiator, the expected polymer molecular weight was estimated and solutions of 5 at three different concentrations that were expected to be in the appropriate range for the Ellman's test were prepared and mixed with Ellman's reagent in the reaction buffer. The reaction of the thiol end groups and Ellman's reagent produces a yellow hue (Fig. 2), which was characterized by a UV-vis absorbance measurement at 412 nm. From the absorbance reading, the amount of thiol groups present were determined and this information, in combination with the known starting concentration of polymer, facilitated the calculation of the average molecular weight of ∼16000 g mol−1 for polymer 5.
:1 ratio of cystamine and di-tert-butyl dicarbonate.17 Modifying the reaction from Fan et al.,18 1 and 2 were reacted to create 3, a tBoc disulphide functional thermal radical initiator depicted in Scheme 1. The structure of this initiator satisfies the requirements laid out above. The azo center will thermally degrade yielding a radical capable of initiating polymerization of acrylic groups and the disulphide group of the initiator will not react with the propagating radicals or monomers. 3 (95.6 mg, 0.13 mmol) was used to initiate the copolymerization of MAA (746 mg, 8.66 mmol) and PBA (405 mg, 2.24 mmol) in a 10% weight/volume solution in 9:1 dioxane:water at 80 °C, forming tBoc/disulphide terminated p-MAA-co-PBA, 4 (seen in Scheme 1). As desired, the hydrophilic block was now terminated with the disulphide which can be cleaved to reveal a thiol group. Polymer 4 was first purified by multiple precipitations into cold diethyl ether to remove any unreacted monomer, which would interfere with the next step of the reaction (cleaving the disulphide) since thiol groups can react with residual acrylate monomers and the thiol groups must be intact for ligation to the hydrophobic maleimide terminated PLA. 4 was characterized by 1H NMR in DMSO-d6. The absence of unreacted acrylic monomers was observed while the tBoc end group of the polymer was visible at 1.37 ppm (Fig. 1). Polymer 4 was then dissolved in a 1:1 acetone:water mixture and the disulphide bond was cleaved using (tris(2-carboxyethyl)phosphine), purified by dialysis to remove the tBoc cysteamine by-product and lyophilized to yield thiol terminated p-MAA-co-PBA, 5 (Scheme 1). 1H NMR suggests both the cleavage and purification were successful as the signal corresponding to the tBoc end group was absent (Fig. 1). 13C NMR also confirmed this (see the ESI†). GPC were run in a variety of solvents (THF, DMF, PBS) to characterize the molecular weight of the hydrophilic block, polymer 4. It was, however, unsuccessful in each case. For this reason, end group analysis using an Ellman's test for free thiols, was used to determine the average molecular weight of the of the hydrophilic polymer block, 5, instead. Ellman's reagent, 5,5′-dithio-bis-(2-nitrobenzoic acid), reacts with sulphhydryl groups in solution producing a measurable yellow colour and is a popular assay for –SH quantification. The test was performed immediately following lyophilization of 5 to limit the ability of the thiol groups to oxidize to disulphides. The published protocol from the Ellman's reagent manufacturer website (ThermoFisher) was followed (see ESI†). Based on the feed ratio of monomer to initiator, the expected polymer molecular weight was estimated and solutions of 5 at three different concentrations that were expected to be in the appropriate range for the Ellman's test were prepared and mixed with Ellman's reagent in the reaction buffer. The reaction of the thiol end groups and Ellman's reagent produces a yellow hue (Fig. 2), which was characterized by a UV-vis absorbance measurement at 412 nm. From the absorbance reading, the amount of thiol groups present were determined and this information, in combination with the known starting concentration of polymer, facilitated the calculation of the average molecular weight of ∼16000 g mol−1 for polymer 5.
 | ||
| Scheme 1 Synthesis of polymers 4, 5 and 6. | ||
 | ||
| Fig. 1 1H NMR in DMSO-d6 of polymers 4, 5 and 6. Full NMR signal assignment of each spectrum can be found in the ESI.† | ||
 | ||
| Fig. 2 Ellman's test of 5 at 11.8 mg mL−1 (left), 5.9 mg mL−1 (middle) and 2.9 mg mL−1 (right). | ||
FTIR spectra were obtained to visualize the thiol generation, however since the stretching signal of thiols are known to be very weak and the thiol is an end group, it was not possible to see the thiol signal in the spectrum of 5 compared to 4. The signal also generally appears from 2500–3400 cm−1, overlapping with the hydroxyl groups of the methacrylic acid.
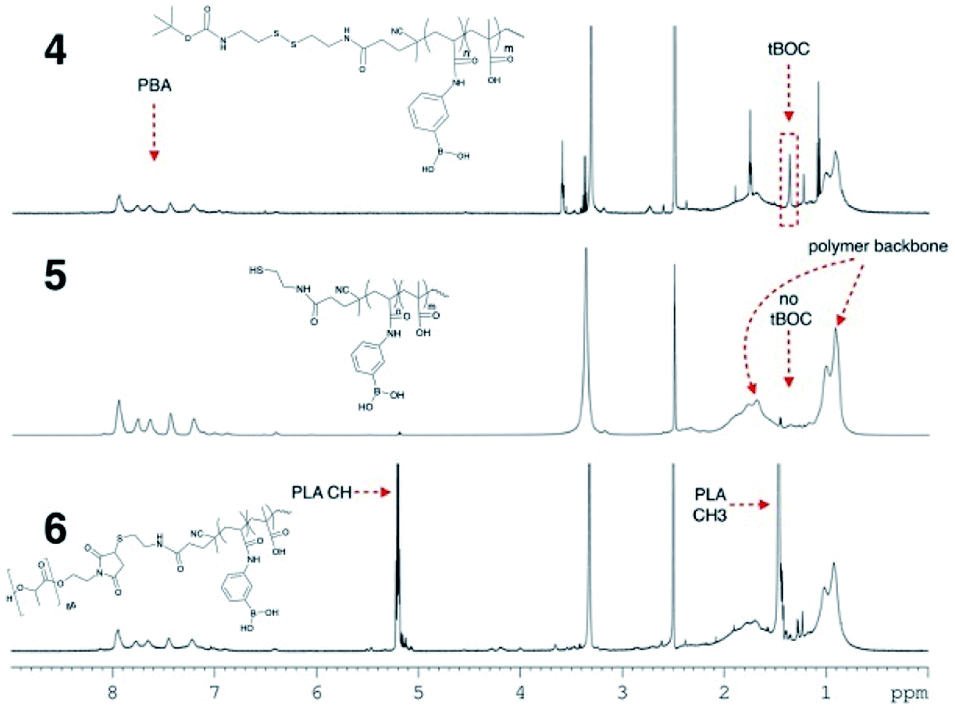
The final reaction step, conjugation of 5 with maleimide terminated PLA, was carried out immediately following lyophilization of 5 since thiols are susceptible to oxidation which would undesirably cause the end groups of the hydrophilic block to join forming a disulphide. 0.84 g of 5 and 1.3 g of maleimide–PLA (6200 g mol−1, determine by 1H NMR using end group analysis) were dissolved in DMF, bubbling with nitrogen to remove any dissolved oxygen. The mixture was left to react stirring at room temperature for 96 h. DMF was chosen as a reaction medium since both polymers are soluble in the solvent and the thiol–maleimide click reaction can proceed in DMF without the need for an additional catalyst. The high polarity of DMF can promote the formation of a thiolate anion spontaneously, kick starting the thiol-Michael addition to the maleimide double bond.19 Excess PLA and a longer reaction time were used during this step to ensure the two large end functional polymers had the opportunity to meet and react to push the reaction to completion. Elevating the reaction temperature could reduce the reaction time necessary for the reaction to reach completion. Following the reaction, the polymer mixture was precipitated into excess cold diethyl ether and vacuum filtered. This yielded both the final polymer, 6, mixed with a small amount of unreacted PLA as observed using 1H NMR. The unreacted PLA was removed by mixing the polymers in DCM, which solubilized PLA, leaving polymer 6 a fine powder. The suspension was vacuum filtered over a Buchner funnel, rinsed with DCM and dried in a vacuum oven at 50 °C for several hours to yield a pure block copolymer, 6. PLA could optionally be recovered through evaporation of the DCM from the filtrate. 6 was characterized by 1H NMR in DMSO-d6 (Fig. 3).
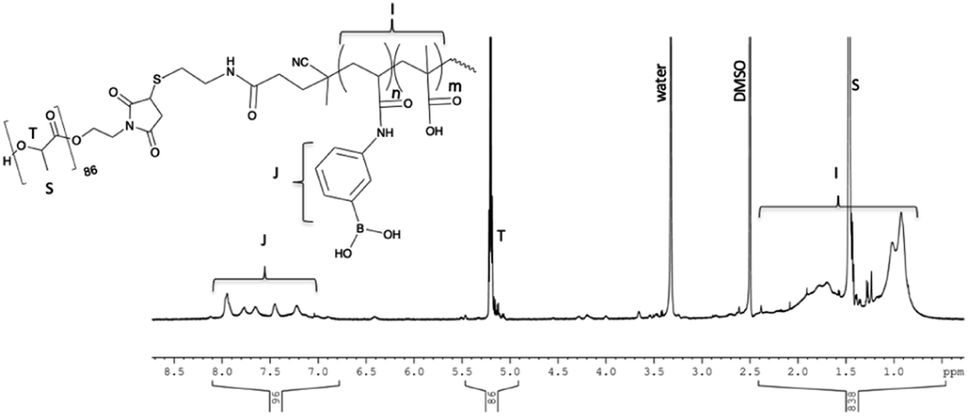 | ||
| Fig. 3 1H NMR of 6, p-PLA-b-(MAA-co-PBA) in DMSO-d6. | ||
Using the known PLA molecular weight as an internal standard, the final molecular weight of the polymer was calculated. The average PLA molecular weight was ∼6192 g mol−1 containing 86 repeating units per maleimide end group. The integration of the signal at 5.2 ppm corresponding to the PLA C–H group was set to 86. The integration of the aromatic protons of PBA from 7–8 ppm was 96 corresponding to a total of ∼24 PBA units in the hydrophilic block. Subtracting the integration of the overlapping PLA-CH3 (3 × 86) and PBA-acrylamide proton signal (3 × 24) from the polymer back bone signal present from 0.5–2.5 ppm and dividing that number by 5 for each unit of the MAA (101 MAA units resulting) gives us a total polymer molecular weight of 19300 g mol−1. The hydrophilic block accounted for 13100 g mol−1 with a 4:1 ratio of MAA:PBA. The calculated molecular weight of the hydrophilic block in the final polymer by NMR was slightly lower than the value calculated using the Ellman's test. While the values are in good agreement, the discrepancy could be in part due to the difference in the methods. NMR measures the average moles of PLA compared to p-MAA-co-PBA in the final product and cannot take into account if a small amount of thiol terminated polymer oxidized during the last reaction step.
The 13C and 1H NMR data of all the polymers are available in the ESI.† The proton and carbon signals of the polymer end groups are weak and some are close together or overlap making it difficult to visualize the shift in the peaks near the thiol and maleimide groups as they react to form the link between the two polymer blocks. 2D Heteronuclear Single Quantum Coherence (HSCQ) experiments show proton–carbon single bond correlations, where the protons lie along the x axis and the carbons are along y axis. This technique was utilized to help assign the signals in the spectra and to confirm thiol–maleimide reaction. Fig. 4 contains the 2D HSQC in DMSO-d6 of the PLA–maleimide, thiol terminated polymer 5 and the final polymer 6. The signal of CH2 next to the thiol of polymer 5 shifts once it reacts with the maleimide from ∼3.25 to 3.15 ppm in the x-1H axis and from ∼43 to 46 ppm in the y-13C axis. The CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH signal of the maleimide observed at 1H – 7.1 ppm and 13C – 135 ppm are not present in the final polymer demonstrating the final polymer contains no unreacted maleimide–PLA.
CH signal of the maleimide observed at 1H – 7.1 ppm and 13C – 135 ppm are not present in the final polymer demonstrating the final polymer contains no unreacted maleimide–PLA.
 | ||
| Fig. 4 2D HSQC in DMSO-d6 of the PLA–maleimide (left), 5 (middle) and the final polymer 6 (right) in the regions of the CH2 near the thiol (top) and the maleimide protons (bottom). | ||
Finally, the ultimate goal of this work was to demonstrate equivalence of the free-radical polymer to its original RAFT version when forming micelles for drug delivery. Using the same solvent evaporation method as previously described,10 6 was used to create micelles from the polymer self-assembly in an aqueous environment (Experimental details in the ESI†). Dynamic light scattering (DLS) demonstrated the micelles were comparable at 85 nm and 59 nm for the free-radical and RAFT versions respectively. The properties of the RAFT and free-radical block copolymer micelles were very similar suggesting the broader polydispersity likely obtained using free-radical polymerization did not have a deleterious effect on the properties of the micelles. Additionally confirming the formation of micelles from the polymer, a hydrophobic therapeutic, cyclosporin A, was encapsulated by the micelles in an aqueous solution at concentrations much higher than the drug's aqueous solubility. Extensive characterization of the free-radical block copolymer micelles, comparison to the analogous RAFT micelles, drug loading, drug release, micelle adhesion to the ocular surface and efficacy of the drug delivery system in an animal model will be the focus of our subsequent full publications.
A versatile method to create block copolymers without the need of controlled radical polymerization techniques has been described. A disulphide functional radical initiator was used to polymerize acrylic monomers. Subsequent cleavage of the disulphide revealed a thiol group capable of conjugating the polymer block to a complementary maleimide functional polymer under mild reaction conditions. The properties of the resultant materials were comparable to a similar polymer synthesized using RAFT. Under optimized conditions, it is possible to use efficient ‘click’ reactions to bring macromolecular structures and polymers of reasonable molecular weight together. This versatile synthetic method can be applied to a variety of monomers and polymers and is an additional tool in the synthesis of block copolymers, without reliance on controlled polymerization methods.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Lina Liu for running the DLS measurements and to the NSERC Idea to Innovation Grant for funding this work.Notes and references
- C. Lee, S. Lee, G. U. Kim, W. Lee and B. J. Kim, Chem. Rev., 2019, 119, 8028–8086 CrossRef CAS PubMed.
- H. T. Sun, J. Zhu, D. Baumann, L. L. Peng, Y. X. Xu, I. Shakir, Y. Huang and X. F. Duan, Nat. Rev. Mater., 2019, 4, 45–60 CrossRef.
- H. Cabral, K. Miyata, K. Osada and K. Kataoka, Chem. Rev., 2018, 118, 6844–6892 CrossRef CAS PubMed.
- X. Li, J. Iocozzia, Y. H. Chen, S. Q. Zhao, X. Cui, W. Wang, H. F. Yu, S. L. Lin and Z. Q. Lin, Angew. Chem., Int. Ed., 2018, 57, 2046–2070 CrossRef CAS PubMed.
- J. F. Lutz, J. M. Lehn, E. W. Meijer and K. Matyjaszewski, Nat. Rev. Mater., 2016, 1, 16024 CrossRef CAS.
- H. B. Feng, X. Y. Lu, W. Y. Wang, N. G. Kang and J. W. Mays, Polymers, 2017, 9, 31 CrossRef PubMed.
- K. Matyjaszewski, J. Saget, J. Pyun, M. Schlögl and B. Rieger, J. Macromol. Sci., Part A: Pure Appl.Chem., 2002, 39, 901–913 CrossRef.
- M. Li, P. De, S. R. Gondi and B. S. Sumerlin, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5093–5100 CrossRef CAS.
- I. Terzic, N. L. Meereboer and K. Loos, Polym. Chem., 2018, 9, 3714–3720 RSC.
- G. Prosperi-Porta, S. Kedzior, B. Muirhead and H. Sheardown, Biomacromolecules, 2016, 17, 1449–1457 CrossRef CAS PubMed.
- Q. H. Wu, J. Yi, S. Y. Wang, D. L. Liu, X. M. Song and G. L. Zhang, Polym. Bull., 2015, 72, 1449–1466 CrossRef CAS.
- F. Xu, T. T. Yan and Y. L. Luo, Macromol. Res., 2011, 19, 1287–1295 CrossRef CAS.
- G. Dorff, M. Hahn, A. Laschewsky and A. Lieske, J. Appl. Polym. Sci., 2013, 127, 120–126 CrossRef CAS.
- A. B. Lowe, Polym. Chem., 2014, 5, 4820–4870 RSC.
- K. Masutani, S. Kawabata, T. Aoki and Y. Kimura, Polym. Int., 2010, 59, 1526–1530 CrossRef CAS.
- H. Wang, Z. Chen, L. Xin, J. Cui, S. Zhao and Y. Yan, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 2175–2185 CrossRef CAS.
- Y. Suga, H. Sunayama, T. Ooya and T. Takeuchi, Chem. Commun., 2013, 49, 8450–8452 RSC.
- W. Fan, W. Shi, W. Zhang, Y. Jia, Z. Zhou, S. K. Brusnahan and J. C. Garrison, Biomaterials, 2016, 103, 101–115 CrossRef CAS PubMed.
- Y. Sun, H. Liu, L. Cheng, S. Zhu, C. Cai, T. Yang, L. Yang and P. Ding, Polym. Int., 2018, 67, 25–31 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental materials and methods, schemes and NMR spectra. See DOI: 10.1039/d1ra06089a |
| This journal is © The Royal Society of Chemistry 2021 |